

Abstract
Rationale:
Staphylococcus-associated glomerulonephritis (SAGN) is a rare immune complex–mediated glomerulonephritis associated with active Staphylococcus infection. We report a case illustrating the importance of clinical history and kidney biopsy findings in establishing the correct diagnosis.
Presenting concerns of the patient:
We report the case of a 64-year-old man with alcohol-associated cirrhosis, type 2 diabetes mellitus, and hypertension who presented to hospital with lower back and abdominal pain, rectal bleeding, a purpuric lower extremity rash, and oliguric acute kidney injury with microscopic hematuria and nephrotic-range proteinuria.
Diagnoses:
Skin biopsy revealed IgA leukocytoclastic vasculitis. Serum cryoglobulins were positive and there was hypocomplementemia with a low C3 level. Magnetic resonance imaging of the lumbar spine revealed septic discitis and epidural abscesses caused by a recent Staphylococcus aureus bacteremia. Kidney biopsy showed IgA-dominant and C3-dominant proliferative glomerulonephritis with subepithelial humps in keeping with SAGN.
Interventions:
Urgent hemodialysis was initiated along with a prolonged course of intravenous cefazolin.
Outcomes:
Remarkably, the patient demonstrated a complete recovery of renal function after 2 months of dialysis dependence and successful treatment of the epidural abscesses.
Lessons learned:
This case shows that SAGN can closely mimic the clinical, laboratory, and histological presentation of Henoch-Schönlein Purpura or cryoglobulinemic vasculitis. Clinical history and kidney biopsy, particularly electron microscopic analysis, are essential to establishing the correct diagnosis to avoid the unnecessary and potentially harmful administration of immunosuppression. Despite the typically poor prognosis of SAGN, this case report illustrates that full renal recovery remains possible with supportive care and eradication of the underlying infection.
Keywords: staphylococcus-associated glomerulonephritis, Henoch-Schönlein Purpura, cryoglobulinemic vasculitis, kidney biopsy
Abrégé
Exposition:
La glomérulonéphrite aigüe post-infection à Staphylococcus (GNAS) est une glomérulonéphrite rare, médiée par un complexe immunitaire et associée à une infection active à Staphylococcus aureus. Nous présentons un cas qui illustre l’importance des antécédents médicaux et des résultats de la biopsie rénale dans la pose d’un diagnostic.
Présentation du cas:
Nous rapportons le cas d’un homme de 64 ans atteint d’une cirrhose alcoolique, de diabète de type 2 et d’hypertension qui s’est présenté à l’hôpital avec des douleurs lombaires et abdominales, un saignement rectal, une éruption purpurique des extrémités inférieures et une insuffisance rénale aigüe oligurique avec hématurie microscopique et protéinurie néphrotique.
Diagnostic:
La biopsie cutanée a révélé une vascularite leucocytoclastique à IgA. Les cryoglobulines sériques étaient positives et une hypocomplémentémie avec un faible taux de C3 a été constatée. L’imagerie par résonnance magnétique du rachis lombal a permis de détecter une discite septique et des abcès périduraux causés par une récente bactériémie à Staphylococcus aureus. La biopsie rénale a quant à elle montré la présence d’une glomérulonéphrite proliférative à IgA et à C3 dominants et de volumineux dépôts sous-épithéliaux en bosse (humps) conformes à une GNAS.
Traitement:
On a amorcé une hémodialyse d’urgence et un traitement prolongé à la céfazoline par voie intraveineuse.
Résultats:
Fait remarquable, après le traitement des abcès périduraux et deux mois de dépendance à l’hémodialyse, la fonction rénale du patient a été complètement rétablie.
Conclusion:
Ce cas démontre que la GNAS peut simuler la présentation clinique, biologique et histologique du purpura d’Henoch-Schönlein ou celle de la vascularite cryoglobulinémique. La revue des antécédents médicaux et l’analyse d’une biopsie rénale (particulièrement par microscopie électronique) sont essentielles pour établir un diagnostic juste et pour éviter l’administration superflue, voire néfaste, d’immunosuppresseurs. Malgré le pronostic sombre normalement associé à la GNAS, ce cas démontre que le rétablissement complet de la fonction rénale demeure possible, pour autant que l’on procède à un traitement symptomatique et que l’on éradique l’infection sous-jacente.
What was known before
Staphylococcus-associated glomerulonephritis (SAGN) is a rare immune complex–mediated glomerulonephritis that can present with oliguric acute kidney injury and nephrotic-range proteinuria.
What this adds
The clinical and histological presentation of SAGN can closely mimic that of Henoch-Schönlein Purpura or cryoglobulinemic vasculitis with cutaneous leukocytoclastic vasculitis, positive serum cryoglobulins, hypocomplementemia, and IgA-dominant glomerulonephritis. Clinical history and kidney biopsy findings are essential to establishing the correct diagnosis. Recovery of renal function is possible with treatment of the underlying infection and avoidance of immunosuppression.
Introduction
Infections caused by the virulent pathogen Staphylococcus aureus are increasing in incidence and often associated with antibiotic resistance and potentially life-threatening complications.1 A rare, but well-recognized, complication of S aureus is an immune complex–mediated glomerulonephritis triggered by a persistent active infection.2 We report the case of a patient with S aureus bacteremia who developed an epidural abscess and oliguric acute kidney injury (AKI) with nephrotic-range proteinuria secondary to staphylococcus-associated glomerulonephritis (SAGN). His clinical presentation was deceptively similar to that of Henoch-Schönlein Purpura (HSP) and cryoglobulinemic vasculitis. We review the epidemiology and clinical features of SAGN with discussion of the essential role for kidney biopsy in establishing the correct diagnosis to prevent the unnecessary and potentially harmful administration of immunosuppression in the setting of Staphylococcus infection.
Case Presentation
A 64-year-old man with no history of renal disease was referred to our Nephrology service due to oliguric AKI. The patient presented to hospital with a 2-day history of decreased urine output, new-onset rash over the lower extremities, intermittent bright red blood per rectum, and lower abdominal tenderness. He had been discharged from another hospital 3 days prior to presentation after an admission for community-acquired pneumonia complicated by methicillin-sensitive Staphylococcus aureus (MSSA) bacteremia which had been treated with a 2-week course of intravenous vancomycin. He had also developed acute worsening of his chronic low back pain and received a lumbar epidural corticosteroid injection while in hospital.
The patient was of South Asian ancestry and previously worked in the manufacturing industry. His medical history also included compensated alcohol-associated cirrhosis Child-Pugh A, resistant hypertension, poorly controlled type 2 diabetes mellitus, obesity, asthma, gastroesophageal reflux disease (GERD), and lumbar spinal stenosis. His medications at the time of admission were acetylsalicylic acid (ASA), valsartan, diltiazem, amlodipine, furosemide, hydrochlorothiazide, metformin, canagliflozin, sitagliptin, insulin lispro, rosuvastatin, theophylline, tiotropium, fluticasone propionate/salmeterol, and salbutamol. He consumed 2 to 3 servings of alcohol per day and was a former smoker with a 25 pack-year smoking history. He denied intravenous or other recreational drug use.
Physical examination revealed an elevated blood pressure of 184/79 mm Hg with otherwise normal vital signs and mental status. Bilateral basilar crackles were detected upon pulmonary auscultation but he appeared otherwise euvolemic. The abdomen was soft and nontender and there was scant red blood on digital rectal examination. There was tenderness to palpation of the lumbar spine with normal tone, power, reflexes, and sensation in the lower extremities. Skin examination revealed multiple patches of nonpalpable purpura in the lower extremities. A Foley catheter was inserted which yielded less than 200 mL of urine over 12 hours.
Laboratory investigations revealed an elevated creatinine of 493 µmol/L from a baseline of 91 µmol/L at the time of discharge 5 days previously from another hospital. There were significant biochemical abnormalities with hyperkalemia (potassium 6.0 mEq/L), hyponatremia (sodium 126 mEq/L), acidemia (bicarbonate 17 mmol/L), and hyperphosphatemia (phosphate 1.78 mmol/L). Urinalysis was remarkable for 1 g/L of albumin and 250 red blood cells (RBC)/µL although no casts were noted on microscopic examination of urine sediment. A urinalysis 6 months ago was negative for protein or microscopic hematuria, which suggests a diagnosis of preexisting diabetic nephropathy or IgA nephropathy. Serum albumin was 30 g/L and there was nephrotic-range proteinuria with a spot urine albumin-creatinine ratio (UACR) 317 mg/mmol and urine protein-creatinine ratio (UPCR) 359 mg/mmol. The most recent UACR done prior to this was more than 1 year ago, at which point it was mildly elevated at 2.6 mg/mmol and was normal 2 years ago. The remaining initial investigations were notable for mild normocytic anemia (hemoglobin 117 g/L), neutrophilia (neutrophil count 10.6 × 109/L), and elevated inflammatory markers with C-reactive protein 91 mg/L and erythrocyte sedimentation rate 60 mm/h. Ultrasound of the abdomen showed a cirrhotic liver with splenomegaly and mild ascites; the kidneys were normal in size and echogenicity with no evidence of hydronephrosis.
A trial of intravenous furosemide failed to improve the oliguria and hyperkalemia so a temporary right femoral vein dialysis catheter was inserted under ultrasound guidance. The patient was urgently initiated on intermittent hemodialysis (IHD) while investigations for the cause of his renal dysfunction were undertaken. There was no clinical evidence of prerenal or postrenal AKI and the time course and systemic symptoms were not in keeping with vancomycin-induced nephrotoxicity. His presentation was therefore felt to be consistent with a rapidly progressive glomerulonephritis (RPGN), particularly given the purpuric rash, microscopic hematuria, and heavy proteinuria. Initial suspicion for endocarditis-associated glomerulonephritis secondary to the recent MSSA bacteremia was discounted by negative serial blood cultures and the absence of valvular vegetations on transthoracic and transesophageal echocardiograms. Consideration was given to other diagnoses including postinfectious glomerulonephritis or immune complex–mediated membranoproliferative glomerulonephritis, which were consistent with the recent infection and presence of hypocomplementemia with slightly low C3 at 0.64 g/L and normal C4. Interestingly, serum cryoglobulin testing was positive for 1 g/L of cryoprecipitate although several repeat samples taken in the following weeks were negative. A work-up for other causes of glomerulonephritis was negative for antinuclear antibody (ANA), anticytoplasmic antibody (ANCA), anti-glomerular basement membrane (GBM) antibody, and rheumatoid factor. Serologies were also negative for hepatitis B virus (HBV), hepatitis C virus (HCV), and human immunodeficiency virus (HIV). Serum and urine protein electrophoresis were unremarkable. Skin biopsy of the purpuric rash revealed a leukocytoclastic vasculitis with positive staining for IgA, IgM, and C3 on immunofluorescence. The presence of IgA raised concern for HSP (also known as IgA vasculitis), particularly with the suggestive history of a recent respiratory tract infection and purpuric rash, abdominal pain, and gastrointestinal bleeding.
Despite the diagnostic uncertainty regarding the etiology of the RPGN, there was initial reluctance to pursue a kidney biopsy or empiric pulsed corticosteroids in light of the patient’s recent infection and medical comorbidities. Furthermore, magnetic resonance imaging (MRI) of the lumbar spine demonstrated septic discitis at the L4-L5 interspace along with epidural abscesses extending from L3 to L5 and S1 to S2 (see Figure 1). The likely etiology of these findings was the recent MSSA bacteremia. The patient was administered a prolonged course of intravenous cefazolin while continuing on thrice-weekly IHD. Following completion of 6 weeks of antibiotic therapy, the patient’s low back pain had significantly diminished, serum complement levels were normalized, and there was radiographic improvement of the epidural abscesses on follow-up MRI.
Figure 1.
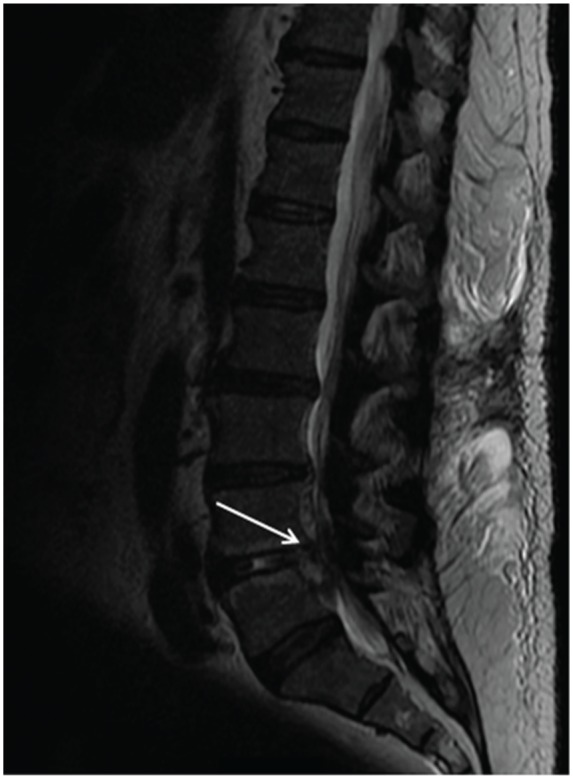
Magnetic resonance imaging of the lumbar spine performed without the administration of intravenous contrast demonstrating septic discitis at the L4-L5 interspace along with epidural abscesses extending from L3 to L5 and S1 to S2.
With this controlled infection no longer posing a contraindication to immunosuppressive therapy, a renal biopsy was performed 7 weeks after presentation to identify treatable etiologies of RPGN, such as HSP or cryoglobulinemic vasculitis. Kidney biopsies are often done and are a recommended procedure to rule out other causes of glomerulonephritis, including C3 nephritis.3 Pathological examination of the biopsy specimen revealed 17 glomeruli of which 4 were globally sclerosed. No features of acute tubular necrosis were found. There was diffuse global hypercellularity with mesangial and endocapillary proliferation as well as infiltrating monocytes and occasional neutrophils, consistent with proliferative glomerulonephritis (see Figure 2). No crescents or necrotizing lesions were identified on light microscopy and there was minimal glomerular sclerosis, suggesting a potential for recovery of renal function. Immunofluorescence revealed diffuse staining with IgA (+2) and C3 (+3) throughout the mesangium and GBM. There was no evidence of interstitial eosinophils, which helped to exclude other diagnosis such as vancomycin-induced nephrotoxicity. There was trace staining for IgG, lambda, and kappa and negative staining for IgM and C1q (see Figure 3). The preliminary interpretation of the light microscopy findings and strong IgA staining on immunofluorescence was thought to be consistent with IgA nephropathy or HSP, prompting the initiation of prednisone at a dose of 1 mg/kg daily. However, electron microscopy performed the following day demonstrated large subepithelial deposits with a hump-like appearance (see Figure 4). The presence of these subepithelial humps and stronger staining for C3 over IgA was finally deemed to be more consistent with the diagnosis of SAGN, and the corticosteroids were immediately discontinued. The diagnosis of SAGN was further supported by the recent MSSA bacteremia, ongoing epidural abscess, and, ultimately, improvement of renal function with eradication of the underlying infection.
Figure 2.
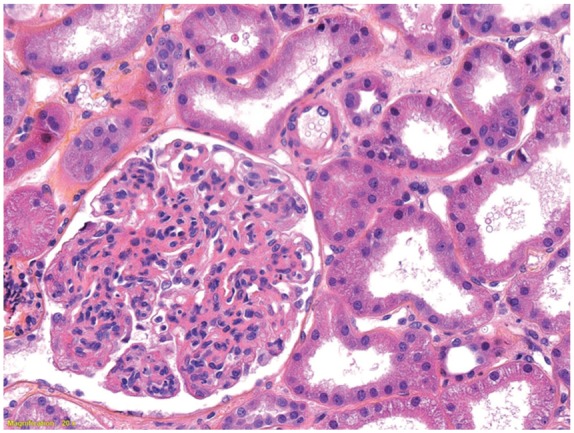
Light microscopy of a renal biopsy demonstrating diffuse global hypercellularity with mesangial and endocapillary proliferation as well as infiltrating monocytes and occasional neutrophils.
Figure 3.

Immunofluorescence of a renal biopsy demonstrating diffuse granular staining for C3 (left) and IgA (right) in the glomerular basement membrane and mesangium.
Figure 4.
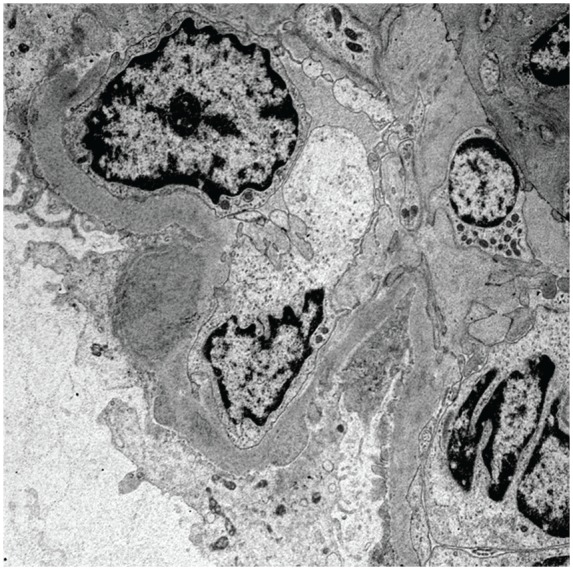
Electron microscopy of a renal biopsy demonstrating a large subepithelial deposit with a hump-like appearance as well as some mesangial and paramesangial electron dense deposits.
The patient showed promising signs of renal recovery with increasing urine output and stabilization of the interdialytic rise in creatinine. Remarkably, 9 weeks after initiation of IHD for RPGN with oliguric AKI, he was successfully transitioned off dialysis and demonstrated full recovery of renal function. Eight months after his initial presentation to hospital, the patient is clinically well and has resumed his usual activities of daily living. At this time, he has some evidence of residual renal injury with ongoing Grade A3 proteinuria (UACR 65 mg/mmol) which could represent irreversible glomerular injury from SAGN, but serum creatinine remains stable at 93 µmol/L.
Discussion
Staphylococcus-associated glomerulonephritis is a rare, recently described immune complex–mediated glomerulonephritis associated with Staphylococcus infection. Unlike postinfectious glomerulonephritis (PIGN) which typically presents 1 to 3 weeks after resolution of a streptococcal skin or respiratory tract infection, SAGN usually arises concurrently with persistent active staphylococcal cellulitis, endocarditis, osteomyelitis, abscess, or infection of an indwelling shunt.4,5 Staphylococcus-associated glomerulonephritis commonly presents with AKI with hypertension, hypocomplementemia, elevated creatinine, microscopic hematuria, and variable proteinuria which reaches the nephrotic range in 21% to 48% of patients.6,7 The pathophysiology of SAGN is poorly understood but is postulated to involve massive T-cell activation by a staphylococcal superantigen that results in cytokine release; polyclonal B-cell overproduction of IgA, IgG, and IgM; and systemic vasculitis with IgA-staphylococcal antigen immune complex formation and deposition.2 This is a rare diagnosis with the largest published review of the literature to date identifying only 83 reported cases of biopsy-proven SAGN.7 Our patient is illustrative of the typical demographic affected by SAGN, with the most common risk factors being male sex (77%), age >65 years old (41%), diabetes mellitus (24%), and absence of previous renal disease (100%).7 Other risk factors include chronic HCV infection, malignancy, cardiac disease, hypertension, intravenous drug use, trauma, and the postoperative state.6,8 Clinicians should consider the diagnosis of SAGN in patients with new-onset renal dysfunction in the setting of an active staphylococcal infection and these underlying risk factors.
A key learning point of this case report is that SAGN can closely mimic the clinical, laboratory, and pathological presentation of HSP and cryoglobulinemic vasculitis. Indeed, our patient presented with cardinal features of HSP including abdominal pain, gastrointestinal bleeding, IgA cutaneous leukocytoclastic vasculitis, and IgA-dominant glomerulonephritis. A purpuric rash and acute glomerulonephritis can also be seen in mixed cryoglobulinemic vasculitis, and positive cryoglobulin testing has been reported in at least one other patient with SAGN in addition to this case.9 Indeed, more than 20% of patients with SAGN present with a leukocytoclastic vasculitis and lower extremity purpuric rash.10 Clinical characteristics were present in our patient that favor the diagnosis of SAGN over HSP including older age, diabetes mellitus, active staphylococcal infection, and presentation with AKI and hypocomplementemia. As in the case of this patient, renal biopsy may ultimately be required to distinguish SAGN from other glomerular diseases, although the histopathological findings of SAGN and IgA vasculitis are often challenging to differentiate.4 Typical microscopic findings of SAGN include mesangioproliferative and/or endocapillary proliferative immune complex glomerulonephritis with crescent formation in 30% to 40% of patients.6,7 Immunofluorescence is similar to HSP and IgA nephropathy with granular IgA and/or C3 immune complex deposition. However, SAGN can be distinguished by stronger immunofluorescent staining for C3 than IgA.4 Subepithelial hump-like deposits can be found on electron microscopy in 31% to 45% of cases of SAGN which also supports this diagnosis over that of IgAN.6,7 The presence of an inflammatory infiltrate with neutrophils can support the diagnosis of SAGN over IgAN, but this is not a universal finding as the largest review of the literature to date showed neutrophilic infiltration in only 24/83 patients with SAGN.7 As in the case of this patient, a renal biopsy can be invaluable in establishing the correct diagnosis and guiding management, particularly when the presentation of SAGN mimics other steroid-responsive disease processes such as HSP or cryoglobulinemic vasculitis. Timely electron micrographic analysis is particularly important in SAGN because of the resemblance to other glomerulopathies on light and immunofluorescence microscopy. The rapid availability of electron micrographs in this case spared the patient from an unnecessary and potentially harmful prolonged course of corticosteroids.
A remarkable feature of this patient’s clinical course was the complete recovery of renal function after a clinically aggressive presentation with oliguric AKI and more than 2 months of dialysis dependence. This is particularly noteworthy given the typically poor prognosis of SAGN which carries a 15% mortality rate, 18% rate of persistent renal dysfunction, and 23% rate of dialysis-dependent end-stage renal disease.7 Older age, elevated serum creatinine, tubulointerstitial scarring, and underlying diabetes mellitus are all predictive of adverse renal outcomes.6,7 As in the case of our patient, recovery from renal injury tends to occur with treatment and resolution of the underlying infection with antibiotics and, in some cases, surgical source control. This differs from other causes of RPGN, which are often empirically treated with pulse dose of steroids in clinical practice, even though there is sparse randomized control trial data supporting this practice.11 Treatment with immunosuppressive agents such as corticosteroids has been reported to be successful in a small number of case reports,10,12 but is generally not advised due to the lack of supportive data and significant risk of sepsis and uncontrolled staphylococcal infection.5,6 The unique clinical course of our patient demonstrates that despite adverse prognostic indicators and prolonged dialysis dependence, complete recovery of renal function remains possible in SAGN with supportive care and successful eradication of the underlying infection without the administration of prolonged immunosuppressive therapy.
Conclusion
Staphylococcus-associated glomerulonephritis is a rare immune complex–mediated glomerulonephritis which can manifest with oliguric AKI and nephrotic-range proteinuria. With positive serum cryoglobulins, hypocomplementemia, abdominal pain, gastrointestinal bleeding, and cutaneous leukocytoclastic vasculitis, this patient’s clinical presentation was deceptively similar to that of HSP or cryoglobulinemic vasculitis. His epidemiological risk factors (eg, older age and diabetes mellitus), history of MSSA bacteremia and epidural abscess, and kidney biopsy findings were essential to establishing the correct diagnosis of SAGN. Although this patient had multiple negative prognostic indicators and more than 2 months of dialysis dependence, his remarkable clinical course demonstrates that full renal recovery is possible with supportive care and treatment of the underlying infection. Clinicians should consider this rare diagnosis in patients who develop RPGN in the setting of active Staphylococcus infection, perform a kidney biopsy if there is diagnostic uncertainty, and avoid immunosuppressive therapies due to the risk of uncontrolled Staphylococcus sepsis.
Footnotes
Ethics Approval and Consent to Participate: The patient provided informed written consent for publication of this case report and accompanying images. Institutional ethics board approval is not required for this type of study.
Consent for Publication: The patient and all contributing authors consented to publication of this case report.
Availability of Data and Materials: Not applicable.
Authors’ Note: These authors contributed equally to this work. Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Author Contributions: TM and RP performed the literature review and wrote the manuscript. LS provided pathology images and edited the manuscript. DN supervised the case report and edited the manuscript.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- 1. Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG., Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28(3):603-661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koyama A, Kobayashi M, Yamaguchi N. Glomerulonephritis associated with MRSA infection: a possible role of bacterial superantigen. Kidney Int. 1995;47(1):207-216. [DOI] [PubMed] [Google Scholar]
- 3. Cattran DC, Feehally J, Cook HT, et al. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO clinical practice guideline for glomerulonephritis. Kidney Inter Suppl. 2012;2:156-162. [Google Scholar]
- 4. Nadasdy T, Hebert LA. Infection-related glomerulonephritis: understanding mechanisms. Semin Nephrol. 2011;31(4):369-375. [DOI] [PubMed] [Google Scholar]
- 5. Glassock RJ, Alvarado A, Prosek J. Staphylococcus-related glomerulonephritis and poststreptococcal glomerulonephritis: why defining “post” is important in understanding and treating infection-related glomerulonephritis. Am J Kidney Dis. 2015;65(6):826-832. [DOI] [PubMed] [Google Scholar]
- 6. Hemminger J, Satoskar A. Staphylococcus infection-associated glomerulonephritis. In: Satoskar A, Nadasdy T. eds. Bacterial Infections and the Kidney. New York, NY: Springer; 2017:37-61. [Google Scholar]
- 7. Wang SY, Bu R, Zhang Q, et al. Clinical, pathological, and prognostic characteristics of glomerulonephritis related to staphylococcal infection. Medicine (Baltimore). 2016;95(15):e3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wehbe E, Salem C, Simon JF, Navaneethan SD, Pohl M. IgA-dominant Staphylococcus infection-associated glomerulonephritis: case reports and review of the literature. NDT Plus. 2011;4(3):181-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Satoskar AA, Nadasdy G, Plaza JA, et al. Staphylococcus infection-associated glomerulonephritis mimicking IgA nephropathy. Clin J Am Soc Nephrol. 2006;1(6):1179-1186. [DOI] [PubMed] [Google Scholar]
- 10. Satoskar AA, Molenda M, Scipio P, et al. Henoch-Schonlein purpura-like presentation in IgA-dominant Staphylococcus infection–associated glomerulonephritis—a diagnostic pitfall. Clin Nephrol. 2013;79(4):302-312. [DOI] [PubMed] [Google Scholar]
- 11. Greenhall G, Salama A. What is new in the management of glomerulonephritis? Clin Kidney J. 2015;8(2):143-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Okuyama S, Wakui H, Maki N, et al. (2008). Successful treatment of post-MRSA infection glomerulonephritis with steroid therapy. Clin Nephrol. 2008;70(4):344-347. [DOI] [PubMed] [Google Scholar]