

Abstract
Imetelstat (GRN163L) is a specific telomerase inhibitor that has demonstrated clinical activity in patients with myeloproliferative neoplasms (MPN) and in patients with solid tumors. The antitumor effects were associated with the development of thrombocytopenia, one of the common side effects observed in patients treated with imetelstat. The events underlying these adverse effects are not apparent. In this report, we investigated the potential mechanisms that account for imetelstat’s beneficial effects in MPN patients and the manner by which imetelstat treatment leads to a reduction in platelet numbers. Using a well-established system of ex vivo megakaryopoiesis, we demonstrated that imetelestat treatment affects normal megakaryocyte (MK) development by exclusively delaying maturation of MK precursor cells. By contrast, additional stages along MPN MK development were affected by imetelstat resulting in reduced numbers of assayable colony-forming unit MK and impaired MK maturation. In addition, treatment with imetelstat inhibited the secretion of fibrogenic growth factors by malignant but not by normal MK. Our results indicate that the delay observed in normal MK maturation may account for imetelstat-induced thrombocytopenia, while the more global effects of imetelstat on several stages along the hierarchy of MPN megakaryopoiesis may be responsible for the favorable clinical outcomes reported in MPN patients.
INTRODUCTION
The Philadelphia chromosome-negative myeloproliferative neoplasms (MPN), including polycythemia vera, essential thrombocythemia (ET) and primary myelofibrosis (MF), are a group of related clonal disorders involving the hematopoietic stem/progenitor cells (HSC/HPC). The mounting complexity of the genetic and epigenetic alterations contributing to MPN pathogenesis and progression poses a major challenge to the development of targeted treatment strategies.1,2 Small-molecule inhibitors of Janus-activated kinase 1/2 (JAK1/2) are presently being used to treat intermediate-/high-risk MF patients resulting in dramatic improvements in systemic symptoms and modest improvements in survival. Several other drugs have recently reported to have beneficial effects in MF and ET patients.1 One such compound is imetelstat, a potent and specific telomerase inhibitor,3,4 which has been evaluated in phase I and II clinical trials in patients with solid tumors and hematological malignancies. Two independent groups have recently reported that imetelstat is active in patients with MF and ET.5,6
Telomerase is a ribonuclear protein complex consisting of a reverse transcriptase catalytic protein subunit (TERT) and a RNA component that act together to extend telomeres at the ends of chromosomes.7 Telomerase is transiently active in normal stem and progenitor cells, is not expressed by normal differentiated somatic tissues but is constitutively upregulated in 85% of human cancers.8
Imetelstat is a 13-mer oligonucleotide that specifically targets the RNA template of human telomerase and is a potent competitive inhibitor of telomerase enzymatic activity.3 Several preclinical studies using in vitro and in vivo model systems have demonstrated the ability of imetelstat to inhibit telomerase activity (TA) and to exert antiproliferative and cytotoxic effects against a wide variety of cancers, including hematological malignancies5,6,9,10 and solid tumors.11–16 However, the dose-limiting toxicity associated with imetelstat administration in patients has been thrombocytopenia.6,17,18
Platelets are produced by bone marrow megakaryocytes (MK) through a complex process termed megakaryopoiesis.19 Normal megakaryopoiesis consists of a succession of events in which MK progenitors initially proliferate and acquire lineage-specific markers, followed by endomitosis-mediated polyploidization (that is, acquisition of greater diploid 2N DNA content) and finally cytoplasmic maturation and platelet production. Any alterations occurring in either of the hierarchical stages of megakaryopoiesis can result in quantitative and/or qualitative platelet disorders. Previously reported in vitro studies have suggested that imetelstat selectively inhibits ET MK colony-forming units (CFU-MK) formation in the absence of exogenous cytokines but not CFU-MK from healthy individuals.20 In this report, we investigated the mechanisms underlying imetelstat’s beneficial effects in MPN patients and the manner by which imetelstat therapy leads to thrombocytopenia.
MATERIALS AND METHODS
Human MK cultures
Human MKs were generated using a two-step liquid culture system of either bone marrow (BM) or peripheral blood (PB) derived CD34+ cells (AllCells, LLC, Emeryville, CA, USA) as previously described.21,22 CFU-MK assays were performed by using the MegaCult System and Detection Kit according to the manufacturer’s instructions (Stem Cell Technologies, Vancouver, BC, Canada). Isolation of CD61+ MK was performed using an Immunomagnetic Selection Kit as per manufacturer’s recommendations (Miltenyi Biotech Inc., Auburn, CA, USA).
MPN PB mononuclear cells (MNCs) were procured from tissue-banked specimens collected after written informed consent was obtained according to guidelines established by the Institutional Review Board of the Icahn School of Medicine at Mount Sinai. PB-MNCs were cultured in MK differentiation media in a manner identical to that used for purified CD34+ cells. PB-MNCs from healthy donors were purchased from All Cells.
Imetelstat sodium (GRN163L) is a 5′ palmitoylated 13-mer thiopho-sphoramidate oligonucleotide composed of the sequence 5′-TAGGGTTAGACAA-3′. Mismatched oligonucleotide (MM1) is a 5′ palmitoylated 13-mer thiophosphoramidate oligonucleotide composed of the sequence 5′-TAGGTGTAAGCAA-3′. Both compounds were provided by Geron Corporation (Menlo Park, CA, USA).
Growth factor profiling
The levels of growth factors (GFs) present in the conditioned media were measured using human ProcartaPlex panels for cytokines (25 plex) and GFs (11 plex) (eBioscience, San Diego, CA, USA) and a Luminex xMAP platform (EMD Millipore, Billerica, MA, USA). The following GFs were assessed: leukemia inhibitory factor, placental GF, stem cell factor, hepatic GF, brain-derived neurotrophic factor (BDNF), fibroblast growth factor (FGF-2), platelet-derived growth factor (PDGF-BB), vascular endothelial growth factor (VEGF-A and VEGF-D), nerve growth factor and endothelial growth factor. Transforming growth factor-β levels were detected using Quanti-kine ELISA assays using the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA).
Statistical analysis
Data are expressed as mean ± s.d. and analyzed using a Student’s unpaired t-test. A P-value ≤ 0.05 was considered statistically significant.
Additional Materials and Methods utilized in this study are described in Supplementary Material and elsewhere.21–23
RESULTS
Imetelstat inhibits malignant but not normal CFU-MK formation We initially tested the effects of imetelstat on the ability of CD34+ cells to generate CFU-MK colonies. Imetelstat (GRN163L) and a mismatched control oligonucleotide referred to as MM1 were tested at previously determined concentrations of 7.5 and 15 μM. PB- or BM-derived primary CD34+ cells from healthy donors were placed in liquid cultures in the presence and absence of either imetelstat or MM1. After 24 h, 625 cells from these cultures were plated in duplicate in MegaCult semisolid media supplemented with cytokines. We found that neither the number nor the size of CFU-MK formed by normal CD34+ cells was affected by exposure to imetelstat (Figure 1a and Supplementary Figure S1A). We next assessed CFU-MK formation by PB-MNCs from patients with ET or MF. After treatment with imetelstat or MM1 for 24 h, MNCs from healthy donors and from MPN patients were plated in MegaCult semisolid media in a manner identical to that used for primary CD34+ cells. Imetelstat-treated PB-MNCs from 13 out of the 15 patients analyzed formed, on average, 60% fewer CFU-MK as compared with PB-MNCs treated with the control oligonucleotide (Figure 1b and Supplementary Table S1). Moreover, in 4 out of the 13 responders, imetelstat not only reduced the number of CFU-MK formed but also reduced CFU-MK size. A representative example illustrate macroscopic and microscopic appearance of CFU-MK (Figure 1c) and differential quantification of small (3–10 MKs), medium (10–50 MKs) and large (450 MKs) CFU-MK formed by MNCs from one MPN patient (Supplementary Figure S1B). Taken together, these results indicate that exposure to imetelstat not only inhibits MPN CFU-MK formation but also limits their proliferative potential. By contrast, neither the number nor the size of CFU-MK formed by PB-MNCs from healthy donors (n = 8) were affected by imetelstat, which confirms the results obtained with purified normal PB-CD34+ cells. These data suggest that imetelstat treatment preferentially affects MPN CFU-MK formation.
Figure 1.
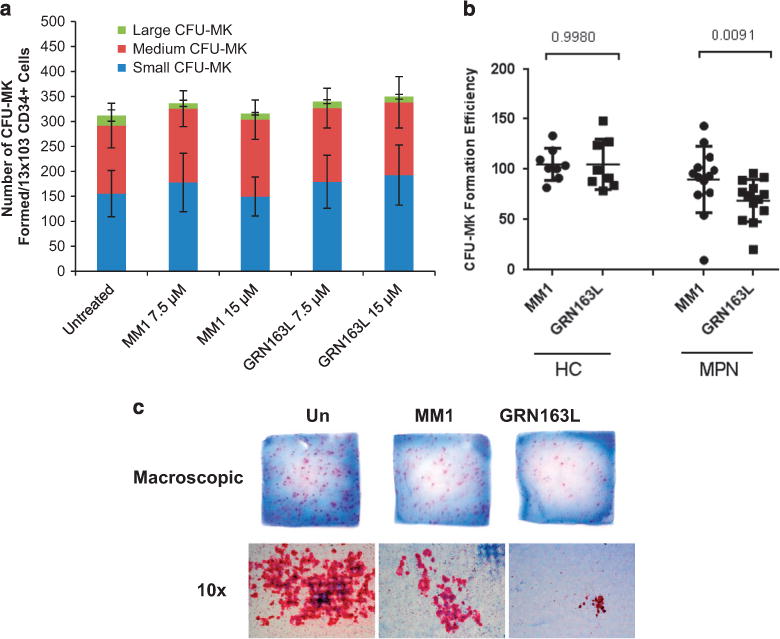
Effects of imetelstat on CFU-MK colony formation. (a) CFU-MK colonies formed by untreated CD34+ cells or by CD34+ cells treated with 7.5 and 15 μM of either mismatched control oligonucleotide (MM1) or imetelstat (GRN163L) were enumerated after 14–16 days of incubation in a collagen-based semisolid media in the presence of thrombopoietin, IL-6 and IL-3. Each column represents the average number of small (3–10 MKs), medium (10–50 MKs) and large (450 MKs) colonies ± s.d. enumerated in three independent experiments. (b) CFU-MK formation by PB-MNCs from HCs (n = 8) and from patients with MPN (n = 15) treated with 15 μM of either mismatched control oligonucleotide (MM1) or imetelstat (GRN163L). The vertical axis indicates CFU-MK clonogenic efficiency after normalization to that of untreated cultures. (c) Representative microphotographs of collagen slabs containing CFU-MK colonies (upper panels) and of individual CFU-MK (lower panels) formed by untreated, MM1- and GRN163L-treated PB-MNCs from one representative MPN patient.
Imetelstat delays normal MK maturation
The ability of normal HPCs to proliferate and commit to the MK lineage was next examined utilizing a well-established, two-step liquid culture system of ex vivo megakaryopoiesis.21–23 In the first step, the cells were expanded in the presence of stem cell factor and thrombopoietin to mimic the early stages of megakaryopoiesis when CD34+ cells proliferate and acquire lineage-specific markers. In the second step, the MKs were allowed to become polyploid, mature and eventually produce platelets in the presence of thrombopoietin only. Cultures generated in the presence of imetelstat for the first 7 days proliferated at a slightly lower rate than the control cultures but the difference did not reach statistical significance. The frequency of CD34+/CD41+ MK precursors generated after 7 days were comparable in the presence and absence of imetelstat. Surprisingly, by day 14, there was a greater proportion of CD34+/CD41+ MK in the cultures generated in the presence of imetelstat as compared with those generated in the untreated or MM1 control cultures (Figure 2a, Supplementary Figures S2A and S3A). Furthermore, untreated and MM1-treated cultures consisted of a heterogeneous MK population containing CD34+/CD41+ MK precursors as well as MK at different stages of maturation, including immature CD34−/CD41+ MK and mature CD41+/CD42+ MK (Supplementary Figure S2). By contrast, in the cultures treated with imetelstat that were predominantly composed of CD34+/CD41+ MK precursors and immature MK, the absolute number of mature CD41+/CD42+ MK was four fold lower (P-value = 0.0376) than that found in untreated or MM1-treated cultures (Figure 2b, Supplementary Figure S2B). These results indicate that imetelstat treatment of normal CD34+ cells did not interfere with MK lineage commitment but rather delayed their maturation. Importantly, we show that this delay in MK maturation was reversible as limited exposure to imetelstat followed by drug removal partially restored the CD34−/CD41+ MK population (that is, there were twice as many CD34−/CD41+ cells in the cultures in which the drug was withdrawn as compared with the cultures exposed continuously to imetelstat; Figure 2c). To better understand these effects, MKs were generated in the absence of imetelstat for the first 7 days and then allowed to undergo terminal maturation in the presence of the drug for 7 additional days. Phenotypical characterization of the cells generated showed that, compared with untreated or MM1-treated cultures, the cultures treated with imetelstat contained a greater proportion of CD34+/CD41+ cells (Figures 3a and b), supporting the observation that drug exposure favors an immature phenotype. By contrast, the overall proportion of mature CD41+/CD42+ MK was comparable in all three conditions (Supplementary Figure S4), implying that terminal maturation by already committed MK was not hindered by treatment. These observations suggested that telomerase may not be required for the later stages of megakaryopoiesis. To test this possibility, we next evaluated hTERT expression and TA at different stages of MK differentiation and after imetelstat treatment.
Figure 2.
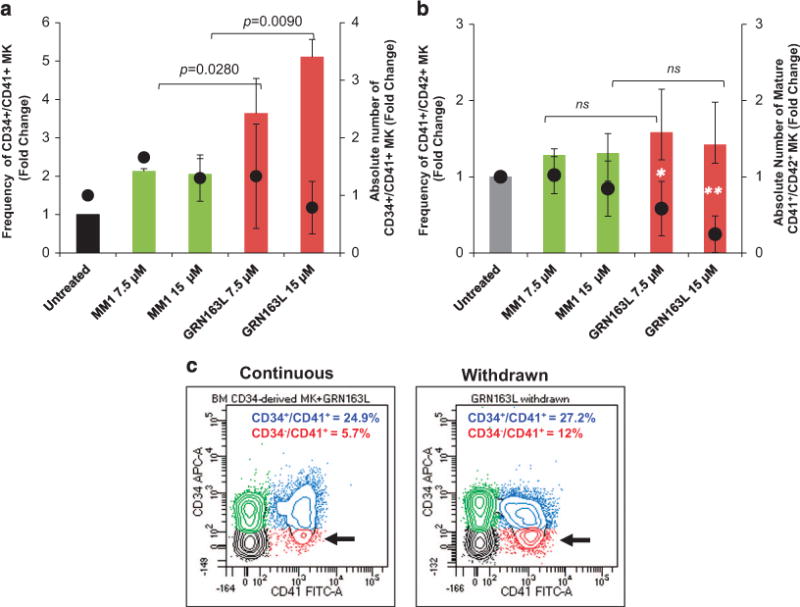
Effects of imetelstat on the normal MK phenotype. (a) MK cultures that were untreated and treated with 7.5 and 15 μM of either MM1 or imetelstat (GRN163L) for 14 days were labeled with FITC-conjugated anti-CD41 and APC-conjugated anti-CD34 antibodies and analyzed by flow cytometry. The columns represent fold changes in the frequency (left y axis) and the absolute number (right y axis) of CD34+/CD41+ MK precursors quantified in the indicated culture conditions. (b) Cells from the same cultures as in panel (a) were labeled with FITC-conjugated anti-CD41 and APC-conjugated anti-CD42b antibodies and analyzed by flow cytometry. The columns represent fold changes in the frequency (left y axis) and the absolute number (right y axis) of mature CD41+/CD42b+ MK quantified in the indicated culture conditions. (c) Representative flow cytometric analyses of MK cultures treated continuously with imetelstat for 9 days (left panel) and of MK cultures in which imetelstat was removed after 6 days (right panel). Immature CD34+/CD41+ and more mature CD34−/CD41+ MK populations are indicated by blue and red colors, respectively. The results in panels (a and b) represent the mean ± s.d. of the frequency and corresponding absolute numbers detected in three independent experiments utilizing CD34+ cells from different healthy donors. NS, not statistically significant.
Figure 3.
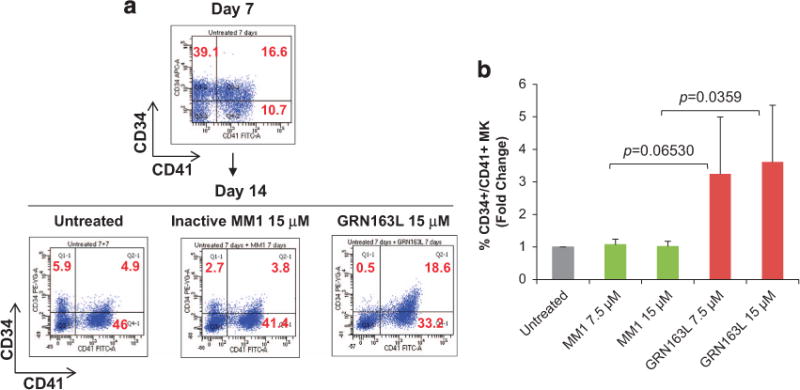
Effects of imetelstat on normal MK maturation. (a) Representative flow cytometric analyses of CD41/CD34 expression in MK cultures that were cultured in the absence of drugs for 7 days (upper panel) and then allowed to mature in the absence (untreated) and presence of 15 μM of either MM1 or imetelstat (GRN163L) for 7 additional days (lower panels). Upper-right quadrants contains CD34+/CD41+ MK precursors. (b) Frequency of CD34+/CD41+ MK precursors quantified in MK cultures that were untreated and treated with 7.5 and 15 μM of either MM1 or imetelstat (GRN163L) as indicated in panel (a). The columns represent the mean ± s.d. of three independent experiments initiated with CD34+ cells from three different healthy donors.
Telomerase expression and activity in MK
Given that telomerase is the direct cellular target of imetelstat, we sought to understand the underlying mechanisms responsible for the observed effects of the drug on MKs. First, we showed that hTERT, the protein component of telomerase, is expressed during the early stages of megakaryopoiesis but declines gradually with MK maturation (Figure 4a). The expression of p21, which is known to be upregulated during MK differentiation,24 was evaluated in parallel to assess the degree of maturation. The direct relationship between the reduction in hTERT expression and MK maturation was confirmed at the transcriptional level by quantitative real-time PCR. hTERT mRNA levels were the greatest during the initial stages of megakaryopoieisis but were markedly downregulated during the later stages (Supplementary Figure S5A). As the enzymatic activity of telomerase depends on both hTERT and its RNA template component, we used a PCR-based assay to measure TA in normal MK. TA was lower in mature MKs (day-11 cultures) than in immature MKs (day-7 cultures) (Figure 4b). These observations suggest that telomerase expression and function are the greatest during the proliferative stages of megakaryopoiesis but decline as MK undergo terminal maturation. We next evaluated hTERT and TA in normal and malignant MK exposed to imetelstat during the proliferative stage of megakaryopoiesis. Treatment of MK cultures derived from normal CD34+ cells with imetelstat resulted in a significant decrease in TA (Figure 4c) and reduced hTERT mRNA levels (Supplementary Figure S5B). hTERT expression was also assessed in untreated and imetelstat-treated cultures from four MPN patients. Imetelstat treatment reduced hTERT mRNA expression in cultures from one patient but did not have an impact on hTERT in cultures derived from the other three patients (Supplementary Figure S5C). By contrast, by utilizing a human MK cell line JAK2V617F+ HEL, which has high baseline levels of telomerase, we confirmed findings reported by others5,12,15 and showed that imetelstat treatment led to a significant decrease in hTERT (Supplementary Figure S5D) and TA (Supplementary Figure S5E). The overall lack of effect observed in the primary MNC MPN cultures could be due to the small sample size and the heterogeneity of these cultures that contain non-dividing MNCs, which may conceal the potential changes in hTERT expression in MK cells.
Figure 4.
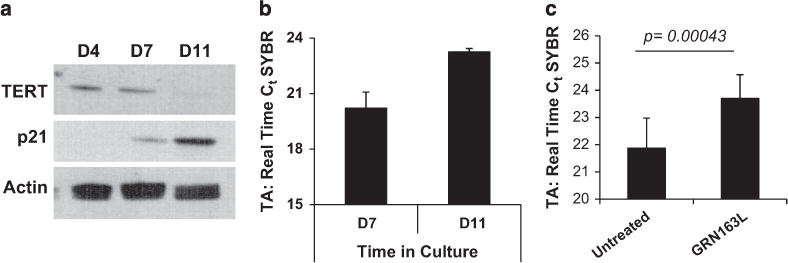
Evaluation of hTERT expression and TA in normal MK. (a) Western blot analyses of protein lysates isolated from MK cultures generated on days 4, 7 and 11 in culture during differentiation and maturation into MK of normal CD34+ cells. After blotting, the membranes were incubated with anti-hTERT and p21 antibodies. Actin expression was utilized as control for protein loading. (b) TA measured in MK cultures on days 7 and 11 during MK maturation. (c) TA measured in primary MK cultures untreated or treated with GRN163L for 7 days. The columns represent the results of duplicate experiments utilizing CD34+ cells from three different normal donors. The y axis in panels (b, c) indicates the number of PCR cycles (Ct) required for the amplification of telomere repeats. A low Ct value indicates a cell extract with high TA, whereas a high Ct value indicates a cell extract with low TA.
Imetelstat prevents MK polyploidization
To determine whether imetelstat affects additional MK-specific events, we examined its effects on normal MK polyploidization and platelet formation. Untreated cultures and cultures treated with the control oligonucleotide MM1 contained large MK with nuclei with numerous lobules, whereas imetelstat-treated cultures contained smaller, hypolobulated MK (Figure 5a). We observed that imetelstat treatment resulted in a 50% reduction of CD41+ MK with 44N DNA content (that is, polyploid) as compared with cultures treated with MM1 (Figure 5b). The inhibitory effects of imetelstat on polyploidization were even more accentuated when immunomagnetically purified MK (day 9) were exposed to the drug. As illustrated in Figure 5c, imetelstat-treated MK lack distinct higher ploidy peaks (that is, 8N, 16N, 32N DNA content) when compared with untreated MK or to those treated with MM1. To further dissect the mechanisms underlying the imetelstat-induced arrest in polyploidization, we evaluated DNA synthesis using bromodeoxyuridine (BrdU) incorporation. Surprisingly, there were twice as many BrdU+ MKs in cultures containing imetelstat (20.1 ± 3.4%) as compared with untreated cultures (9.8 ± 2.7%) or those treated with MM1 (11.05 ± 5.1%) (Figure 6a). A more careful analysis, however, indicated that the fraction of low ploidy BrdU+ MKs (gate P2 in Figure 6b) was the major contributor to the increase in the overall number of BrdU+ MK cells detected in imetelstat-treated cultures. These observations suggest that imetelstat treatment does not interfere with DNA synthesis but rather halts MK endomitosis at the 2N and 4N stage. Moreover, the increased BrdU incorporation observed in imetelstat-containing cultures reflects the observed predominance of MK precursors and immature MK, which retain the ability to undergo DNA synthesis.
Figure 5.
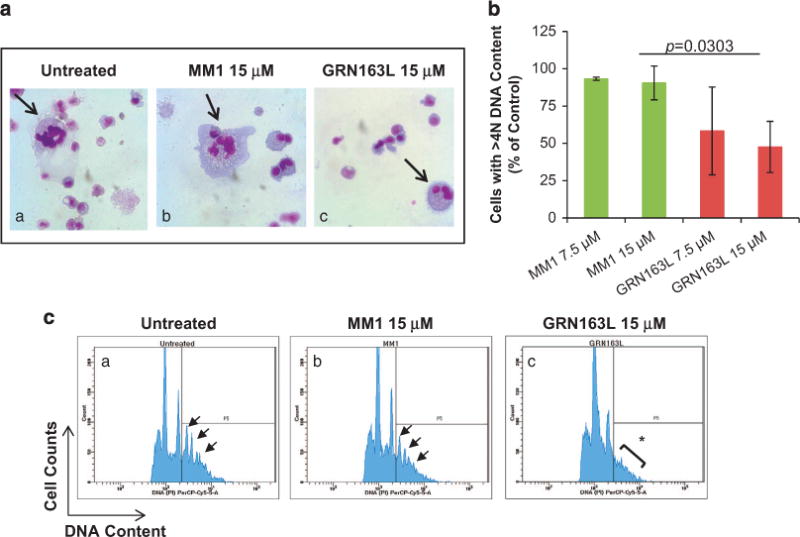
Effects of imetelstat on normal MK polyploidization. (a) Representative Wright-Giemsa stained cytospin preparations of cells from control cultures (untreated) and from cultures incubated for 14 days in the presence of 15 μM MM1 or GRN163L. Large, multilobulated MK are present in untreated cultures and in cultures treated with MM1 (arrows in panels (a, b), whereas smaller hypolobulated MK are present in GRN163L-treated cultures (arrow in panel (c)). Microphotographs were obtained using an Olympus BX40 microscope with a dry 40 × /0.75 objective. (b) Quantification of MK ploidy detected after 14 days of incubation in control cultures and in cultures generated in the presence of 7.5 and 15 μM of MM1 or GRN163L. The numbers represent the fraction of CD41+ MK with DNA content 44N normalized to that detected in untreated cultures. (c) CD41+ MK immunomagnetically isolated from cultures generated in the absence of drugs for 9 days were treated with either MM1 or GRN163L and allowed to undergo polyploidization for three additional days. Representative histograms illustrating DNA content (x axis) of untreated and MM1-treated MK indicate the presence of distinct ploidy peaks ranging from 4N to 32N DNA content (arrows in panel (a, b)); such ploidy peaks are absent in imetelstat-treated MK (asterisk in panel (c)).
Figure 6.
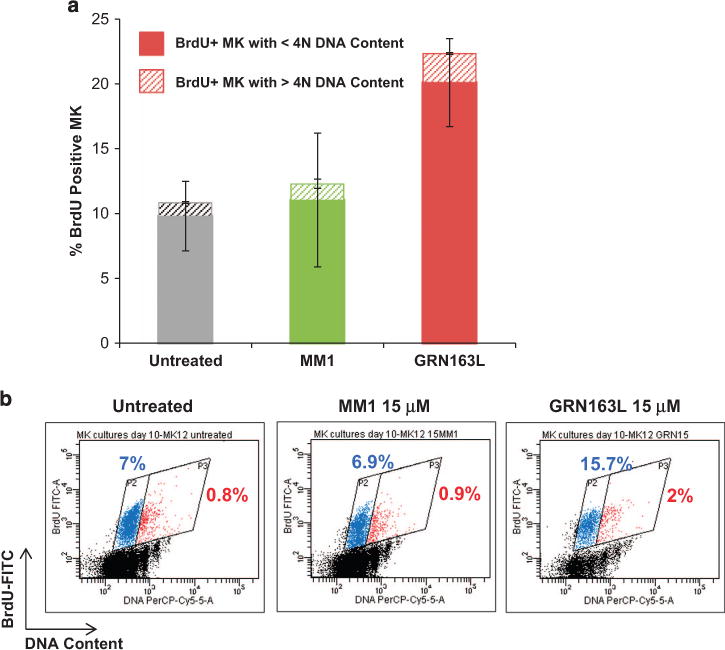
Effects of imetelstat on DNA synthesis during endomitosis. (a) BrdU incorporation ability of MK cultures grown in the absence (untreated) and presence of 15 μM MM1 or GRN163L. The columns indicate the fractions of BrdU positive MK with low (solid) and high (pattern) ploidy levels, that is, o4N and 44N DNA content, respectively. (b) Representative dot plot histograms of BrdU-labeled MK generated in untreated cultures and in cultures exposed to 15 μM of MM1 or GRN163L for 48 h during MK polyploidization. BrdU incorporation, indicating active DNA synthesis, is represented on the y axis while DNA content as evaluated by 7-AAD staining is represented on the x axis. The fraction of BrdU-positive MK with o4N DNA content and with 44N DNA content are indicated in the blue (P2) and red (P3) gates, respectively.
We also evaluated the effects of imetelstat on the ability of normal MK to extend proplatelets or produce platelets and found that drug treatment did not influence these processes ex vivo (Supplementary Material and Supplementary Figure S6). The finding that the number of culture-derived platelets in imetelstat-treated cultures was comparable to that detected in control cultures suggested that immature MK, which were predominant in the presence of the drug, were able to generate platelet-like particles. This was confirmed by the mean fluorescence intensity of the thiazole orange-labeled platelets, reflecting an increased ribonucleotide content characteristic of ‘younger’ platelets, which was significantly increased in the presence of imetelstat (Supplementary Figures S6B and D). Platelet release by immature MKs has been previously reported in disease conditions associated with increased platelet demand.25,26
In summary, our ex vivo studies demonstrate that imetelstat affects normal megakaryopoiesis by hindering maturation and polyploidization of MK precursors resulting in their accumulation at the expense of their more mature progeny, which would eventually diminish the pool of mature polyploid MK capable of producing platelets.
Imetelstat treatment inhibits MPN megakaryopoiesis ex vivo
To evaluate the effects of imetelstat on malignant megakaryopoiesis, MKs were generated from PB-MNCs from patients with ET and MF. Owing to the limited number of MPN CD34+ cells that can be isolated from the small clinical samples available, the cultures were initiated with PB-MNCs. PB-MNCs from healthy donors were studied in parallel and served as controls. After 7 days, the total number of viable cells was reduced by the addition of either imetelstat or control MM1 to both normal and MPN MNC cells as compared with untreated cultures (Figure 7a). In order to determine the direct effects of imetelstat on MPN MK within these MNC collections, the same cultures were analyzed phenotypically. The percentages of CD34+/CD41+ and CD34−/ CD41+ MKs was first detected in each culture condition (Supplementary Figures S7A and B) and used to calculate the absolute numbers of total MK precursors and more mature MK, respectively. Treatment of normal MNCs with imetelstat did not affect the absolute numbers of CD34+/CD41+ MK precursors generated when compared with untreated condition but decreased the number of MK precursors generated from MPN MNCs (Figure 7b). More importantly, MPN cultures treated with imetelstat contained significantly fewer CD34−/CD41+ MK than the untreated control or those treated with MM1, whereas the number of CD34−/CD41+ MK generated from healthy donors were not significantly affected (Figure 7c). Of note, we found that the inhibitory effects of the inactive form of the drug observed in both healthy control (HC) and MPN cultures is off-target as detailed in Supplementary Material and Supplementary Figure S8. The specific inhibition of terminal MPN MK maturation by imetelstat, however, was confirmed by flow cytometry, showing that fully mature CD41+/CD42b+ MK were either absent or reduced in MPN MNCs cultures treated with the drug (Supplementary Figure S7C).
Figure 7.
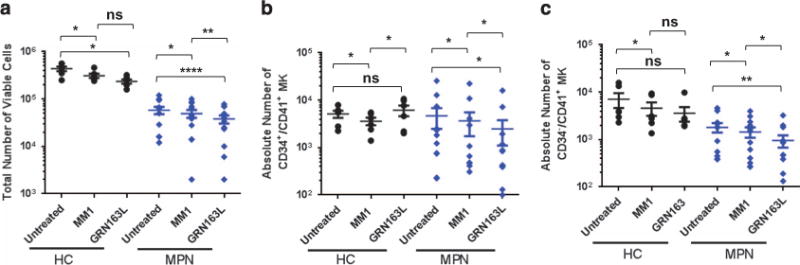
Effects of imetelstat on MPN megakaryopoiesis. (a) Total number viable cells in untreated or drug-treated (MM1 or GRN163L) MK cultures initiated with PB-MNCs from HCs (n = 4) and from MPN patients (n = 12). (b) Quantification of the absolute number of CD34+/CD41+ MK precursors generated by PB-MNCs from HCs (n = 5) or MPN patients (n = 12) after 7 days in cultures untreated and treated with MM1 or GRN163L. (c) Quantification of the absolute number of CD34−/CD41+ MK generated after 7 days in cultures treated with control oligonucleotide (MM1) or GRN163L by PB-MNCs from HCs (n = 6) or MPN patients (n = 12). NS, not statistically significant. *P-value ≤ 0.05, **P-value ≤ 0.005, ****P-value ≤ 0.00005.
The ability of imetelstat to inhibit MPN MK was then compared among the MPN-derived MK cultures; there were no statistically significant differences between MF- and ET-derived cultures or between the different genotypes (that is, JAK2V617V vs other mutations or triple negative) (Supplementary Figure S9). Although the small sample size does not allow definitive conclusions, a trend toward a greater reduction in CFU-MK formation ability was observed in cultures derived from ET patients when compared with those derived from MF patients. MKs generated from three patients with the JAK2V617F mutation were further analyzed by allele-specific PCR27 to evaluate the effects of imetelstat on the genotype of MPN MK. As compared with untreated cultures, treatment with 15 μM imetelstat but not the MM1 reduced the JAK2V617F allele burden in MK derived from two out of the three patients analyzed (Supplementary Table S2).
Imetelstat has preferential activity on malignant but not on normal MK
We next explored the cellular and molecular consequences of imetelstat-mediated effects on normal and malignant megakaryopoiesis. Apoptosis was assessed during the initial and later stages of megakaryopoiesis in cultures derived from normal CD34+ cells exposed to imetelstat. We did not observe a statistically significant difference in the fraction of Annexin V-positive cells in cultures treated with imetelstat when compared with untreated cultures or those treated with the mismatched control MM1 (6 ± 0.1% Annexin-positive cells in untreated cultures, 7 ± 0.8% in cultures treated with 15 μM MM1 and 10.1 ± 1.6% in cultures treated with 15 μM imetelstat; P-values ⩾ 0.05). MK cultures generated by PB-MNCs from healthy donors and from patients with MPN were also assessed for apoptosis. The percentage of Annexin V-positive cells within the CD41+ MK population was determined in cultures from HCs (n = 4) and from MPN patients (n = 8) generated in the absence and presence of 15 μM mismatched MM1 or imetelstat. Interestingly, exposure of both healthy and MPN cultures to imetelstat did not result in a significant increase in the fraction of apoptotic MK when compared with cultures treated with MM1. MM1- and imetelstat-treated HC cultures had 6.8+1.9% and 10.7 ± 3.9% Annexin V+/ CD41+ cells, respectively (P-value = 0.1029); MM1- and imetelstat-treated MPN cultures had 32.3 ± 18.6% and 35.2 ± 26.2% Annexin V+/CD41+ cells, respectively (P-value = 0.3095). These findings suggested that the same imetelstat concentration that affects MK maturation does not induce apoptosis of primary MK.
Considering that malignant MKs contribute to MPN marrow fibrosis and disease progression by elaborating cytokines,28,29 we evaluated the ability of normal and MPN-derived MK to elaborate fibrogenic factors ex vivo. Thus we tested a panel of GFs by multiplex immunoassays to measure their levels in media conditioned (CM) by MK from healthy donors and MPN MNCs in the absence or presence of imetelstat. Of the 12 proteins tested (complete list provided in Materials and methods section), 4 showed statistically significant changes between MM1 and imetelstat-treated cultures: BDNF, FGF-2, PDGF-BB, and VEGF-A, all of which are known to have a role in promoting fibrosis.30–33 In the absence of treatment, these GF were secreted at higher levels in MPN cultures as compared with cultures containing cells from healthy donors (Figure 8a). Yet, in the presence of imetelstat, the levels of these proteins were significantly lower in MPN-derived cultures than those found in cultures from healthy donors (Figure 8b). For instance, the levels of FGF and PDGF were decreased by 25% and 65%, respectively (P-values ≤ 0.005) in MPN cultures treated with imetelstat as compared with those treated with MM1 control. By contrast, imetelstat did not significantly change PDGF levels in cultures derived from healthy donors and induced only a modest decrease in FGF levels (P-value = 0.0496). Similarly, the levels of VEGF-A were reduced by imetelstat treatment in MPN but not in normal MK cultures. Finally, the drug decreased the amount of BDNF by 55% in CM from MPN MK and only by 15% in CM from normal MK cultures. Of note, MK and platelets are the major sources of BDNF found in the human serum.34 Although a role of BDNF in MPN-related fibrosis has not been reported, it has been demonstrated that it contributes to fibroblast proliferation and the development of pulmonary fibrosis.33 Interestingly, we did not observe significant changes in the levels of transforming growth factor-β measured in MPN MK cultures following imetelatat treatment. Taken together, these results indicate that the levels of fibrogenic factors secreted by primary MPN MK ex vivo are diminished following treatment with imetelstat, providing a possible explanation for the ability of this drug to reduce BM fibrosis in MPN patients.
Figure 8.
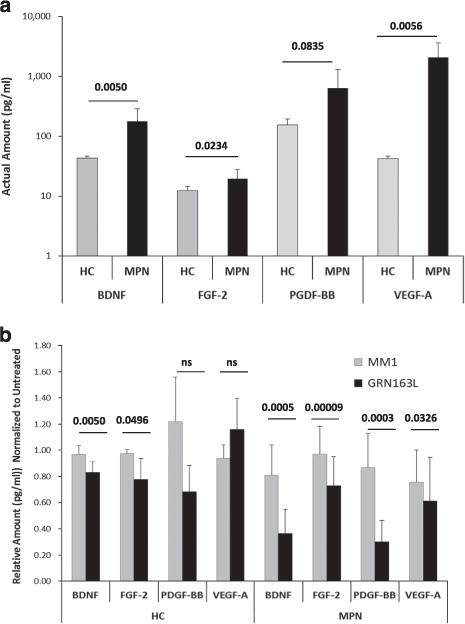
Quantitative analysis of the GFs present in MK conditioned media. Media conditioned (CM) by MK cultures generated from untreated HCs and MPN PB-MNCs (a) and by MK cultures treated with MM1 or GRN163L for 7 days (b) were analyzed by multiplex immunoassays to measure the secreted levels of BDNF, FGF-2, PDGF-BB and VEGF-A. The results represent the mean ± s.d. measured in CM generated by cultures from HCs (n = 4) and MPN patients (n = 8). NS, not statistically significant.
DISCUSSION
Thrombocytopenia is one of the major dose-limiting toxicities associated with imetelstat administration to patients with hematological malignancies and solid tumors.6,17,18 At the same time, imetelstat treatment resulted in remarkable clinical and molecular responses in ET and MF patients.5,6 In this study, we provide novel insights into the role of telomerase inhibition in normal and MPN-related megakaryopoiesis.
Initially, we demonstrated that, upon imetelstat exposure, normal CD34+ HPC were capable of generating CFU-MK and committing to MK lineage but had limited ability to become fully mature MK. This was reflected by a delayed loss of CD34 expression, which resulted in the accumulation of CD34+/CD41+ MK precursors at the expense of more mature CD34−/CD41+ and fully mature CD41+/CD42b+ MK (Figures 2 and 3). This delay in maturation, however, was reversible as drug removal was able to restore the CD34−/CD41+ MK population.
In order to become polyploid, MK precursors cease to divide but continue DNA synthesis in the absence of physical cell division.35 In this study, we showed that imetelstat prevented MK polyploidization (Figure 5) but did not interfere with DNA synthesis during endomitosis. The latter process was evaluated by using BrdU incorporation assays, which revealed that the proportion of BrdU-positive MK was greater in imetelstat-treated cultures as compared with controls (Figure 6a). Interestingly, this increase was accounted for by the fraction of BrdU-positive MK with diploid DNA content, which was twice as frequent in cultures treated with imetelstat as compared with both untreated and mismatched oligonucleotide MM1-treated controls (Figures 6a and b). This apparently surprising result correlated with the accumulation of MK precursors and immature MK (that is, two cell populations actively synthesizing DNA) in imetelstat-treated cultures. It is known that interfering with telomere function in diploid eukaryotic cells triggers DNA damage leading to cell death.36 We found that normal MKs exposed to imetelstat did not develop significant degrees of apoptosis. We speculate that human MK may respond to telomerase dysfunction not by undergoing apoptosis but by arresting polyploidization. This would explain not only the reduced MK ploidy levels but also the survival of imetelstat-treated cultures enriched in diploid and low ploidy MKs. Thus imetelstat prevents these low ploidy MK precursors to undergo further polyploidization and maturation, which results in reduced number of mature MK.
Our results, schematically summarized in Supplementary Figure S10, indicate that imetelstat affects normal and MPN megakaryopoiesis by acting at different stages of MK development. The inhibition of MK maturation observed in normal MK cultures exposed to imetelstat would result in a diminished pool of platelet-forming mature MK, which would eventually lead to reduced platelet numbers in patients with normal BM function. Notably, we found that imetelstat-induced maturation defect of normal MK is reversible after drug removal, suggesting that interruption of treatment in patients can restore platelet counts. By contrast, we demonstrate that imetelstat selectively inhibits MPN megakaryopoiesis by acting at additional stages of MK development. First, exposure to imetelstat inhibited CFU-MK formation by MPN but not by normal HPCs (Figure 1b). These results confirm the observations by Baerlocher and colleagues who reported reduced endogenous CFU-MK formation by ET but not by normal CD34+ cells exposed to imetelstat in vitro and in vivo.5,20 Moreover, preferential inhibition of malignant HSC/HPC by imetelstat was reported in a study of splenic CD34+ cells from patients with MF37 and in a preclinical model of acute myeloid leukemia (AML).10 By using patient-derived AML xenografts, Bruedigam et al.38 have recently demonstrated robust efficacy of imetelstat against AML cells while relatively preserving normal myeloid and HSC.
Second, the effects of the drug on MK differentiation were more accentuated in MPN MK cultures; there were fewer CD34+/CD41+ MK precursors in drug-treated MPN cultures and the generation of more terminally differentiated CD34−/CD41+ and CD42b+/CD41+ MK was significantly inhibited as compared with controls (Figure 7). Given that malignant MKs are major contributors to the development of fibrosis in MPN,28,29 we measured several fibrogenic GFs in the CM generated by normal and MPN-derived MK cultures. The levels of three GFs known to be involved in promoting fibrosis either directly (PDGF, FGF) or indirectly (VEGF)30–32 were considerably downregulated by imetelstat in cultures of MPN but not of normal MKs (Figure 8). The levels of BDNF, a neurotrophic factor mainly synthesized by MK and released from platelets in humans, was reduced by imetelstat treatment to a greater extent in MPN-derived CM as compared with normal donors-derived CM. It has been reported that BDNF stimulates lung fibroblast proliferation and contributes to the development of pulmonary fibrosis,33 but its role in promoting marrow fibrosis in MF has yet to be explored. Nonetheless, the reduced secretion of these fibrogenic factors in MPN MK cultures exposed to imetelstat not only correlates with the drug’s ability to inhibit MK maturation but also provides a potential explanation for the reduced marrow fibrosis observed in MPN patients treated with imetelstat.6 Given that MNC-derived MK cultures are contaminated by other myeloid cells such as monocytes/macrophages, it is possible that the frequency and secretory profile of these cells could also be affected by imetelstat treatment. Although the presence of non-MK cells was not evaluated in this study, in addition to GF, a panel of secreted cytokines were also assessed in the CM from MK-biased cultures. Of the 25 cytokines evaluated, only interleukin (IL)-4, IL-6, IL-10, IL-17A and IL-1RA were found at detectable but very low levels across HC and MPN cultures. Yet, there was no difference in the levels of these cytokines between untreated and imetelstat-treated conditions (data not shown). Moreover, tumor necrosis factor-α and IL-1, which are the major cytokines secreted by macrophages, were not detected in either HC or MPN cultures.
Our findings, corroborated with the observations by others, strongly suggest that by acting at the MPN HSC/HPC level and at the different stages of MPN megakaryopoiesis, imetelstat may diminish the pool of malignant MKs thus explaining, at least in part, the clinical benefits of the drug observed in the phase I/II studies in MPN patients. We show that the inhibitory effects of imetelstat on either MPN CFU-MK formation or MK generation in liquid culture occur irrespective of their mutational status or MPN type (Supplementary Table S1 and Supplementary Figure S9). Recent clinical studies have also shown that both JAK2V617F and CALR mutation burdens were each reduced by imetelstat treatment.5,6,37 It has been suggested that TA during ex vivo culture of hematopoietic cells from AML, chronic myeloid leukemia and MPN patients is different than that found in their unmanipulated counterparts.39 It is therefore conceivable that the inhibitory actions of imetelstat observed ex vivo in MPN hematopoietic cells may not entirely reflect those occurring in patients treated with imetelstat. Furthermore, as recently reported by Tefferi et al.,6 the clinical response to imetelstat may be also determined by the mutational status and/or the presence of secondary mutations.
In a clinical trial of imetelstat in MPN patients, significant changes were not observed between baseline and posttreatment telomere length among patients who had a clinical response.6 The companion study, however, reports that telomere length did not predict response to imetelstat but TA was reduced in average by 36% in the six patients analyzed.5 In this study, we demonstrate that imetelstat inhibits telomerase in normal MK but it reduced hTERT levels in only one of the four MPN patients analyzed (Figure 4 and Supplementary Figure S5). Although these observations could be attributed to the reduced sample size and the different cell fractions analyzed, it has been suggested that imetelstat’s actions in MPN might be due to off-target effects.40 Alternatively, the response to imetelstat could be influenced by the baseline levels of telomerase, which are reportedly elevated in MPN patients,41,42 and/or by the epigenetic makeup of the TERT promoter as has been recently shown in AML/myelodysplastic syndromes.43 Taken together, these observations indicate that TA inhibition might not be the only mode of action of imetelstat and the exact molecular mechanisms underlying its effects on normal and malignant MK require further investigation.
In summary, we report that ex vivo MPN megakaryopoiesis is selectively inhibited by imetelstat owing to its ability to decrease clonogenicity, prevent MK maturation and reduce the secretion of fibrogenic factors. By contrast, imetelstat affected normal mega-karyopoiesis by solely delaying MK precursor maturation, which was reversible upon drug withdrawal. Thus the inhibitory actions of imetelstat on MPN HSC/HPC combined to the more accentuated inhibition of MPN MK maturation may explain the differential effects observed between normal and MPN MK. These data point to the potential mechanisms by which imetelstat exerts its inhibitory effects on malignant MK in patients with MPN and serve as an explanation for the thrombocytopenia which occurs in patients with MKs that are not involved by a malignant process.
Supplementary Material
Acknowledgments
We thank Dr Fei Huang from Janssen Research & Development, LLC for the critical discussion of the manuscript. Research funding for this work was provided by Geron Corporation and Janssen Research & Development, LLC. Janssen Biotech, Inc. has entered into an exclusive worldwide license and collaboration agreement to develop and commercialize imetelstat for oncology, including hematological myeloid malignancies. CI-R has received research funding for this work from Geron Corporation and Janssen Research & Development, LLC.
Footnotes
CONFLICT OF INTEREST
KE is a former employee of Geron Corporation. The other authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS GM performed research, analyzed data and discussed results. TK performed research and analyzed data. FY performed research. KE analyzed data and discussed results. JDC revised the manuscript. RH designed the study, interpreted results and wrote the manuscript. CI-R designed the study, analyzed data, interpreted results and wrote the manuscript.
Supplementary Information accompanies this paper on the Leukemia website (http://www.nature.com/leu)
References
- 1.Mascarenhas J, Hoffman R. Myeloproliferative neoplasms: new translational therapies. Mt Sinai J Med. 2010;77:667–683. doi: 10.1002/msj.20225. [DOI] [PubMed] [Google Scholar]
- 2.Tefferi A, Pardanani A. Myeloproliferative neoplasms: a contemporary review. JAMA Oncol. 2015;1:97–105. doi: 10.1001/jamaoncol.2015.89. [DOI] [PubMed] [Google Scholar]
- 3.Roth A, Harley CB, Baerlocher GM. Imetelstat (GRN163L)—telomerase-based cancer therapy. Recent Results Cancer Res. 2010;184:221–234. doi: 10.1007/978-3-642-01222-8_16. [DOI] [PubMed] [Google Scholar]
- 4.Ruden M, Puri N. Novel anticancer therapeutics targeting telomerase. Cancer Treat Rev. 2013;39:444–456. doi: 10.1016/j.ctrv.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 5.Baerlocher GM, Oppliger Leibundgut E, Ottmann OG, Spitzer G, Odenike O, McDevitt MA, et al. Telomerase inhibitor imetelstat in patients with essential thrombocythemia. N Engl J Med. 2015;373:920–928. doi: 10.1056/NEJMoa1503479. [DOI] [PubMed] [Google Scholar]
- 6.Tefferi A, Lasho TL, Begna KH, Patnaik MM, Zblewski DL, Finke CM, et al. A pilot study of the telomerase inhibitor imetelstat for myelofibrosis. N Engl J Med. 2015;373:908–919. doi: 10.1056/NEJMoa1310523. [DOI] [PubMed] [Google Scholar]
- 7.Blackburn EH. Telomeres and telomerase: their mechanisms of action and the effects of altering their functions. FEBS Lett. 2005;579:859–862. doi: 10.1016/j.febslet.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 8.Mocellin S, Pooley KA, Nitti D. Telomerase and the search for the end of cancer. Trends Mol Med. 2013;19:125–133. doi: 10.1016/j.molmed.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Brennan SK, Wang Q, Tressler R, Harley C, Go N, Bassett E, et al. Telomerase inhibition targets clonogenic multiple myeloma cells through telomere length-dependent and independent mechanisms. PLoS One. 2010;5:e12487, 1–8. doi: 10.1371/journal.pone.0012487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruedigam C, Bagger FO, Heidel FH, Paine Kuhn C, Guignes S, Song A, et al. Telomerase inhibition effectively targets mouse and human AML stem cells and delays relapse following chemotherapy. Cell Stem Cell. 2014;15:775–790. doi: 10.1016/j.stem.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burchett KM, Yan Y, Ouellette MM. Telomerase inhibitor Imetelstat (GRN163L) limits the lifespan of human pancreatic cancer cells. PLoS One. 2014;9:e85155. doi: 10.1371/journal.pone.0085155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hochreiter AE, Xiao H, Goldblatt EM, Gryaznov SM, Miller KD, Badve S, et al. Telomerase template antagonist GRN163L disrupts telomere maintenance, tumor growth, and metastasis of breast cancer. Clin Cancer Res. 2006;12:3184–3192. doi: 10.1158/1078-0432.CCR-05-2760. [DOI] [PubMed] [Google Scholar]
- 13.Joseph I, Tressler R, Bassett E, Harley C, Buseman CM, Pattamatta P, et al. The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010;70:9494–9504. doi: 10.1158/0008-5472.CAN-10-0233. [DOI] [PubMed] [Google Scholar]
- 14.Koziel JE, Herbert BS. The telomerase inhibitor imetelstat alone, and in combination with trastuzumab, decreases the cancer stem cell population and self-renewal of HER2+ breast cancer cells. Breast Cancer Res Treat. 2015;149:607–618. doi: 10.1007/s10549-015-3270-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marian CO, Cho SK, McEllin BM, Maher EA, Hatanpaa KJ, Madden CJ, et al. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin Cancer Res. 2010;16:154–163. doi: 10.1158/1078-0432.CCR-09-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marian CO, Wright WE, Shay JW. The effects of telomerase inhibition on prostate tumor-initiating cells. Int J Cancer. 2010;127:321–331. doi: 10.1002/ijc.25043. [DOI] [PubMed] [Google Scholar]
- 17.Thompson PA, Drissi R, Muscal JA, Panditharatna E, Fouladi M, Ingle AM, et al. A phase I trial of imetelstat in children with refractory or recurrent solid tumors: a Children’s Oncology Group Phase I Consortium Study (ADVL1112) Clin Cancer Res. 2013;19:6578–6584. doi: 10.1158/1078-0432.CCR-13-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiappori AA, Kolevska T, Spigel DR, Hager S, Rarick M, Gadgeel S, et al. A ran-domized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann Oncol. 2015;26:354–362. doi: 10.1093/annonc/mdu550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaushansky K. Thrombopoiesis. Semin Hematol. 2015;52:4–11. doi: 10.1053/j.seminhematol.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Brunold C, Braschler T, Go N, Ninomoto J, Kashani H, Stuart M, et al. Imetelstat, a potent telomerase inhibitor, inhibits the spontaneous growth of CFU-Meg in vitro from essential thrombocythemia patients but not from healthy individuals. Blood. 2011;118 Abstract 3843. [Google Scholar]
- 21.Iancu-Rubin C, Gajzer D, Mosoyan G, Feller F, Mascarenhas J, Hoffman R. Pano-binostat (LBH589)-induced acetylation of tubulin impairs megakaryocyte maturation and platelet formation. Exp Hematol. 2012;40:564–574. doi: 10.1016/j.exphem.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iancu-Rubin C, Gajzer D, Tripodi J, Najfeld V, Gordon RE, Hoffman R, et al. Down-regulation of stathmin expression is required for megakaryocyte maturation and platelet production. Blood. 2011;117:4580–4589. doi: 10.1182/blood-2010-09-305540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iancu-Rubin C, Mosoyan G, Glenn K, Gordon RE, Nichols GL, Hoffman R. Activation of p53 by the MDM2 inhibitor RG7112 impairs thrombopoiesis. Exp Hematol. 2014;42:137–145e5. doi: 10.1016/j.exphem.2013.11.012. [DOI] [PubMed] [Google Scholar]
- 24.Baccini V, Roy L, Vitrat N, Chagraoui H, Sabri S, Le Couedic JP, et al. Role of p21 (Cip1/Waf1) in cell-cycle exit of endomitotic megakaryocytes. Blood. 2001;98:3274–3282. doi: 10.1182/blood.v98.12.3274. [DOI] [PubMed] [Google Scholar]
- 25.Koike Y, Miyazaki K, Higashihara M, Kimura E, Jona M, Uchihashi K, et al. Clinical significance of detection of immature platelets: comparison between percentage of reticulated platelets as detected by flow cytometry and immature platelet fraction as detected by automated measurement. Eur J Haematol. 2010;84:183–184. doi: 10.1111/j.1600-0609.2009.01364.x. [DOI] [PubMed] [Google Scholar]
- 26.Dusse LM, Freitas LG. Clinical applicability of reticulated platelets. Clin Chim Acta. 2015;439:143–147. doi: 10.1016/j.cca.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 27.Lu M, Xia L, Li Y, Wang X, Hoffman R. The orally bioavailable MDM2 antagonist RG7112 and pegylated interferon alpha 2a target JAK2V617F-positive progenitor and stem cells. Blood. 2014;124:771–779. doi: 10.1182/blood-2013-11-536854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ciurea SO, Merchant D, Mahmud N, Ishii T, Zhao Y, Hu W, et al. Pivotal contributions of megakaryocytes to the biology of idiopathic myelofibrosis. Blood. 2007;110:986–993. doi: 10.1182/blood-2006-12-064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vannucchi AM, Migliaccio AR, Paoletti F, Chagraoui H, Wendling F. Pathogenesis of myelofibrosis with myeloid metaplasia: lessons from mouse models of the disease. Semin Oncol. 2005;32:365–372. doi: 10.1053/j.seminoncol.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Kreipe H, Busche G, Bock O, Hussein K. Myelofibrosis: molecular and cell biological aspects. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S21. doi: 10.1186/1755-1536-5-S1-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaudhary NI, Roth GJ, Hilberg F, Muller-Quernheim J, Prasse A, Zissel G, et al. Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur Respir J. 2007;29:976–985. doi: 10.1183/09031936.00152106. [DOI] [PubMed] [Google Scholar]
- 32.Mesa RA, Hanson CA, Rajkumar SV, Schroeder G, Tefferi A. Evaluation and clinical correlations of bone marrow angiogenesis in myelofibrosis with myeloid meta-plasia. Blood. 2000;96:3374–3380. [PubMed] [Google Scholar]
- 33.Ricci A, Graziano P, Bronzetti E, Saltini C, Sciacchitano S, Cherubini E, et al. Increased pulmonary neurotrophin protein expression in idiopathic interstitial pneumonias. Sarcoidosis Vasc Diffuse Lung Dis. 2007;24:13–23. [PubMed] [Google Scholar]
- 34.Chacon-Fernandez P, Sauberli K, Colzani M, Moreau T, Ghevaert C, Barde YA. Brain-derived neurotrophic factor in megakaryocytes. J Biol Chem. 2016;291:9872–9881. doi: 10.1074/jbc.M116.720029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravid K, Lu J, Zimmet JM, Jones MR. Roads to polyploidy: the megakaryocyte example. J Cell Physiol. 2002;190:7–20. doi: 10.1002/jcp.10035. [DOI] [PubMed] [Google Scholar]
- 36.Longhese MP. DNA damage response at functional and dysfunctional telomeres. Genes Dev. 2008;22:125–140. doi: 10.1101/gad.1626908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Hu C, Li Y, Qiu J, Lam M, Eng K, et al. Effects of imetelstat on CD34+ cells of patients with myelofibrosis. Blood. 2014;124 Abstract 1879. [Google Scholar]
- 38.Bruedigam C, Wackrow B, Song A, Porter A, Lee S, Moore A, et al. The preclinical efficacy of a novel telomerase inhibitor, imetelstat, in AML - a randomized trial in patient-derived xenografts. Blood. 2016 ASH Annual Meeting; Abstract 578:202. [Google Scholar]
- 39.Engelhardt M, Mackenzie K, Drullinsky P, Silver RT, Moore MA. Telomerase activity and telomere length in acute and chronic leukemia, pre- and post-ex vivo culture. Cancer Res. 2000;60:610–617. [PubMed] [Google Scholar]
- 40.Armanios M, Greider CW. Treating myeloproliferation—on target or off? N Engl J Med. 2015;373:965–966. doi: 10.1056/NEJMe1508740. [DOI] [PubMed] [Google Scholar]
- 41.Ruella M, Salmoiraghi S, Risso A, Carobbio A, Buttiglieri S, Spatola T, et al. Telomere shortening in Ph-negative chronic myeloproliferative neoplasms: a biological marker of polycythemia vera and myelofibrosis, regardless of hydroxycarbamide therapy. Exp Hematol. 2013;41:627–634. doi: 10.1016/j.exphem.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 42.Spanoudakis E, Bazdiara I, Pantelidou D, Kotsianidis I, Papadopoulos V, Margaritis D, et al. Dynamics of telomere’s length and telomerase activity in Philadelphia chromosome negative myeloproliferative neoplasms. Leuk Res. 2011;35:459–464. doi: 10.1016/j.leukres.2010.07.042. [DOI] [PubMed] [Google Scholar]
- 43.Zhao X, Tian X, Kajigaya S, Cantilena CR, Strickland S, Savani BN, et al. Epigenetic landscape of the TERT promoter: a potential biomarker for high risk AML/MDS. Br J Haematol. 2016;175:427–439. doi: 10.1111/bjh.14244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.