

Abstract
Delivery of miniaturized dystrophin genes via adeno-associated viral vectors is one leading approach in development to treat Duchenne muscular dystrophy. Here we directly compared the functionality of five mini- and micro-dystrophins via skeletal muscle-specific transgenic expression in dystrophin-deficient mdx mice. We evaluated their ability to rescue defects in the microtubule network, passive stiffness and contractility of skeletal muscle. Transgenic mdx mice expressing the short dystrophin isoform Dp116 served as a negative control. All mini- and micro-dystrophins restored elevated detyrosinated α-tubulin and microtubule density of mdx muscle to values not different from C57BL/10, however, only mini-dystrophins restored the transverse component of the microtubule lattice back to C57BL/10. Passive stiffness values in mdx muscles expressing mini- or micro-dystrophins were not different from C57BL/10. While all mini- and micro-dystrophins conferred significant protection from eccentric contraction-induced force loss in vivo and ex vivo compared to mdx, removal of repeats two and three resulted in less protection from force drop caused by eccentric contraction ex vivo. Our data reveal subtle yet significant differences in the relative functionalities for different therapeutic constructs of miniaturized dystrophin in terms of protection from ex vivo eccentric contraction-induced force loss and restoration of an organized microtubule lattice.
Introduction
Loss of dystrophin protein results in the X-linked recessive disease Duchenne muscular dystrophy [DMD; (1,2)]. As a structural protein, dystrophin links the actin cytoskeleton to the transmembrane dystrophin-glycoprotein complex [DGC; (3–7)]. Without dystrophin, the sarcolemma lacks stability and becomes damaged during muscle contraction (8–10). Striated muscle of DMD patients presents with fibrosis, inflammation and atrophy (11,12). Muscle weakness leads to loss of ambulation and eventual death from cardiac or respiratory failure in the second to third decade of life (13). Current therapies such as ventilator support (14) and corticosteroids (15) prolong life but do not provide a cure for the 1 in 5000 boys born with DMD (16).
The mdx mouse is the most studied model for DMD (17,18). Exhibiting a lack of dystrophin protein, mdx mice display fibrosis, inflammation and loss of muscle strength similar to DMD patients, albeit proportionally later in the mouse lifespan (19,20). Loss of dystrophin in mdx mice also impacts the microtubule cytoskeleton. Wild-type skeletal muscle exhibits a subsarcolemmal microtubule lattice organized into a perpendicular array of longitudinal and transverse elements that become disorganized in mdx muscle along with densification of longitudinal microtubules and loss of the transverse microtubules (4,5,21,22). Dystrophin is proposed to act as a guide to position microtubules transversely along the Z-disk and M-line and its loss can lead to lattice disorganization (4,23). Mice lacking ankyrin-B, β2-spectrin, dynactin-4 or obscurin also present with dystrophin mislocalization and disorganized microtubules (24–26), while transgenic expression of nearly full-length dystrophin on the mdx background is able to restore microtubule organization back to normal (5).
We have shown that disorganization and densification of the microtubule lattice correlates with a loss of torque production in anterior crural muscles exposed to eccentric contractions in vivo (5). Other groups have analyzed tubulin modifications in mdx tissue such as the post-translation and post-polymerization α-tubulin detyrosination modification associated with stable microtubules (27,28). α-Tubulin detyrosination is increased in mdx skeletal (22,29) and cardiac muscle (30), and is thought to stiffen the cytoskeleton leading to mechanically stimulated production of reactive oxygen species (ROS) and increased cytosolic calcium (31). The increased cytoskeletal stiffness may rely upon detyrosinated α-tubulin crosslinks with desmin intermediate filaments (32) which are also elevated at the protein level in mdx mice (29,33,34).
It remains unclear how dystrophin may influence microtubule organization in vivo. While we previously showed that spectrin like repeats 20–24 of dystrophin were required for microtubule binding activity in vitro, transgenic expression of a dystrophin construct substituted with the equivalent but microtubule non-binding repeats of utrophin was still able to restore lattice density and organization of mdx muscle in vivo (5). Here, we attempted to gain insight into the dystrophin domains necessary for normal microtubule lattice density and organization by measuring these properties in several previously reported transgenic mdx mouse lines expressing truncated dystrophin constructs (35–38). Most truncations removed variable portions of the central rod region, which is often deleted in a milder dystrophinopathy known as Becker muscular dystrophy [BMD (39,40)], but retained the N-terminal actin binding (NT) and cysteine-rich (CR) domains because they are critical for binding to actin and to the DGC, respectively (41–43). Because we previously found that transgenic expression of a nearly full-length dystrophin lacking only the C-terminal domain (CT) failed to reduce the elevated passive stiffness of mdx muscle back to C57BL/10 levels (5), we also measured mechanical function across the lines expressing miniaturized dystrophins inclusive or exclusive of the CT domain. Our data show that both the NT and the CR domains are required to significantly improve microtubule organization beyond that measured in mdx. Truncated dystrophins sharing only the NT and CR domains were also fully rescued for microtubule density to C57BL/10 levels and significantly protected anterior crural muscles from increased torque and force loss after in vivo and ex vivo eccentric contractions, respectively. However, complete rescue of microtubule organization to C57BL/10 appears to require domains in addition to the NT and CR, and potentially spectrin-like repeats 20–24.
Results
This study made use of five available transgenic mdx lines expressing truncated dystrophin constructs (35–38). DysΔH2-R15-mdx expressed a mini-dystrophin that models a Becker-like internal deletion, while DysΔR4–23/ΔCT-mdx, flµDys-mdx (DysΔR4–23/ΔCT flanked by loxP sites, see Materials and Methods), and DysΔR2–15/ΔR18–23/ΔCT-mdx expressed micro-dystrophins with larger deletions (Fig. 1A). An additional Becker-like truncated dystrophin, DysΔH2-R19/ΔCT-mdx, has been partly characterized previously (5) and was analyzed here for additional cytoskeleton and physiological pathologies. The sixth line, Dp116-mdx, served as a transgenic control which expressed DP116, a short dystrophin isoform endogenously expressed from an internal promoter in Schwann cells (44). DysΔH2-R15-mdx, DysΔH2-R19/ΔCT-mdx, DysΔR4–23/ΔCT-mdx and Dp116-mdx transgenic models have been previously confirmed to express dystrophin of the expected molecular weight at levels near or above wild-type dystrophin protein expression (5,35–38). Transverse extensor digitorum longus (EDL) sections stained for dystrophin confirmed continuous sarcolemmal dystrophin localization in C57BL/10 as well as the transgenic mini-dystrophins, micro-dystrophins and Dp116-mdx (Fig. 1B).
Figure 1.
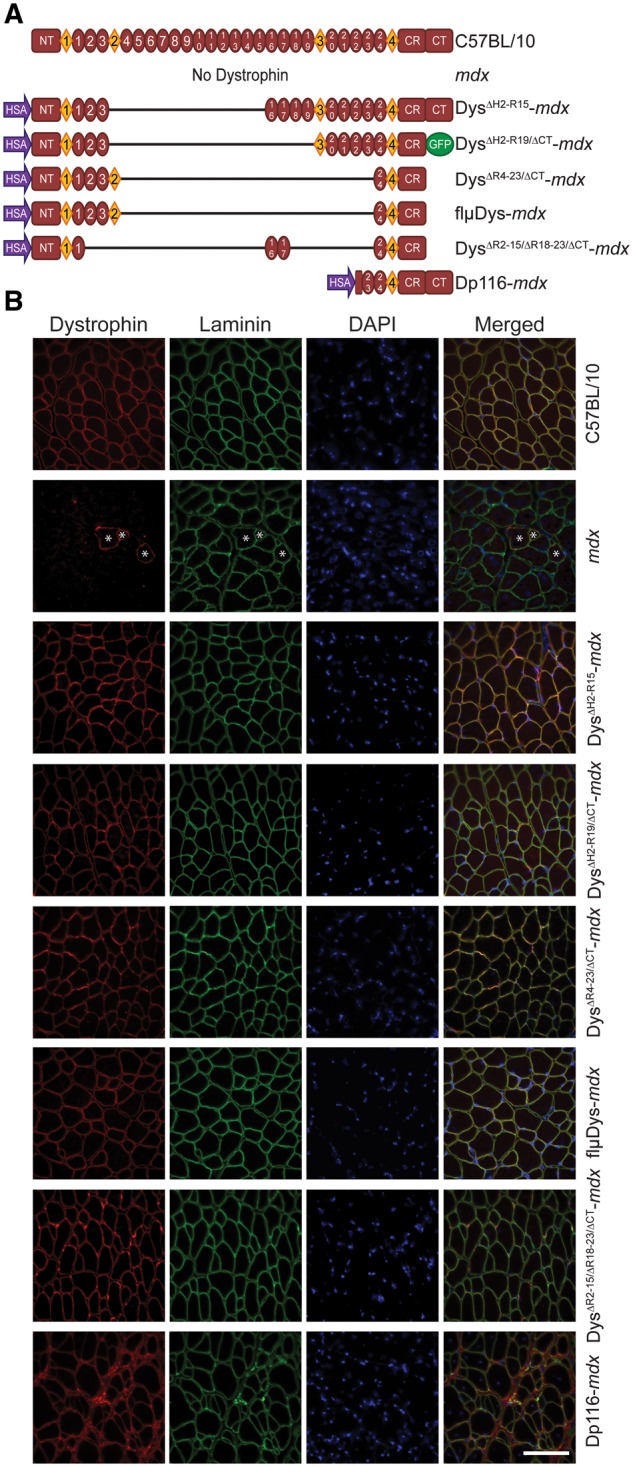
Schematic representation of transgenic dystrophin constructs. (A) Full-length dystrophin in C57BL/10, absence of dystrophin in mdx and skeletal muscle-specific truncated dystrophins on the mdx background. HSA, human skeletal α-actin promoter; NT, N-terminus; diamonds represent hinge regions; ovals represent spectrin-like repeats; CR, cysteine-rich domain; CT, C-terminus. (B) Sarcolemmal localization of C57BL/10 and transgenic dystrophin. Dystrophin protein in mdx mice was present only in revertant fibers (denoted with *). Laminin outlines all individual fibers. Dystrophin (red), laminin (green), nuclei (DAPI). Scale bar is 100 µm.
Immunofluorescence microscopy of α-tubulin in EDL fibers from C57BL/10, mdx and the transgenic lines was used to assess the microtubule lattice (Fig. 2A). Planar density of the subsarcolemmal lattice revealed that DysΔH2-R15-mdx, DysΔR4–23/ΔCT-mdx, flµDys-mdx and DysΔR2–15/ΔR18–23/ΔCT-mdx were all restored to densities not different from C57BL/10 (Fig. 2B). In contrast, Dp116-mdx microtubule density remained significantly elevated above C57BL/10 levels (Fig. 2B). Microtubule lattice organization was assessed via the angle of microtubules within the subsarcolemmal lattice relative to the fiber long axis as measured by the texture detection technique (TeDT) program (45). Disorganized mdx fibers lacked transverse microtubules while C57BL/10 organized fibers had a large proportion of transverse microtubules at 90° from the muscle fiber axis. The two mdx lines expressing mini-dystrophins, DysΔH2-R15-mdx and DysΔH2-R19/ΔCT-mdx (5), were both highly organized and not different from C57BL/10 microtubule organization (Fig. 2C). The three micro-dystrophins, DysΔR4–23/ΔCT-mdx, flµDys-mdx and DysΔR2–15/ΔR18–23/ΔCT-mdx, were all significantly less organized than C57BL/10 although each none-the-less afforded significant protection over mdx (Fig. 2C). Dp116-mdx showed a lack of transverse microtubules that was not significantly different from mdx. Western blot analysis showed that all five mini- and micro-dystrophin transgenic models had C57BL/10 levels of detyrosinated α-tubulin while Dp116-mdx remained elevated above C57BL/10 (Fig. 3A). Dp116-mdx also had elevated desmin protein levels as did DysΔR4–23/ΔCT-mdx and DysΔR2–15/ΔR18–23/ΔCT-mdx (Fig. 3B).
Figure 2.

Effect of mini- and micro-dystrophin expression on microtubule lattice organization and densification. (A) Subsarcolemmal microtubules were organized into a precise perpendicular lattice in EDL fibers from C57BL/10 and DysΔH2-R15-mdx; an intermediate lattice in DysΔR4–23/ΔCT-mdx, flμDys-mdx and DysΔR2–15/ΔR18–23/ΔCT-mdx; and a disorganized lattice in mdx and Dp116-mdx. Images are representative of those obtained for n≥10 fibers from n≥5 mice for each genotype except Dp116-mdx where n=3. Scale bar is 20 μm. (B) Microtubule density significantly varied across genotypes by one-way ANOVA (P<0.001). Mdx and Dp116-mdx exhibited significantly more dense microtubule lattices compared to C57BL/10 by Dunnett’s post-test (*P<0.001). The four mini-/micro-Dys lines were all restored to densities not different from density in C57BL/10 mice. (C) Transverse microtubules (near 90°) were not significantly different between DysΔH2-R15-mdx and C57BL/10. DysΔR4–23/ΔCT-mdx, flμDys-mdx and DysΔR2–15/ΔR18–23/ΔCT-mdx all exhibited significantly less transverse microtubules than C57BL/10, but also significantly more than mdx. Dp116-mdx was not different from mdx, which had significantly less transverse microtubules than all other lines. Angle of microtubule intersection varied by genotype (interaction P<0.001) in a two-way ANOVA. Bonferroni post-hoc t-test analysis concluded that DysΔR4–23/ΔCT-mdx, flμDys-mdx and DysΔR2–15/ΔR18–23/ΔCT-mdx were significantly less than C57BL/10 at *P<0.001 from 84 to 92°. Dp116-mdx and mdx were significantly less than C57BL/10 at #P<0.001 from 76 to 100°. The colors of the microtubule directionality traces match the symbol color-to-genotype scheme used in Figure 2B. Error bars are mean±SEM.
Figure 3.
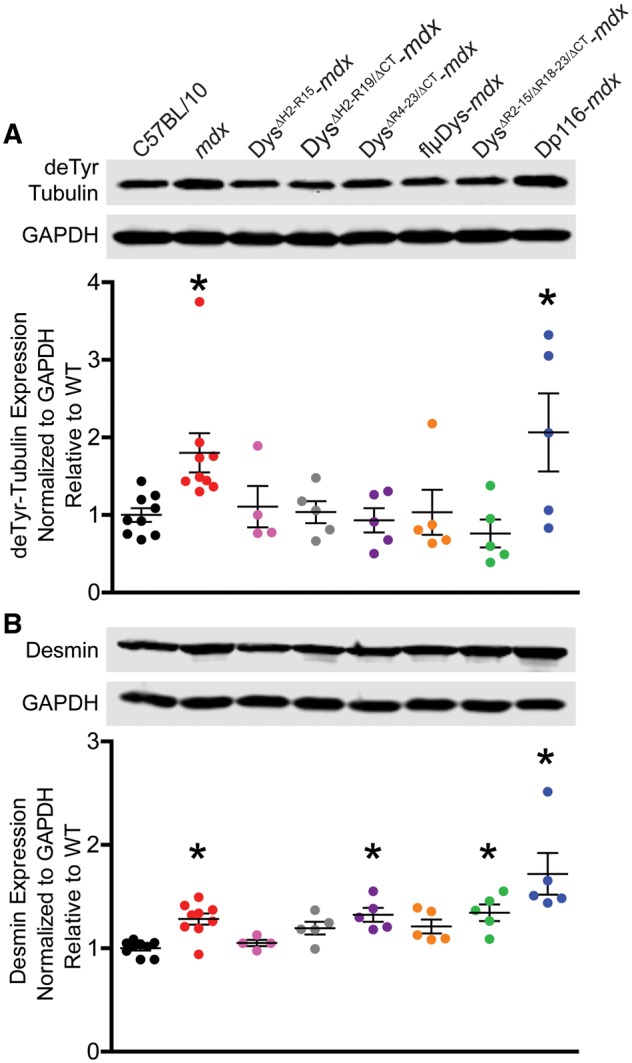
Effect of mini- and micro-dystrophin expression on detyrosinated α-tubulin and desmin immunoreactivity. (A) All mini- and micro-dystrophin transgenic mdx exhibited C57BL/10 levels of detyrosinated α-tubulin while mdx and Dp116-mdx were significantly elevated above C57BL/10 (one-way ANOVA; P=0.006). Dunnett’s post-test to compare each line with C57BL/10 considered significant at *P<0.05. (B) Desmin protein levels were significantly different across lines by one-way ANOVA (P<0.001). By Dunnett’s post-tests, both mini-dystrophin transgenics and flμDys-mdx restored desmin protein levels to not different from C57BL/10 while mdx (*P<0.05), DysΔR4–23/ΔCT-mdx (*P<0.05), DysΔR2–15/ΔR18–23/ΔCT-mdx (*P<0.05) and Dp116-mdx (*P<0.001) remained significantly elevated. Error bars are mean±SEM.
As each of the mini- and micro-dystrophins improved microtubule organization in the mdx mouse (Fig. 2C), we expected that each line would also protect against eccentric contraction-induced force drop as compared to mdx. Because the physiology of DysΔH2-R15-mdx, DysΔR4–23/ΔCT-mdx, DysΔR4–23/ΔCT-mdx, DysΔR2–15/ΔR18–23/ΔCT-mdx and Dp116-mdx muscle were previously characterized in different studies with slightly different protocols (5,35,36,46,47), we reassessed here to directly compare the lines in our standard protocols (5). First, we measured in vivo isometric torque of the anterior crural muscles (tibialis anterior, EDL and extensor hallucis longus muscles). There were no differences in maximal isometric torque of the anterior crural muscles among C57BL/10, mdx and the six transgenic mdx mice (Fig. 4A), including when normalized to body mass (data not shown). These data suggest that torque generating capacity is independent of microtubule lattice organization. Following eccentric contractions of the anterior crural muscles, we measured no difference in force drop between C57BL/10 and the mini- and micro-dystrophin transgenic mdx models (Fig. 4B and C), while Dp116-mdx and mdx mice lost significantly more force from contractions 10 through 70, indicating that subsarcolemmal microtubule lattice disorganization contributes to in vivo eccentric contraction-induced force drop as previously observed (29).
Figure 4.
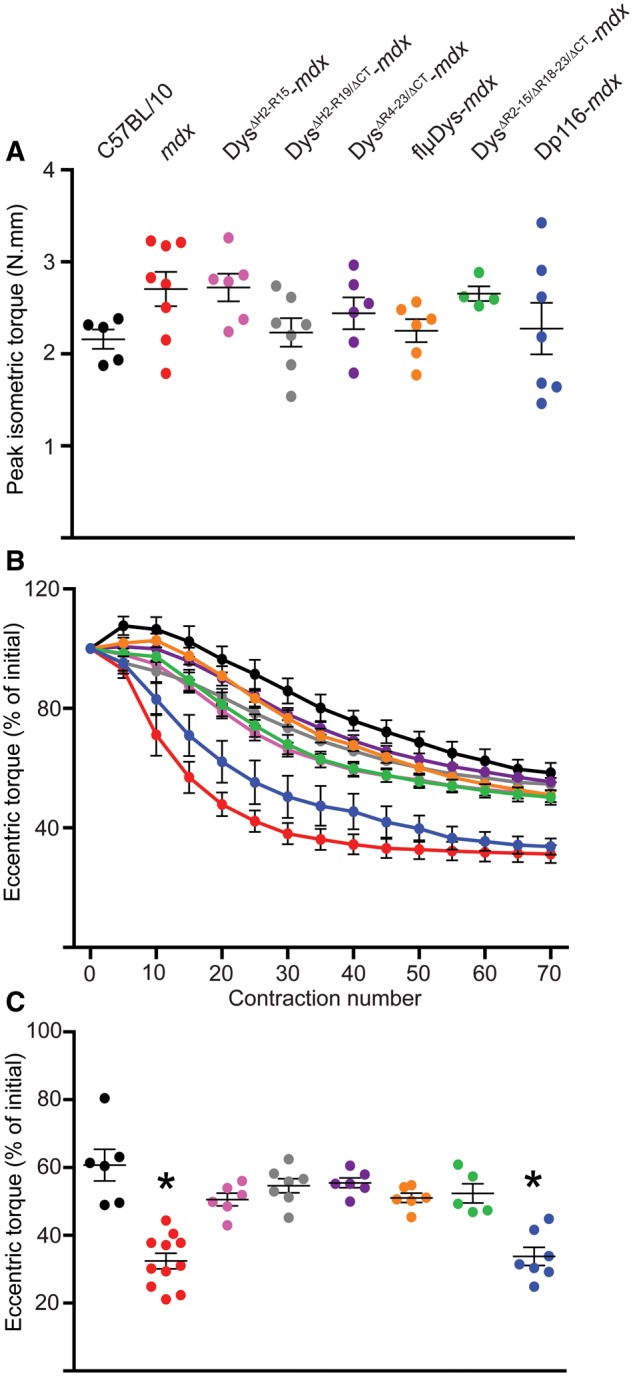
Effect of mini- and micro-dystrophin expression on eccentric contraction-induced torque drop of the anterior crural muscles in vivo. (A) Peak isometric torque of the anterior crural muscles was not different between C57BL/10, mdx and any of the transgenic mdx mice (one-way ANOVA; P=0.167). (B) Mdx and Dp116-mdx showed significantly increased susceptibility to eccentric contractions 10 through 70 while none of the transgenics expressing mini- or micro-dystrophin were different from C57BL/10. The interaction between contraction number and genotype was significant by two-way ANOVA (interaction P<0.001). (C) Final eccentric torque at contraction 70 as a percentage of contraction 1 for each mouse line (one-way ANOVA; P<0.001). Both one-way and two-way ANOVAs used Bonferroni post-hoc t-tests to confirm individual differences with post-hoc *P<0.05 considered significant. *Statistically different from C57BL/10. Error bars are mean±SEM. The symbol color-to-genotype scheme in Figure 4A was also utilized for Figure 4B and C.
As expected, isolated EDL muscles from mdx mice generated ∼40% less specific force than C57BL/10 (Fig. 5A), and except for Dp116-mdx, EDL muscles of all lines had specific forces that were not different from C57BL/10 (Fig. 5A). Repeated eccentric contractions caused >80% eccentric force loss in mdx and Dp116-mdx EDL muscles. In contrast, DysΔH2-R15-mdx, DysΔH2-R19/ΔCT-mdx and DysΔR4–23/ΔCT-mdx muscles were completely rescued while DysΔR2–15/ΔR18–23/ΔCT-mdx was intermediate between and significantly different from C57BL/10 and mdx muscles (Fig. 5B and C). In apparent contrast to our findings with DysΔR2–15/ΔR18–23/ΔCT-mdx, it was recently shown that adeno-associated virus (AAV) delivery of a highly similar micro-dystrophin construct rescued eccentric contraction-induced force loss in the more severely-affected DBA/2J-mdx to levels not different from DBA/2J controls (48). In our eccentric contraction assay, force loss data for EDL muscles from DBA/2J-mdx and DBA/2J mice (Supplementary Material, Fig. S1) recapitulated those reported previously (48). However, we observed that EDL muscles from DBA/2J mice exhibited force losses that were not significantly different from DysΔR2–15/ΔR18–23/ΔCT-mdx, but greater than those measured in C57BL/10 (Supplementary Material, Fig. S1). Because we previously found that transgenic mdx mice expressing a nearly full-length dystrophin lacking only the CT (DysΔ71–78-mdx) exhibited significantly elevated passive stiffness (5), we measured the force required to lengthen non-contracting EDL muscles (passive stiffness) in each of the transgenic lines in this study. We found that both mdx and Dp116-mdx mice had elevated passive stiffness compared to C57BL/10, while all mini- and micro-dystrophin transgenic mdx exhibited levels of passive stiffness not different from C57BL/10 (Fig. 5D), regardless of whether they expressed the CT. A complete accounting of the measured physiological parameters for EDL muscles is presented in Table 1 and Supplementary Material, Table S1.
Figure 5.

Effect of mini- and micro-dystrophin expression on eccentric contraction-induced force drop in isolated EDL muscles. (A) mdx and Dp116-mdx muscles generated lower specific isometric tetanic force compared to C57BL/10 and all mini- and micro-dystrophin constructs (one-way ANOVA; P<0.001). (B) mdx and Dp116-mdx had significantly greater eccentric force losses compared to C57BL/10 and all mini- and micro-dystrophin constructs. DysΔR2–15/ΔR18–23/ΔCT-mdx had an intermediate eccentric force loss that was significantly different from C57BL/10 from contractions 6 through 10. The interaction between contraction number and genotype was significant by two-way ANOVA (interaction P<0.001). (C) Final eccentric force at contraction 10 as a percentage of contraction 1 for each mouse line (one-way ANOVA; P<0.001). (D) mdx and Dp116-mdx muscles had significantly higher passive stiffness compared to C57BL/10 and all mini- and micro-dystrophin constructs (one-way ANOVA; P<0.001). Both one-way and two-way ANOVAs used Bonferroni post-hoc t-tests to confirm individual differences with post-hoc P<0.05 considered significant. *Statistically different from C57BL/10. Error bars are mean±SEM. The symbol color-to-genotype scheme in Figure 5C and D was also utilized for Figure 5A and B.
Table 1.
Physiological parameters of isolated EDL muscles used in ex vivo force measurements
| Parameter | C57BL/10 | mdx | DysΔH2-R15-mdx | DysΔH2-R19/ΔCT-mdx | DysΔR4–23/ΔCT-mdx | flµDys-mdx | DysΔR2–15/ΔR18–23/ΔCT-mdx | Dp116-mdx | P-value |
|---|---|---|---|---|---|---|---|---|---|
| Body mass (g) | 36.2±2.2 | 36.4±1.3 | 36.0±0.7 | 40.4±1.4 | 31.6±0.7d | 37.4±1.6 | 31.3±1.7d | 32.2±0.7d | <0.001 |
| EDL mass (mg) | 13.1±0.3 | 18.3±0.8a | 12.3±0.4gd | 12.5±0.1gd | 11.9±0.3gd | 12.6±0.4gd | 10.7±0.3agd | 20.0±0.7a | <0.001 |
| Lo (mm) | 12.9±0.1 | 13.0±0.2 | 12.8±0.3 | 13.9±0.1 | 12.9±0.2 | 12.8±0.5 | 12.6±0.2 | 13.9±0.2 | 0.051 |
| CSA (mm2) | 2.2±0.1 | 3.2±0.2a | 2.1±0.1gd | 1.9±0.1adg | 2.0±0.1deg | 2.1±0.1deg | 1.8±0.1adg | 3.1±0.01a | <0.001 |
| Po (mN) | 480±8.9 | 408±13ag | 349±21ag | 378±24ag | 428±11abcdeg | 363±7.3ag | 380±12ag | 354±29a | <0.001 |
| Specific Po (N/cm2) | 21.5±0.5 | 13.1±0.5a | 17.8±0.8bg | 20.3±0.9bg | 20.4±0.9bg | 19.2±0.8bg | 19.7±0.8bg | 11.6±1.1ab | <0.001 |
| ΔP0 (%) | 1.2±0.4 | 91.0±1.2a | 1.0±1.7 | 7.0±1.6 | 5.9±0.8 | 2.0±1.8 | 36.7±4.5abg | 88.6±3.7a | <0.001 |
| Peak eccentric force (mN) | 795±15 | 614±17a | 644±27a | 642±38a | 704±24a | 594±9.7a | 601±32a | 532±36a | <0.001 |
| Eccentric force loss (%) | 4.6±0.5 | 87.0±1.1a | 6.2±4.4 | 11.8±1.5 | 13.7±1.5 | 8.3±2.8 | 35.4±4.2abg | 80.0±4.3a | <0.001 |
| Active stiffness (N/m) | 935±36 | 671±17a | 675±26a | 784±46a | 781±35a | 694±19a | 674±28a | 613±30a | <0.001 |
| Peak twitch (mN) | 144±6.5 | 104±4.7a | 89.9±6.1a | 117±4.5ace | 101±7.0ac | 92.2±5.5a | 116±3.7ace | 110±6.3a | <0.001 |
| Twitch force development time (ms) | 18.0±0.3 | 18.7±0.5 | 19.1±0.2g | 19.5±0.2g | 19.5±0.9g | 18.3±0.3 | 19.5±0.2g | 17.1±0.5 | <0.001 |
| Twitch ½ relaxation time (ms) | 16.0±0.5 | 20.4±1.1ag | 22.4±0.5ag | 22.3±1.3ag | 19.5±1.6g | 20.7±0.9ag | 19.5±0.5g | 15.2±0.6 | <0.001 |
| Tetanic maximal rate of contraction (mN/s) | 15.7±0.6 | 12.2±0.6a | 10.8±0.4a | 13.4±0.1c | 12.1±0.4a | 12.1±0.4a | 12.0±0.4a | 12.3±0.7a | <0.001 |
| Tetanic maximal rate of relaxation (mN/s) | 23.2±0.6 | 19.1±1.1a | 16.7±1.3a | 16.8±0.9a | 20.8±0.6f | 18.0±0.6a | 16.5±0.9a | 16.7±1.5a | <0.001 |
Values are mean±SEM.
Lo=optimal muscle length, CSA=physiological cross sectional area, Po=maximal isometric tetanic force. ΔPo=change in Po after eccentric contractions. n≥4 for each line. Statistics were performed using one-way ANOVA with Bonferroni post-hoc t-test analyses with P<0.05 considered significant.
Significantly different from C57BL/10.
Significantly different from mdx.
Significantly different from DysΔH2-R15-mdx.
Significantly different from DysΔH2-R19/ΔCT-mdx.
Significantly different from flµDys-mdx.
Significantly different from DysΔR2–15/ΔR18–23/ΔCT-mdx.
Significantly different from Dp116-mdx.
Discussion
AAV-mediated gene therapy for DMD has been under intense investigation (49,50). However, due to AAV capacity constraints, miniaturized dystrophins have been a focus of continued development and characterization. Recent evidence has shown that microtubules underpin several pathophysiological and biochemical perturbations associated with the dystrophic skeletal muscle pathology of mdx mice including stretch activated ROS production, calcium ion fluctuations and possibly susceptibility to eccentric contractions (5,22,29,31). We sought to analyze the ability of mini- and micro-dystrophins to prevent dystrophic microtubule pathology.
Through our quantitative comparison of microtubule perturbations, we showed that the combined presence of the NT and the CR domains of dystrophin are necessary to organize the subsarcolemmal microtubule lattice in skeletal muscle of mdx mice and restore microtubule density to C57BL/10 levels. Mini-dystrophin DysΔH2-R15-mdx restored microtubule organization to that no different from C57BL/10. DysΔH2-R19/ΔCT-mdx, a second mini-dystrophin, was previously shown to completely rescue microtubule organization as well (5). All micro-dystrophins tested also significantly improved organization. We are not the first to show micro-dystrophin rescue of microtubule lattice organization as rAAV-mediated delivery of DysΔR4–23/ΔCT to mdx mice was previously reported to restore organization (21). However, quantification was not performed in that publication and here we show this construct, and the other micro-dystrophins (DysΔR2–15/ΔR18–23/ΔCT-mdx and flµDys-mdx), are only capable of intermediate rescue of microtubule organization. While the constructs analyzed here demonstrate the requirement of both the NT and CR domains for proper microtubule organization and density, it is possible a truncated dystrophin containing the NT in the absence of the CR would provide microtubule rescue as this particular combination has not been assessed. However, mice expressing dystrophin lacking the CR have been shown incapable of restoring the DGC or rescuing mdx pathology (51) and are hypothesized to be incapable of restoring wild-type microtubules. Based on comparison between the complete microtubule organization of the mini-dystrophins and the partial restoration by micro-dystrophins, we speculate that spectrin-like repeats 20–24 also contribute to microtubule organization. Interestingly, the dystrophin R20–24 region is inclusive of domains that have been previously associated with microtubule binding in vitro (29). Microtubule density is not influenced by the R20–24 region of dystrophin, and based on the results presented here, inclusion of NT and CR appears sufficient for restoration of C57BL/10 microtubule density.
A link between aberrant mdx microtubules and force loss was demonstrated in a previous study reporting partial protection of skeletal muscle from in vivo eccentric contraction-induced force loss through modulation of α-tubulin detyrosination level (31). Here, we report complete protection from in vivo eccentric contraction-induced force loss in mini- and micro-dystrophin transgenics. It is also important to note that the five mini- and micro-dystrophin transgenics that were protected from force loss, also exhibited detyrosinated α-tubulin reduced to C57BL/10 levels; data that support a connection between elevated detyrosinated α-tubulin and susceptibility to force loss. However, the Fiona-mdx mouse which expresses full-length transgenic utrophin on the mdx background has C57BL/10 levels of detyrosinated α-tubulin (29) and still suffers from in vivo eccentric contraction-induced force loss (5). While we believe that microtubules influence force loss, it is worth noting that mini- or micro-dystrophin expression improves an array of mdx perturbations beyond microtubules which may also modulate force loss.
Detyrosinated α-tubulin has also been linked to increased cytoskeletal stiffness and reduced cardiomyocyte contractility (31,32). Detyrosinated α-tubulin was shown to interact with the desmin intermediate filament network to form crosslinks which led to an overall increase in cytoskeletal stiffness (32). We have previously noted elevated detyrosinated α-tubulin, desmin and passive stiffness in the DysΔ71–78-mdx mouse (29), which lacks the CT and therefore suggested a possible role for the CT in regulating passive stiffness. However, we now show that passive stiffness remains significantly elevated in Dp116-mdx, which contains the CT, but is restored to C57BL/10 levels in multiple mini- and micro-dystrophin transgenic lines lacking the CT. Our data agree with previous characterization demonstrating reduced muscle stiffness in DysΔH2-R15-mdx, DysΔR4–23/ΔCT-mdx and DysΔR2–15/ΔR18–23/ΔCT-mdx as compared to mdx (46). We do, however, find additional support for the co-requirement of elevated detyrosinated α-tubulin and desmin for increased passive stiffness. In both mdx and Dp116-mdx we measured increased immunoreactivity of detyrosinated α-tubulin and desmin protein that segregated with the elevated passive stiffness only exhibited by these two lines. DysΔR4–23/ΔCT-mdx and DysΔR2–15/ΔR18–23/ΔCT-mdx, which exhibit C57BL/10 levels of passive stiffness, also have elevated desmin protein but remain not different than C57BL/10 for detyrosinated α-tubulin. As passive stiffness is a robust phenotype of dystrophic muscle (52–55), correction or normalization to C57BL/10 levels has been identified as clinically important and more work on the pathomechanism is needed to determine all contributing factors (56,57).
Susceptibility of dystrophic muscle to eccentric contractions was first characterized in 1993 (58,59) and has become a foundational efficacy test for various therapeutic and pharmacological therapies. We confirm that all miniaturized dystrophins studied rescue in vivo and ex vivo eccentric contraction-induced force drop as previously described (5,35,36), but present new evidence that removal of the R2–3 region of dystrophin impacts susceptibility to eccentric contraction-induced force drop. Biophysical characterization of dystrophin repeats identified a phospholipid binding domain associated with R1–3 (60,61). Binding is thought to occur through electrostatic and hydrophobic interactions (60) involving tryptophan residues (62) and anionic phospholipid phosphatidylserine (63). Others have previously speculated that the interactions associated with R1–3 might impact mini-dystrophin functionality (61,64). Because DysΔR2–15/ΔR18–23/ΔCT-mdx lacking spectrin-like repeats 2 and 3 exhibits a small yet significant force loss following ex vivo eccentric contractions, our data support a role for the dystrophin R1–3 phospholipid binding domain in modulating radial force transmission and mechanical vulnerability. Indeed, there are multiple DMD and BMD patients that exhibit large in-frame deletions that include loss of R1–3 and flanking regions, as well as a few documented BMD cases that are attributed to deletions solely within R1–3 of dystrophin (65,66). Additionally, while mini- and micro-dystrophin transgenic expression has been shown to prevent Evans blue dye infiltration into skeletal muscle of basally active mice (36,67) we do not know if these transgenes leave the mice susceptible to sarcolemmal damage during and following a bout of eccentric contractions.
In summary, our studies support important roles for the NT and CR domains of dystrophin in restoration of mdx microtubule lattice organization and density toward C57BL/10. Our results also suggest microtubule organization modulates susceptibility to eccentric contraction-induced force drop in dystrophin-deficient skeletal muscle, and that spectrin-like repeats R1–3 are required for complete protection. In conclusion, our data support further study of dystrophin R1–3 and R20–24 as they pertain to eccentric contraction-induced force loss and microtubule organization, respectively.
Materials and Methods
Mice—all animals were housed and treated in accordance with the standards set by the University of Minnesota Institutional Animal Care and Use Committee. C57BL/10 (wild-type), mdx and DBA/2J mice used in this study were obtained from The Jackson Laboratory. DBA/2J-mdx mice were a kind gift from Dr. Michael Kyba at the University of Minnesota. Generation of DysΔH2-R15-mdx (35), DysΔH2-R19/ΔCT-mdx (36), DysΔR4–23/ΔCT-mdx (37), DysΔR2–15/ΔR18–23/ΔCT-mdx (37) and Dp116-mdx (38) mice were previously reported. Transgenic mice expressing a floxed-μDysΔR24-R23/ΔCT transgene (flμDys) regulated by the human α-skeletal actin promoter were generated at the University of Washington transgenic animal core laboratory. Full details will be described elsewhere (Ramos et al., manuscript in preparation). All transgenic dystrophin lines were backcrossed onto the mdx (C57BL/10ScSn-Dmdmdx/J) mouse strain obtained from The Jackson Laboratory. All mice used in this study were adult males (3–6 months of age).
Immunofluorescence—EDL muscles were cryopreserved in melting isopentane. Ten micrometer transverse sections were cut and fixed in −20°C acetone. Sections were washed with phosphate buffered saline (PBS) and blocked with goat serum or bovine serum albumin followed by Rodent Block M (Biocare Medical). Primary antibodies including 1:1000 anti-dystrophin N-terminus (Rb2, described in Rybakova et al., 1996), 1:400 anti-dystrophin C-terminus (Dys2, Leica) and 1:2000 anti-laminin (L0663, Sigma Aldrich) were incubated overnight in a humidified chamber at 4°C. Slides were washed with 0.1% triton in PBS. Secondary antibody incubation occurred for 1 h at room temperature with goat anti-rat Alexa Fluor 488 and goat anti-rabbit Alexa Fluor 568 or donkey anti-mouse Alexa Flour 568. Slides were washed with 0.1% triton in PBS and sealed using Prolong Gold antifade reagent with DAPI (Cell Signaling Technology). A Deltavision PersonalDV deconvolution microscope acquired 20× images that are representative of n = 2 mice per genotype.
Western blot analysis—gastrocnemius muscles were mechanically disrupted with mortar and pestle in liquid nitrogen. Ground tissue was lysed with 1% sodium dodecyl sulfate in 1×PBS with protease inhibitors (100 uM Aprotinin, 0.79 mg/ml benzamide, 10 nM E-64, 10 uM leupeptin, 0.1 mg/ml pepstatin, 1 mM phenylmethylsulfonyl fluoride; Sigma-Aldrich). Protein lysate was clarified by centrifugation and concentration measured by A280 absorbance. Forty microgram (desmin blots) or 50 µg (detyrosinated tubulin blots) of total protein was loaded on a 10% polyacrylamide gel for 1.25 h at 150 V. Protein was transferred to polyvinylidene fluoride membrane (MilliporeSigma) at 0.8 A for 1.5 h. Membranes were blocked in 5% non-fat milk in PBS for 1 h. Primary antibodies included 1:10 000 anti-deTyr-tubulin [gift from Dr. Gregg Gundersen (27)], 1:1000 anti-Desmin (DE-U-10, Sigma Aldrich) and 1:10 000 anti-GAPDH (71.1, Sigma Aldrich) were incubated overnight at 4°C. Secondary antibodies, DyLight 680 and 800 (1:10 000, Cell Signaling), were incubated for 1 h at room temperature. Membranes were imaged and band density quantified with a Licor Odyssey Infrared Imaging System and accompanying software. Western blots were representative of n ≥ 4 for each genotype.
Muscle Fiber Imaging—EDL muscles were harvested from perfusion-fixed mice and immunostained with 1:200 anti-α-tubulin (DM1A; Sigma Aldrich). Perfusion media [50 mM pipes, 5 mM EGTA, 2 mM MgSO4, 10% (vol/vol) DMSO and 0.1% triton X-100, pH 6.8] contained 4% (wt/vol) paraformaldehyde. Muscles were further post-fixed in 4% (wt/vol) paraformaldehyde in PBS (PBS: 8 mM NaH2PO4, 42 mM Na2HPO4 and 150 mM NaCl, pH 7.5) for 24 h while rotating. Single muscle fibers or bundles of two or three muscle fibers were mechanically teased apart and incubated with anti-α-tubulin overnight at 4°C with rotation. Secondary staining with goat anti-mouse secondary antibody coupled to Alexa Fluor 568 at 1:250 (Invitrogen) for 2 h at room temperature was used for visualization. Fibers were mounted on slides using Prolong Gold antifade reagent with DAPI (Cell Signaling Technology) to visualize nuclei. Fibers were imaged on the Olympus FluoView FV1000 with the 60× oil immersion objective using the accompanying software. ImageJ sum slices projection was used to combine Z-stacks of 2–3 sarcolemmal images. Brightness and contrast were adjusted using ImageJ. Images are representative of n ≥3 mice per genotype and n ≥ 10 fibers per mouse.
Quantitative microtubule lattice analysis—directionality: using a previously developed directionality analysis program [TeDT (45)], microtubule lattice directionality was calculated for all mouse lines, minimally n≥ 3 mice per genotype and n ≥ 10 fibers per mouse. Microtubule directionality proportions were normalized to extend 0 and 1 (relative HD). Density: density of the microtubule lattice was calculated as a fraction of pixels above threshold for all images obtained for directionality analysis. The researcher was blinded to mouse genotype for the duration of the density analysis.
In vivo muscle torque measurements—mice were anesthetized with isoflurane and tested for peak isometric torque generation of the anterior crural muscles. A total of 70 eccentric contractions were initiated through percutaneous stimulation of the common peroneal nerve. The foot was passively rotated about the ankle from 0 to 19° dorsiflexion followed by 38° of plantarflexion at 2000°/s using an optimized voltage. Each eccentric contraction was separated by 10 s. Eccentric torque is plotted as a percentage of torque generated on the first eccentric contraction.
Ex vivo EDL force measurements—mice were anaesthetized with 75 mg/kg pentobarbital and analyzed for susceptibility to eccentric contractions. EDL muscles were anchored to a force transducer and incubated in Krebs–Ringer bicarbonate buffer. Baseline contractile force measurements and passive stiffness were conducted as previously described (68). For eccentric contractions, EDL muscles were passively shortened to 95% resting length and then maximally stimulated for 200 ms while the muscle was simultaneously lengthened to 105% resting length at 0.5 lengths/s. Each contraction was separated by 3 min to avoid fatigue (69).
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
The authors would like to thank Drs. Wenhua Liu and Evelyn Ralston (National Institute of Arthritis and Musculoskeletal and Skin Diseases) for providing the microtubule directionality analysis program and Dr. Michael Kyba (University of Minnesota) for DBA/2J-mdx mice.
Conflict of Interest statement. J.S.C and D.D. are members of the scientific advisory board for Solid Biosciences and equity holders of Solid Biosciences. The Chamberlain and Duan labs have each received research support from Solid Biosciences.
Funding
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases [R01 AR042423 to J.M.E.], the National Institute of Neurological Disorders and Stroke [R01 NS90634 to D.D.], the National Institute on Aging Training Program for Functional Proteomics of Aging [T32 AG029796 to D.M.N.] and the Jackson Freel DMD Research Fund [D.D.].
References
- 1. Hoffman E.P., Brown R.H., Kunkel L.M., Avner P., Amar L., Arnaud D., Hanauer A., Cambrou J., Bridges L.R., Brockdorff N. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 51, 919–928. [DOI] [PubMed] [Google Scholar]
- 2. Koenig M., Beggs A.H., Moyer M., Scherpf S., Heindrich K., Bettecken T., Meng G., Müller C.R., Lindlöf M., Kaariainen H. (1989) The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am. J. Hum. Genet., 45, 498–506. [PMC free article] [PubMed] [Google Scholar]
- 3. Rybakova I.N., Patel J.R., Ervasti J.M. (2000) The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol., 150, 1209–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prins K.W., Humston J.L., Mehta A., Tate V., Ralston E., Ervasti J.M. (2009) Dystrophin is a microtubule-associated protein. J. Cell Biol., 186, 363–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belanto J.J., Mader T.L., Eckhoff M.D., Strandjord D.M., Banks G.B., Gardner M.K., Lowe D.A., Ervasti J.M. (2014) Microtubule binding distinguishes dystrophin from utrophin. Proc. Natl. Acad. Sci. U. S. A., 111, 5723–5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ervasti J.M. (2007) Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta Mol. Basis Dis., 1772, 108–117. [DOI] [PubMed] [Google Scholar]
- 7. Ervasti J.M., Campbell K.P. (1993) A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol., 122, 809–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mokri B., Engel A.G. (1975) Duchenne dystrophy: electron microscopic findings pointing to a basic or early abnormality in the plasma membrane of the muscle fiber. Neurology, 25, 1111–1120. [DOI] [PubMed] [Google Scholar]
- 9. Straub V., Rafael J.A., Chamberlain J.S., Campbell K.P. (1997) Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J. Cell Biol., 139, 375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramaswamy K.S., Palmer M.L., van der Meulen J.H., Renoux A., Kostrominova T.Y., Michele D.E., Faulkner J.A. (2011) Lateral transmission of force is impaired in skeletal muscles of dystrophic mice and very old rats. J. Physiol., 589, 1195–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vohra R.S., Lott D., Mathur S., Senesac C., Deol J., Germain S., Bendixen R., Forbes S.C., Sweeney H.L., Walter G.A.. et al. (2015) Magnetic resonance assessment of hypertrophic and pseudo-hypertrophic changes in lower leg muscles of boys with Duchenne muscular dystrophy and their relationship to functional measurements. PLoS One, 10, e0128915.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Spencer M.J., Tidball J.G. (2001) Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul. Disord., 11, 556–564. [DOI] [PubMed] [Google Scholar]
- 13. Emery A.E.H., Muntoni F., Quinlivan R.C.M. (2015) Duchenne Muscular Dystrophy, 4th edn.Oxford University Press, Oxford, United Kingdom. [Google Scholar]
- 14. Bach J.R., Martinez D. (2011) Duchenne muscular dystrophy: continuous noninvasive ventilatory support prolongs survival. Respir. Care, 56, 744–750. [DOI] [PubMed] [Google Scholar]
- 15. Ricotti V., Ridout D.A., Scott E., Quinlivan R., Robb S.A., Manzur A.Y., Muntoni F., Manzur A., Muntoni F., Robb S.. et al. (2011) Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J. Neurol. Neurosurg. Psychiatry, 21, 705–705. [DOI] [PubMed] [Google Scholar]
- 16. Mendell J.R., Shilling C., Leslie N.D., Flanigan K.M., Al-Dahhak R., Gastier-Foster J., Kneile K., Dunn D.M., Duval B., Aoyagi A.. et al. (2012) Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol., 71, 304–313. [DOI] [PubMed] [Google Scholar]
- 17. Bulfield G., Siller W.G., Wight P.A., Moore K.J. (1984) X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. U. S. A., 81, 1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sicinski P., Geng Y., Ryder-Cook A.S., Barnard E.A., Darlison M.G., Barnard P.J. (1989) The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science, 244, 1578–1580. [DOI] [PubMed] [Google Scholar]
- 19. Muntoni F., Mateddu A., Marchei F., Clerk A., Serra G. (1993) Muscular weakness in the mdx mouse. J. Neurol. Sci., 120, 71–77. [DOI] [PubMed] [Google Scholar]
- 20. Lefaucheur J.P., Pastoret C., Sebille A. (1995) Phenotype of dystrophinopathy in oldMDX mice. Anat. Rec., 242, 70–76. [DOI] [PubMed] [Google Scholar]
- 21. Percival J.M., Gregorevic P., Odom G.L., Banks G.B., Chamberlain J.S., Froehner S.C. (2007) rAAV6-microdystrophin rescues aberrant Golgi complex organization in mdx skeletal muscles. Traffic, 8, 1424–1439. [DOI] [PubMed] [Google Scholar]
- 22. Khairallah R.J., Shi G., Sbrana F., Prosser B.L., Borroto C., Mazaitis M.J., Hoffman E.P., Mahurkar A., Sachs F., Sun Y.. et al. (2012) Microtubules underlie dysfunction in Duchenne muscular dystrophy. Sci. Signal., 5, ra56.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Oddoux S., Zaal K.J., Tate V., Kenea A., Nandkeolyar S.A., Reid E., Liu W., Ralston E. (2013) Microtubules that form the stationary lattice of muscle fibers are dynamic and nucleated at golgi elements. J. Cell Biol., 203, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ayalon G., Davis J.Q., Scotland P.B., Bennett V. (2008) An ankyrin-based mechanism for functional organization of dystrophin and dystroglycan. Cell, 135, 1189–1200. [DOI] [PubMed] [Google Scholar]
- 25. Ayalon G., Hostettler J.D., Hoffman J., Kizhatil K., Davis J.Q., Bennett V. (2011) Ankyrin-B interactions with spectrin and dynactin-4 are required for dystrophin-based protection of skeletal muscle from exercise injury. J. Biol. Chem., 286, 7370–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Randazzo D., Giacomello E., Lorenzini S., Rossi D., Pierantozzi E., Blaauw B., Reggiani C., Lange S., Peter A.K., Chen J.. et al. (2013) Obscurin is required for ankyrinB-dependent dystrophin localization and sarcolemma integrity. J. Cell Biol., 200, 523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gundersen G.G., Kalnoski M.H., Bulinski J.C. (1984) Distinct populations of microtubules: tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell, 38, 779–789. [DOI] [PubMed] [Google Scholar]
- 28. Gundersen G.G., Khawaja S., Bulinski J.C. (1987) Postpolymerization detyrosination of alpha-tubulin: a mechanism for subcellular differentiation of microtubules. J. Cell Biol., 105, 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Belanto J.J., Olthoff J.T., Mader T.L., Chamberlain C.M., Nelson D.M., McCourt P.M., Talsness D.M., Gundersen G.G., Lowe D.A., Ervasti J.M. (2016) Independent variability of microtubule perturbations associated with dystrophy in the mdx mouse. Hum. Mol. Genet., 25, 4951–4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prosser B.L., Ward C.W., Lederer W.J. (2011) X-ROS signaling: rapid mechano-chemo transduction in heart. Science, 333, 1440–1445. [DOI] [PubMed] [Google Scholar]
- 31. Kerr J.P., Robison P., Shi G., Bogush A.I., Kempema A.M., Hexum J.K., Becerra N., Harki D.A., Martin S.S., Raiteri R.. et al. (2015) Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat. Commun., 6, 8526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robison P., Caporizzo M.A., Ahmadzadeh H., Bogush A.I., Chen C.Y., Margulies K.B., Shenoy V.B., Prosser B.L. (2016) Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science, 352, 6284, aaf0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Doran P., Martin G., Dowling P., Jockusch H., Ohlendieck K. (2006) Proteome analysis of the dystrophin-deficient MDX diaphragm reveals a drastic increase in the heat shock protein cvHSP. Proteomics, 6, 4610–4621. [DOI] [PubMed] [Google Scholar]
- 34. Rayavarapu S., Coley W., Cakir E., Jahnke V., Takeda S., Aoki Y., Grodish-Dressman H., Jaiswal J.K., Hoffman E.P., Brown K.J.. et al. (2013) Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell. Proteomics, 12, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lai Y., Thomas G.D., Yue Y., Yang H.T., Li D., Long C., Judge L., Bostick B., Chamberlain J.S., Terjung R.L.. et al. (2009) Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J. Clin. Invest, 119, 624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li S., Kimura E., Ng R., Fall B.M., Meuse L., Reyes M., Faulkner J.A., Chamberlain J.S. (2006) A highly functional mini-dystrophin/GFP fusion gene for cell and gene therapy studies of Duchenne muscular dystrophy. Hum. Mol. Genet., 15, 1610–1622. [DOI] [PubMed] [Google Scholar]
- 37. Li D., Yue Y., Lai Y., Hakim C.H., Duan D. (2011) Nitrosative stress elicited by nNOSµ delocalization inhibits muscle force in dystrophin-null mice. J. Pathol., 223, 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Judge L.M., Haraguchiln M., Chamberlain J.S. (2006) Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J. Cell Sci., 119, 1537–1546. [DOI] [PubMed] [Google Scholar]
- 39. Beggs A.H., Hoffman E.P., Snyder J.R., Arahata K., Specht L., Shapiro F., Angelini C., Sugita H., Kunkel L.M. (1991) Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am. J. Hum. Genet., 49, 54–67. [PMC free article] [PubMed] [Google Scholar]
- 40. Angelini C., Fanin M., Freda M.P., Martinello F., Miorin M., Melacini P., Siciliano G., Pegoraro E., Rosa M., Danieli G.A. (1996) Prognostic factors in mild dystrophinopathies. J. Neurol. Sci., 142, 70–78. [DOI] [PubMed] [Google Scholar]
- 41. Way M., Pope B., Cross R.A., Kendrick-Jones J., Weeds A.G. (1992) Expression of the N-terminal domain of dystrophin in E. coli and demonstration of binding to F-actin. FEBS Lett., 301, 243–245. [DOI] [PubMed] [Google Scholar]
- 42. Chung W., Campanelli J.T. (1999) WW and EF hand domains of dystrophin-family proteins mediate dystroglycan binding. Mol. Cell Biol. Res. Commun., 2, 162–171. [DOI] [PubMed] [Google Scholar]
- 43. Ishikawa-Sakurai M., Yoshida M., Imamura M., Davies K.E., Ozawa E. (2004) ZZ domain is essentially required for the physiological binding of dystrophin and utrophin to -dystroglycan. Hum. Mol. Genet., 13, 693–702. [DOI] [PubMed] [Google Scholar]
- 44. Imamura M., Araishi K., Noguchi S., Ozawa E. (2000) A sarcoglycan-dystroglycan complex anchors Dp116 and utrophin in the peripheral nervous system. Hum. Mol. Genet., 9, 3091–3100. [DOI] [PubMed] [Google Scholar]
- 45. Liu W., Ralston E. (2014) A new directionality tool for assessing microtubule pattern alterations. Cytoskeleton, 71, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hakim C.H., Duan D. (2013) Truncated dystrophins reduce muscle stiffness in the extensor digitorum longus muscle of mdx mice. J. Appl. Physiol., 114, 482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Judge L.M., Arnett A.L.H., Banks G.B., Chamberlain J.S. (2011) Expression of the dystrophin isoform Dp116 preserves functional muscle mass and extends lifespan without preventing dystrophy in severely dystrophic mice. Hum. Mol. Genet., 20, 4978–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hakim C.H., Wasala N.B., Pan X., Kodippili K., Yue Y., Zhang K., Yao G., Haffner B., Duan S.X., Ramos J.. et al. (2017) A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of Duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev., 6, 216–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chamberlain J.R., Chamberlain J.S. (2017) Progress toward gene therapy for Duchenne muscular dystrophy. Mol. Ther., 25, 1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Duan D. (2016) Dystrophin gene replacement and gene repair therapy for Duchenne muscular dystrophy in 2016: an interview. Hum. Gene Ther. Clin. Dev., 27, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gardner K.L., Kearney J.A., Edwards J.D., Rafael-Fortney J.A. (2006) Restoration of all dystrophin protein interactions by functional domains in trans does not rescue dystrophy. Gene Ther., 13, 744–751. [DOI] [PubMed] [Google Scholar]
- 52. Hakim C.H., Grange R.W., Duan D. (2011) The passive mechanical properties of the extensor digitorum longus muscle are compromised in 2- to 20-mo-old mdx mice. J. Appl. Physiol., 110, 1656–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cornu C., Goubel F., Fardeau M. (2001) Muscle and joint elastic properties during elbow flexion in Duchenne muscular dystrophy. J. Physiol., 533, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cornu C., Goubel F., Fardeau M. (1998) Stiffness of knee extensors in Duchenne muscular dystrophy. Muscle Nerve, 21, 1772–1774. [DOI] [PubMed] [Google Scholar]
- 55. Lacourpaille L., Hug F., Guével A., Péréon Y., Magot A., Hogrel J.-Y., Nordez A. (2015) Non-invasive assessment of muscle stiffness in patients with Duchenne muscular dystrophy. Muscle Nerve, 51, 284–286. [DOI] [PubMed] [Google Scholar]
- 56. Smith L.R., Barton E.R. (2014) Collagen content does not alter the passive mechanical properties of fibrotic skeletal muscle in mdx mice. AJP Cell Physiol., 306, C889–C898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chapman M.A., Pichika R., Lieber R.L. (2015) Collagen crosslinking does not dictate stiffness in a transgenic mouse model of skeletal muscle fibrosis. J. Biomech., 48, 375–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U. S. A., 90, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moens P., Baatsen P.H.W.W., Maréchal G. (1993) Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil., 14, 446–451. [DOI] [PubMed] [Google Scholar]
- 60. Legardinier S., Hubert J.F., Bihan O., Le, Tascon C., Rocher C., Raguénès-Nicol C., Bondon A., Hardy S., Rumeur E.L. (2008) Sub-domains of the dystrophin rod domain display contrasting lipid-binding and stability properties. Biochim. Biophys. Acta Proteins Proteomics, 1784, 672–682. [DOI] [PubMed] [Google Scholar]
- 61. Zhao J., Kodippili K., Yue Y., Hakim C.H., Wasala L., Pan X., Zhang K., Yang N.N., Duan D., Lai Y. (2016) Dystrophin contains multiple independent membrane-binding domains. Hum. Mol. Genet., 25, 3647–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Le Rumeur E., Fichou Y., Pottier S., Gaboriau F., Rondeau-Mouro C., Vincent M., Gallay J., Bondon A. (2003) Interaction of dystrophin rod domain with membrane phospholipids: evidence of a close proximity between tryptophan residues and lipids. J. Biol. Chem., 278, 5993–6001. [DOI] [PubMed] [Google Scholar]
- 63. Le Rumeur E., Pottier S., Da Costa G., Metzinger L., Mouret L., Rocher C., Fourage M., Rondeau-Mouro C., Bondon A. (2007) Binding of the dystrophin second repeat to membrane di-oleyl phospholipids is dependent upon lipid packing. Biochim. Biophys. Acta Biomembr., 1768, 648–654. [DOI] [PubMed] [Google Scholar]
- 64. Vié V., Legardinier S., Chieze L., Le Bihan O., Qin Y., Sarkis J., Hubert J.-F., Renault A., Desbat B., Le Rumeur E. (2010) Specific anchoring modes of two distinct dystrophin rod sub-domains interacting in phospholipid Langmuir films studied by atomic force microscopy and PM-IRRAS. Biochim. Biophys. Acta Biomembr., 1798, 1503–1511. [DOI] [PubMed] [Google Scholar]
- 65. Flanigan K.M., Dunn D.M., von Niederhausern A., Soltanzadeh P., Gappmaier E., Howard M.T., Sampson J.B., Mendell J.R., Wall C., King W.M.. et al. (2009) Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum. Mutat., 30, 1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tuffery-Giraud S., Béroud C., Leturcq F., Yaou R.B., Hamroun D., Michel-Calemard L., Moizard M.-P., Bernard R., Cossée M., Boisseau P.. et al. (2009) Genotype-phenotype analysis in 2, 405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum. Mutat., 30, 934–945. [DOI] [PubMed] [Google Scholar]
- 67. Harper S.Q., Hauser M.A., DelloRusso C., Duan D., Crawford R.W., Phelps S.F., Harper H.A., Robinson A.S., Engelhardt J.F., Brooks S.V.. et al. (2002) Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med., 8, 253–261. [DOI] [PubMed] [Google Scholar]
- 68. Moran A.L., Warren G.L., Lowe D.A. (2005) Soleus and EDL muscle contractility across the lifespan of female C57BL/6 mice. Exp. Gerontol., 40, 966–975. [DOI] [PubMed] [Google Scholar]
- 69. Lowe D.A., Warren G.L., Hayes D.A., Farmer M.A., Armstrong R.B. (1994) Eccentric contraction-induced injury of mouse soleus muscle: effect of varying [Ca2+]o. J. Appl. Physiol., 76, 1445–1453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.