

Summary
At the root of most fatal malignancies are aberrantly activated transcriptional networks that drive metastatic dissemination. Although individual metastasis-associated genes have been described, the complex regulatory networks presiding over the initiation and maintenance of metastatic tumours are still poorly understood. There is untapped value in identifying therapeutic targets that broadly govern coordinated transcriptional modules dictating metastatic progression. Here we reverse engineered and interrogated a breast cancer-specific transcriptional interaction network (interactome) to define transcriptional control structures causally responsible for regulating genetic programs underlying breast cancer metastasis in individual patients. Our analyses confirmed established pro-metastatic transcription factors and uncovered TRIM25 as a key regulator of metastasis-related transcriptional programs. Further, in vivo analyses established TRIM25 as a potent regulator of metastatic disease and poor survival outcome. Our findings suggest that identifying and targeting keystone proteins, like TRIM25, can effectively collapse transcriptional hierarchies necessary for metastasis formation, thus representing an innovative cancer intervention strategy.
Keywords: ARACNe, MARINa, VIPER, breast, cancer, metastasis, transcription, TRIM25, EFP
Graphical Abstract
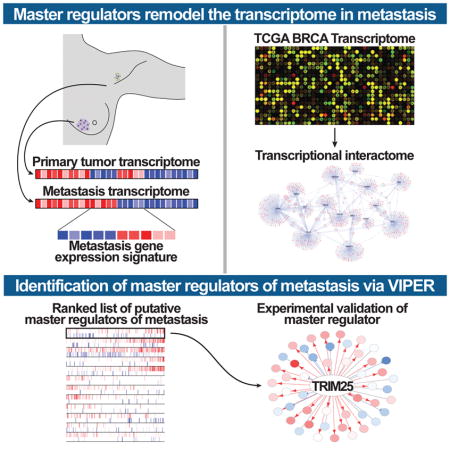
Introduction
Metastatic progression is the primary underlying cause of breast cancer-associated death (Spano et al., 2012). Although both functional and genomic studies have identified important genetic aberrations associated with risk of malignant progression (Navin, 2015), the fundamental drivers and the associated complex mechanisms of breast cancer metastasis remain poorly mapped and poorly understood. Specifically, several pathways have been identified as contributors to metastatic disease (Hanahan and Weinberg, 2011) but identification and dissection of the regulatory machinery that is necessary and sufficient to implement metastatic progression (metastasis checkpoints) remains a critically unanswered question in cancer research.
To systematically and quantitatively address this question, we approached metastatic progression as a transition between two cellular states defined by the differential gene expression signature of these states in the same patient, using patient-matched primary and metastatic samples. Then we investigated the specific transcriptional regulators responsible for initiating this transition – and ultimately for maintaining the stability of the metastatic state – based on their mechanistic ability to regulate differentially expressed genes (i.e. the transition signature). We have shown that the specific transcriptional regulators that determine such cancer-related state transitions can be efficiently and systematically elucidated by interrogating tumor-specific transcriptional networks (henceforth interactomes) with representative differential gene expression signatures, using the Virtual Inference of Protein activity by Regulon Enrichment analysis (VIPER) algorithm (Alvarez et al., 2016), which further extends the Master Regulator Inference algorithm (MARINa) (Lefebvre et al., 2010) to the analysis of single samples. These validated analyses have helped elucidate master regulator (MR) proteins whose concerted, aberrant activity is both necessary and sufficient for tumor phenotype implementation and maintenance, including tumor initiation (Aytes et al., 2014; Carro et al., 2010), progression (Aytes et al., 2014; Lefebvre et al., 2010), and drug resistance (Piovan et al., 2013; Rodriguez-Barrueco et al., 2015).
To implement this approach, we first assembled a breast cancer specific regulatory network, using the Algorithm for the Reconstruction of Accurate Cellular Networks (ARACNe) (Basso et al., 2005; Margolin et al., 2006b). Then, we identified differential gene expression signatures representing same-patient cell state transitions from primary tumors to lymph node metastases, using both ER-positive (ER+) and triple-negative (TNBC) breast cancer samples. Finally, we used the VIPER algorithm to prioritize transcriptional regulators that are the most likely causal determinant of these metastasis-related signatures. Using this strategy, we uncovered a hierarchy of transcriptional regulators controlling metastatic progression, revealing both previously unknown and well-established pro-metastatic transcription factors, thus confirming the potential value of the proposed methodology.
Results
Identifying keystone transcriptional regulators of breast cancer metastasis
In order to identify the transcriptional machinery driving breast cancer metastasis (Figure 1A), we first characterized the transcriptional signature representative of breast carcinoma metastatic progression (MET gene expression signature or MET-GES). This was achieved by differential expression analysis of patient-matched primary tumors and lymph node metastases from 20 ER+ and 11 TNBC breast cancer patients (Figure S1A and Table S1). Critically, two-tail gene set enrichment analysis (GSEA2) of these signatures revealed strong concordance across the two subtypes, suggesting the existence of common metastatic progression mechanisms (Figures S1B–S1D), independent of hormonal state.
Figure 1.

Transcriptome analysis of matched primary tumors and lymph-node metastasis in ER+ and TNBC. (A) Schematic regulators of the metastasis phenotype (MRs) in breast carcinoma, representing the strategy used to generate VIPER inferred candidate master regulators of the metastatic state. (B) VIPER inferred master master regulators for metastatic progression in breast carcinoma, ER+ tumors, and TNBC tumors. The genome wide expression signature is represented in the x-axis and by the color scale on top of the figures, where genes were rank sorted from the most downregulated in the metastasis (leftmost in cyan) to the ones upregulated (rightmost in red). Each transcription factor (TF) regulon is indicated by the red and blue vertical bars, which denote the genes whose expression is induced (red) or repressed (blue) by each TF according to the regulatory model. The enrichment of each regulon on the gene expression signature is indicated by the Normalized Enrichment Score (NES), whose color scale is indicated below the figures, and the associated p value. (C) Heatmap depicting the VIPER-inferred protein activity for the top 20 regulatory proteins selected by their consistent dysregulation in metastases when compared to matched primary tumors. (D) Regulatory relationship between TRIM25 and the putative target transcripts: blue: repression, red: induction, grey: undetermined. The color of the targets are proportional to their differential expression in the METs vs. the primary tumors, red: up-regulated, blue: down-regulated in the METs. See also Figure S1 and S2.
To identify the genes that causally implement the MET-GES – thus representing candidate causal determinants of breast carcinoma metastatic progression – we utilized the VIPER algorithm (Aytes et al., 2014; Carro et al., 2010). To assemble a breast carcinoma specific interactome, we analyzed 851 TCGA breast carcinoma gene expression profiles using the ARACNe algorithm (Basso et al., 2005; Margolin et al., 2006a). ARACNe is an information theory-based approach to infer mechanistic interactions between transcription factors (TFs) and target genes based on large sets of gene expression data, which has proven very effective in assembling interactomes for VIPER analysis (Alvarez et al., 2016; Basso et al., 2010; Carro et al., 2010; Kushwaha et al., 2015; Lefebvre et al., 2010). The ARACNe-inferred breast cancer interactome included 1,748 TFs, regulating 18,783 target genes through 365,634 transcriptional interactions. A schematic representation of our pipeline is represented in Figure 1A.
Finally, we inferred the regulatory proteins that are candidate drivers of the MET-GES by VIPER analysis of the breast carcinoma interactome. The algorithm prioritizes the regulatory proteins that are the most likely determinants of an observed differential expression signature – and thus of the associated cell state transition – by assessing the enrichment of their direct targets (regulons) in differentially expressed signature genes (i.e., in genes that are over or under expressed during metastatic progression, in this case). Thus, the ARACNe-inferred regulon of each regulatory protein (Table S2) is used as a highly multiplexed, endogenous reporter for its role in physically controlling metastatic progression. We used the statistical significance, estimated by sample permutation analysis, to rank-sort the list of putative master regulators (MRs) of the metastatic phenotype in ER+ breast cancer and TNBC (Table S3). Given the strong concordance between ER+ and TNBC MET-GES, and between candidate MRs of the two subtypes (Figures S1B–S1D), we inferred the MRs of metastatic progression of breast carcinoma regardless of subtype by VIPER analysis of a subtype-agnostic MET-GES. This generated a single ranked list of breast cancer metastatic progression candidate MRs (BRCA), independent of hormonal status (Figure 1B and Table S3). From these analyses, TRIM25 emerged by far as the strongest candidate MR (p= 3.25 × 10−5), A schematic summary of these data is shown in Figure S1E. Of the top candidate MRs, most are largely unexplored metastasis-associated genes, ranking significantly higher than well-known pro-metastatic TFs such as c-MYC (#585), ETV4 (#1443) and SMAD4 (#1447). Nevertheless, the high ranking of breast cancer-specific metastasis associated TFs such as PRRX-1 (#6, p = 6.8 × 10−4) further corroborates the accuracy of our approach. Importantly, top candidate MR genes were not highly differentially expressed following metastatic progression. In fact, none of the top 15 candidate MR genes were in the top 100 most differentially expressed genes, and therefore, could not have been identified without direct analysis of their regulatory context. This further emphasizes the distinct advantage of the proposed methodology. As previously shown (Chen et al., 2014), regulatory proteins with the highest probability to directly regulate the signature of interest are more likely to be critically necessary to implement the associated cell state transition because they represent the most proximal integrators (i.e., regulatory bottlenecks) of genetic and epigenetic determinants of the associated phenotype. This suggests that TRIM25 may be a more universal determinant of metastatic progression in breast cancer as compared to previously identified genes that appear further down in the prioritized MR list.
We based the analysis on an average gene expression signature of metastatic progression across all ER+ and TNBC METs. Thus, while it is effective in identifying mechanisms that are common across most of the patients, it fails to capture mechanisms that may be patient-specific. We thus extended our analysis at the single patient level by inferring the transcriptional regulators driving each patient’s metastatic progression with the VIPER algorithm (Alvarez et al., 2016). This analysis also identified TRIM25 as the most consistently activated regulator in these metastases (Figure 1C), further supporting its role as a key regulator of the metastatic progression in both ER+ and TNBC tumors.
TRIM25 is a global regulator of breast cancer metastasis signature
To functionally test these hypotheses, we performed siRNA-mediated knock-down for a selected set of 14 MRs in 2 ER+ (MCF7 and T-47D) and 2 TNBC (BT549 and MDA-MB-231) cell lines and analyzed subsequent changes in gene expression using RNA-seq. The MR genes selected for validation represent the top most differentially activated TFs in each of the patient-derived signatures, including the breast carcinoma BRCA MET-GES, the ER+ MET-GES and the TNBC MET-GES (Figures 1B, 1C, S2A, and Table S3). MRs were prioritized by stepwise linear regression with exhaustive selection to first identify the best combination of MRs controlling the metastatic progression signature and then to compute the fraction of metastasis signature genes regulated by each MR, using GSEA leading-edge analysis (Subramanian et al., 2005). The best linear combination of n MRs, where n is the highest number of MRs for which all coefficients of the model were different than zero at Bonferroni’s corrected p< 0.001, was selected by maximizing the variance explained by the model (Figures S2B, S2C and Table S5). Employing a complementary approach, we repeated this analysis by separately comparing the expression between metastasis and primary tumor for each individual patient. Stepwise linear regression with exhaustive search was applied to each gene expression signature, and then integrated by the Stouffers’s method. Results were very similar using either method (Table S5).
The identification of established metastasis-associated genes – such as PRRX-1, FOXQ1, and PAX5 (O’Brien et al., 2011; Ocana et al., 2012; Ratkaj et al., 2010; Zhang et al., 2011) – demonstrates the robustness of our methodology toward elucidating metastatic progression drivers. Based on these analyses, TRIM25 emerged as the strongest (i.e., most statistically significant) MR candidate using both TNBC cell lines and patient breast tumor derived signatures (Figures S2B and S2C). TRIM25 also significantly regulated the ER+ MET-GES in 1 ER+ cell line (Table S5). Critically, siRNA-mediated silencing of TRIM25 strongly affected 50% of the top 20 VIPER inferred candidate MRs (Table S4), demonstrating that TRIM25 can coordinate multiple downstream transcriptional programs to implement the metastatic progression transcriptional signature. The predicted regulatory relationship between TRIM25 and target transcripts is shown in Figure 1D.
TRIM25 expression is not correlated with estrogen signaling
TRIM25 was originally named, ‘estrogen responsive finger protein’ (EFP), and has since been described as an estrogen-responsive gene and a co-factor for estrogen signaling. Therefore, the identification of TRIM25 as the most statistically significant candidate metastatic progression MR in TNBC was surprising. Yet, when we compared TRIM25 expression in ER+ and TNBC tumors from TCGA, it was actually significantly higher in the latter (Figure S2D). Consistent with these findings, we did not observe estrogen-dependent expression of TRIM25 in multiple high-throughput studies that test the effects of estrogen on gene expression (Carroll et al., 2006; Creighton et al., 2006; Kamalakaran et al., 2005; Lin et al., 2004). To further investigate the relationship between TRIM25 and estrogen signaling, we generated a Spearman’s correlation matrix using RNA-seq expression data from 1100 breast cancer tumors, comparing TRIM25 expression with 10 established estrogen-responsive genes (Figure S2E). As expected, Estrogen receptor 1 (ESR1) expression correlated strongly with all ten estrogen-responsive genes. Yet, despite being expressed in ER+ tumors, expression of TRIM25 was not correlated with the expression of ESR1 or of other estrogen responsive genes. For comparison, we also included the Y box binding protein 1 (YBX1) in these analyses, a gene specifically associated with estrogen-negative tumors. While the analysis revealed significant negative correlation between YBX1 expression and the expression of estrogen responsive genes, as well as ESR1, no correlation was detected with TRIM25 expression. Taken together, these results suggest that TRIM25 expression can be regulated by factors independent of estrogen or estrogen signaling, especially in TNBCs.
TRIM25 mediates breast cancer metastasis in human xenograft models
To test the effect of TRIM25 on the metastatic potential of TNBC cells, we engineered human MDA-MB-231 cells, derived from a TNBC tumor, to express either ‘high’ (TRIM25high) or ‘low’ (TRIM25low) levels of TRIM25 (Figure S3A), and labelled them with Luc-GFP or Luc-RFP. Distinctly labeled TRIM25high and TRIM25low cells were mixed 1:1, injected into the lateral tail vein of athymic nude mice, and monitored for the development of lung metastases via bioluminescence imaging. When lung metastases were visible, mice were sacrificed and their lungs were first imaged via ex vivo fluorescence imaging and spectral unmixing for a global picture of the relative abundance of TRIM25high vs. TRIM25low cells (Figure 2A). Lungs were then sectioned and percent content of TRIM25high vs. TRIM25low cells was accurately quantified via immunofluorescence (Figure 2B and 2C). We found that TRIM25high cells constituted the overwhelming majority of metastatic burden in the lungs compared to TRIM25low cells (Figures 2A–2C). We confirmed these results in parallel experiments, in which TRIM25high and TRIM25low fluorophores were switched.
Figure 2.

TRIM25 affects the metastatic potential of TNBC cells. (A) Representative images of lungs isolated from athymic nude mice injected in the lateral tail vein with MDA-MB-231 cells infected with indicated constructs. Representative images of IVIS ex vivo fluorescence imaging with spectral unmixing (left, scale bar represents 5mm) and immunofluorescence (right, scale bar represents 100μm). (B) Quantification of (A) for each mouse separately. Numbers (1–5) denote different mice, A and B denote 1 left or 1 right lung lobe respectively. (C) Quantification of (A) by group. Data are represented as box and whiskers plot (min to max, centred value at median), n represents 8–10 biological replicates as indicated; p value represents pairwise t-test (two-tailed) for red vs. green. (D) Representative image and quantification of primary tumors isolated from NOD scid gamma mice injected in the mammary fat pad with MDA-MB-231 cells infected with indicated constructs. Scale bar represents 1mm. (E) Quantification of the lungs from (D) for each mouse separately. Numbers 1–5 denote different mice, A and B denote 1 left or 1 right lung lobe respectively. (F) Quantification of the lungs from (D) by group. Data are represented as box and whiskers plot (min to max, centred value at the median), n represents 8–10 biological replicates as indicated; p value represents pairwise t-test (two-tailed) for red vs. green. (G–H) Quantification and representative images of lungs from athymic nude mice injected in the lateral tail vein with CAL-51 cells infected with indicated constructs. Data are mean ± s.e.m., n=5 mice per group. Scale bar represents 50μm. See also Figure S3.
Intriguingly, increased expression of TRIM25 did not affect proliferation or invasion of MDA-MB-231 cells in vitro (Figures S3B and S3C). We then used a primary culture generated from a patient-derived xenograft (PDX) model of TNBC to test the effect of TRIM25 perturbation on primary tumor growth in vivo. Consistent with the in vitro assays, we found that neither TRIM25 knockdown nor overexpression altered primary tumor growth in the mammary fat pad of NOD scid gamma (NSG) mice (Figures S3D–S3F). Similarly, when we injected the aforementioned fluorescently labelled TRIM25high:TRIM25low cell mixture into the mammary fat pad of NSG mice, we found that TRIM25 did not affect cellular proliferation at the orthotopic site, with TRIM25high and TRIM25low cells showing uniform representation in primary tumors > 1cm in diameter (Figure 2D). Notably, when these mice became symptomatic and lungs were harvested, we found that TRIM25high cells metastasized to the lung more efficiently than TRIM25low cells, despite representing a similar fraction in corresponding primary tumors (Figures 2E and 2F). Interestingly, perturbation of TRIM25 expression in MDA-MB-231 cells injected into the mammary fat pad of NSG mice in limiting dilution experiments revealed that TRIM25 knock-down resulted in decreased seeding potential, suggesting TRIM25 may be involved in promoting self-renewal (a stem-like phenotype) (Figure S3G). We further explored the relationship between TRIM25 and metastasis in an additional TNBC cell line CAL-51. TRIM25 knock-down in CAL-51 cells injected into the lateral tail vein of athymic nude mice resulted in significantly decreased lung metastases (Figures 2G and S3H). Lungs were stained with anti-human COX4 to clearly differentiate human CAL51 metastases from mouse lung tissue (Figure 2G). Overexpression of TRIM25 in CAL51 cells resulted in a ~2fold increase in lung metastases (p=0.10; Figures 2H and S3H). This increase was not due to an increase in extravasation (Figure S3I). Together these data demonstrate that TRIM25 is a potent regulator of TNBC metastasis.
The E3 ubiquitin ligase activity of TRIM25 in promoting metastasis
In order to determine whether the E3 ubiquitin ligase domain of TRIM25 was required for TRIM25 to promote metastasis, first we employed a clustered regularly interspaced short palindromic repeats (CRISPR) strategy to knock-out TRIM25 (TKO) in 2 TNBC cell lines (Figures 3A and S4A), then we expressed either full-length TRIM25 (TRIM25) or a RING-deficient TRIM25 (TRIM25ΔR) in these cells (Figures 3B and 3C). The lack of E3 ubiquitin ligase activity was confirmed in TRIM25ΔR cells (Figure S4B). Tail vein injected CAL-51-TKO cells expressing either TRIM25 or TRIM25ΔR rescued the metastasis phenotype to a similar extent, suggesting that the E3 ubiquitin ligase activity of TRIM25 is not required (at least entirely) for promoting metastasis in this context (Figure 3D). In contrast, orthotopic injection of BT549-TKO cells expressing TRIM25ΔR only partially rescued the seeding potential of TNBCs that express full length TRIM25 (Figure 3E) suggesting that the E3 ubiquitin ligase domain of TRIM25 may, in part, contribute to tumorigenicity and therefore metastatic potential. Interestingly, the expression of TRIM25 or TRIM25ΔR does not alter the localization of TRIM25 in TNBCs via immunofluorescence (Figure S4C). Consistent with the role of TRIM25 in promoting metastasis, expression of TRIM25 or TRIM25ΔR (Figure S4D) in an already highly metastatic TNBC line did not significantly affect primary tumor growth or spontaneous lung metastasis (Figures 3F and 3G). Further, injection of these cells into the lateral tail vein of nude mice revealed no differences in metastatic seeding in the lung (Figure 3H). Together these data suggest TRIM25 promotes metastasis through multiple mechanisms, with at least one being independent of its E3 ligase activity.
Figure 3.

E3 ubiquitin ligase activity of TRIM25 in TNBC cell metastasis. (A) CRISPR mediated knock-out of TRIM25 in BT549 and CAL-51 TNBC cells. Gene schematic indicates sites targeted by sgRNAs as well as sequencing results from a single cell clone of either BT549 or CAL-51 (B) Immunoblot of BT549-TKO cells infected with indicated constructs. (C) Immunoblot of CAL-51-TKO cells infected with indicated constructs. (D) Quantification of metastatic burden in the lungs of NSG mice injected with CAL-51-TKO cells infected with indicated constructs. Representative images of lungs stained with anti-human COX4 are shown. n=5 mice per group. Scale bar represents 50μm. (E) Tumorigenicity of BT549-TKO cells infected with indicated constructs and injected into the mammary fat pad of NSG mice. Representative bioluminescent images are show. Number of injections and tumor take is indicated. (F) Average tumor volume of MDA-MB-231 cells infected with indicated constructs and injected into the mammary fat pad of nude mice. n=5 mice per group. (G) Spontaneous lung metastasis of MDA-MB-231 cells infected with indicated constructs and injected into the mammary fat pad of athymic nude mice. Representative images of lungs are shown, n=5 mice per group. Scale bar represents 50μm. (H) In vivo lung metastasis of MDA-MB-231 cells infected with indicated constructs injected into the mammary fat pad of athymic nude mice. Representative images of lungs are shown, n=5 mice per group. Scale bar represents 50μm. See also Figure S4.
TRIM25-mediated transcriptional regulation of the MET-GES
Since TRIM25 contains a hallmark zinc finger B-box DNA binding domain (Figure S5A), we hypothesized that it regulates the BRCA metastatic signature, at least in part, as a bona fide transcription factor. Cell fractionation of ER+ breast cancer and TNBC cells revealed that TRIM25 is present in the cytoplasmic, nuclear and chromatin fractions (Figures 4A and S5B). Immunofluorescence revealed TRIM25 was most abundantly localized to the perinuclear region (Figure 4B). To investigate the DNA binding properties of TRIM25, we performed chromatin immunoprecipitation sequencing (ChIP-seq) in the TNBC cell lines BT549 and MDA-MB-231. TRIM25 ChIP antibody was validated according to ENCODE guidelines (Landt et al., 2012) and a representative IP is shown in Figure S5C. The distribution of ChIP-seq peaks (genome ontology) is shown in Figures 4C and 4D. Interestingly, TRIM25 primarily associated with DNA at CpG islands and gene promoters. Several ChIP targets were selected for validation by qPCR (Figure S5D). When we examined the metastasis signature genes by plotting TRIM25 ChIP-seq reads (-input) for the top 876 genes (p<0.05) from the TNBC MET-GES, we observed that TRIM25 mainly localizes to the transcriptional start site (TSS) of these genes (Figures 4E–4H).
Figure 4.

TRIM25-mediated transcriptional regulation of the MET-GES. (A) Immunoblot for TRIM25 in MDA-MD-231 cells that have been fractionated into cytoplasmic, nuclear and chromatin fractions. HSP90, SP1 and H3K9me3 were used as control of fractionation efficiency. (B) Immunofluoresence of TRIM25 in MDA-MB-231 cells. Scale bar represents 10μm. (C–D) Distribution of TRIM25 ChIP-seq peaks in BT549 (C) and MDA-MB-231 (D) cells. Bars show the ratio enrichment of TRIM25 ChIP reads over input mapping to each type of genomic location. (E–H) Alignment plots of TRIM25 ChIP-seq reads (minus input) from BT549 (E–F) and MDA-MB-231 (G–H) cells compared to 876 genes from MET-GES. (I) iGSEA analysis of ranked TRIM25 ChIP-seq TSS peaks from BT549 and MDA-MB-231 cells for enrichment on the siTRIM25 gene expression signature for each matching cell line. (J–K) GSEA of top (iGSEA infered) TRIM25 ChIP-seq TSS peaks in BT549 (J) and MDA-MB-231 (K) cells for enrichment on siTRIM25 gene expression signature. (L–M) GSEA of top (iGSEA infered) TRIM25 ChIP-seq TSS peaks in BT549 (L) and MDA-MB-231 (M) cells for enrichment on MET-GES. See also Figure S5.
To further refine the identity of genes transcriptionally regulated by TRIM25, we performed iterative GSEA analysis (iGSEA) while incrementally increasing the number of top TRIM25-bound genes, based on TSS peak scores, compared to the cell line data in which TRIM25 was silenced (GES) as negative control. The TRIM25-bound gene set was determined as the smallest set for which a maximum GSEA enrichment score could be obtained (Figure 4I). These TRIM25-bound genes were significantly enriched in genes that were upregulated following siRNA mediated silencing of TRIM25 (siTRIM25-GES) both in BT549 and in MDA-MB-231 cells. This was consistent with TRIM25 functioning, at least in part, as a transcriptional repressor (Figures 4J and 4K). Specific overlap is shown in Figures S5E, S5F and Table S4. Further, iGSEA inferred TRIM25-bound genes were significantly enriched among the downregulated MET-GES genes for both BT549 and MDA-MB-231 cells, again suggesting that TRIM25 may function as a transcriptional repressor to regulate the MET-GES (Figures 4L and 4M). Specific overlap is represented in Figures S5G, S5H and Table S4.
TRIM25 mediates MET-GES regulation at the post-transcriptional level
TRIM25 is also an established RNA-binding protein (Choudhury et al., 2014). Therefore, we assessed whether TRIM25 physically interacts with the mRNA of metastasis-associated genes, and whether this binding was associated with their expression levels. To test this hypothesis, we performed RNA immunoprecipitation sequencing (RIP-seq) in BT549 and MDA-MB-231 cells to identify TRIM25-bound RNA species, which were scored and ranked (see methods) (Table S4). We randomly chose 3 positive and 3 negative RIP-seq target genes to confirm by qRT-PCR (Figure S6A). Following iGSEA analysis (Figure 5A), as described in the previous section, we detected a strong enrichment of TRIM25-bound RNAs among genes that were elevated upon knockdown of TRIM25 in both MDA-MB-231 and BT549 cells (Figures 5B and 5C), suggesting that TRIM25 negatively regulates the abundance of these transcripts. The specific overlap is represented in Figures S6B and S6C. Similarly, we found enrichment of the top iGSEA inferred TRIM25-bound RNAs among genes that were reduced in the MET-GES, again suggesting that TRIM25 may negatively regulate the abundance of these MET-GES genes (Figures 5D and 5E). Specific overlap is represented in Figures S6D, S6E and Table S4.
Figure 5.

TRIM25-mediated post-transcriptional regulation of the MET-GES. (A) iGSEA analysis of ranked TRIM25 RIP-seq TSS peaks from BT549 and MDA-MB-231 cells for enrichment on siTRIM25 gene expression signature in matching cell lines. (B–C) GSEA of TRIM25 RIP-seq peaks in BT549 (B) and MDA-MB-231 (C) cells for enrichment on siTRIM25 gene expression signature in matching cell lines. (D–E) GSEA of the top TRIM25 bound genes (mRNA) in BT549 (D) and MDA-MB-231 (E) cells for enrichment on MET-GES. (F) Venn diagram illustrating the overlap between the inferred TRIM25 transcriptionally and post-transcriptionally regulated genes. (G) Overlap between MSigDB pathways associated with TRIM25 transcriptionally regulated genes and post-transcriptionally regulated genes (FDR p value<0.05). Gene sets for each biological process (centre ovals) are shown. See also Figure S6.
TRIM25 regulates the MET-GES through transcriptional and post-transcriptional mechanisms
Next, we proceeded to examine the relationship between transcriptional and post-transcriptional regulation of gene expression, as mediated by TRIM25. First, to identify the core set of genes transcriptionally regulated by TRIM25 in TNBC, we integrated the TRIM25 ChIP-seq results with gene expression changes resulting from TRIM25 knockdown in cell lines. While the former indicates binding of TRIM25 to promoter regions, the later provides clues about causal regulatory effects on transcription. Confirming our analysis and the ARACNe-inferred regulon of TRIM25, these core genes were significantly enriched in genes downregulated in the MET-GES (Figure S6F). In parallel, we identified a core set of genes post-transcriptionally regulated by TRIM25 in TNBC by integrating the TRIM25 RIP-seq results with gene expression profiles following siRNA mediated silencing of TRIM25 in TNBC cell lines. This second core gene set, which is post-transcriptionally regulated by TRIM25, was also significantly enriched in genes downregulated in the MET-GES (Figure S6G). Remarkably, we also found significant overlap between the transcriptional and post-transcriptional TRIM25-regulated core gene-sets (Figure 5F), suggesting that TRIM25 regulates the expression of these genes at both transcriptional and post-transcriptional levels, thus implementing a classic feed-forward loop structure.
To gain insight into the potentially distinct biological processes mediated by TRIM25 transcriptionally vs. post-transcriptionally, we performed pathway enrichment analysis using KEGG, Biocarta and Reactome pathways from the MSigDB database (Subramanian et al., 2005), on these core gene sets. We found a striking overlap between processes regulated by TRIM25 transcriptionally and post-transcriptionally (Figure 5G and Table S6), suggesting that TRIM25 may control these biological processes through tight transcriptional and post-transcriptional mechanisms.
TRIM25 promotes tumorigenicity and a stemness phenotype
To assess which cellular processes associated with metastasis are modulated by TRIM25, we first inferred the highest confidence MET-GES genes transcriptionally and post-transcriptionally regulated by TRIM25 as the genes in the leading-edge when computing the enrichment of the core set of genes transcriptionally or post-transcriptionally regulated by TRIM25 on the MET-GES (Figure S7A). Next, we performed Gene Ontology (GO) analysis on these gene sets. Enriched biological processes were dominated by developmental processes (Figure 6A), suggesting that TRIM25 may mediate metastases by affecting differentiation.
Figure 6.

TRIM25 regulates the MET-GES and modulates tumorigenicity and stemness. (A) GO analysis of core MET-GES genes transcriptionally or post-transcriptionally regulated by TRIM25. (B) Bioluminescence imaging and tumor take rate 100 days after orthotopic injection of BT549 cells infected with indicated shRNA into the mammary fat pad of NSG mice; n=4–8 mice as indicated. (C) GSEA for oncogenic signatures associated with differentially expressed genes from RNA-seq after TRIM25 knockdown in BT549 and MDA-MB-231 cells. Normalized enrichment score of the ESC_J1_Up_Late.V1_Up signature (Left). (Right) representative gene expression changes from this signature. (D) Schematic representation of the 7/9 pluripotency factors and the location of their respective binding sites (number of base pairs from the transcriptional start site) at the TRIM25 promoter. (E) Representative images of ESCs infected with indicated treatments are shown. Scale bar represents 500μm. (F) qRT-PCR quantification of differentiation marker genes in ESCs infected with TRIM25 shRNA compared to scramble control. Data represents mean ± s.e.m., n=4. (G–H) qRT-PCR quantification of NANOG, POU5F1, and SOX2 in (G) BT549-TKO and (H) CAL-51-TKO cells infected with indicated constructs. Data represents mean ± s.e.m. n=3. See also Figure S7
Next we tested whether altering more than one master regulator, that together more broadly cover the MET-GES, is sufficient to enhance tumor implantation in an otherwise poorly tumorigenic breast cancer cell line, BT549. We found that modulation of TRIM25 and zinc finger protein 581 (ZNF581) in combination yielded the most substantial coverage of the MET GES (61%) compared to all other pairs of candidates (Figures S2B, S2C and Table S5). We therefore overexpressed TRIM25 and knocked down ZNF581 in BT549 cells (Figure S7B) and performed a limiting dilution assay in the mammary fat pads of NSG mice. Concomitant TRIM25 overexpression and ZNF581 knockdown significantly enhanced tumorigenicity (Figure 6B) without affecting proliferation in vitro (Figure S7C), indicative of an expanded tumor initiating cell population with this combined genetic alteration. These data suggest that broad coverage of our metastasis gene expression signature could perpetuate the metastasis phenotype.
To gain further insight into the specific effect of TRIM25 on the phenotype of TNBC cells, we characterized the global transcriptional changes downstream of TRIM25. GSEA was performed on genes identified by RNA-seq to be differentially expressed upon knockdown of TRIM25 in BT549 and MDA-MB-231 cells. Of the 189 MSigDB oncogenic gene signatures tested, we found that the strongest enrichment was a signature related to differentiation of embryonic stem cells (ESCs), suggesting that TRIM25 may be involved in the maintenance of stemness (Figure 6C and Table S7). This is particularly interesting as TRIM25 was previously found to be the target of 7 of 9 pluripotency factors in ESCs (Figure 6D) (Kim et al., 2008) and TRIM25 expression is significantly decreased during embryonic differentiation (Kwon et al., 2013).
We functionally tested this correlative relationship using human ESCs (hESCs). To determine whether loss of TRIM25 could induce differentiation, we lentivirally expressed short hairpins against TRIM25. Strikingly, TRIM25 depletion induced changes in hESC morphology consistent with differentiation (Figure 6E). qRT-PCR of RNA isolated 4 days post-transfection revealed a considerable increase in mesoderm, endoderm and ectoderm marker genes after TRIM25 knockdown, suggesting that TRIM25 is required to maintain a stem cell state (Figure 6F). Recently, Akrap and colleagues used single-cell RNA-sequencing to identify markers of functionally enriched stem and progenitor cells in breast cancer. In both primary breast tumors and TNBC cell lines POU5F1, NANOG and SOX2 were major determinants of a quiescent CSC-like cell phenotype with high-stemness and tumorgenicity (Akrap et al., 2016). Using our CRISPR TRIM25 knockout cell models we found that expression of either TRIM25 or TRIM25ΔR in TNBC-TKO cells increased the expression of POU5F1, NANOG and SOX2 (Figures 6G and 6H) suggesting that TRIM25 regulates the core genes involved in maintaining quiescent CSC-like phenotypes. Together, these results suggest TRIM25 promotes metastasis through increasing the stemness and tumorigenicity of breast cancer cells, enhancing their capacity for colonization, survival, and outgrowth at the secondary site.
Across 32 cancer types from TCGA, TRIM25 is rarely deleted and is most frequently amplified in breast cancer (Figures 7A and 7B). TRIM25 amplification is associated with a significant increase in mRNA expression (Figure 7C). Kaplan-Meier log-rank test using RNA-seq data consisting of 1038 breast cancer tumors from TCGA revealed that elevated TRIM25 expression is significantly associated with poor overall survival (Figure 7D). Further, PREdiction of Clinical Outcomes from Genomic profiles (PRECOG) analysis of TRIM25 confirmed that TRIM25 expression is significantly associated with poor outcome in breast cancer (Figure 7E). In fact, TRIM25 was most strongly associated with poor outcome in breast cancer over any other cancer type. There were no significant differences based on TRIM25 expression or predicted protein activity in normal breast tissue compared to primary tumors (Figures 7F and 7G). However, there was a significant difference in the activity of TRIM25 protein between primary tumors and metastases (Figure 7H). In addition, although the TCGA dataset only contains 7 matched pairs of primary tumors and metastases, 6/7 metastases had higher TRIM25 expression than their respective primary tumors (Figure 7I). TRIM25 immunohistochemistry in primary breast tumors, lymph-node and distant metastases revealed similar TRIM25 localization suggesting localization of TRIM25 is not overtly altered during metastatic progression (Figure 7J). Together, these data supports a role of TRIM25 as a driver of poor outcome in breast cancer.
Figure 7.

TRIM25 is frequently amplified in breast cancer and is associated with poor overall survival. (A) Frequency of TRIM25 copy number alterations (CNA) in 32 cancer types from the TCGA. CS, cell carcinoma; pRCC, papillary renal cell carcinoma; PCPG, pheochromocytoma and paraganglioma; ccRCC, clear cell renal cell carcinoma; GBM, glioblastoma multiforme; AML, acute myleiod leukemia; chRCC, chromophobe renal cell carcinoma; DLBC, diffuse large B-cell lymphoma; ACC, adenoid cystic carcinoma. (B) Mutation frequency of TRIM25 in 32 cancer types from the TCGA. Cancers not represented on graph have no known mutations. (C) TRIM25 expression in TRIM25 diploid vs.TRIM25 amplified tumors (TCGA). (D) Kaplan-Meier survival analysis based on TRIM25 expression (RNA-seq v2 RSEM) from 1,038 breast cancer patients from TCGA. TRIM25 ‘High’ is defined by a z-score>1, TRIM25 ‘Low’ is all other patients. HR, hazard ratio. (E) PREdiction of Clinical Outcomes from Genomic profiles (PRECOG) analysis of TRIM25. (F) TRIM25 expression in breast tumors vs normal breast tissue. (G) Predictive TRIM25 relative activity in breast tumors vs normal breast tissue. (H) Predictive TRIM25 relative activity in primary breast cancer tumors vs. metastases. (I) TRIM25 expression in primary breast cancer tumors vs. matched metastases (TCGA IDs indicated). (J) Immunohistochemistry of TRIM25 in primary breast tumor, lymph-node and distant (bone) metastases. Scale bar represents 50μm.
Discussion
Context-specific regulatory networks, inferred by unbiased reverse engineering algorithms, provide a framework for identifying the proteins causally associated with specific phenotypic states (Lefebvre et al., 2010). Here we reverse engineered a breast cancer-specific regulatory network to uncover a transcriptional hierarchy driving breast cancer metastasis. Importantly, we discovered that TRIM25, multifunctional transcription factor, lies at the epicentre of this hierarchy. Altered TRIM25 activity causes coordinated broad changes in the metastatic program, affecting the expression of many metastasis effectors simultaneously, and promoting the metastatic phenotype.
Multifunctional proteins such as TRIM25 challenge a conventional ‘one protein–one function’ paradigm. Many transcription factors that bind double stranded DNA with sequence specificity also bind RNA (Cassiday and Maher, 2002). Similarly, several RNA-binding E3 ubiquitin ligases have recently been discovered to direct cellular regulatory pathways by affecting both protein and mRNA stability (Cano et al., 2010). Interestingly, several of these RNA-binding E3 ubiquitin ligases contain a B-box zinc finger DNA-binding domain analogous to TRIM25. Here, we demonstrate that TRIM25 has the potential to control cellular phenotypes by interacting with DNA in addition to RNA (Streich et al., 2013) targets. Moreover, our data show that TRIM25 represses many targets at both levels, representing a transcriptional/post-transcriptional feed-forward topology (Lefebvre et al., 2010; Mangan and Alon, 2003). This complex regulatory architecture and diversified functionality has not yet been described for TRIM25.
Although we focused our studies on the role of TRIM25 in TNBC, VIPER analysis identified this regulator protein as the most statistically significant metastatic progression MR both in TNBC and in ER+ tumors, suggesting that it may play an equally relevant role in ER+ breast cancer as well. Indeed, it has been proposed that TRIM25 may perpetuate the estrogen signaling cascade in the absence of estrogen receptor, resulting in the evolution of estrogen-independence and resistance to hormone therapy (Nalepa and Harper, 2002). A similar phenomenon was recently observed in prostate cancer whereby the glucocorticoid receptor (GR) substitutes for the androgen receptor (AR) resulting in resistance to anti-androgen therapy (Arora et al., 2013). However, here we show data supporting an estrogen-independent role of TRIM25. Further, in a parallel and ongoing study, we did not find TRIM25 as differentially expressed or differentially active TFs in letrozole-resistant breast cancer patients (data not shown). Therefore, the phenotypes observed in studies that link TRIM25 with tumorigenicity and survival of estrogen-dependent cells in the absence of estrogen (Urano et al., 2002), may have been the result of TRIM25 promoting the stemness and tumor-initiating phenotypes described here, thus suggesting that its role in metastatic progression may be fully independent of hormonal status.
The dynamic nature of metastasizing cancer cells cannot simply be explained by the accumulation of progressive genetic changes in the primary tumor (Monteiro and Fodde, 2010). The capacity to resist cytotoxic or targeted therapy, to undergo multiple profound phenotypic changes, to overcome the challenges of foreign microenvironments, and to maintain the ability to eventually recapitulate the heterogeneous composition of the primary tumor at distant organ sites is not typical behavior for a differentiated epithelial cell. Rather, this is likely achieved by stem-like cells that maintain active DNA repair, express a variety of drug transporters, resist apoptosis and pathotropism, and have highly dynamic cellular plasticity that supports multiple hallmarks of cancer (Lawson et al., 2015). Our data suggests that collapsing transcriptional hierarchies that regulate the balance between differentiation and self-renewal, by targeting keystone proteins like TRIM25, is critical to affect the coordinated transcriptomic reprogramming that is required for metastasis, and may represent promising future targets for cancer intervention.
Experimental Procedures
Primary human breast cancer and matched lymph node metastases
Paired primary tumor and lymph node metastases were collected during surgical resection. All tissue was used with informed consent and banked at Memorial Sloan-Kettering Cancer Center (MSKCC) in accordance with IRB approval. All samples were snap-frozen and stored at −80 °C until use. All samples were H&E stained and independently reviewed by a breast cancer pathologist. Tumors were micro-dissected to obtain >70% purity. RNA was isolated from fresh frozen samples using the RNeasy Plus mini prep kit (Qiagen, Valencia, CA, USA). Nucleic acid quality was determined with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Primary tumor and matched metastasis pairs, for which RNA of sufficient quantity and quality was available, were analyzed on Affymetrix GeneChip Human Genome U133 2.0 Array (Affymetrix, Santa Clara, CA, USA) at the genomics core at Memorial Sloan Kettering Center for Molecular Oncology (Manhattan, NY, USA) according to the manufacturer’s protocol.
Animal studies
All animal work was done in accordance with the Institutional Animal Care and Use Committee at MSKCC. Athymic Nu/Nu mice were obtained from Harlan Laboratories (Indianapolis, IN, USA) and NOD-scid IL2Rgnull (NSG) mice were obtained from Jackson Laboratories and (Bar Harbor, ME, USA) and randomized. MDA-MB-231 cells were first infected with ‘GREEN’ pHIV-Luc-ZsGreen (Addgene, Cambridge, MA, USA) or ‘RED’ pHIV-Luc-dsRED vectors. Cells were then infected with indicated constructs (Figure S3A). All animals were injected between 5 and 7 weeks of age. Single-cell suspensions of the indicated cells were mixed 1:1 with serum-free media for intravenous injections or 1:1 with Matrigel (BD Biosciences, Franklin Lakes, NJ, USA) for orthotopic injections. TRIM25high (TRIM25 overexpression) and TRIM25low (TRIM25 knock-down) cells with opposing fluorescent labels were mixed 1:1 and injected (5×105 cells) into the lateral tail vein of athymic nude mice or 1×10^6 cells into the mammary fat pad of NSG mice. The development primary tumors or lung metastases was monitored using bioluminescence imaging BLI (IVIS, Xenogen, Alameda, CA, USA). When lung metastases were visible, mice were sacrificed and their lungs were imaged via ex vivo fluorescence imaging and spectral unmixing. Lungs were then sectioned and stained with anti-RFP antibody (Abcam, Cambridge, MA, USA). Lung tissues were blindly scanned using a Carl Zeiss Axioimager Z1 microscope (Zeiss, Oberkochen, Germany) equipped with a Tissue Gnostics slide scanning platform. TissueQuest analysis software was used for automated quantification of RFP+ and GFP+ tumor cells per total number of DAPI+ cells within a full cross section of the primary tumor. For lung tissue analysis, two lobes per animal (one left and one right) were used for red:green ratio calculations. Two-tailed pairwise t-test was used to statistically compare relative red vs. green. Animals that were ill or died due to circumstances unrelated to the experiment were excluded from the study.
CAL-51 cells (1×106) were injected into the lateral tail vein of 4-6week old athymic nude mice. Lungs were harvested 8 weeks post injection. CAL-51 TKO cells (1×106) were injected into the lateral tail vein of 4–6week old NSG mice. Lungs were harvested 8 weeks post injection. BT549-TKO (2×106) cells were injected into the mammary fat pad of 4–6week old NSG mice and monitored to tumor take by bioluminescence imaging (IVIS). MDA-MB-231 cells (EV, TRIM25, ΔTRIM25 − 1x 106) were injected into the mammary fat pad of 4–6 week old NSG mice. Primary tumors and lungs were harvested 8 weeks post injection. MDA-MB-231 (EV, TRIM25, ΔTRIM25 − 5x 105) were injected into the lateral tail vein of 4–6week old athymic nude mice. Lungs were harvested 8 weeks post injection.
Next generation sequencing data
TCGA RNA-seq, amplification, deletion, mutation, and RPPA data was downloaded from the MSKCC cbio portal (http://www.cbioportal.org/) (Cerami et al., 2012; Gao et al., 2013). Clinical TCGA breast cancer data was downloaded from the Broad Institute GDAC Firehose (https://gdac.broadinstitute.org). PREdiction of Clinical Outcomes from Genomic Profiles (PRECOG) data was generated using the online PRECOG tool at https://precog.stanford.edu/. Spearman’s correlation matrix was generated using corrplot 0.73 and PerformanceAnalytics 1.4.3541 in R.
RNA-seq analysis of cell lines
Total RNA was isolated from cell lines using the RNeasy Plus mini prep kit (Qiagen, Valencia, CA, USA). Nucleic acid quality was determined with the Agilent 2100 Bioanalyzer. RNA Sequencing was also performed at the New York Genome Center (Manhattan, NY, USA) using a HiSeq 2500 Ultra-High-Throughput Sequencing System (Illumina, San Diego, CA, USA). Raw reads in the fastq format were aligned to Human Genome HG19 using the RNA-seq STAR aligner version 2.4.0d (http://www.ncbi.nlm.nih.gov/pubmed/23104886, http://www.ncbi.nlm.nih.gov/pubmed/26334920) as recommended by user manual downloaded along with the software. STAR aligner was chosen for mapping accuracy and speed (http://www.nature.com/nmeth/journal/v10/n12/full/nmeth.2722.html).
Mapped reads for each sample were counted for each gene in annotation files in GTF format (gencode.v19.annotation.gtf available for download from GENECODE website (http://www.gencodegenes.org/releases/19.html)) using the FeatureCounts read summarization program (http://www.ncbi.nlm.nih.gov/pubmed/?term=24227677) following the user guide (http://bioinf.wehi.edu.au/subread-package/SubreadUsersGuide.pdf). Individual count files were merged to generate the raw-counts matrix by an in-house R script, normalized to account for differences in library size and the variance was stabilized by fitting the dispersion to a negative-binomial distribution as implemented in the DESeq R package (http://bioconductor.org/packages/release/bioc/html/DESeq.html)(Anders and Huber, 2010).
ChIP-seq and RIP-seq sequencing was performed at the Memorial Sloan Kettering Center for Molecular Oncology (Manhattan, NY, USA) using a HiSeq 2500 Ultra-High-Throughput Sequencing System (Illumina, San Diego, CA, USA). Raw sequencing data were aligned to the hg37 genome build using the Burrows-Wheeler Aligner (BWA) version 0.7.10 (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2705234/). Further indel realignment, base-quality score recalibration and duplicate-read removal were performed using the Genome Analysis Toolkit (GATK) version 3.2.2 (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2928508/) following raw reads alignments guidelines (http://www.nature.com/ng/journal/v43/n5/pdf/ng.806.pdf). Peaks were called from the resulting bam files by Model-based Analysis of ChIP-seq (MACS2) version 2.1.0 (http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3868217/) following the manual at https://github.com/taoliu/MACS/.
As a first filtering step, resulting raw peaks were filtered by pval<0.01. Next, for each peak, RPKM (Reads Per Kilobase of transcript per Million mapped reads) was calculated for IP (immuno-precipitated) and INPUT (for ChIP-seq) bam files. Only peaks with RPKM INPUT/RPKM IP ratio greater than 0.5 were reported to final results.
Statistical analysis
Kaplan–Meier method and log rank test was performed using the Prism 6 Software (GraphPad Software, La Jolla, CA, USA). Heatmaps were generated using Partek® Genomics Suite® software, version 6.6 Copyright ©; 2014 (Partek Inc., St. Louis, MO, USA). We performed a minimum 3 replicates per experiment to ensure adequate sample size for statistical analyses. The Cancer Cell Line Encyclopedia (CCLE) was used to eliminate candidate MR from potential functional validation based on the lack of expression in breast cancer cell lines (http://www.broadinstitute.org/ccle/home) (Barretina et al., 2012).
Enrichment of Oncogenic signatures on the TRIM25 silencing signature was performed by Gene Set Enrichment Analysis (Subramanian et al., 2005). Oncogenic signatures gene sets v5.0 were downloaded from http://software.broadinstitute.org/gsea/downloads. Differentially expressed genes (FDR p<0.01, Table S4) between siSCR (control) vs. siTRIM25 were pre-ranked based on z-score and the GSEA was implemented using JAVA downloaded from the Broad Institute (http://software.broadinstitute.org/gsea). Two-tail GSEA (GSEA2) analysis was performed as previously described (Kruithof-de Julio et al., 2011) using an in-house implementation in R.
Differential expression analysis after MR gene silencing was performed by comparing the silenced samples vs. the scramble shRNA and mock controls independently using the moderated Student t-test as implemented in the limma package from Bioconductor (Ritchie et al., 2015). Gene expression signatures based on the scramble and mock controls were integrated using the Stouffer’s approach: , where Z1 and Z2 are Z-scores estimated by the moderated Student’s t-test. Lineal regression with exhaustive selection was performed with the package leaps for R-system (https://cran.r-project.org/web/packages/leaps). The approach selected the lineal combination of n MRs that best predicts the subtype-matched MET-GES. We selected n as the highest number of MRs for which all coefficients of the model were different than zero at Bonferroni’s corrected p < 1 × 10−3. The proportion of the MET-GES modulated by each MR was inferred as the proportion of MET genes in the leading edge when computing the enrichment of the MET-GES on each MR silencing gene expression signature by GSEA.
Assembly of context-specific regulatory models and Master Regulator analysis
A breast carcinoma context-specific network model of transcriptional regulation was assembled with the ARACNe Algorithm, based on 851 RNA-seq expression profiles obtained from The Cancer Genome Atlas. ARACNe was ran with 100 bootstraps, p value threshold of 10−8 and 0 DPI tolerance, generating a network of 1,748 transcription factors associated with 18,783 target genes by 459,569 interactions. The regulatory models were generated from the ARACNe results using the VIPER package from Bioconductor (http://bioconductor.org/packages/release/bioc/html/viper.html).
The gene expression signatures for 20 ER+ and 11 TNBC metastasis (MET-GES) were computed with paired Student’s t-test by comparing their profiles against the matching primary tumor ones. Then, the enrichment of each regulatory protein regulon on the MET-GESs was inferred by the VIPER algorithm (Alvarez et al., 2016; Aytes et al., 2014) as implemented in the VIPER package for R available from Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/viper.html). Statistical significance was estimated by permuting the samples uniformly at random 1,000 times.
For single patient-based analysis, gene expression signatures were computed by comparing each MET expression profile with the matching primary tumor expression profile. A null model for statistical testing was generated by permuting the samples uniformly at random 1,000 times. The most consistent master regulators across all tumors were prioritized according to the average dysregulation (NES) divided by its standard deviation.
Database accession numbers
Supplementary Material
Differentially expressed genes between primary tumors and matched metastases (MET-GES)
The ARACNe Inferred Network
VIPER inferred regulators of the MET-GES
Transcriptional and post-transcriptional regulation of TRIM25 target genes
Linear regression of select TF knockdown vs. MET-GES
MSigDB pathways associated with TRIM25 transcriptionally regulated or post-transcriptionally regulated genes
GSEA results from MSigDB c6.all.v5.0.symbols oncogenic signatures after siTRIM25 in TNBC cells
Acknowledgments
We thank Agnes Viale and the MSKCC genomics core for technical assistance. We thank members of the Chan and Califano labs for helpful discussions. L.A.W. was supported by The Canadian Institutes of Health Research PDF Award MFE-127325. This work was supported by a Department for Defense Era of Hope Grant (BC120568) (T.A.C.), the STARR Cancer Consortium (T.A.C.), the Geoffrey Beene Cancer Center (T.A.C.), and the MSKCC Metastasis Research Center (T.A.C.). NIDDK (1 DP2 DK098093-01) and CADC pilot grant (S. C. and S. M). Memorial Sloan Kettering Cancer Center Core Grant P30 CA008748 to the Microchemistry and Proteomics Core.
Footnotes
Author Contributions
T.A.C. and A.C. conceived the study. L.A.W., T.A.C., M.J.A., E.Y.S., and M.R. designed the experiments. L.A.W. E.Y.S., M.R., S.M., M.D., and V.K.R. performed the experiments. M.J.A., L.A.W., V.M., A.D., and S.T. conducted the bioinformatics analysis. L.A.W., M.J.A., E.Y.S., M.R. S.M., T.A.C. analyzed the data. L.T.V. provided the clinical samples. L.A.W., M.J.A., A.C. and T.A.C. wrote the manuscript. All authors reviewed and edited the final manuscript.
References
- Akrap N, Andersson D, Bom E, Gregersson P, Stahlberg A, Landberg G. Identification of Distinct Breast Cancer Stem Cell Populations Based on Single-Cell Analyses of Functionally Enriched Stem and Progenitor Pools. Stem Cell Reports. 2016;6:121–136. doi: 10.1016/j.stemcr.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, Califano A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet. 2016;48:838–847. doi: 10.1038/ng.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome biology. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aytes A, Mitrofanova A, Lefebvre C, Alvarez MJ, Castillo-Martin M, Zheng T, Eastham JA, Gopalan A, Pienta KJ, Shen MM, et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer cell. 2014;25:638–651. doi: 10.1016/j.ccr.2014.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso K, Margolin AA, Stolovitzky G, Klein U, Dalla-Favera R, Califano A. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005;37:382–390. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- Basso K, Saito M, Sumazin P, Margolin AA, Wang K, Lim WK, Kitagawa Y, Schneider C, Alvarez MJ, Califano A, et al. Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood. 2010;115:975–984. doi: 10.1182/blood-2009-06-227017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano F, Miranda-Saavedra D, Lehner PJ. RNA-binding E3 ubiquitin ligases: novel players in nucleic acid regulation. Biochemical Society transactions. 2010;38:1621–1626. doi: 10.1042/BST0381621. [DOI] [PubMed] [Google Scholar]
- Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, Sulman EP, Anne SL, Doetsch F, Colman H, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–325. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Cassiday LA, Maher LJ., 3rd Having it both ways: transcription factors that bind DNA and RNA. Nucleic acids research. 2002;30:4118–4126. doi: 10.1093/nar/gkf512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Alvarez MJ, Talos F, Dhruv H, Rieckhof GE, Iyer A, Diefes KL, Aldape K, Berens M, Shen MM, et al. Identification of Causal Genetic Drivers of Human Disease through Systems-Level Analysis of Regulatory Networks. Cell. 2014;159:402–414. doi: 10.1016/j.cell.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury NR, Nowak JS, Zuo J, Rappsilber J, Spoel SH, Michlewski G. Trim25 Is an RNA-Specific Activator of Lin28a/TuT4-Mediated Uridylation. Cell reports. 2014;9:1265–1272. doi: 10.1016/j.celrep.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creighton CJ, Cordero KE, Larios JM, Miller RS, Johnson MD, Chinnaiyan AM, Lippman ME, Rae JM. Genes regulated by estrogen in breast tumor cells in vitro are similarly regulated in vivo in tumor xenografts and human breast tumors. Genome biology. 2006;7:R28. doi: 10.1186/gb-2006-7-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Kamalakaran S, Radhakrishnan SK, Beck WT. Identification of estrogen-responsive genes using a genome-wide analysis of promoter elements for transcription factor binding sites. The Journal of biological chemistry. 2005;280:21491–21497. doi: 10.1074/jbc.M409176200. [DOI] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, Wang J, Orkin SH. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruithof-de Julio M, Alvarez MJ, Galli A, Chu J, Price SM, Califano A, Shen MM. Regulation of extra-embryonic endoderm stem cell differentiation by Nodal and Cripto signaling. Development. 2011;138:3885–3895. doi: 10.1242/dev.065656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushwaha R, Jagadish N, Kustagi M, Tomishima MJ, Mendiratta G, Bansal M, Kim HR, Sumazin P, Alvarez MJ, Lefebvre C, et al. Interrogation of a context-specific transcription factor network identifies novel regulators of pluripotency. Stem Cells. 2015;33:367–377. doi: 10.1002/stem.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon SC, Yi H, Eichelbaum K, Fohr S, Fischer B, You KT, Castello A, Krijgsveld J, Hentze MW, Kim VN. The RNA-binding protein repertoire of embryonic stem cells. Nature structural & molecular biology. 2013;20:1122–1130. doi: 10.1038/nsmb.2638. [DOI] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome research. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson DA, Bhakta NR, Kessenbrock K, Prummel KD, Yu Y, Takai K, Zhou A, Eyob H, Balakrishnan S, Wang CY, et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature. 2015;526:131–135. doi: 10.1038/nature15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Rajbhandari P, Alvarez MJ, Bandaru P, Lim WK, Sato M, Wang K, Sumazin P, Kustagi M, Bisikirska BC, et al. A human B-cell interactome identifies MYB and FOXM1 as master regulators of proliferation in germinal centers. Mol Syst Biol. 2010;6:377. doi: 10.1038/msb.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Strom A, Vega VB, Kong SL, Yeo AL, Thomsen JS, Chan WC, Doray B, Bangarusamy DK, Ramasamy A, et al. Discovery of estrogen receptor alpha target genes and response elements in breast tumor cells. Genome biology. 2004;5:R66. doi: 10.1186/gb-2004-5-9-r66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A. 2003;100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, Califano A. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC bioinformatics. 2006a;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin AA, Wang K, Lim WK, Kustagi M, Nemenman I, Califano A. Reverse engineering cellular networks. Nature protocols. 2006b;1:662–671. doi: 10.1038/nprot.2006.106. [DOI] [PubMed] [Google Scholar]
- Monteiro J, Fodde R. Cancer stemness and metastasis: therapeutic consequences and perspectives. European journal of cancer. 2010;46:1198–1203. doi: 10.1016/j.ejca.2010.02.030. [DOI] [PubMed] [Google Scholar]
- Nalepa G, Harper JW. Efp: a ring of independence? Nature medicine. 2002;8:661–662. doi: 10.1038/nm0702-661. [DOI] [PubMed] [Google Scholar]
- Navin NE. The first five years of single-cell cancer genomics and beyond. Genome research. 2015;25:1499–1507. doi: 10.1101/gr.191098.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien P, Morin P, Jr, Ouellette RJ, Robichaud GA. The Pax-5 gene: a pluripotent regulator of B-cell differentiation and cancer disease. Cancer research. 2011;71:7345–7350. doi: 10.1158/0008-5472.CAN-11-1874. [DOI] [PubMed] [Google Scholar]
- Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer cell. 2012;22:709–724. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- Piovan E, Yu J, Tosello V, Herranz D, Ambesi-Impiombato A, Da Silva AC, Sanchez-Martin M, Perez-Garcia A, Rigo I, Castillo M, et al. Direct reversal of glucocorticoid resistance by AKT inhibition in acute lymphoblastic leukemia. Cancer cell. 2013;24:766–776. doi: 10.1016/j.ccr.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratkaj I, Stajduhar E, Vucinic S, Spaventi S, Bosnjak H, Pavelic K, Kraljevic Pavelic S. Integrated gene networks in breast cancer development. Functional & integrative genomics. 2010;10:11–19. doi: 10.1007/s10142-010-0159-2. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic acids research. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Barrueco R, Yu J, Saucedo-Cuevas LP, Olivan M, Llobet-Navas D, Putcha P, Castro V, Murga-Penas EM, Collazo-Lorduy A, Castillo-Martin M, et al. Inhibition of the autocrine IL-6-JAK2-STAT3-calprotectin axis as targeted therapy for HR-/HER2+ breast cancers. Genes & development. 2015;29:1631–1648. doi: 10.1101/gad.262642.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano D, Heck C, De Antonellis P, Christofori G, Zollo M. Molecular networks that regulate cancer metastasis. Seminars in cancer biology. 2012;22:234–249. doi: 10.1016/j.semcancer.2012.03.006. [DOI] [PubMed] [Google Scholar]
- Streich FC, Jr, Ronchi VP, Connick JP, Haas AL. Tripartite motif ligases catalyze polyubiquitin chain formation through a cooperative allosteric mechanism. The Journal of biological chemistry. 2013;288:8209–8221. doi: 10.1074/jbc.M113.451567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano T, Saito T, Tsukui T, Fujita M, Hosoi T, Muramatsu M, Ouchi Y, Inoue S. Efp targets 14-3-3 sigma for proteolysis and promotes breast tumour growth. Nature. 2002;417:871–875. doi: 10.1038/nature00826. [DOI] [PubMed] [Google Scholar]
- Zhang H, Meng F, Liu G, Zhang B, Zhu J, Wu F, Ethier SP, Miller F, Wu G. Forkhead transcription factor foxq1 promotes epithelial-mesenchymal transition and breast cancer metastasis. Cancer research. 2011;71:1292–1301. doi: 10.1158/0008-5472.CAN-10-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Differentially expressed genes between primary tumors and matched metastases (MET-GES)
The ARACNe Inferred Network
VIPER inferred regulators of the MET-GES
Transcriptional and post-transcriptional regulation of TRIM25 target genes
Linear regression of select TF knockdown vs. MET-GES
MSigDB pathways associated with TRIM25 transcriptionally regulated or post-transcriptionally regulated genes
GSEA results from MSigDB c6.all.v5.0.symbols oncogenic signatures after siTRIM25 in TNBC cells