

Abstract
Introduction
Vascular smooth muscle cells (VSMCs) play an important role in the development and progression of atherosclerosis and vascular injuries in terms of proliferation and migration. Therefore, the aim of this study was to investigate the anti-migratory and proliferative effects of naked gold nanoparticles (AuNPs) on VSMCs.
Materials and methods
One set of physically synthesized AuNPs (pAuNPs) and three sets of chemically synthesized AuNPs (cAuNPs) were tested.
Results and discussion
Among them, the pAuNPs were found to significantly and markedly inhibit platelet-derived growth factor (PDGF)-induced VSMC migration. Transmission electron microscopy revealed that the pAuNPs were ingested and aggregated in the cytoplasm at an early stage of treatment, while the viability of VSMCs was not affected within 24 hours of treatment. The pAuNP treatment enhanced cellular mitochondrial activity but inhibited basal and PDGF-induced VSMC proliferation, as determined by MTT, WST-1, and BrdU cell proliferation assays. Furthermore, the pAuNPs did not interfere with PDGF signaling or matrix metalloproteinase-2 expression/activity. Unlike the cAuNPs, the pAuNPs could markedly reduce VSMC adhesion to collagen, which was supported by the findings that the pAuNPs could inhibit collagen-induced tyrosine protein and focal adhesion kinase (FAK) phosphorylation and actin cytoskeleton reorganization during cell adhesion. The in vitro effects of the pAuNPs were confirmed in the in vivo rat balloon-injured carotid artery model by diminishing the proliferating VSMCs.
Conclusion
Taken together, the present study provides the first evidence that naked pAuNPs can reduce VSMC migration and compromise cell adhesion by affecting FAK and tyrosine-protein activation. The pAuNPs also have an inhibitory effect on PDGF-induced VSMC proliferation and can reduce proliferating/migrating VSMC expression in vivo.
Keywords: adhesion, AuNP, cardiovascular disease, FAK, platelet-derived growth factor, VSMC, TEM
Introduction
Cardiovascular diseases (CVDs) are diseases that involve heart or blood vessels, mainly myocardial infarction, stroke, and pulmonary embolism, and are the leading cause of death worldwide.1 Among the causes of CVDs, atherosclerosis is a major contributing factor. However, even with access to the highest technology and most recently available secondary prevention therapies, currently the burden of recurrent events after acute coronary syndromes remains unacceptable.2 Thus, atherosclerosis remains high on the list of global challenges to long and healthy lives.3
Vascular smooth muscle cells (VSMCs) play an important role in the development and progression of atherosclerosis4 and vascular restenosis.5 Under pathological conditions, the subendothelial accumulation of leukocytes from the bloodstream initiates the development of atherosclerotic plaques, which is also characterized by the migration of VSMCs from the medial to the intima layer of the artery.6 The presence of a large number of intimal VSMCs, which, for example, form a fibrous cap, has been considered evidence that VSMC migration from media plays an important role in atherogenesis.4 Data from multiple studies support that platelet-derived growth factor (PDGF) may contribute to VSMC accumulation in atherosclerosis. Recently, clinical imaging studies have identified one of several features of atherosclerotic plaque instability leading to rupture. A thin or fragmented fibrous cap comprises smooth muscle α-actin-positive cells presumed to be derived from VSMCs.4
A previous study has shown that PDGFs play a prominent role in the migration of VSMCs into the neointima following acute injury and in atherosclerosis and participate in the pathogenesis of CVDs.6 PDGFR-β mRNA and protein are increased in lesions of atherosclerosis, but their expression is primarily limited to VSMCs.7,8 Recent studies have also indicated that PDGF is required for phenotypic switching of cultured VSMCs.9–11
Gold-based compounds have long been used for therapeutic purposes, such as for treating arthritis and cancer. Production of naked gold nanoparticles (AuNPs) typically involves one of two methods, namely physical or chemical. Most commercially available colloidal gold solutions are obtained by chemical reduction and usually have a particle size between 5 and 20 nm. In contrast, AuNPs produced by physical methods usually have a particle size ranging from 0.5 to 50 nm. In general, chemically synthesized gold contains unreacted tetrachloroauric acid and semi-reacted monovalent and trivalent gold ions, whereas physically synthesized gold contains no added dispersing agents.12
Most studies on AuNPs have been conducted on polyethylene glycol (PEG)-coated or conjugated AuNPs rather than naked or unmasked AuNPs.13 For example, PEG-AuNPs bind to arthritic synovial fluid vascular endothelial growth factor (VEGF), exert antiangiogenic activities, and reduce macrophage infiltration and proinflammatory cytokine levels, resulting in the attenuation of inflammation and arthritis.14 AuNPs conjugated with heparin polysaccharides reduce basic fibroblast growth factor (bFGF)-induced angiogenesis in vivo.15 Moreover, ginkgolide A-AuNPs inhibit VSMC proliferation and migration in vitro and have been shown to reduce neointimal hyperplasia in a mouse model;16 moreover, curcumin-AuNPs ameliorate tumor necrosis factor (TNF)-induced intercellular adhesion molecule-1 expression by lung epithelial cells.17 However, naked AuNPs have been shown to bind to certain growth factors, such as VEGF and bFGF, resulting in the inhibition of endothelial cell (EC) proliferation in vitro and angiogenesis in vivo.18 In addition, naked AuNPs can upregulate the expression of proinflammatory genes, such as TNF and interleukins, in in vitro macrophages.19 Recently, we reported that naked physically synthesized AuNPs (pAuNPs) induce heme oxygenase-1 in human vascular ECs,20 compromise TNF-α-induced EC adhesion molecule expression through the NF-κB and protein degradation pathways, and can reduce neointima formation in a rat carotid balloon injury model.21 Vascular injury not only involves EC inflammation that attracts leukocyte transmigration but also involves changes in VSMC behaviors. Therefore, we hypothesized that naked pAuNPs may affect VSMC functions and behaviors.
In this study, the effects of naked pAuNPs and chemically synthesized AuNPs (cAuNPs) on PDGF-induced VSMC migration and proliferation were evaluated. One set of pAuNPs and one set of cAuNPs were found to significantly inhibit PDGF-induced migration. Because of the marked inhibitory effect of pAuNPs on VSMC migration, the mechanism of action of the particles was further investigated in this study.
Materials and methods
AuNPs
Four sets of AuNPs were used in this study; the main particles were pAuNPs obtained from Gold NanoTech Inc. (Taipei, Taiwan), and the three other sets were cAuNPs. The three types of cAuNPs were 3–5 nm 1-mercapto-(triethylene glycol)methyl ether functionalized AuNPs (3-EG AuNPs), (1-mercaptoundec-11-yl)tetra(ethyleneglycol) functionalized AuNPs (4-EG AuNPs) (Nanoprobes Inc., Yaphank, NY, USA) (gifts from our colleague, Dr Han-Min Chen, Department of Life Science), and 5 nm standard unconjugated AuNPs (std AuNPs) (Cytodiagnostics, Burlington, Ontario, Canada). The pAuNPs had an average size of 3.38 nm.21 The pAuNPs and 3-EG and 4-EG AuNPs were dispersed in ddH2O, whereas the std AuNPs were dispersed in 0.1 mg/mL citrate. Based on the atomic radius (0.16 nm) and weight of Au (196.96 amu), the molar concentrations of 2 and 5 ppm pAuNPs used in this study were estimated to be 1.08 and 2.70 nM, respectively. The conversion is detailed in the supplementary materials of our previous study.21
Materials
Bovine serum albumin (BSA) and collagen (type I) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). AM (2′,7′-Bis-(2-Carboxyethyl)-5-(and-6)-Carboxyfluorescein/acetoxymethyl ester (BCECF/AM) and fluorescein isothiocyanate (FITC)-conjugated phalloidin were obtained from Molecular Probes (Eugene, OR, USA). Antibodies (Abs) directed against PLCγ1, p-PLCγ1 (Tyr783), p-p38 MAPK (Thr180/Tyr182), p-PI-3K p85 (Tyr458)/p55 (Tyr199), p-Akt (Ser473), total Akt, p-focal adhesion kinase (pFAK) (Tyr397), and phospho-PKC (including the p-PKC and PKC isoforms) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). The Abs raised against phospho-ERK1/2 (Thr202/Tyr204) and PDGFR-β were obtained from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Recombinant PDGF-BB (PDGF) and total ERK1/2 were purchased from R&D Systems, Inc. (Minneapolis, MN, USA). The Abs raised against phosphotyrosine (4G10), p-PDGFRβ (Tyr716), p-FAK (Tyr397), and α-tubulin were obtained from EMD Millipore (Billerica, MA, USA).
Preparation of VSMCs
The animal experimental procedures were approved by the Fu-Jen Animal Experiment Committee and followed the National Institutes of Health guidelines for the care and use of laboratory animals. Rat aortic smooth muscle cells (SMCs) were isolated and characterized as previously described22 and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum, penicillin (100 units/mL), streptomycin (100 μg/mL), and fungizone (250 ng/mL) (Thermo Fisher Scientific, Waltham, MA, USA). Three to six passage cells were used in this study. Unless otherwise indicated, cells reaching 80%–90% confluency were starved and synchronized in DMEM at 37°C overnight and then subjected to further analysis.
Determination of subcellular location of AuNPs
The subcellular distribution of the pAuNPs was analyzed by transmission electron microscopy (TEM) using a JEM-1400 (JEOL, Tokyo, Japan) operated at 110 kV. Cells were fixed in 4% paraformaldehyde (PAF) and 2.5% glutaraldehyde, rinsed in 0.1 M phosphate-buffered saline (PBS, pH 7.2), and post-fixed in 1% osmium tetroxide for 2 hours. The specimens were dehydrated in ethanol by gradually replacing the water with an increasing concentration of ethanol. After reaching 100% ethanol, the specimens were infiltrated with a plastic monomer using Spurr’s resin kit. The specimens were sectioned to a thickness of 75 nm with an ultra-microtome (EM-UC7; Leica Microsystems, Wetzlar, Germany).
Cell migration assay
A cell migration assay with VSMCs was performed by using a Transwell apparatus (8.0 μm pore size; Corning Incorporated, Corning, NY, USA). The lower face of the polycarbonate filter (Transwell insert) was coated with type I collagen (10 μg/mL) for 30 minutes in a laminar flow hood. The lower chamber was filled with serum-free and PDGF-containing medium. VSMCs (2.5×105 cells/mL) were applied to the upper chamber in the presence of vehicle or AuNPs (2–5 ppm). After 3 hours of incubation, all nonmigrant cells were removed from the upper face of the Transwell membrane with a cotton swab, and migrant cells were fixed and stained with 0.5% toluidine blue in 4% PAF. Migration was determined by morphological microscopy and quantified by directly counting the number of stained cells per ×100 field (high-power field [HPF]) under a phase-contrast microscope (Nikon Eclipse Ti-E®; Nikon Corporation, Tokyo, Japan) and imaged.
Lactate dehydrogenase (LDH) release, MTT, WST-1, and BrdU incorporation assays
Cell viability was determined by the LDH release assay. Cellular mitochondrial activity was determined by the MTT (Sigma-Aldrich Co.) and WST-1 (Hoffman-La Roche Ltd., Basel, Switzerland) assays. Cell proliferation was performed by using a Cell Proliferation ELISA, BrdU colorimetric kit (Hoffman-La Roche Ltd.) according to the manufacturer’s protocol. LDH release in the culture medium was determined by the CytoTox 96® Non-Radioactive Cytotoxicity Assay Kit (Promega Corporation, Madison, WI, USA). The MTT and WST-1 assays were based on cellular mitochondrial activity. For the BrdU incorporation assay, 60%–70% confluent VSMCs grown in 96-well tissue culture microplates were starved overnight and then treated with the pAuNPs for the indicated time intervals, followed by labeling by the addition of BrdU for an additional 2 hours. After removing the labeling medium, the cells were fixed, and the DNA was denatured. The anti-BrdU-POD Ab was added, then bound to the BrdU incorporated into the newly synthesized cellular DNA. The reaction product was quantified by measuring the absorbance using an ELISA reader.
Cell lysate preparation and Western blot analysis
Cell lysate was prepared as previously described.23 Total proteins were separated by electrophoresis on sodium dodecyl sulfate polyacrylamide gels (SDS-PAGE), electroblotted onto polyvinylidene difluoride membranes, and then probed using a primary mAb. Immunoblots were detected by a chemiluminescence reagent (EMD Millipore). For some experiments, membranes were stripped with a striping buffer (62.5 mM Tris-HCl, pH 6.7, 2% SDS, and 100 mM β-mercaptoethanol), washed, and reprobed with Abs for α-tubulin level or the corresponding total proteins and developed as described above.
Determination of matrix metalloproteinase-2 (MMP-2) activity and expression
Cells were seeded in 24-well plates and treated with PBS or PDGF (10 ng/mL) in the presence of vehicle or pAuNPs (2–5 ppm) for 16 hours. The conditioned media were collected and analyzed immediately by gelatin zymography. Briefly, samples were subjected to SDS-PAGE in a gel containing 1% gelatin and then incubated in the development buffer (5 mM CaCl2, 1% Triton-X 100, 50 mM Tris-HCl, pH 7.6) at 37°C for 24 hours. After incubation, the gel was stained with 1% Coomassie Brilliant Blue R250 to visualize MMP-2.
Cell adhesion assay
VSMCs pretreated with vehicle or AuNPs for 30 minutes were added to 96-well plates precoated with collagen (10 μg/mL) in the presence of PBS or PDGF for an additional 3 hours. After a brief wash, the adherent cells were fixed with 1% PAF, and cell adhesion was analyzed by phase-contrast/immunofluorescence microscopy, MTT assay, and fluorometry. To measure cell adhesion by the MTT assay, the cells were incubated with MTT (0.5 mg/mL) for 2 hours, followed by the addition of dimethyl sulfoxide. The absorbance was then measured spectrophotometrically in a Sunrise Microplate Reader at 550 nm (Tecan Trading AG, Zürich, Switzerland). To measure cell adhesion by immunofluorescence microscopy and fluorometry, the cells were labeled with BCECF/AM (10 μg/mL), whose excitation and emission wavelengths are 488 and 525 nm, respectively. After cell adhesion, a brief wash was performed to remove unattached cells, the extent of cell adhesion was analyzed by fluorescence microscopy under a Nikon Eclipse Ti-S fluorescence microscope (Nikon Corporation) and quantitatively measured by fluorometry on a Wallac Victor 3 1420 multilabel counter (PerkinElmer Inc., Waltham, MA, USA).
Immunofluorescence microscopy
Cells were adhered to collagen-precoated coverslips in the presence of vehicle or pAuNPs for 1.5 hours. The adherent cells were fixed by 4% PAF for 20 minutes and then treated with 0.2% Triton X-100 for 15 minutes. After being blocked with 3% BSA for 30 minutes, cells were incubated with anti-phosphotyrosine (4G10) or pFAK Ab overnight and then incubated with FITC-conjugated secondary Ab for an additional 1 hour. Alternatively, cells were incubated with FITC-conjugated phalloidin 1 hour followed by incubation with 4′,6-diamidino-2-phenylindole (DAPI) (1:3,000; Thermo Fisher Scientific). After the coverslips were mounted, cells were analyzed under a Nikon Eclipse Ti-S fluorescence microscope (Nikon Corporation).
Histological examination of carotid artery in rats
Vascular injury was induced by balloon catheter inflation of the common carotid artery of Wistar rats;21 the experiment was approved by the Institutional Animal Care and Use Committee of Fu-Jen Catholic University and followed the National Institutes of Health guidelines for the care and use of laboratory animals. The rats were anesthetized by intra-peritoneal administration of pentobarbital (50 mg/kg), and a 2-Fr Fogarty arterial embolectomy balloon catheter was inserted into the right carotid artery to cause vascular injury. Vehicle (ddH2O) or pAuNPs (0.125 mg/kg per day) (~5 rats per group) were orally administered to the rats 3 days before and 14 days after the vascular injury. Rats were euthanized, and the left and right carotid arteries were removed, fixed, and embedded. The tissues were sectioned to a thickness of 5 μm. Immunohistochemistry was performed to identify α-smooth muscle actinin (α-SMA) and proliferating cell nuclear antigen (PCNA) with their respective Abs (LifeSpan BioSciences, Inc., Seattle, WA, USA) in the paraffin-embedded aortas as previously described.21 The secondary Abs were used at a dilution of 1:250. After additional washing, the slides were stained with one-step 3-amino-9-ethylcarbazole (Biogenex, Fremont, CA, USA) for 5–30 minutes. Sections were washed with tap water and mounted with 100% glycerol.
Statistical analysis
Data are expressed as means ± standard error of mean. The means of two groups of data were compared using the unpaired, two-tailed Student’s t-test, whereas multiple groups were compared by one-way analysis of variance (ANOVA).
Results
Effect of AuNPs on PDGF-induced VSMC migration
To investigate the effect of AuNPs on VSMC migration, a bidirectional Transwell system was used, and PDGF was taken as a chemoattractant for VSMC migration. One set of pAuNPs and three sets of cAuNPs (3-EG and 4-EG AuNPs and std AuNPs) were tested for their effect on VSMC migration. The experiment was divided into two groups (Figure 1A and B), as the pAuNPs and EG AuNPs were dispersed in ddH2O and the std AuNPs were dispersed in citrate. As shown in Figure 1, treatment of cells with PDGF for 3 hours effectively increased VSMC migration by a factor of ~2–2.5. Migration was significantly inhibited by the pAuNPs. However, it was not inhibited by the 3-EG and 4-EG AuNPs (Figure 1A) and was only slightly inhibited by the std AuNPs (Figure 1B). The pAuNPs inhibited migration in a concentration-dependent manner, and PDGF-induced VSMC migration was nearly blocked at a concentration of 5 ppm. In contrast, only slight inhibition was observed for the std AuNPs at the same concentration. Therefore, the pAuNPs were used as the main material investigated throughout this study.
Figure 1.

Effect of AuNPs on PDGF-induced VSMC migration.
Notes: The upper chamber, with VSMCs added in the presence of the indicated AuNPs or their corresponding vehicle ddH2O in (A) and 0.1 mg/mL citrate in (B), was assembled with the lower chamber filled with medium in the presence of vehicle, PDGF (10 ng/mL), and/or the indicated AuNPs. After incubation for 3 hours, VSMCs that had migrated to the underside of the filter membrane were photographed (left panels) and counted in HPF (magnification, 100×) under a phase-contrast light microscope (right panels). The results shown are representative of three to five independent experiments. **P<0.01, and ***P<0.001 versus PDGF+vehicle. Scale bar =100 μm.
Abbreviations: 3-EG AuNPs, 1-mercapto-(triethylene glycol)methyl ether functionalized AuNPs; 4-EG AuNPs, (1-mercaptoundec-11-yl)tetra(ethyleneglycol) functionalized AuNPs; AuNPs, gold nanoparticles; HPF, high-power field; pAuNPs, physically synthesized gold nanoparticles; PDGF, platelet-derived growth factor; std AuNPs, standard unconjugated AuNPs; VSMC, vascular smooth muscle cell; NS, nonsignificant; veh, vehicle.
Subcellular location of pAuNPs at an early stage of treatment and effect of pAuNP treatment on VSMC morphology
To trace the subcellular location of the pAuNPs at an early stage of treatment, a TEM analysis was performed. Figure 2A shows that black spots with strong contrast and sharp margins located along the cell peripheries and cytoplasm were found in the VSMCs treated with the pAuNPs at 0.5 and 3 hours of treatment. These black spots were considered aggregates of the pAuNPs due to their high-density metal characteristics. Some of the spots appeared to have been endocytosed by the cells. In contrast, these sharp spots were not found in the control cells (0 hours). Gray spots with coarse margins were observed and might have been high-molecular-weight proteins, such as glycogen. Next, the VSMC morphology after pAuNP treatment was further analyzed by phase-contrast microscopy. Cell morphology was examined at 3, 16, 24, and 48 hours after the pAuNP treatment. Figure 2B shows that no apparent morphological changes and cell death were observed at 3–24 hours of treatment. However, a slight decrease in cell number was observed at 48 hours of treatment. Taken together, our results show that the pAuNPs entered the intracellular space of VSMCs after co-incubation at a very early stage. No distinct change in cell morphology after the pAuNP treatment was noted, but a slight decrease in VSMC number was observed when the treatment was prolonged to 48 hours.
Figure 2.

TEM and morphological analysis of VSMCs treated with pAuNPs.
Notes: VSMCs were incubated with pAuNPs for the indicated time intervals. (A) The subcellular distribution of the pAuNPs was analyzed by TEM. Scale bar =0.5 μm. (B) Cell morphology was analyzed by phase-contrast microscopy. Scale bar =100 μm.
Abbreviations: TEM, transmission electron microscopy; pAuNPs, physically synthesized gold nanoparticles; VSMC, vascular smooth muscle cell; ER, endoplasmic reticulum; M, mitochondria; N, nucleus.
Effect of pAuNPs on VSMC viability, mitochondrial activity, and proliferation
A previous study reported that AuNPs from the same source with an average size of 5.9 nm cause cytotoxicity in human chronic myelogenous leukemia cells.24 To examine whether the pAuNPs affect VSMC viability, the LDH release and MTT assays were performed. As shown in Figure 3A, incubation of cells with the pAuNPs over a period of 24 hours did not appreciably affect LDH release in the medium. However, an increase in LDH release was observed when the incubation time was prolonged to 48 hours, indicating that the pAuNPs did not cause cytotoxicity before 24 hours of treatment but did induce cytotoxicity at 48 hours (ANOVA, P<0.001). Surprisingly, the MTT assay revealed inversely enhanced mitochondrial activity in the VSMCs challenged with the pAuNPs and PDGF for 24 and 48 hours (Figure 3B). A similar but more pronounced enhancing effect was found by using WST-1 as a measuring reagent (Figure 3C). WST-1 has been widely and recently used as it is claimed to have soluble cleavage products and to be more stable than MTT and XTT for measuring mitochondrial activity to represent cell viability and growth/proliferation.25
Figure 3.

Effect of pAuNPs on viability, mitochondrial activity, and proliferation of VSMCs.
Notes: Cells were treated with PBS or PDGF (10 ng/mL) in the presence of vehicle or pAuNPs for the indicated time intervals. After incubation, cells were subjected to analysis by the LDH release (A), MTT (B), WST-1 (C), and BrdU incorporation (D) assays (n=3–5). *P<0.05, **P<0.01, and ***P<0.001 compared with each vehicle control. ##P<0.05 compared with basal control level. The arrows indicate a decrease in BrdU incorporation into DNA. Multiple groups were compared by one-way ANOVA.
Abbreviations: pAuNPs, physically synthesized gold nanoparticles; VSMC, vascular smooth muscle cell; PBS, phosphate-buffered saline; PDGF, platelet-derived growth factor; LDH, lactate dehydrogenase; ANOVA, analysis of variance.
Next, we examined whether the pAuNPs affect VSMC proliferation by the BrdU incorporation assay. Figure 3D shows that the addition of PDGF-BB (10 ng/mL) to VSMCs caused marked cell proliferation at 16 hours, but proliferation decreased thereafter. At 48 hours of incubation, PDGF nearly lost its enhancing activity on VSMC proliferation in contrast to serum (open bars) (ANOVA, P<0.001). It was found that basal and PDGF-induced BrdU incorporation into DNA were reduced by the pAuNPs in a concentration-dependent manner within 24 hours of incubation, particularly at 16 and 24 hours (arrows). However, the particles did not affect serum-induced BrdU incorporation. Taken together, our results demonstrate that the pAuNPs do not affect cell viability over 24 hours of incubation but induce cytotoxicity at 48 hours of incubation. It is strongly suspected that the pAuNPs can enhance cellular mitochondrial activity in VSMCs. In addition, the particles inhibit basal and PDGF-induced VSMC proliferation.
Effect of pAuNPs on PDGF signaling
As VSMC migration and proliferation by PDGF are mediated through the signal transduction pathways,26–28 we further determined the effect of pAuNPs on PDGF signaling. Figure 4 shows that treatment of cells with PDGF induced protein kinase activation in VSMCs, including PDGF receptor β (PDGFRβ), phospholipase Cγ (PLCγ), PI-3K/Akt, MAPKs, protein kinase Cμ, ζ, θ (PKCμ, ζ, θ), and CREB. However, coincubation of cells with pAuNPs did not significantly inhibit the activation of these protein kinases by PDGF, as determined by Western blotting (left panels) and quantitative densitometry (right panels).
Figure 4.

Effect of pAuNPs on PDGF signaling in VSMCs.
Notes: Cells were preincubated with Veh or pAuNPs (2 and 5 ppm) for 30 minutes, followed by PDGF (10 ng/mL) stimulation for (A) 10 minutes or (B) the indicated duration. After incubation, cells were collected, and protein phosphorylation and expression were determined by Western blotting (n=3). Similar data were quantitatively analyzed by densitometry. The phosphorylation level of each protein was calculated by the ratio of the expression level of phosphorylated protein to the expression level of the corresponding total protein. Data are expressed as fold changes relative to the basal level (untreated with PDGF). There is no significance between PDGF+vehicle control and each pAuNP treatment group.
Abbreviations: pAuNPs, physically synthesized gold nanoparticles; VSMC, vascular smooth muscle cell; PDGF, platelet-derived growth factor; Veh, vehicle.
Effect of pAuNPs on MMP-2 expression and activity
Next, we examined the effect of pAuNPs on MMP expression and activity, which have been shown to be responsible for matrix degradation during migration. We previously showed that MMP-2 is a dominant MMP expressed in VSMCs.29 Therefore, MMP-2 was used as a main target in this analysis. As shown in Figure 5, pAuNPs had no effect on MMP-2 activity or expression in the presence or absence of PDGF, as assayed by zymography and Western blotting, respectively.
Figure 5.
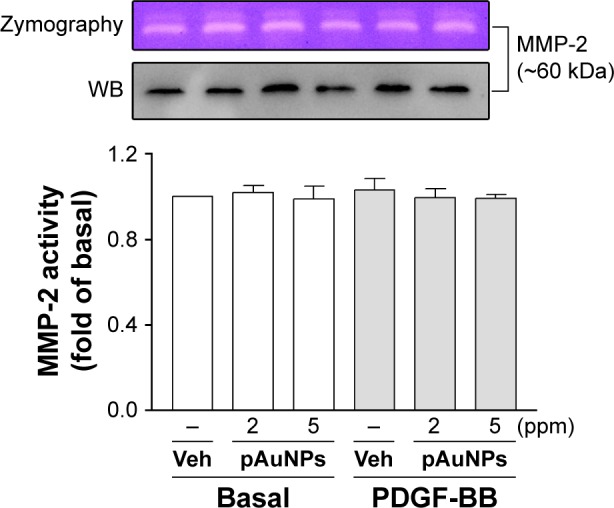
Gelatin zymography and Western blotting of MMP-2 expression and activity.
Notes: VSMCs were treated with Veh or pAuNPs (2 and 5 ppm) in the presence or absence of PDGF (10 ng/mL) at 37°C for 16 hours. The media were removed, centrifuged, and then analyzed by gelatin zymography and WB. The quantitation of MMP-2 activity based on similar zymographic results is shown in the lower panel (n=3).
Abbreviations: pAuNPs, physically synthesized gold nanoparticles; VSMC, vascular smooth muscle cell; PDGF, platelet-derived growth factor; MMP-2, matrix metalloproteinase-2; Veh, vehicle; WB, Western blotting.
pAuNPs inhibit VSMC adhesion to collagen
To further elucidate the possible mechanism of action of pAuNPs in inhibiting VSMC migration, we determined VSMC adhesion to extracellular matrix (ECM). Figure 6 shows that VSMCs could adhere to collagen and spread in the absence of PDGF, but a slight increase in the number of adherent cells was observed with PDGF stimulation (Figure 6A). Treatment of cells with pAuNPs resulted in a significant reduction in VSMC adhesion to the collagen-coated plate in a concentration-dependent manner, whether in the absence or presence of PDGF. The number of adherent cells markedly decreased, and some of those cells were unable to spread on collagen and became rounded. However, the cAuNPs (3-EG and 4-EG AuNPs) did not affect VSMC adhesion, and the std AuNPs slightly inhibited PDGF-induced VSMC adhesion, as assayed by morphological analysis and the MTT assay (Figure 6B and C). To further confirm this result, immunofluorescence microscopy and a quantitative analysis by fluorometry were performed; the results showed that the pAuNPs inhibited VSMC appreciably and the std AuNPs slightly inhibited VSMC adhesion (Figure 6D).
Figure 6.

Effect of AuNPs on VSMC adhesion to collagen.
Notes: Cells were treated with vehicle or the indicated AuNPs for 30 minutes and allowed to adhere to collagen-precoated dishes in the presence or absence of PDGF (10 ng/mL) for 1 hour. VSMC adhesion was analyzed by (A–C) microscopy, MTT reduction, and (D) immunofluorescence microscopy and fluorometry (n=2–3). *P<0.05, **P<0.01, and ***P<0.001 compared with vehicle control. Scale bar =100 μm.
Abbreviations: 3-EG AuNPs, 1-mercapto-(triethylene glycol)methyl ether functionalized AuNPs; 4-EG AuNPs, (1-mercaptoundec-11-yl)tetra(ethyleneglycol) functionalized AuNPs; AuNPs, gold nanoparticles; pAuNP, physically gold nanoparticle; std AuNPs, standard unconjugated AuNPs; VSMC, vascular smooth muscle cell.
Focal adhesions, the best characterized cell–matrix adhesions, are composed of clusters of integrin receptors associated with large complexes of signaling proteins, including talin, vinculin, src, and focal adhesion kinase (FAK), linked to the actin cytoskeleton.30 To further explore the effect of pAuNPs on cellular signaling during VSMC–ECM adhesion, cells were allowed to adhere onto a collagen-precoated dish in the presence of vehicle or pAuNPs for the indicated time intervals. It was found that FAK was activated in a time-dependent manner. A similar activation profile could be observed on a protein that migrated at ~125 kDa using a phosphotyrosine Ab as a detection Ab, indicating that this is a tyrosine phosphorylated protein. The pAuNP treatment significantly inhibited FAK and protein (~125 kDa) tyrosine phosphorylation (Figure 7A). Interestingly, the activation of a protein that migrated at 70~90 kDa, possibly src, syk, or PI-3K, was also inhibited by the pAuNP treatment (Figure 7A, upper panel, arrow). In line with these observations, immunofluorescence microscopy indicated that protein tyrosine and FAK phosphorylation were pronounced at the cell periphery in the control cells but appeared to be diffuse in the pAuNP-treated cells (Figure 7B, upper panels). Moreover, actin cytoskeleton reorganization in response to cell–ECM interactions, presented as the formation of stress fibers across the cells, was observed in the control cells but disappeared in some pAuNP-treated cells. Nucleus staining by DAPI and phase imaging indicated cell locations (Figure 7B, lower panels).
Figure 7.

Effect of pAuNPs on collagen-induced protein tyrosine and FAK phosphorylation and actin cytoskeleton reorganization in VSMCs.
Notes: (A) Cells treated with vehicle or pAuNPs were allowed to adhere to collagen-precoated dishes for the indicated durations. Then, the cells were analyzed by Western blotting, and protein phosphorylation was quantified by densitometry (n=3–4). *P<0.05 compared with control. (B) Suspended VSMCs pretreated with vehicle (control) or pAuNPs (5 ppm) for 0.5 hours were allowed to adhere to collagen-precoated glass coverslips for an additional 1 hour. The cells were analyzed by immunofluorescence microscopy using anti-phosphotyrosine, p-FAK Ab, phalloidin-FITC, and DAPI as described in the Materials and methods section. Arrows indicate that stress fibers were formed in the control cells but disappeared in some pAuNP-treated cells. Scale bar =100 μm.
Abbreviations: pAuNPs, physically synthesized gold nanoparticles; VSMC, vascular smooth muscle cell; FAK, focal adhesion kinase; Ab, antibody; FITC, fluorescein isothiocyanate; DAPI, 4′,6-diamidino-2-phenylindole; Veh, vehicle.
Expression pattern of VSMCs in balloon-injured carotid artery
We previously demonstrated that pAuNPs reduce monocyte adhesion to TNF-α-stimulated human vascular ECs and neointima formation in the rat carotid balloon injury model.21 To identify the expression pattern of SMCs in control and balloon-injured rats, cross sections of paraffin-embedded arteries were stained with α-SMA (a specific marker of SMCs) and PCNA (a marker for proliferating cells). As indicated in Figure 8, both control and balloon-injured carotid arteries obtained from control (untreated) rats showed positive staining for α-SMA but negative staining for PCNA staining in the areas of the vascular media layer, suggesting the presence of SMCs but a lack of proliferation in the vascular medial layers. In response to balloon injury, both α-SMA-positive and PCNA-positive staining were observed in the neointima and some areas in the medial layer of artery, indicating that proliferating SMCs were developed during injury. However, positive staining for both the markers was apparently reduced in the arteries of the pAuNP-treated rats.
Figure 8.

Effect of pAuNPs on the expression pattern of VSMCs in rat balloon-induced carotid artery.
Notes: Representative photomicrographs of α-SMA- and PCNA-stained sections of uninjured and balloon-injured carotid arteries of rats fed with or without the pAuNPs (five rats per group). The expression pattern of (proliferating) VSMCs in the sections was determined by immunohistochemistry using the Ab specific to α-SMA and PCNA. Red staining indicates positive regions. Arrows indicate proliferating VSMCs in the injured carotid arteries. Scale bar =100 μm. The images are representative of three independent experiments.
Abbreviations: pAuNPs, physically synthesized gold nanoparticles; PCNA, proliferating cell nuclear antigen; VSMC, vascular smooth muscle cell; α-SMA, α-smooth muscle actinin; M, media layer; I, intima; Ab, antibody.
Discussion
Although VSMC migration is a difficult process to quantify in human atherosclerosis, in rodents, (neo)intimal VSMCs must have arisen by migration from media or the transdifferentiation of invading myeloid cells from the lumen.4 VSMC migration is generally involved in vascular repair, and PDGF and its receptors are involved in the development of intimal lesions in acute vascular injury.31 Some studies have shown that functional AuNPs conjugated with ginkgolide A can reduce VSMC proliferation and migration in vitro and neointimal hyperplasia in a mouse model,16 and the use of epigallocatechin-3-gallate-conjugated AuNPs is considered a potential alternative approach to prevent restenosis.32 However, the effects of naked AuNPs on the behaviors of VSMCs have been less commonly investigated. In this study, we compared four types of AuNPs, including one set of pAuNPs and three sets of cAuNPs. We demonstrated that naked pAuNPs could mitigate PDGF-induced VSMC migration and adhesion. Moreover, the pAuNPs enhanced cellular mitochondrial activity and tended to inhibit proliferation in VSMCs. In contrast, the naked cAuNPs (3-EG and 4-EG AuNPs) and std AuNPs showed no or only slight activity of this type.
Regarding how the pAuNPs affect PDGF-induced VSMC migration, we suggest that the particles mainly act by affecting cell adhesion. As shown in Figure 4, the pAuNPs did not affect PDGF-induced intracellular kinase phosphorylation, including that of PDGFRβ, PLCγ, PI-3K/Akt, MAPKs, PKC isoforms, and CREB. These kinases have been demonstrated to be involved in cell growth and migration.33 It has been reported that AuNPs can inhibit VEGF/bFGF and IL-1β signaling.18,34,35 In addition, AuNPs have been shown to block protein–protein interactions to regulate their functions. For example, AuNPs could directly bind to heparin-binding growth factors, presumably through cysteine residues of the heparin-binding domain, to inhibit angiogenesis.36 Our unpublished data indicate that the pAuNPs used in this study could also reduce VEGF-induced vascular EC migration. However, direct interaction of the pAuNPs with PDGF and involvement of MMP-2 are unlikely to be due to the lack of inhibition of PDGF signaling and the lack of an effect of the pAuNPs on MMP-2 expression and activity, respectively (Figures 4 and 5). Therefore, the inhibition of cell adhesion to ECM and collagen-induced signaling seemed to be the main mechanism of action of the pAuNPs. This conclusion is supported by the observation that the pAuNPs reduced cell adhesion to collagen in the presence or absence of PDGF (Figure 6).
In response to integrin engagement, several tyrosine kinases, including FAK and src, are activated, leading to the activation of Rac and cdc42. This activation ultimately promotes actin assembly and is required for cell adhesion.30,37 In this study, collagen-induced tyrosine phosphorylation at 125 kDa and FAK phosphorylation at Tyr397 (~125 kDa) were significantly and markedly inhibited by the pAuNPs (Figure 7A). In parallel, immunofluorescence microscopy indicated that protein tyrosine and FAK phosphorylation were more pronounced at the cellular peripheries in the control cells but were relatively weak in the pAuNP-treated cells. In line with these observations, the reorganization of the actin cytoskeleton, manifested as the formation of stress fibers within cells, also apparently decreased in the presence of pAuNPs (Figure 7B). As integrin–ECM interaction requires extracellular metal ions, such as Mn2+, Mg2+, or Ca2+,38 whether the pAuNPs could interfere with this process, including integrin expression and/or affinity in VSMCs, merits further study.
According to the findings shown in Figure 3B and C, the pAuNPs enhanced MTT and WST-1 reduction at 16, 24, and 48 hours, especially in the presence of PDGF. A similar but relatively weak effect on human vascular ECs has also been observed in our laboratory.21 It has been reported that the increase in cellular ATP content and MTT reduction possibly results from an increase in mitochondrial activity and cells undergoing death.39 A comparison of the increases in LDH release and MTT and WST-1 reduction reveals that an increase in MTT and WST-1 reduction may proceed before cells undergo death, as a significant increase in LDH release was observed at 48 hours of treatment (Figure 3A). MTT and WST-1 are reagents generally used for measuring cell viability/proliferation in a similar manner, based on the cleavage of the tetrazolium salt to formazan by cellular mitochondrial dehydrogenases. Therefore, it is strongly suspected that the pAuNP treatment could enhance VSMC “mitochondrial activity.” However, whether the increase in mitochondrial activity has functions other than simply reflecting cell death is an interesting question and remains to be investigated. Our TEM analysis indicated that the pAuNPs could be detected at the cell peripheries and in the cytoplasm of VSMCs at an early stage of treatment (Figure 2B), suggesting that the pAuNPs had entered VSMCs. According to a previous report40 and the models suggested by Sultana et al,41 internalized AuNPs can be transported, transformed, and ultimately liberated into the cytoplasm, leading to local accumulation and alteration of physiological functions of cells. Therefore, we can conclude that the endocytosed pAuNPs actually compromise VSMC functions. Interestingly, compared with our previous findings, we could observe more pAuNP aggregates in the cytoplasm of the VSMCs than in vascular ECs.21 This finding might be explained by several studies that have shown that VSMCs can ingest LDL during atherogenesis42 and uptake of cholesterol, causing VSMCs to transdifferentiate into macrophages.43
Enhanced cellular uptake, low cytotoxicity, and flexibility in synthesis and functionalization support the potential of “functional AuNPs” as an effective platform for drug and nucleic acid delivery to cells.44 Our present study and previous study reveal that “naked pAuNPs” can exhibit anti-migratory activity toward VSMCs as well as HO-1-inducing and anti-inflammatory activities toward vascular ECs, indicating that this type of AuNP has a vascular protective effect. It is noteworthy to explain why pAuNPs and cAuNPs exhibit differential activities toward VSMCs. In this study, it is unlikely that inhibition resulted from the stabilizing/dispersing agent in the AuNP solution because the dispersing agent for the pAuNPs was ddH2O. In addition, the in vitro effects could be confirmed by in vivo observation. Both α-SMA-positive and PCNA-positive staining in the neointima and some areas in the medial layer of artery, representing proliferating (and possibly migrating) VSMCs in the injured artery, were markedly reduced in the balloon-injured arteries of the pAuNP-treated rats (Figure 8). Figure 3B shows that the pAuNPs had an inhibitory effect on VSMC proliferation. Because the pAuNPs were administered to rats in vivo for 17 days, the anti-adhesive, migratory, and proliferative effects of the pAuNPs on VSMCs are strongly suspected to contribute to the particles’ in vivo inhibitory effect on the rat balloon-injured artery model.
In conclusion, we provide the first evidence that “naked” pAuNPs can inhibit VSMC migration and adhesion and enhance VSMC mitochondrial activity. We also demonstrated that in vivo application of the pAuNPs to rats can reduce α-SMA and PCNA staining in the neointima and some areas in the medial layer of artery. These effects elicited by the pAuNPs might contribute to the in vivo vascular protective effects of particles.
Acknowledgments
This work was supported by research grants (106-2320-B-030-001-MY3 and 103-SKH-FJU-03) from Ministry of Science Technology and Shin Kong Wu Ho-Su Memorial Hospital (Taipei, Taiwan), respectively. Parts of the results of this article were presented at the 46th Scientific Meeting of the Japan Atherosclerosis Society, Tokyo, Japan, 2014.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473(7347):317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 2.Jernberg T, Hasvold P, Henriksson M, Hjelm H, Thuresson M, Janzon M. Cardiovascular risk in post-myocardial infarction patients: nationwide real world data demonstrate the importance of a long-term perspective. Eur Heart J. 2015;36(19):1163–1170. doi: 10.1093/eurheartj/ehu505. [DOI] [PubMed] [Google Scholar]
- 3.Libby P, Bornfeldt KE, Tall AR. Atherosclerosis: successes, surprises, and future challenges. Circ Res. 2016;118(4):531–534. doi: 10.1161/CIRCRESAHA.116.308334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118(4):692–702. doi: 10.1161/CIRCRESAHA.115.306361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marx SO, Totary-Jain H, Marks AR. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv. 2011;4(1):104–111. doi: 10.1161/CIRCINTERVENTIONS.110.957332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340(2):115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 7.Rubin K, Tingstrom A, Hansson GK, et al. Induction of B-type receptors for platelet-derived growth factor in vascular inflammation: possible implications for development of vascular proliferative lesions. Lancet. 1988;1(8599):1353–1356. doi: 10.1016/s0140-6736(88)92177-0. [DOI] [PubMed] [Google Scholar]
- 8.Ross R, Masuda J, Raines E, et al. Localization of PDGF-B protein in macrophages in all phases of atherogenesis. Science. 1990;248(4958):1009–1012. doi: 10.1126/science.2343305. [DOI] [PubMed] [Google Scholar]
- 9.Deaton RA, Gan Q, Owens GK. Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am J Physiol Heart Circ Physiol. 2009;296(4):H1027–H1037. doi: 10.1152/ajpheart.01230.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209(1):13–22. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. 2012;95(2):156–164. doi: 10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Magnusson M, Deppert K, Malm J-O, Bovin J-O, Samuelson L. Gold nanoparticles: production, reshaping, and thermal charging. J Nanopart Res. 1999;1(2):243–251. [Google Scholar]
- 13.Khan HA, Abdelhalim MAK, Alhomida AS, Al-Ayed MS. Effects of naked gold nanoparticles on proinflammatory cytokines mRNA expression in rat liver and kidney. Biomed Res Int. 2013;2013:590730. doi: 10.1155/2013/590730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsai C-Y, Shiau A-L, Chen S-Y, et al. Amelioration of collagen-induced arthritis in rats by nanogold. Arthritis Rheum. 2007;56(2):544–554. doi: 10.1002/art.22401. [DOI] [PubMed] [Google Scholar]
- 15.Melissa MK, Ashavani K, Shaymaa M, et al. Gold and silver nano-particles conjugated with heparin derivative possess anti-angiogenesis properties. Nanotechnology. 2009;20(45):455104. doi: 10.1088/0957-4484/20/45/455104. [DOI] [PubMed] [Google Scholar]
- 16.Weakley SM, Wang X, Mu H, et al. Ginkgolide A-gold nanoparticles inhibit vascular smooth muscle proliferation and migration in vitro and reduce neointimal hyperplasia in a mouse model. J Surg Res. 2011;171(1):31–39. doi: 10.1016/j.jss.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yen F-L, Tsai M-H, Yang C-M, et al. Curcumin nanoparticles ameliorate ICAM-1 expression in TNF-α-treated lung epithelial cells through p47 phox and MAPKs/AP-1 pathways. PLoS One. 2013;8(5):e63845. doi: 10.1371/journal.pone.0063845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukherjee P, Bhattacharya R, Wang P, et al. Antiangiogenic Properties of Gold Nanoparticles. Clin Cancer Res. 2005;11(9):3530–3534. doi: 10.1158/1078-0432.CCR-04-2482. [DOI] [PubMed] [Google Scholar]
- 19.Yen H-J, Hsu S-H, Tsai C-L. Cytotoxicity and immunological response of gold and silver nanoparticles of different sizes. Small. 2009;5(13):1553–1561. doi: 10.1002/smll.200900126. [DOI] [PubMed] [Google Scholar]
- 20.Lai TH, Shieh JM, Tsou CJ, Wu WB. Gold nanoparticles induce heme oxygenase-1 expression through Nrf2 activation and Bach1 export in human vascular endothelial cells. Int J Nanomedicine. 2015;10:5925–5939. doi: 10.2147/IJN.S88514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lai T-H, Chung C-H, Chen B-H, et al. Gold nanoparticles compromise TNF-a-induced endothelial cell adhesion molecule expression through NF-kB and protein degradation pathways and reduce neointima formation in a rat carotid balloon injury model. J Biomed Nanotechnol. 2016;12(12):2185–2201. doi: 10.1166/jbn.2016.2315. [DOI] [PubMed] [Google Scholar]
- 22.Lo HM, Hung CF, Tseng YL, Chen BH, Jian JS, Wu WB. Lycopene binds PDGF-BB and inhibits PDGF-BB-induced intracellular signaling transduction pathway in rat smooth muscle cells. Biochem Pharmacol. 2007;74(1):54–63. doi: 10.1016/j.bcp.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 23.Chen CP, Hung CF, Lee SC, Lo HM, Wu PH, Wu WB. Lycopene binding compromised PDGF-AA/-AB signaling and migration in smooth muscle cells and fibroblasts: prediction of the possible lycopene binding site within PDGF. Naunyn Schmiedebergs Arch Pharmacol. 2010;381(5):401–414. doi: 10.1007/s00210-010-0501-1. [DOI] [PubMed] [Google Scholar]
- 24.Tsai Y-Y, Huang Y-H, Chao Y-L, et al. Identification of the nanogold particle-induced endoplasmic reticulum stress by omic techniques and systems biology analysis. ACS Nano. 2011;5(12):9354–9369. doi: 10.1021/nn2027775. [DOI] [PubMed] [Google Scholar]
- 25.Berridge MV, Herst PM, Tan AS. Tetrazolium dyes as tools in cell biology: new insights into their cellular reduction. Biotechnol Annu Rev. 2005;11:127–152. doi: 10.1016/S1387-2656(05)11004-7. [DOI] [PubMed] [Google Scholar]
- 26.Tallquist M, Kazlauskas A. PDGF signaling in cells and mice. Cytokine Growth Factor Rev. 2004;15(4):205–213. doi: 10.1016/j.cytogfr.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Ginnan R, Singer HA. PKC-δ-dependent pathways contribute to PDGF-stimulated ERK1/2 activation in vascular smooth muscle. Am J Physiol Cell Physiol. 2005;288(6):C1193–C1201. doi: 10.1152/ajpcell.00499.2004. [DOI] [PubMed] [Google Scholar]
- 28.Campbell M, Trimble ER. Modification of PI3K- and MAPK-dependent chemotaxis in aortic vascular smooth muscle cells by protein kinase CβII. Circ Res. 2005;96(2):197–206. doi: 10.1161/01.RES.0000152966.88353.9d. [DOI] [PubMed] [Google Scholar]
- 29.Lo HM, Wu MW, Pan SL, Peng CY, Wu PH, Wu WB. Chrysin restores PDGF-induced inhibition on protein tyrosine phosphatase and reduces PDGF signaling in cultured VSMCs. J Nutr Biochem. 2012;23(6):667–678. doi: 10.1016/j.jnutbio.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 30.Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011;3(9):a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sano H, Sudo T, Yokode M, et al. Functional blockade of platelet-derived growth factor receptor-beta but not of receptor-alpha prevents vascular smooth muscle cell accumulation in fibrous cap lesions in apolipoprotein E-deficient mice. Circulation. 2001;103(24):2955–2960. doi: 10.1161/01.cir.103.24.2955. [DOI] [PubMed] [Google Scholar]
- 32.Khoobchandani M, Katti K, Maxwell A, Fay W, Katti K. Laminin receptor-avid nanotherapeutic EGCg-AuNPs as a potential alternative therapeutic approach to prevent restenosis. Int J Mol Sci. 2016;17(3):316. doi: 10.3390/ijms17030316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heldin CH, Ostman A, Ronnstrand L. Signal transduction via platelet-derived growth factor receptors. Biochim Biophys Acta. 1998;1378(1):F79–F113. doi: 10.1016/s0304-419x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- 34.Kim D, Lee J, Johnson AL. Vascular endothelial growth factor and angiopoietins during hen ovarian follicle development. Gen Comp Endocrinol. 2016;232:25–31. doi: 10.1016/j.ygcen.2015.11.017. [DOI] [PubMed] [Google Scholar]
- 35.Karthikeyan B, Kalishwaralal K, Sheikpranbabu S, Deepak V, Haribalaganesh R, Gurunathan S. Gold nanoparticles downregulate VEGF- and IL-1β-induced cell proliferation through Src kinase in retinal pigment epithelial cells. Exp Eye Res. 2010;91(5):769–778. doi: 10.1016/j.exer.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Bhattacharya R, Mukherjee P. Biological properties of “naked” metal nanoparticles. Adv Drug Deliv Rev. 2008;60(11):1289–1306. doi: 10.1016/j.addr.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 37.DeMali KA, Wennerberg K, Burridge K. Integrin signaling to the actin cytoskeleton. Curr Opin Cell Biol. 2003;15(5):572–582. doi: 10.1016/s0955-0674(03)00109-1. [DOI] [PubMed] [Google Scholar]
- 38.Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol. 2011;3(3):a004994. doi: 10.1101/cshperspect.a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leite PEC, Pereira MR, Santos CADN, Campos APC, Esteves TM, Granjeiro JM. Gold nanoparticles do not induce myotube cytotoxicity but increase the susceptibility to cell death. Toxicol In Vitro. 2015;29(5):819–827. doi: 10.1016/j.tiv.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 40.Shukla R, Bansal V, Chaudhary M, Basu A, Bhonde RR, Sastry M. Biocompatibility of gold nanoparticles and their endocytotic fate inside the cellular compartment: a microscopic overview. Langmuir. 2005;21(23):10644–10654. doi: 10.1021/la0513712. [DOI] [PubMed] [Google Scholar]
- 41.Sultana S, Djaker N, Boca-Farcau S, et al. Comparative toxicity evaluation of flower-shaped and spherical gold nanoparticles on human endothelial cells. Nanotechnology. 2015;26(5):055101. doi: 10.1088/0957-4484/26/5/055101. [DOI] [PubMed] [Google Scholar]
- 42.Libby P. Inflammation in atherosclerosis. Nature. 2002;420(6917):868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 43.Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci U S A. 2003;100(23):13531–13536. doi: 10.1073/pnas.1735526100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acharya S, Hill RA. High efficacy gold-KDEL peptide-siRNA nanoconstruct-mediated transfection in C2C12 myoblasts and myotubes. Nanomedicine. 2014;10(2):329–337. doi: 10.1016/j.nano.2013.07.015. [DOI] [PubMed] [Google Scholar]