

Abstract
Many organisms can adapt to changes in the solute content of their surroundings (i.e., the osmolarity). Hyperosmotic shock causes water efflux and a concomitant reduction in cell volume, which is countered by the accumulation of osmolytes. This volume reduction increases the crowded nature of the cytoplasm, which is expected to affect protein stability. In contrast to traditional theory, which predicts that more crowded conditions can only increase protein stability, recent work shows that crowding can destabilize proteins through transient attractive interactions. Here, we quantify protein stability in living Escherichia coli cells before and after hyperosmotic shock in the presence and absence of the osmolyte, glycine betaine. The 7-kDa N-terminal src-homology 3 domain of Drosophila signal transduction protein drk is used as the test protein. We find that hyperosmotic shock decreases SH3 stability in cells, consistent with the idea that transient attractive interactions are important under physiologically relevant crowded conditions. The subsequent uptake of glycine betaine returns SH3 to the stability observed without osmotic shock. These results highlight the effect of transient attractive interactions on protein stability in cells and provide a new explanation for why stressed cells accumulate osmolytes.
Keywords: glycine betaine, osmotic shock, osmolytes, protein NMR, protein stability
Introduction
Life on earth has adapted to a vast range of ionic environments, from pure water to 6 M NaCl, by regulating the concentration of solutes called osmolytes [1,2]. A large increase in extracellular salt concentration causes water efflux, which reduces the cell volume and increases the concentration of macromolecules within the already crowded cytoplasm [3,4]. This hyperosmotic shock is expected to affect protein stability. Traditional theory predicts that more crowded conditions can only stabilize proteins. Here, we test this idea by measuring protein stability in hyperosmotically stressed cells. We find that increasing the crowded nature of the cytoplasm decreases protein stability, consistent with recent studies showing that proteins can be destabilized by transient attractive interactions between the crowding molecules and the test protein [5–13].
Cells adapt to the loss of water by synthesizing or accumulating osmoprotecting solutes [14]. The bacterium Escherichia coli accumulates osmolytes such as glycine betaine to concentrations of nearly 1 M [15]. These solutes are known protein stabilizers in vitro [16], and it has been suggested that the accumulation of osmolytes by stressed cells prevents protein aggregation [14]. Here, we directly measure the effect of glycine betaine on protein stability in living E. coli and show that increasing the glycine betaine concentration in cells returns the stability lost due to hyperosmotic stress. These results provide a new explanation for why stressed cells accumulate osmolytes.
The protein used in these experiments is the 7-kDa N-terminal SH3 domain of Drosophila signal transduction protein drk (SH3). This metastable protein exists in a simple, reversible two-state equilibrium between its folded state and its unfolded ensemble [17] such that both forms are present at comparable concentrations under non-denaturing conditions. SH3 can be labeled with a fluorine atom on its sole tryptophan at position 36, allowing the application of 19F NMR spectroscopy [10,18]. Exchange between the folded form and the unfolded ensemble is slow compared to the difference in the NMR frequencies of the fluorine label in the two states, enabling the quantification of the modified standard-state free energy of unfolding by integrating the resonances: . The ability to measure SH3 stability both in vitro and in living cells [10] makes this protein useful for studying protein folding under stressed conditions in cells in the presence and absence of glycine betaine.
Results and discussion
Quantification of protein stability in cells
Increasing the external osmolarity of E. coli by adding 0.3 M NaCl to the external media causes water efflux, reducing the cell volume by ~35% and increasing the concentrations of macromolecules [3]. The qualitative conclusions that hyperosmotic stress destabilizes SH3 are easy to see by examining the areas under the peaks marked F and U in Fig. 1. When osmotically stressed E. coli cultures are provided with betaines, the stressed cells rapidly accumulate these compounds to maintain the turgor pressure and prevent dehydration [19]. More specifically, 1 mM glycine betaine in the media under hyperosmotic conditions results in a cytoplasmic concentration of 0.68 ± 0.07 m [20]. The qualitative conclusion that the accumulation of glycine betaine restores the stability is also easy to see by inspecting Fig. 1.
Fig. 1.
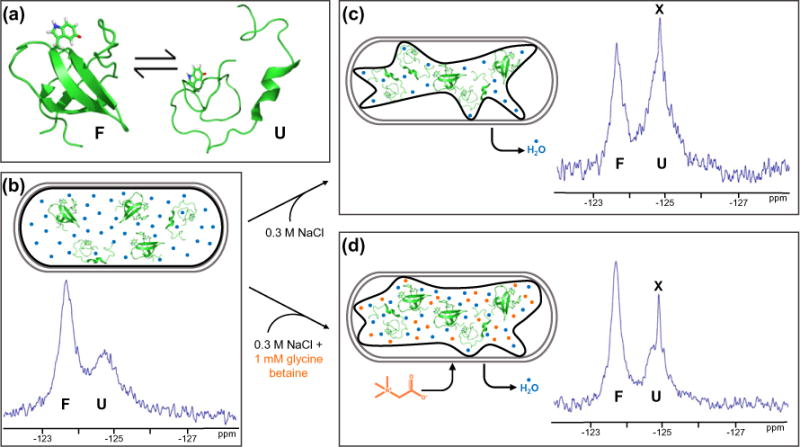
Glycine betaine reverses the destabilizing effect of hyperosmotic shock. (a) SH3 exists in an equilibrium between its folded state (PDB ID: 2A36) and an unfolded ensemble with a free energy of unfolding near zero under non-denaturing conditions. Tryptophan 36 with fluorine at position 5 is highlighted in red. Protein stability was measured in live E. coli cells under three conditions at 298 K. (b) Both the unfolded and folded forms are populated in cells under normal osmotic conditions. Gray outlines represent the cell wall. Black outlines represent the cytoplasmic membrane. Blue circles represent H2O. (c) Hyperosmotic shock caused by adding 0.3 M NaCl to the media destabilizes SH3. (d) Adding 1 mM glycine betaine to the 0.3 M NaCl causes the uptake of glycine betaine, returning SH3 to the stability observed without osmotic shock. Orange circles represent glycine betaine. Leakage of fluorine-containing metabolites (X) occurs upon hyperosmotic shock (Fig. S1).
SH3 aggregation does not complicate our analysis because the protein aggregates only under highly acidic (pH 2) conditions [21,22]. For experiments conducted with 0.3 M NaCl, the cells cannot adapt because glycine betaine is not synthesized by E. coli grown under osmotic shock in minimal media lacking precursors such as choline [15]. Trehalose and glutamic acid are the major organic osmolytes in E. coli grown in minimal medium under osmotic stress in the absence of betaines [15], but this is not a problem in our studies because cells were not grown at high osmolarity. Furthermore, trehalose and glutamic acid are not detected when shocked cells are given glycine betaine [15].
To quantify stabilities, we had to remove the contribution of two fluorine containing molecules whose resonances overlap with that of the unfolded ensemble—free 5-fluoroindole and truncated SH3, which we collectively refer to as X. The truncated protein is soluble, lacks nine C-terminal residues (Fig. S2), and is unable to fold (Fig. S3). The fluorine atom on the truncated protein and on 5-fluoroindole experiences an environment more similar to that of the unfolded ensemble of intact SH3 than that experienced by folded SH3. In addition, weak attractive interactions in cells cause resonance broadening [10]. For these reasons, it is unsurprising that the chemical shifts of these two species overlap with the shift of the unfolded ensemble (Fig. S3). As described next, we removed the contribution of X via the cell lysate spectrum.
The in-cell spectrum is acquired first (Fig. 2b). The areas under the peaks are proportional to the populations of the species. The downfield resonance corresponds to only the folded SH3. The area under the folded peak in the in-cell spectrum is proportional to the concentration of the folded state. The upfield peak comprises resonances of the unfolded ensemble, free 5-fluoroindole, and the truncated protein (U+X). The sum of these two integrals (∫(U+X)-IC + ∫FIC) is proportional to the total concentration of all fluorine-containing species. To determine which fraction of this total comprises the unfolded form now just remains. We begin by writing an expression for the fraction of folded SH3 among all fluorine-containing species by dividing the area under the folded state peak (∫FIC) by the sum of the areas of the two peaks:
| (1) |
Fig. 2.
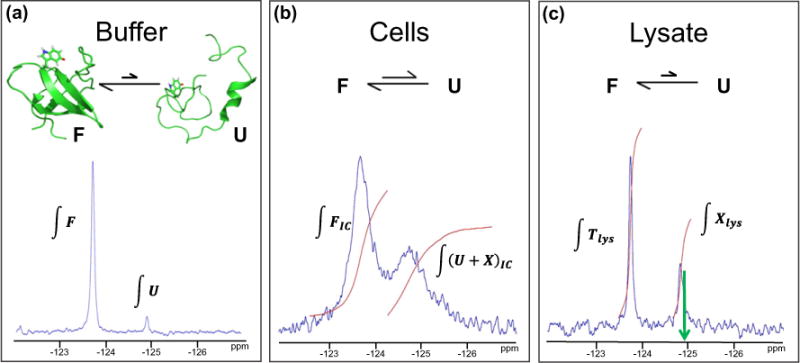
Correcting for other fluorine-containing species (X) in cells. (a) In D2O-containing buffer at 298 K, N95% of purified SH3 is in the folded state (F) and b5% is in the unfolded ensemble (U). The areas of the resonances (∫F) and (∫U) are proportional to the population of F and U. (b) The folded state is favored in D2O-suspended cells at 298 K, but a small population of SH3 remains unfolded because of the attractive interactions in cells. The upfield peak in cells comprises resonances from U and X. Therefore, the area under the upfield peak is proportional to the sum of the populations of unfolded SH3 and the fluorine-containing metabolites (∫(U+X)IC). (c) In the clarified lysate of these cells at 298 K, the folding equilibrium shifts such that N95% of SH3 exists in the folded form. Therefore, the downfield peak represents the total concentration of SH3 in the experiment (∫Tlys). The upfield peak represents only the fluorine-containing metabolites (∫Xlys). The green arrow indicates the chemical shift of unfolded SH3.
The cells are then lysed, and a spectrum of the clarified lysate is acquired (Fig. 2c). It is important to realize that the volume of cells in the sample comprises only about 50% of the total volume; the remainder of the volume comprises the buffer surrounding the cells [23]. The attractive interactions that destabilize SH3 in cells are attenuated in the lysate because the buffer that surrounded the cells dilutes the lysate. This attenuation results in a shift in equilibrium toward the folded state. In other words, N95% of SH3 that was unfolded in cells is folded in the lysate. If present, however, unfolded intact SH3 would show itself as a peak with a chemical shift indicated in Fig. 2c by the green arrow. In addition, previous experiments show individual peaks for metabolites and unfolded SH3 in the lysate under slightly different conditions [10]. Thus, in the clarified lysate, the downfield lysate resonance represents total intact SH3 (Tlys). Equation (2) gives the fraction of fluorine-containing species comprising intact SH3:
| (2) |
Approximately 60% of all fluorine-containing species comprise intact SH3. The remaining 40% comprise the truncated form and other fluorine-containing metabolites.
Equations (1) and (2) use the areas to obtain the fraction of all fluorine-containing species that is folded SH3 and the fraction that is intact SH3, respectively. The fraction of intact SH3 minus the fraction of folded SH3 yields the fraction of all fluorine-containing species that is unfolded and intact SH3 in cells (UIC,frac):
| (3) |
Multiplying UIC,frac by the sum of the two peaks in the in-cell spectrum gives the population of unfolded SH3 in cells. Equation (4), where R is the gas constant and T is the absolute temperature, gives the modified standard-state free energy of unfolding in cells:
| (4) |
Integration of both the raw spectra and the deconvoluted spectra shows no significant differences in stabilities (Table S1). These calculations contain the inherent assumption that the intact protein in the clarified lysate does not interact significantly with other cellular components, and, therefore, the equilibrium between the folded state and the unfolded ensemble is the same as it is in buffer. In support of this assumption, a 19F spectrum of uncentrifuged lysed cells shows a resonance for unfolded SH3 as a shoulder to the Xlys peak (Fig. S4). These two peaks are also broader than those in clarified lysate, suggesting increased interactions with the biomolecules in the uncentrifuged lysate.
Osmotic shock destabilizes SH3 in cells
The increase in the concentration of macromolecules caused by the osmotic-stress-induced decrease in cellular volume increases hard-core interactions. According to hard-core repulsion-based theories, this should stabilize proteins [24], but we observe a ~1 kcal/mol decrease in SH3 stability (Fig. 1c and Table 1). This result is another example of crowding-induced protein destabilization via transient attractive interactions [5–13].
Table 1.
Free energies of unfolding at 298 K with standard deviations of the mean from three trials
| Condition | , (kcal/mol) |
|---|---|
| Buffer | |
| D2O (pH 7.8) | 1.5 ± 0.1 |
| D2O + 0.3 M NaCl | 1.7 ± 0.2 |
| D2O + 0.3 M NaCl +1 mM glycine betaine | 1.8 ± 0.1 |
| H2O (pH 7.8) | 0.63 ± 0.03 |
| H2O (pH 7.8), 0.68 m glycine betaine | 1.1 ± 0.1 |
| Cells | |
| D2O | 1.6 ± 0.2 |
| D2O + 0.3 M NaCl | 0.54 ± 0.08 |
| D2O + 0.3 M NaCl +1 mM glycine betaine | 1.5 ± 0.2 |
Glycine betaine stabilizes SH3 in osmotically shocked cells
Adding 1 mM glycine betaine to the medium of cells stressed with 0.3 M NaCl stabilizes SH3 compared to hyperosmotic shock alone (Fig. 1d, Table 1, and Table S2), returning (1.5 ± 0.2 kcal/mol) to the value from unstressed cells (1.6 ± 0.2 kcal/mol). Our results show the physiological importance of glycine betaine for protecting protein stability in living cells and are consistent with our previous in vitro data showing that glycine betaine mitigates the destabilizing effects of cytosol on chymotrypsin inhibitor 2 [25].
It is well known that glycine betaine stabilizes proteins in vitro [16,26–31]. Therefore, we also examined the effect of the estimated physiological concentration of glycine betaine in stressed cells, 0.68 m, on purified SH3 in vitro. We conducted this experiment in H2O instead of D2O, because the protein is less stable in H2O (Table 1 and Fig. S5), allowing us to measure more accurately the population of the unfolded ensemble. In buffer, 0.68 m glycine betaine increases SH3 stability by 0.5 ± 0.1 kcal/mol. In stressed cells, this concentration of glycine betaine increases the stability by 1.0 ± 0.2 kcal/mol. This result shows that glycine betaine has a synergistic effect in cells. The reason for the synergism is unclear but may be related to the fact that the osmolyte stabilizes many proteins in the cell, not just SH3.
In summary, our results show that increasing the concentration of macromolecules in a cell via osmotic shock destabilizes proteins, and the accumulation of glycine betaine ameliorates the destabilization. The protein destabilization effect of osmotic stress has not been reported quantitatively in living cells, although our observation is consistent with studies showing that transient attractive interactions destabilize proteins [5–10]. Most importantly, our data provide a new explanation for osmolyte accumulation in cells. Although it was known that osmolyte accumulation by osmotically stressed E. coli maintains proper turgor pressure and allows return to more normal growth rates [32], the effect of glycine betaine on protein stability in living cells had not been reported. The fact that less stable proteins are more likely to aggregate provides an explanation for the suggestion that osmolyte accumulation helps prevent protein aggregation [14,33,34]. In summary, our results provide an additional explanation for the ubiquity of osmolytes in nature; they relieve the destabilizing effect of osmotic stress.
Materials and Methods
In-cell NMR [10]
The plasmid containing the gene encoding the drkN SH3 protein was transformed into BL21-Gold(DE3) cells (Agilent) by heat shock. A single colony was used to inoculate 5 mL of Lenox broth (10 g/L tryptone, 5 g/L yeast extract, 5 g/L NaCl) supplemented with 100 μg/mL ampicillin. The culture was incubated at 37 °C with shaking (New Brunswick Scientific Innova I26, 225 rpm). After 8 h, 50 μL of the culture was used to inoculate 50 mL of supplemented M9 minimal media [50 mM Na2HPO4, 20 mM KH2PO4, 9 mM NaCl, 4 g/L glucose, 1 g/L NH4Cl, 0.1 mM CaCl2, 2 mM MgSO4, 10 mg/L thiamine, 10 mg/L biotin, and 150 mg/L ampicillin (pH 7.4)]. The culture was shaken overnight at 37 °C.
The next morning, the culture was diluted to 100 mL with supplemented M9 minimal media, and 5-fluoroindole, dissolved in dimethyl sulfoxide, was added to a final concentration of 0.1 g/L [35]. The culture was shaken for an additional 30 min at 37 °C. Expression was induced with IPTG (1 mM final concentration). After 45 min, cells were pelleted at 1000g and resuspended in 100 mL of fresh M9 minimal media. For osmotic shock experiments, 0.3 M NaCl or 0.3 M NaCl and 1 mM glycine betaine were added to the M9 buffer. Expression was again induced with 1 mM IPTG. After 45 min, the cells were pelleted at 1000g and washed three times with in-cell NMR buffer [200 mM Hepes, 100 mM bis-tris propane, 150 μg/mL ampicillin, and 50 μg/mL chlor-amphenicol (pH 7.8)] prepared in 99.9% D2O. Ampicillin selects for plasmid containing cells, and chloramphenicol stops protein expression. Stated pH values for D2O-containing buffers are pH meter readings and uncorrected for the isotope effect. For osmotic shock experiments, 0.3 M NaCl or 0.3 M NaCl and 1 mM glycine betaine were added to the in-cell NMR buffer. Cell pellets were gently resuspended in 200 μL in-cell NMR buffer and loaded into shaped NMR tubes with 0.25 mm glass wall (Bruker) to increase signal-to-noise in samples containing high concentrations of salt. Cell slurries were ~50% cells and ~50% buffer by volume.
A 19F NMR spectrum of the cell slurry was acquired. After the experiment, the sample was gently pelleted, and a spectrum of the twofold-diluted supernatant was acquired to assess and correct for protein leakage, which was only observed under stressed conditions (Fig. S1). The pellet was resuspended in 400 μL of in-cell NMR buffer plus protease inhibitors and lysed by sonication (Fisher Scientific Sonic Dismembrator Model 500, 10% amplitude, 30 s, 67% duty cycle). The sonicated sample was centrifuged for 10 min at 16,000g, and the supernatant was used to obtain lysate spectra.
Protein expression and purification of drkN SH3
Transformation and growth were performed as described above, except 100-mL overnight cultures were prepared by inoculating supplemented M9 minimal media with 100 μL of the 8-h culture. The next morning, the overnight cultures were added to 900 mL of supplemented M9 media. The cultures were shaken at 37 °C until an optical density at 600 nm (OD600) of 0.6 was reached. Then, 60 mg of 5-fluoroindole dissolved in 250 μL of dimethyl sulfoxide was added. The cultures were shaken for an additional 30 min, after which expression was induced with IPTG to a final concentration of 1 mM. After 2 h, cells were pelleted at 1000g at 10 °C for 30 min. Cells were resuspended in 50 mM Tris (pH 7.5) containing protease inhibitors (Sigma-Aldrich P-2714, containing AEBSF, aprotinin, bestatin, E-64, EDTA, and leupeptin) and frozen at −80 °C.
Cells were thawed at room temperature and lysed by sonication (15% amplitude, 15 min, 67% duty cycle) in an ice bath. Cell debris was removed by centrifugation at 16,000g for 30 min at 10 °C, and the supernatant was passed through a 0.45-μm filter.
The purification of drkN SH3 involved three chromatography steps using a GE AKTA FPLC. The first step was anion exchange chromatography [GE Q Sepharose column, 1.6 cm × 10 cm, 2.5–22.5% gradient, 50 mM Tris wash/50 mM Tris 2 M NaCl eluent buffer (pH 7.5)]. SH3 binds weakly and was eluted at 15% of the gradient. Protease inhibitors were added to the fractions containing drkN SH3, which were then passed through a 0.22-μm filter. The next step was size-exclusion chromatography [GE Superdex 75 column, eluted with 50 mM Na2HPO4, 20 mM KH2PO4, and 9 mM NaCl (pH 7.4)].
One-dimensional 19F spectra after the size-exclusion step frequently showed that the protein was apparently destabilized by 200–300 cal/mol compared to “more stable” batches. We determined by using mass spectrometry that this SH3 was contaminated with truncated SH3 missing its nine C-terminal residues (Supplementary Fig. S3). NMR experiments indicated that the 19F resonance from truncated protein exactly overlapped that of the intact unfolded peak, which skewed the stability. Further one- and two-dimensional experiments indicated that truncated SH3 cannot fold (Supplementary Fig. S2).
To solve this problem, we added a final chromatography step, hydrophobic interaction chromatography [GE HiTrap Phenyl HP, 100%–0% gradient, 50 mM sodium phosphate, 1.5 M (NH4)2SO4, wash to 50 mM sodium phosphate (pH 7.0)], which separated truncated SH3 from the full-length protein. Purified full-length SH3 was buffer exchanged into 17 MΩ cm−1 H2O at 5 °C using a GE PD-10 desalting column. The sample was flash-frozen in an ethanol/CO2(s) bath and lyophilized for 12 h (Labconco FreeZone).
NMR
In vitro samples were prepared by adding 1 mg of purified SH3 to NMR buffer [50 mM acetic acid/sodium acetate, Hepes, and bis-tris propane (pH 7.8)] prepared in 99.9% D2O. Stated pH values for D2O-containing buffers are pH meter readings and uncorrected for the isotope effect. Methods for acquiring 19F spectra were similar to those used previously [10]. Spectra were acquired on a Bruker Avance III HD spectrometer with a QCI cryoprobe operating at a Larmor frequency of 470 MHz and running TopSpin Version 3.2. Sodium 2,2-dimethyl-2-silapentane-5-sulfonate (Cambridge Isotope Laboratories) was added to in vitro samples and used to reference the spectra via the Xi factor for 19F. In-cell samples were not referenced because we were concerned with protein stability and only needed the areas under the resonances. Spectra were acquired at 25 °C. The D2O in the sample was used to lock the spectrometer. A sweep width of either 70 ppm or 20 ppm was used, and the number of scans varied from 64 to 128 for in vitro experiments and 128 to 256 for in-cell experiments.
Data processing
Data were processed and analyzed with TopSpin. Free induction decays of 75,000 points each were subjected to a 15-Hz line broadening function before zero filling to 260,000 points followed by Fourier transformation. Resonances were integrated using two methods. Spectra were manually integrated to obtain resonance areas or were deconvoluted by fitting each peak to a Lorentzian function followed by automatic integration using TopSpin.
Mass spectrometry
The sample was resuspended in 500 μL of a 50:50 acetonitrile: 0.01% formic acid mixture to a final concentration of 10 μM and directly infused into a Thermo LTQ-FT-ICR mass spectrometer with an ESI source operating in positive-ion mode. Data were acquired in high-resolution mode over 250 scans and deconvoluted using MagTran, selecting for six species in a mass range from 10 Da to 100,000 Da.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (MCB 1410854 and CHE 1607359). We thank Greg Young for spectrometer maintenance, William Monteith for initial studies of stability under hyperosmotic conditions, Brandie Ehrmann for assistance with mass spectrometry, and Elizabeth Pielak for helpful comments on the manuscript.
Abbreviations
- SH3
N-terminal SH3 domain of Drosophila signal transduction protein drk
Appendix A. Supplementary Data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2017.03.001.
Footnotes
Edited by J. Buchner
References
- 1.Wood JM. Osmosensing by bacteria: signals and membrane-based sensors, Microbiol. Mol Biol Rev. 1999;63:230–262. doi: 10.1128/mmbr.63.1.230-262.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yancey PH, Clark ME, Hand SC, Bowlus RD, Somero GN. Living with water stress: evolution of osmolyte systems. Science. 1982;217:1214–1222. doi: 10.1126/science.7112124. [DOI] [PubMed] [Google Scholar]
- 3.Cayley DS, Guttman HJ, Record MT., Jr Biophysical characterization of changes in amounts and activity of Escherichia coli cell and compartment water and turgor pressure in response to osmotic stress. Biophys J. 2000;78:1748–1764. doi: 10.1016/s0006-3495(00)76726-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zimmerman SB, Trach SO. Estimation of macromolecule concentrations and excluded volume effects for the cytoplasm of Escherichia coli. J Mol Biol. 1991;222:599–620. doi: 10.1016/0022-2836(91)90499-v. [DOI] [PubMed] [Google Scholar]
- 5.Monteith WB, Cohen RD, Smith AE, Guzman-Cisneros E, Pielak GJ. Quinary structure modulates protein stability in cells. Proc Natl Acad Sci U S A. 2015;112:1739–1742. doi: 10.1073/pnas.1417415112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danielsson J, Mu X, Lang L, Wang H, Binolfi A, Theillet FX, et al. Thermodynamics of protein destabilization in live cells. Proc Natl Acad Sci U S A. 2015;112:12,402–12,407. doi: 10.1073/pnas.1511308112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebbinghaus S, Dhar A, McDonald D, Gruebele M. Protein folding stability and dynamics imaged in a living cell. Nat Methods. 2010;7:319–323. doi: 10.1038/nmeth.1435. [DOI] [PubMed] [Google Scholar]
- 8.Inomata K, Ohno A, Tochio H, Isogai S, Tenno T, Nakase I, et al. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature. 2009;458:106–109. doi: 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- 9.Ignatova Z, Krishnan B, Bombardier JP, Marcelino AMC, Hong J, Gierasch LM. From the test tube to the cell: exploring the folding and aggregation of a beta-clam protein. Biopolymers. 2007;88:157–163. doi: 10.1002/bip.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith AE, Zhou LZ, Gorensek AH, Senske M, Pielak GJ. In-cell thermodynamics and a new role for protein surfaces. Proc Natl Acad Sci U S A. 2016;113:1725–1730. doi: 10.1073/pnas.1518620113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu I, Mori T, Ando T, Harada R, Jung J, Sugita Y, et al. Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. elife. 2016;5:e19274. doi: 10.7554/eLife.19274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Sarkar M, Smith AE, Krois AS, Pielak GJ. Macromolecular crowding and protein stability. J Am Chem Soc. 2012;134:16,614–16,618. doi: 10.1021/ja305300m. [DOI] [PubMed] [Google Scholar]
- 13.Cohen RD, Pielak GJ. Electrostatic contributions to protein quinary structure. J Am Chem Soc. 2016;138:13,139–13,142. doi: 10.1021/jacs.6b07323. [DOI] [PubMed] [Google Scholar]
- 14.Cayley S, Record MT. Roles of cytoplasmic osmolytes, water, and crowding in the response of Escherichia coli to osmotic stress: biophysical basis of osmoprotection by glycine betaine. Biochemistry. 2003;42:12,596–12,609. doi: 10.1021/bi0347297. [DOI] [PubMed] [Google Scholar]
- 15.Larsen PI, Sydnes LK, Landfald B, Strøm AR. Osmo-regulation in Escherichia coli by accumulation of organic osmolytes: betaines, glutamic acid, and trehalose. Arch Microbiol. 1987;147:1–7. doi: 10.1007/BF00492896. [DOI] [PubMed] [Google Scholar]
- 16.Santoro MM, Liu YF, Khan SMA, Hou LX, Bolen DW. Increased thermal-stability of proteins in the presence of naturally-occurring osmolytes. Biochemistry. 1992;31:5278–5283. doi: 10.1021/bi00138a006. [DOI] [PubMed] [Google Scholar]
- 17.Zhang O, Forman-Kay JD. Structural characterization of folded and unfolded states of an SH3 domain in equilibrium in aqueous buffer. Biochemistry. 1995;34:6784–6794. doi: 10.1021/bi00020a025. [DOI] [PubMed] [Google Scholar]
- 18.Evanics F, Bezsonova I, Marsh J, Kitevski JL, Forman-Kay JD, Prosser RS. Tryptophan solvent exposure in folded and unfolded states of an SH3 domain by 19F and 1H NMR. Biochemistry. 2006;45:14,120–14,128. doi: 10.1021/bi061389r. [DOI] [PubMed] [Google Scholar]
- 19.Cayley S, Record MT, Jr, Lewis BA. Accumulation of 3-(n-morpholino)propanesulfonate by osmotically stressed Escherichia coli K-12. J Bacteriol. 1989;171:3597–3602. doi: 10.1128/jb.171.7.3597-3602.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis BA, Cayley S, Padmanabhan S, Kolb VM, Brushaber V, Anderson CF, et al. Natural abundance 14N and 13C NMR of glycine betaine and trehalose as probes of the cytoplasm of Escherichia coli K12. J Magn Reson. 1990;90:612–617. [Google Scholar]
- 21.Guijarro JI, Sunde M, Jones JA, Campbell ID, Dobson CM. Amyloid fibril formation by an SH3 domain. Proc Natl Acad Sci U S A. 1998;95:4224–4228. doi: 10.1073/pnas.95.8.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Laureto PP, Taddei N, Frare E, Capanni C, Costantini S, Zurdo J, et al. Protein aggregation and amyloid fibril formation by an SH3 domain probed by limited proteolysis. J Mol Biol. 2003;334:129–141. doi: 10.1016/j.jmb.2003.09.024. [DOI] [PubMed] [Google Scholar]
- 23.Barnes CO, Monteith WB, Pielak GJ. Internal and global protein motion assessed with a fusion construct and in-cell NMR spectroscopy. Chembiochem. 2011;12:390–391. doi: 10.1002/cbic.201000610. [DOI] [PubMed] [Google Scholar]
- 24.Sarkar M, Li C, Pielak GJ. Soft interactions and crowding. Biophys Rev. 2013;5:187–194. doi: 10.1007/s12551-013-0104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarkar M, Pielak GJ. An osmolyte mitigates the destabilizing effect of protein crowding. Protein Sci. 2014;23:1161–1164. doi: 10.1002/pro.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Felitsky DJ, Cannon JG, Capp MW, Hong J, Van Wynsberghe AW, Anderson CF, et al. The exclusion of glycine betaine from anionic biopolymer surface: why glycine betaine is an effective osmoprotectant but also a compatible solute. Biochemistry. 2004;43:14,732–14,743. doi: 10.1021/bi049115w. [DOI] [PubMed] [Google Scholar]
- 27.Knapp S, Ladenstein R, Galinski EA. Extrinsic protein stabilization by the naturally occurring osmolytes β-hydroxyectoine and betaine. Extremophiles. 1999;3:191–198. doi: 10.1007/s007920050116. [DOI] [PubMed] [Google Scholar]
- 28.Guinn EJ, Pegram LM, Capp MW, Pollock MN, Record MT. Quantifying why urea is a protein denaturant, whereas glycine betaine is a protein stabilizer. Proc Natl Acad Sci U S A. 2011;108:16,932–16,937. doi: 10.1073/pnas.1109372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canchi DR, Garcia AE. Cosolvent effects on protein stability. Annu Rev Phys Chem. 2013;64:273–293. doi: 10.1146/annurev-physchem-040412-110156. [DOI] [PubMed] [Google Scholar]
- 30.Bruzdziak P, Panuszko A, Stangret J. Influence of osmolytes on protein and water structure: a step to understanding the mechanism of protein stabilization. J Phys Chem B. 2013;117:11,502–11,508. doi: 10.1021/jp404780c. [DOI] [PubMed] [Google Scholar]
- 31.Auton M, Rosgen J, Sinev M, Holthauzen LM, Bolen DW. Osmolyte effects on protein stability and solubility: a balancing act between backbone and side-chains. Biophys Chem. 2011;159:90–99. doi: 10.1016/j.bpc.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Record MT, Jr, Courtenay ES, Cayley DS, Guttman HJ. Responses of E. coli to osmotic stress: large changes in amounts of cytoplasmic solutes and water. Trends Biochem Sci. 1998;23:143–148. doi: 10.1016/s0968-0004(98)01196-7. [DOI] [PubMed] [Google Scholar]
- 33.Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 34.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 35.Crowley PB, Kyne C, Monteith WB. Simple and inexpensive incorporation of 19F-Tryptophan for protein NMR spectroscopy. Chem Commun. 2012;48:10681–10683. doi: 10.1039/c2cc35347d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.