

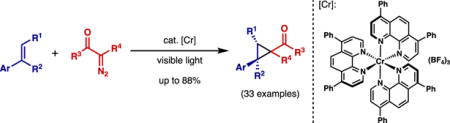
Abstract
The chromium photocatalyzed cyclopropanation of diazo reagents with electron rich alkenes is described. The transformation occurs under mild conditions and features specific distinctions from traditional diazo-based cyclopropanations (e.g., avoiding β-hydride elimination, chemoselectivity considerations, etc.). The reaction appears to work most effectively using chromium catalysis, and a number of decorated cyclopropanes can be accessed in generally good yields.
Graphical abstract
Cyclopropanes are important structures in organic chemistry.1 Their rigid structure provides a well-defined spatiality that can be decorated with features that can impart function, and they are prevalent in natural and unnatural bioactive molecules.2 The cyclopropane ring strain also allows them to serve as useful 3C components in the construction of larger ring systems.3 There are several synthetic methods toward accessing cyclopropanes from alkenes,4 including the transition-metal catalyzed reaction with diazo compounds.5 This method, most commonly using Rh or Cu catalysis, has proven widely effective. That said, orthogonal approaches using these reactants may present strategic synthetic alternatives.
The use of photocatalysis6 for the formation of cyclopropanes is relatively underexplored (Scheme 1). For UV light-mediated diazo decomposition, alkene cyclopropanation can be favorable using triplet sensitization.7 Pérez-Prieto and Stiriba have described diarylketone catalysts for sensitized cyclopropanations.8 In terms of metal catalyst systems, Stephenson and coworkers described an atom-transfer process of bromomalonate across an alkene via iridium catalysis, which upon treatment with base affords a cyclopropane product.9 Guo and coworkers reported a Ru(bpy)32+-catalyzed method that combines dibromomalonates and benzylidenemalononitriles;10 this process occurs via the formation of the α-bromomalonate anion in situ. A Ru-catalyzed method using diiodomethane to achieve radical additions to styrene derivatives was recently described by Suero and coworkers.11
Scheme 1.
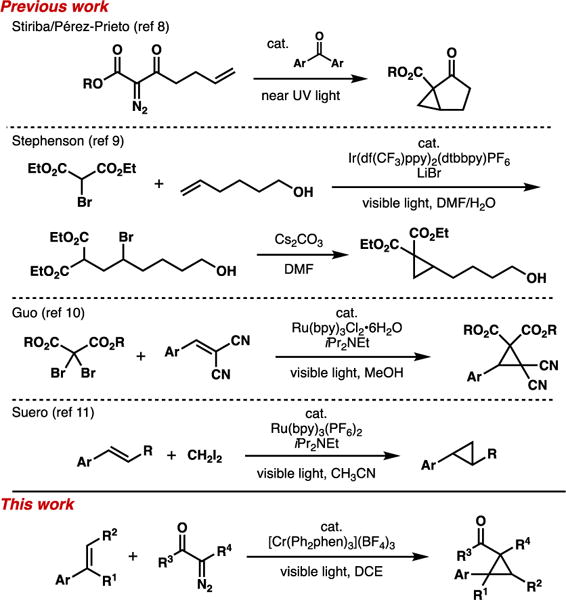
Approaches to cyclopropane synthesis using photocatalysis.
Bauld had previously shown that aminium oxidants were capable of inducing radical cation-based cyclopropanations between alkenes and diazo reagents.12 We were curious whether this transformation could be achieved using photocatalytic oxidative systems. Toward the design of sustainable synthetic methods, we have been interested in the application of photocatalysts based on first-row transition metals to supplant the traditionally employed Ru and Ir systems. We have recently reported on the use of poly-pyridinyl and -phenanthrolinyl ligated Cr photocatalysts for mediating (4+2)-cycloadditions.13 Herein, we describe a (2+1)-cycloaddition between electron-rich alkenes and diazo reagents promoted by these chromium complexes. Importantly, we highlight the orthogonality of catalytic activation of this process (alkene oxidation vs. diazo decomposition), which we anticipate will render this method useful in synthetic chemistry.
We began our studies by investigating the proposed cycloaddition between trans-stilbene and ethyl diazoacetate using the catalysts in Figure 1. We first utilized the reaction conditions developed in the original Cr-catalyzed report (1 mol % [Cr(Ph2phen)3](BF4)3, NUV irradiation, CH3NO2) with 5 equivalents of the diazoacetate (Table 1, enry 1). To our delight, the cycloaddition occurred, and cyclopropane 3aa was formed in good yield, validating the hypothesis that these oxidizing catalysts could induce this process. Byproducts 4aa and 5aa were also detected in small quantities. We found that lowering the equivalents of the diazoacetate to 1.1 equiv and using visible light were comparably effective to the original conditions. Other photooxidizing chromium catalysts we have explored were able to promote the transformation, albeit in lower yields (Table 1, entries 4-6). Additionally, when oxidizing systems based on Ru(II) photocatalysts were employed, the yields were poor (Table 1, entries 7-10). A range of solvents were evaluated, and we ultimately found that dichloroethane was optimal for the transformation, affording the highest yield of cyclopropane 3aa with minimal formation of byproducts 4aa and 5aa (Table 1, entry 15).14 Control experiments (Table 1, entries 16-18) confirmed the importance of both the light and the photocatalyst to reactivity.
Figure 1.
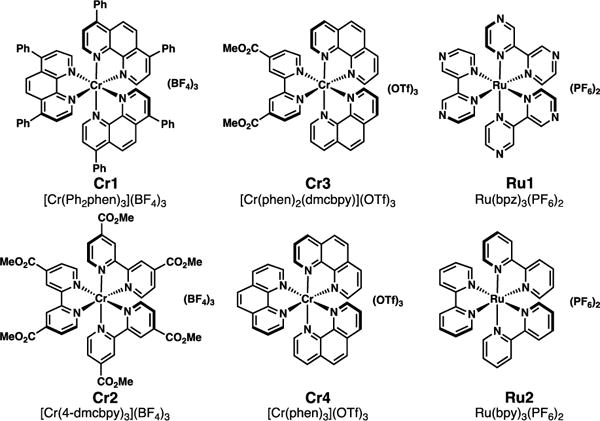
Photocatalysts evaluated.
Table 1.
Optimization of Cr-catalyzed cyclopropanation.
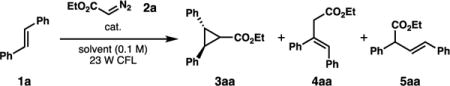
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| entry | cat. (mol%) | solvent | 2a equiv | time (h) | NMR yield (%)a | ||
| 3aa | 4aa | 5aa | |||||
| 1b | Cr1 (1) | CH3NO2 | 5 | 24 | 60 | 8 | 7 |
| 2 | Cr1 (1) | CH3NO2 | 5 | 24 | 61 | 10 | 8 |
| 3 | Cr1 (1) | CH3NO2 | 1.1 | 40 | 60 | 8 | 11 |
| 4 | Cr2 (1) | CH3NO2 | 1.1 | 40 | 5 | 1 | 1 |
| 5 | Cr3 (1) | CH3NO2 | 1.1 | 40 | 40 | 8 | 7 |
| 6 | Cr4 (1) | CH3NO2 | 1.1 | 40 | 50 | 9 | 8 |
| 7 | Ru1 (1) | CH3NO2 | 1.1 | 24 | 3 | 0 | 0 |
| 8 | Ru1 (1) | CH3NO2 | 5 | 14 | 9 | 4 | 5 |
| 9 | Ru2 (5) + MV (15)c | CH3NO2 | 5 | 21 | 12 | 2 | 2 |
| 10 | Ru2 (5) | CH3NO2 | 5 | 21 | 0 | 0 | 0 |
| 11 | Cr1 (1) | CHCl3 | 1.1 | 24 | 18 | 1 | 2 |
| 12 | Cr1 (1) | acetone | 1.1 | 24 | 57 | 5 | 8 |
| 13 | Cr1 (1) | CH2Cl2 | 1.1 | 24 | 73 | 2 | 7 |
| 14 | Cr1 (1) | CH3CN | 1.1 | 24 | 67 | 8 | 10 |
| 15 | Cr1 (1) | DCE | 1.1 | 24 | 73 | 0 | 5 |
| 16 | none | DCE | 1.1 | 14 | 0 | 0 | 0 |
| 17 | CrCl3 (10) | DCE | 1.1 | 49 | 0 | 0 | 0 |
| 18d | Cr1 (1) | DCE | 1.1 | 49 | 0 | 0 | 0 |
Determined using veratraldehyde as a standard.
Near UV light (bulbs at 300, 350, and 419 nm) used instead of 23 W CFL.
MV: methyl viologen2+·(PF6)2.
Reaction performed in dark (foil wrapped).
With optimized conditions for cyclopropanation in hand, we established the alkene scope (Scheme 2). A range of stilbenes were competent reactants, and the products were accessed in generally high yields. Aryl alkyl alkenes were also participatory in this cycloaddition. Although the yields were overall good, there was in general little diastereoselectivity as the cyclopropanes were observed as approx. 1:1 mixtures with a couple of exceptions. For compounds 3ia-3ma, the reactivity pattern somewhat mirrors that of the (4+2)-cycloaddition; the pendant oxygenated group if electron rich (i.e., alcohol or ether) will hamper reactivity due to donation into the presumed radical cation intermediate.15 Curiously, an alkyl tosylate also rendered the reaction unproductive (3na).16 Gratifyingly, trisubstituted alkenes were reactive in this chemistry (e.g., 3sa, 3ta), which had not been the case for the (4+2) process. Tetrasubstituted olefins unfortunately proved unreactive.
Scheme 2. Cyclopropanation - alkene scope.
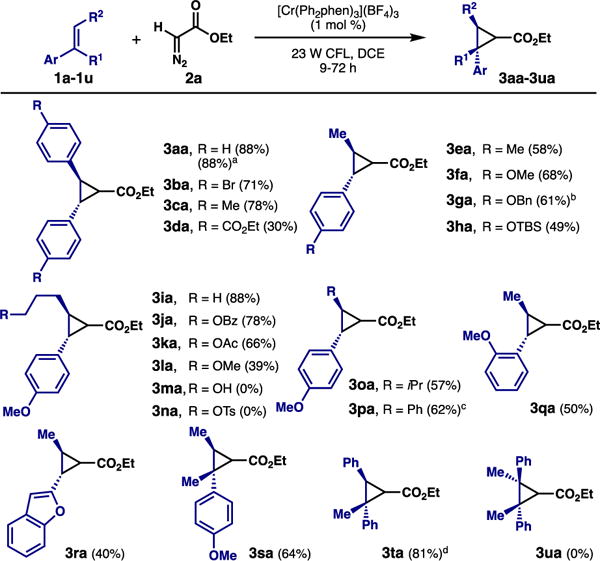
a Gram scale experiment. b Reaction performed with needle outlet open to air. c Product observed in 3.0:1 dr. d Product observed in 4.1:1 dr.
The alkene reactivity observed appears to correlate well with respective reduction potentials. As seen in Figure 2, a window of competent reactivity was observed for alkenes with reduction potentials in the range E1/2 = +1.11-1.80 V (vs. SCE).17,18 Stilbene derivatives 1v (E1/2 = +0.99 V) and 1w (E1/2 = +1.94 V) were both unproductive in the cycloaddition. This spectrum of reactivity is reflective of the presumed radical cation mechanism pathway, necessitating an appropriate oxidation capacity in order to achieve proper activity.
Figure 2.
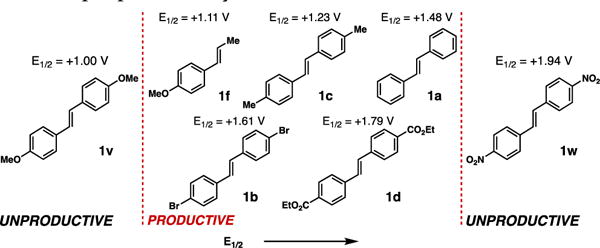
Alkene reactivity range based on reduction potential.
The chemoselectivity of the cycloaddition, dictated by electronics, is a unique and important attribute of this transformation. The more commonly-employed cyclopropanations of diazo compounds using metal catalysis (i.e., Rh or Cu catalysts) may be governed by other factors, such as favorable intramolecularity. Scheme 3 is illustrative. Trans-anethole (1f) and diazoacetate 2b were combined under both Cu-catalyzed cyclopropanation conditions and our standard Cr photocatalytic conditions. The Cu-catalyzed reaction19 afforded a mixture of both possible cyclopropane products (3fb and 6) in low conversion.20 Alternatively, with Cr catalysis the intermolecular addition product (3fb) was observed exclusively. Additionally, the cyclopropanation of compound 7 was successful, establishing that intramolecular processes can occur on the condition that it is electronically feasible. We anticipate this type of chemoselectivity can be strategically employed in complex synthetic settings, a distinguishing feature of this Cr-catalyzed cyclopropanation.
Scheme 3.
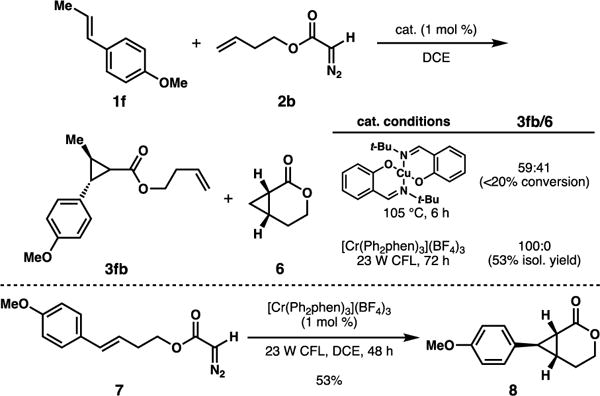
Alkene chemoselectivity.
Different diazo esters and arylketones were also generally effective (Scheme 4). Notably, α-alkyl diazo esters were competent partners, with no observation of β-hydride elimination, typically a common phenomenon in diazo decomposition cyclopropanations.21 The mechanistic pathway differentiation of this process enables cyclopropanation to predominate. Additionally, the mechanism ensures that diazo dimerization is avoided, and thus no slow addition protocols were necessary.22
Scheme 4. Cyclopropanation - diazo scope.
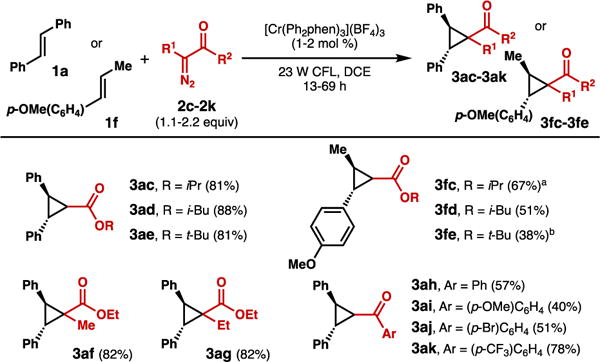
a Product observed in 1.4:1 dr. b Product observed in 1.5:1 dr.
A different outcome was observed in the reaction between trans-anethole and diazopropionate 2f. Expected cyclopropane 3ff was indeed the main product, but byproduct dihydrofuran 9ff was also observed in measurable yield (Scheme 5). Resubjecting the cyclopropane product to the photocatalyst conditions also generated the dihydrofuran in high yield.23 Control experiments indicated that the chromium catalyst and light were both required for this rearrangement to occur.24
Scheme 5.
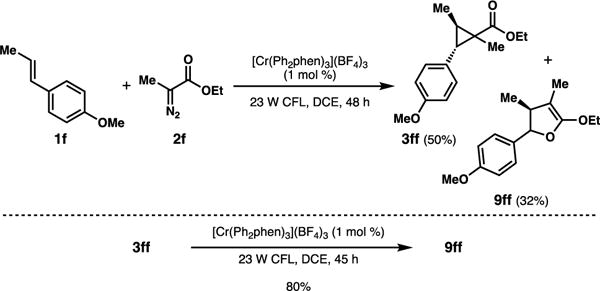
Cyclopropanation and rearrangement.
Additionally, when a competition experiment was performed with both diazoacetate 2a and diene 10, the cyclopropane product was the predominant observed species, highlighting the increased nucleophilicity of 2a in intercepting the radical cation.
Our proposed mechanism for the cycloaddition is consistent with the previous processes described by Bauld (Scheme 7, shown for formation of cyclopropane 3aa).12b The excited state Cr(III) catalyst induces alkene single electron oxidation.25 The resulting radical cation undergoes nucleophilic attack by the diazo compound. Loss of N2 affords a “long-bonded” radical cation intermediate 14.12b At this stage, electron transfer from either the reduced-state Cr complex or another equivalent of alkene yields the cyclopropane product. Turnover occurs either via reentry of the Cr complex into the photocatalytic cycle, or through radical propagation by the electron transfer from the alkene component.26 Byproducts 4aa and 5aa would arise from intermediate 13 when N2 is lost via phenyl or hydrogen migration, respectively.27 The observation of differential reactivities between this process and our reported (4+2)-cycloadditions (alkene competencies, O2 dependencies) indicate that a more thorough analysis of the mechanism will be necessary for full elucidation.
Scheme 7.
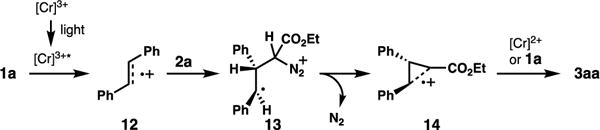
Proposed mechanism.
In summary, a radical cation cycloaddition between alkenes and diazo species using Cr catalysis and visible light is described. The formation of polysubstituted cyclopropanes via this process may offer an attractive alternative to the more commonly employed Rh or Cu complexes. The chemoselectivity aspects are a distinguishing characteristic of this transformation, and by virtue of this no syringe pump mode of addition is necessary. Current efforts are dedicated toward enriching our mechanistic understanding and expanding the suite of cycloadditions mediated by these Cr photocatalysts; these endeavors will be later disclosed.
Supplementary Material
Scheme 6.
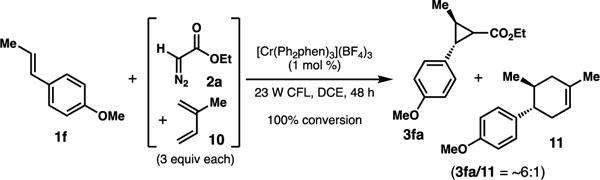
Competition experiment between diazo and diene.
Acknowledgments
This research was carried out by the Catalysis Collaboratory for Light-activated Earth Abundant Reagents (C-CLEAR), which is supported by the NSF and EPA through the Network for Sustainable Molecular Design and Synthesis program (NSF-CHE-1339674). FJS is supported by the NSF Graduate Research Fellowship Program under Grant No. 038550-02. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF. We acknowledge collaborators Niels Damrauer (Univ. Colorado), Anthony Rappé (Colorado St. Univ.), and Matthew Shores (Colorado St. Univ.) and their groups for enriching discussions.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, compound characterization data, and spectra (PDF)
Author Contributions
The manuscript was written through contributions of all authors./All authors have given approval to the final version of the manuscript.
References
- 1.(a) Patai S, Rappoport Z, editors. The Chemistry of the Cyclopropyl Group. Wiley & Sons; New York: 1987. [Google Scholar]; b de Meijere A, editor. Small Ring Compounds in Organic Synthesis VI. Vol. 207 Springer; Berlin, Germany: 2000. [Google Scholar]
- 2.(a) Faust R. Angew Chem Int Ed. 2001;40:2251. doi: 10.1002/1521-3773(20010618)40:12<2251::aid-anie2251>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]; (b) Wessjohann LA, Brandt W, Thiemann T. Chem Rev. 2003;103:1625. doi: 10.1021/cr0100188. [DOI] [PubMed] [Google Scholar]; (c) Brackmann F, de Meijere A. Chem Rev. 2007;107:4493. doi: 10.1021/cr078376j. [DOI] [PubMed] [Google Scholar]
- 3.For select reviews, see:; (a) Rubin M, Rubina M, Gevorgyan V. Chem Rev. 2007;107:3117. doi: 10.1021/cr050988l. [DOI] [PubMed] [Google Scholar]; (b) Reissig H-U, Zimmer R. Chem Rev. 2003;103:1151. doi: 10.1021/cr010016n. [DOI] [PubMed] [Google Scholar]; (c) Wong HNC, Hon MY, Tse CW, Yip YC, Tanko J, Hudlicky T. Chem Rev. 1989;89:165. [Google Scholar]
- 4.For reviews, see:; (a) Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem Rev. 2003;103:977. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]; (b) Kimimura A. In: Methods and Applications of Cycloaddition Reactions in Organic Syntheses. Nishiwaki N, editor. John Wiley & Sons; Hoboken, N J: 2014. p. Pt I-1. [Google Scholar]
- 5.(a) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides. Wiley; New York: 1998. [Google Scholar]; (b) Davies HML, Antoulinakis EG. Org React. 2001;57:1. [Google Scholar]
- 6.For select reviews on photocatalysis, see:; (a) Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xuan J, Xiao W-J. Angew Chem Int Ed. 2012;51:6828. doi: 10.1002/anie.201200223. [DOI] [PubMed] [Google Scholar]; (c) Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n. [DOI] [PubMed] [Google Scholar]; (d) Angnes RA, Li Z, Correia CRD, Hammond GB. Org Biomol Chem. 2015;13:9152. doi: 10.1039/c5ob01349f. [DOI] [PubMed] [Google Scholar]; (e) Yoon TP. ACS Catal. 2013;3:895. doi: 10.1021/cs400088e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ando W, Imai I, Migita T. J Org Chem. 1972;37:3596. [Google Scholar]; (b) Moss RA, Joyce MA. J Am Chem Soc. 1977;99:1262–1264. [Google Scholar]; (c) Fien J, Kirmse W. Angew Chem Int Ed. 1998;37:2232. doi: 10.1002/(SICI)1521-3773(19980904)37:16<2232::AID-ANIE2232>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 8.(a) Pastor-Pérez L, Wiebe C, Pérez-Prieto J, Stiriba S-E. J Org Chem. 2007;72:1541. doi: 10.1021/jo062426n. [DOI] [PubMed] [Google Scholar]; (b) Pastor-Pérez L, Barriau E, Frey H, Pérez-Prieto J, Stiriba S-E. J Org Chem. 2008;73:4680. doi: 10.1021/jo800254f. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen JD, Tucker JW, Konieczynksa MD, Stephenson CRJ. J Am Chem Soc. 2011;133:4160. doi: 10.1021/ja108560e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang Y, Qian R, Zheng X, Zeng Y, Sun J, Chen Y, Ding A, Guo H. Chem Commun. 2015;51:54. doi: 10.1039/c4cc08203f. [DOI] [PubMed] [Google Scholar]
- 11.del Hoyo AM, Herraiz AG, Suero MG. Angew Chem Int Ed. 2017;56:1610. doi: 10.1002/anie.201610924. [DOI] [PubMed] [Google Scholar]
- 12.(a) Stufflebeme G, Lorenz KT, Bauld NL. J Am Chem Soc. 1986;108:4234. [Google Scholar]; (b) Bauld NL, Stufflebeme G, Lorenz KT. J Phys Org Chem. 1989;2:585. [Google Scholar]; (c) Yueh W, Bauld NL. J Am Chem Soc. 1995;117:5671. [Google Scholar]
- 13.(a) Stevenson SM, Shores MP, Ferreira EM. Angew Chem Int Ed. 2015;54:6506. doi: 10.1002/anie.201501220. [DOI] [PubMed] [Google Scholar]; (b) Higgins RF, Fatur SM, Shepard SG, Stevenson SM, Boston DJ, Ferreira EM, Damrauer NH, Rappé AK, Shores MP. J Am Chem Soc. 2016;138:5451. doi: 10.1021/jacs.6b02723. [DOI] [PubMed] [Google Scholar]; (c) Stevenson SM, Higgins RF, Shores MP, Ferreira EM. Chem Sci. 2017;8:654. doi: 10.1039/c6sc03303b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The alkene byproducts were only observed in the diaryl alkene-based cyclopropanations in <10% yield, and were readily removed by column chromatography.
- 15.(a) Gassman PG, Bottorff KJ. Tetrahedron Lett. 1987;28:5449. [Google Scholar]; (b) Gassman PG, Bottorff KJ. J Am Chem Soc. 1987;109:7547. [Google Scholar]; (c) Mizuno K, Tamai T, Nishiyama T, Tani K, Sawasaki M, Otsuji Y. Angew Chem Int Ed Engl. 1994;33:2113. [Google Scholar]; (d) Asaoka T, Kitazawa T, Wada T, Inoue Y. J Am Chem Soc. 1999;121:8486. [Google Scholar]; (e) Hamilton DS, Nicewicz DA. J Am Chem Soc. 2012;134:18577. doi: 10.1021/ja309635w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.For this specific reaction, sulfonyloxy transfer to the diazoacetate was observed instead.
- 17.Kubota T, Uno B, Matsuhisa Y, Miyazaki H, Kano K. Chem Pharm Bull. 1983;31:373. [Google Scholar]; (b) ref 12b.
- 18.The reduction potential of [Cr(Ph2phen)3]3+ (E*1/2 = +1.33 V vs. SCE in CH3NO2) is within a viable range for alkene oxidation competency. We note that these values should only be used as a general guide, as they are sensitive to the conditions of measurement (solvent, etc.).
- 19.(a) Corey EJ, Myers AG. Tetrahedron Lett. 1984;25:3559. [Google Scholar]; (b) Sacconi L, Ciampolini M. J Chem Soc. 1964:276–280. [Google Scholar]
- 20.Attempts at Rh catalysis were less effective than those with Cu.
- 21.For an elegant solution using Rh catalysis, see; Panne P, DeAngelis A, Fox JM. Org Lett. 2008;10:2987. doi: 10.1021/ol800983y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Other potential carbene precursors, such as tosylhydrazones, triazoles, sulfur and iodonium ylides, as well as donor-donor and acceptor-acceptor diazo species, were ineffective.
- 23.Although this ring expansion (acylcyclopropane → dihydrofuran) is more commonly observed with ketones and aldehydes, there are isolated reports of this process with donor-acceptor based cyclopropylesters promoted by Lewis acid and/or heat. See:; (a) Abdallah H, Grée R, Carrié R. Tetrahedron. 1985;41:4339. [Google Scholar]; (b) Granger K, Snapper ML. Eur J Org Chem. 2012:2308. [Google Scholar]; (c) Yadav VK, Balamurugan R. Org Lett. 2001;3:3717. doi: 10.1021/ol0163169. [DOI] [PubMed] [Google Scholar]; (d) Saalfrank RW, Gündel J, Roßmann G, Hanek M, Rost W, Peters K, Von Schnering HG. Chem Ber. 1990;123:1175. [Google Scholar]
- 24.Dihydrofuran formation was not observed for the stilbene-based cyclopropanes.
- 25.Ethyl diazoacetate (E1/2 = +2.10 V)12b is outside of the range of oxidation capability.
- 26.In our (4+2) studies, we noted an important O2 dependency that was consistent with a photocatalyst turnover pathway. We do not observe a similar dependency for this cycloaddition.
- 27.In a singular example, Bauld reported a similar methyl migration. See ref 12b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.