

Summary
Krüppel-like factors (KLFs) are deoxyribonucleic acid–binding transcriptional factors that regulate various pathways that control metabolism and other cellular mechanisms. Various KLF isoforms have been associated with cellular, organ, or systemic metabolism. Altered expression or activation of KLFs has been linked to metabolic abnormalities, such as obesity and diabetes, as well as with heart failure. This review article summarizes the metabolic functions of KLFs, as well as the networks of different KLF isoforms that jointly regulate metabolism in health and disease.
Key Words: diabetes, heart failure, metabolism, obesity, Krüppel-like factors
Abbreviations and Acronyms: APOE, apolipoprotein E; ATP, adenosine triphosphate; BAT, brown adipose tissue; BCAA, branched chain amino acid; C/EBP, CCAAT/enhancer-binding protein; CBP, cAMP response element binding protein binding protein; CREB, cAMP response element binding protein; CtBP, C-terminal binding protein; DIO, diet-induced obesity; Dlk1, delta-like noncanonical notch ligand 1; DNA, deoxyribonucleic acid; FA, fatty acid; FGF, fibroblast growth factor; GLUT4, glucose transporter type 4; HDAC, histone deacetylases; KLF, Krüppel-like factor; MODY, maturity-onset diabetes of the young; PEPCK, phosphoenolpyruvate carboxykinase; PGC, PPARγ coactivator 1; PPAR, peroxisome proliferator-activated receptor; RNA, ribonucleic acid; SCD1, stearoyl–coenzyme A desaturase 1; SK1, sphingosine kinase-1; SREBP-1c, sterol regulatory element-binding protein-1c; SUMO, small ubiquitin-like modifier; T2DM, type 2 diabetes mellitus; TG, triglyceride; TGF, transforming growth factor; WAT, white adipose tissue
Central Illustration
Krüppel-Like Factor Biology
Krüppel-like factors (KLFs) are zinc finger proteins with the ability to bind CACCC or GT box deoxyribonucleic acid (DNA) elements and act as either transcriptional activators or repressors. The regions of KLFs that do not participate in DNA binding are highly divergent and participate in protein–protein interactions. The name “Krüppel-like factor” was derived from the homologous Drosophila protein Krüppel, which means “cripple” and is associated with development. The founding member of the mammalian KLF family was discovered in red blood cell lineage and plays a major role in β-globin expression and erythrocyte development. The KLF family has 18 members thus far, with a broad range of expression profile among several tissues. KLF isoforms have been associated with regulation of metabolic pathways and energetic homeostasis in various organs, such as the liver, adipose tissue, heart, skeletal muscle, lungs, and myeloid cells. The present review article summarizes studies in cell systems and animal models (Table 1) that implicate KLFs in the regulation of organ and systemic metabolism, as well as their role in the pathophysiology of metabolic diseases. Thus far, no metabolic functions have been attributed to KLF12, KLF17, or KLF18.
Table 1.
Animal Models With Genetic Variations of KLFs
| Animal Model | Phenotype (Ref. #) | |
|---|---|---|
| KLF1 | Klf1–/– | Lethal E14, deficit in β-globin expression and erythropoiesis, severe anemia 135, 136 |
| Klf1–/– and Rb+/-;Klf1+/– | Cell cycle perturbation via control of E2f2 (S-phase entry) 130, 131 | |
| Klf1–/– and Klf1+/– | Transcriptional regulation of macrophage-expressed Dnase2α gene (red blood cell maturation) (138) | |
| KLF2 | Tea-TCR-Klf2 Tg | Restrainment of CD4+ cell differentiation T follicular helper (217) |
| Klf2–/– | Lethal E12.5–14.5, defective blood vessel, severe bleeding (218) | |
| Splenic and lymph node T cells, abnormal cell surface phenotype (219) | ||
| Embryonic globin genes expression affected but not adult globin genes (144) | ||
| Splenomegaly, increased splenic B-cell subsets (220) | ||
| Altered B-cell tissue distribution and expression of trafficking molecules (221) | ||
| Endothelial-Klf2–/– | Cardiac failure, vascular abnormalities (222) | |
| Cerebral cavernous malformations, vascular lesions, enlarged capillaries, irregular structure (223) | ||
| Glomerular endothelial cell injury, diabetic nephropathy (215) | ||
| Hematopoietic Klf2–/– | Restriction of chemokine receptor expression patterns, regulation of T-cell migration (224) | |
| T-cell-Klf2–/– | Unrestrained cytokine production, bystander chemokine receptor upregulation (225) | |
| Fetal liver Klf2–/– chimeras | Regulation of thymocyte and T-cell migration (226) | |
| Klf2–/–; APOE–/– | Increased atherosclerosis (161) | |
| Klf2/–/; Ldlr–/– | Increased atherosclerosis (162) | |
| KLF3 | Klf3–/– | Reduced adipocyte size and less white adipose tissue (87) |
| Resistance to diet-induced obesity and glucose intolerance, increased adipolin (90) | ||
| Impaired silencing of embryonic globin expression during development (139) | ||
| KLF3-H275R | Homozygotes: Lethal E14.5–16.5, severe cardiac defects Heterozygotes: Perinatal death, thick myocardial wall, enlarged chambers (227) |
|
| KLF4 | Klf4–/– | Death shortly after birth, dehydration due to skin barrier permeability 228, 229 |
| Cardiomyocyte-Klf4–/– | Mild increase of cardiac mass (153), aging-related cardiomyopathy, pressure overload cardiac hypertrophy (56) | |
| Smooth muscle-Klf4–/– | Reduced atherosclerosis lesion size, increased plaque stability and cap thickness (165) | |
| Endothelial-Klf4Tg; APOE-/– | Reduced atherosclerosis (163) | |
| Endothelial-Klf4–/–; APOE–/– | Increased atherosclerosis (163) | |
| KLF5 | Klf5–/– | Homozygotes: Lethal E8.5 Heterozygotes: Decreased arteria wall thickening, angiogenesis, cardiac hypertrophy, interstitial fibrosis (230) |
| Cardiac fibroblast Klf5–/– | Resistance in pressure overload–mediated cardiac hypertrophy (154) | |
| Lung-Klf5–/– | Impaired surfactant protein and lipid production, lung structural abnormalities (231) | |
| Intestine-Klf5–/– | Altered crypt architecture and intestine morphology (232) | |
| Hematopoetic-Klf5–/– | Splenomegaly, increased peripheral white blood cells (233) | |
| Cardiomyocyte-Klf5–/– | Dilated cardiomyopathy associated with energetic deficiency (59) | |
| Klf5+/– | Lower white adipose tissue mass after birth that is abrogated later (83) | |
| Resistance to diet-induced obesity despite increased food intake (51) | ||
| KLF6 | Klf6–/– | Lethal E12.5, hematopoiesis defects (234) |
| Klf6+/– | Reduced fibrosis in response to Ang II overload (158) | |
| Cardiomyocyte-Klf6–/– | Reduced fibrosis in response to Ang II overload (158) | |
| Cardiac fibroblast-Klf6–/– | Lack of protection to Ang II–induced fibrosis (158) | |
| Hepatocyte-Klf6–/– | Reduced glucokinase and insulin sensitivity (117) | |
| Reduced hepatic PPARα protein (187) | ||
| KLF7 | Klf7–/– | Lethal within the first 3 days of life, low respiratory rate, cyanosis, defects of the olfactory and optic nerves, impaired axon growth and abnormal dendrite organization (235) |
| Neuronal Klf7 chimera | Increased neuronal regeneration capacity after axon injury (236) | |
| KLF8 | Klf8–/– | Viable but reduced lifespan (139) |
| KLF9 | Klf9–/– | Reduces activity in rotarod and contextual fear–conditioning tests (237) |
| KLF10 | Klf10–/– | Hyperglycemia, increased hepatic glucose production, increased plasma triglycerides, loss of circadian expression of hepatic lipid and carbohydrate metabolism genes (105) |
| Impaired insulin secretion (pancreas) (119) | ||
| KLF11 | Klf11–/– | Decreased circulating insulin, increased peripheral insulin sensitivity, dysregulation of genes involved in lipid metabolism, resistance to diet-induced obesity (126) |
| KLF12 | — | — |
| KLF13 | Klf13–/– | Splenomegaly and reduced number of circulating erythrocytes (86) |
| KLF14 | Liver-Klf14–/– | Lower plasma HDL level (169) |
| KLF15 | Klf15–/– | Fasting hypoglycemia, reduced expression of hepatic and skeletal muscle catabolism genes, loss of circadian expression of hepatic metabolism genes, loss of bile acid synthetic enzymes 67, 114, 119, 197 |
| Protection against hepatic insulin resistance and fatty liver under high-fat feeding (196) | ||
| Fatigue after endurance exercise, reduced skeletal muscle fatty acid oxidation gene expression (66) | ||
| Cardiac lipid oxidation deficit, susceptibility to pressure overload–induced cardiac hypertrophy 60, 155 | ||
| Adipose-KLF15 Tg | Insulin resistance and protection from weight gain, improved glucose tolerance due to increased insulin synthesis by pancreas (171) | |
| KLF16 | — | — |
| KLF17 | — | — |
| KLF18 | — | — |
| Double k/o | Klf3–/–; Klf8–/– | Lethal E14.5 (139) |
Ang = angiotensin; APOE = apolipoprotein E; CD = cluster of differentiation; E = embryonic day; HDL = high-density lipoprotein; KLF = Krüppel-like factor; k/o = knockout; PPAR = peroxisome proliferator activated receptor; Tg = transgenic.
KLFs have 3 conserved zinc finger motifs at the C-terminal domain of 23-25 amino acids, which bind on GC-rich sequences with a preference for the CACCC sequence (1). The zinc finger domains also contain nuclear localization signals 2, 3, 4, 5, 6. Although the C-terminal domain of the KLFs contains primarily nuclear localization signals and the DNA-binding region, the N-terminal domain bears regions that interact with other proteins.
Post-translational modifications of KLFs, as well as the proteins that KLFs interact with, alter their transcriptional activity or subcellular localization. KLFs with structural homology, particularly in the N-terminal domains, share functional similarity. Depending on their structural features and transcriptional roles, KLFs have been divided into 3 major groups (Figure 1) (1). Group 1 includes KLF3, KLF8, and KLF12, which interact with transcriptional repressors. Group 2 comprises KLF1, KLF2, KLF4, KLF5, KLF6, and KLF7, which function primarily as transcriptional activators although interaction with transcriptional repressors has also been reported. Finally, group 3 includes KLF9, KLF10, KLF11, KLF13, KLF14, and KLF16, which have mostly been described as transcriptional repressors. KLF15 and KLF17 have not been categorized in any of these groups because little is known about their protein interaction motifs. Various KLF members of group 2, such as KLF1, KLF2, KLF4, KLF5, and KLF6, as well as KLF13 from group 3, bind to acetyl-transferases, such as cAMP response element-binding protein (CREB) binding protein (CBP), p300, and p300/CBP-associated factor 7, 8, 9, 10, 11. This interaction leads to acetylation of KLFs that stimulates their transcriptional activity 12, 13 or acetylation of histones followed by chromatin remodeling that ignites transcription in regions that are targeted by KLFs 14, 15, 16, 17. Accordingly, binding of KLFs, such as KLF1, KLF4, KLF5, and KLF11, with histone deacetylases (HDACs) suppresses transcriptional activity due to deacetylation of either KLFs 18, 19 or histones 20, 21. Another mechanism of transcriptional repression by KLFs involves interaction with the transcriptional repressors Sin3A (22), C-terminal binding protein (CtBP)1, and CtBP2 23, 24, 25, which in turn recruit HDACs (26), methyltransferases (27), and other silencing complexes such as polycomb proteins (28) and Ikaros (29).
Figure 1.

Transcriptional Coregulators of KLF Isoforms
KLF isoforms can be divided into 3 groups based on their interaction motifs for transcriptional co-activators and co-repressors. Group 1 includes KLFs which contain the CtBP binding motif. Group 2 includes KLFs which contain the Sin3A interaction domain. Group 3 includes KLFs which interact with acetyl-transferases. KLF = Krüppel-like factor; CBP = cAMP response element binding protein; CtBP = C-terminal binding protein; HDAC = deacetylases; NF-κB = nuclear factor–κB; P/CAF = p300/CBP-associated factor; PGC-1 = PPARγ coactivator 1.
Post-translational modifications of KLFs alter their subcellular localization, interactions with other proteins, and transcriptional activity. Histone acetyltransferases mediate acetylation of KLFs, which generally promotes binding of KLFs on DNA and transcriptional activation 7, 8, 9, 10, 11, 12. Phosphorylation also modulates transcriptional activity by regulating protein–protein interaction of KLFs with other transcriptional regulators, including either activators 14, 30, 31, 32, 33, 34, 35, 36 or repressors (37). The stability of KLFs is regulated by ubiquitination that is mediated by ubiquitin ligases, such as WWP1, Itch, and FBXO22, which eventually activate the proteasome pathway 38, 39, 40, 41, 42, 43, 44, 45, 46. KLFs undergo modification by the small ubiquitin-like modifier (SUMO) peptide. SUMOylation may either promote 47, 48, 49 or suppress 50, 51 KLF transcriptional activity by promoting nuclear translocation (47), degradation (50), or interaction with transcriptional corepressors (51). Thus, KLFs may either activate or suppress gene transcription depending on post-translational modifications that affect stability of the protein or interaction with transcriptional coregulators.
KLF Biology in Organs of Metabolic Interest
Heart
The heart is a major regulator of systemic metabolism; it consumes a large amount of lipids and glucose to produce the 5 to 6 kg of adenosine triphosphate (ATP) that is needed for pumping approximately 7,000 l of blood per day. To meet this energetic demand, the heart relies primarily on fatty acid (FA) β-oxidation, which supplies approximately 70% of the heart’s ATP (52). Therefore, a robust regulatory network is in place to coordinate cardiac and systemic metabolism. Disruption of this network results in altered uptake, transport, and use of FAs in the heart and systemic metabolic changes. To illustrate the consequences of disrupting the network, deletion of the triglyceride (TG) hydrolytic enzyme lipoprotein lipase in cardiomyocytes reduces uptake of circulating TG, leading to hypertriglyceridemia (53). Similarly, expression of lipoprotein lipase only in the heart normalizes TG levels in mice with lipoprotein lipase deletion in skeletal muscle and adipose (54). As such, changes in the energy demands of the heart affect cardiac function and may also influence the systemic metabolic balance.
Various members of the KLF family interact with critical regulators of cardiac mitochondrial function and FA oxidation and are therefore necessary for maintaining the metabolic function of the heart (Figure 2). Mitochondrial biogenesis is regulated in large part by the coactivator peroxisome proliferator-activated receptor (PPAR)γ coactivator 1 (PGC-1), which interacts with nuclear receptors, including estrogen-related receptor, PPARα, and nuclear respiratory factor, to regulate genes critical to mitochondrial function (55). KLF4 forms a transcriptional complex with estrogen-related receptor and PGC-1, in which KLF4 is required for optimal activity of the estrogen-related receptor–PGC-1 complex (56). Concordantly, cardiac-specific Klf4–/– mice develop heart dysfunction with aging or pressure overload associated with reduced ATP production, increased reactive oxygen species production, and mitochondrial dysfunction. Furthermore, embryonic deletion of cardiac KLF4 results in increased mortality likely resulting from impaired mitochondrial biogenesis. In smooth muscle cells, AMPK, a well-described energy-sensing kinase and regulator of PGC-1α, directly activates KLF4 expression (57). The extent that the AMPK–KLF4 axis functions in cardiomyocyte energy homeostasis is to be determined.
Figure 2.

KLF Isoforms Associated With Cardiac Lipid Metabolism
Summary of KLF isoforms and metabolic pathways involved in cardiomyocyte biology. A detailed description of the pathways involved in included in the text. Figure was produced using Servier Medical Art (http://www.servier.com/). BCAA = branched chain amino acid; ERR = estrogen-related receptor; KLF = Krüppel-like factor; PPAR = peroxisome proliferator-activated receptor; other abbreviations as in Figure 1.
Genes necessary for FA oxidation are regulated by the nuclear receptor PPARα, which activates expression of genes involved in lipid uptake, lipid transport, and β-oxidation (58). Three members of the KLF family promote FA oxidation in the heart through their interaction with PPARα. KLF4 activates expression of PPARα through direct interaction with the Ppara promoter. Consistently, KLF4 overexpression results in increased expression of FA oxidation gene expression, whereas KLF4 knockdown disrupts the effects of PPARα agonist WY16463 (56). Likewise, cardiomyocyte KLF5 was recently implicated as a regulator of cardiac metabolism, where it promotes FA oxidation by directly activating transcription of PPARα (59). Cardiac Ppara and its target genes are markedly downregulated in the hearts of cardiomyocyte-specific Klf5–/– mice, resulting in reduced cardiac FA oxidation, reduced ATP production, and triglyceride accumulation. These mice develop cardiac dysfunction around 8 months of age, eventually progressing to dilated cardiomyopathy, likely resulting from the metabolic deficit.
KLF15 induces expression of genes necessary for lipid metabolism, which are reduced in Klf15–/– hearts (60). KLF15 interacts physically with PPARα, which may account for the lipid oxidation deficits observed in Klf15–/– hearts (61). Importantly, regulation of PPARα target genes requires KLF15 because silencing Klf15 reduces the cardiomyocyte response to PPARα agonists. Circadian expression of cardiac KLF15 is implicated in physiological metabolic adaptations of mouse hearts to the sleep–wake cycle by regulating the circadian expression of catabolic genes (62). By oscillating expression of metabolic genes, KLF15 allows the heart to meet metabolic demands during the day and repair damages resulting from cellular metabolism at night.
Skeletal muscle
As in the heart, skeletal muscle requires a large amount of ATP to facilitate contraction. Substrates of skeletal muscle metabolism are numerous and include creatine phosphate, glycogen stores, circulating glucose, and free FAs, which are differentially used during rest, strenuous exercise, or exercise of long duration (63). Furthermore, skeletal muscle cells are subclassified as type 1, or slow-twitch, and type 2, or fast-twitch, fibers, with type 1 muscle fibers using primarily oxidative phosphorylation and type 2 fibers requiring anaerobic glucose metabolism (64). FA oxidation is the preferred source of energy for skeletal muscle during endurance exercise. PPARδ is a key regulator of FA oxidation–related gene expression in skeletal muscle and acts as a mediator of muscle fiber type and adaptation to endurance exercise (65). Association of PPARδ function with skeletal muscle KLF5 is important for its function (Figure 3). Under basal conditions, SUMOylated KLF5 forms a complex with PPARδ and corepressors, and inhibits genes involved in FA oxidation and energy uncoupling, including Cpt1b, uncoupling protein (Ucp)2, and Ucp3 expression (51). Treatment with PPARδ agonist results in deSUMOylation of KLF5 and association with transcriptional activation complexes, thereby promoting FA oxidation.
Figure 3.

KLF Isoforms That Control Metabolism in Skeletal Muscles
Summary of KLF isoforms and metabolic pathways involved in skeletal muscle biology. A detailed description of the pathways involved in included in the text. Figure was produced using Servier Medical Art (http://www.servier.com/). GLUT4 = glucose transporter type 4; other abbreviations as in Figures 1 and 2.
KLF15 also regulates lipid use in skeletal muscle during prolonged exercise (66). Klf15 expression is increased after endurance exercise (Figure 3), and Klf15–/– mice fatigue more rapidly when subjected to treadmill running, likely resulting from impaired lipid oxidation in type 1 fibers. After KLF15 knockdown in cultured myotubules, expression of genes required for lipid use is acutely decreased. Interestingly, these changes occur despite a 3-fold induction of Ppargc1a expression and without changes in PPARα or PPARδ expression. It is therefore likely that KLF15 is a critical component of the PGC-1–PPAR complex in skeletal muscle, as has previously been described in the heart (61).
Insulin-dependent glucose uptake, which is important for type 2 muscle fibers, results from translocation of preformed glucose transporter type 4 (GLUT4)-containing vesicles to the cell membrane. Upon insulin stimulation, these vesicles fuse with the plasma membrane, and GLUT4 transporters are exposed to the outer surface of the cells, which facilitates uptake of glucose. Skeletal muscle KLF15 physically interacts with MEF2A and directly promotes expression of GLUT4 (Figure 3) (63). Disruption of the KLF15-binding site within the Glut4 promoter prevents MEF2A-mediated induction of GLUT4, suggesting that protein–protein interaction between these proteins is a critical event for their synergistic coordination for binding on the Glut4 promoter. The relevance of GLUT4 regulation by KLF15 in regulating insulin sensitivity or to the progression of metabolic disease has not been investigated.
Increased expression of KLF15 is an important event for the skeletal muscle response to fasting (Figure 3) 66, 67, 68. In the fasted state, KLF15 increases expression of acetyl–coenzyme A synthetase 2, which is a mitochondrial matrix enzyme responsible for producing acetyl–coenzyme A from acetate and coenzyme A (68) that is then channeled to the Krebs cycle. KLF15 also mediates protein catabolism in the skeletal muscle, which is required for providing amino acids that are used for gluconeogenesis in the liver in a process known as the alanine cycle 67, 69. In the fasted state, circulating levels of alanine are increased, resulting from catabolism of branched chain amino acids (BCAAs) in the skeletal muscle. Circulating alanine can be converted to pyruvate and used for gluconeogenesis in the liver 70, 71. Consistently, fasted Klf15–/– mice exhibit profound hypoglycemia and reduced expression of skeletal muscle protein catabolic gene expression, including the mitochondrial enzyme branched chain aminotransferase 2 67, 69.
Adipose tissue
Adipose tissue is critical for energy homeostasis and in general fulfills 3 different tasks. First, it is involved in TG synthesis and storage. Second, during fasting or increased energy demands, adipose TGs are hydrolyzed, and FAs are released into circulation. Third, it functions as an endocrine organ producing adipokines, such as leptin, adiponectin, resistin, visfatin, and many others. Adipokines are responsible for the regulation of lipid and carbohydrate metabolism, appetite, and energy expenditure 72, 73. Consequently, dysfunction of these processes is tightly linked to metabolic disorders, including the metabolic syndrome and obesity. In mammals, there are 2 major types of adipose tissue that differ in anatomy and function: white adipose tissue (WAT) and brown adipose tissue (BAT) (74). The main function of adipocytes from WAT is the storage of TGs during energy intake and the release of FAs when energy is required. The major function of adipocytes from BAT is dissipating energy via heat production (thermogenesis), which is mediated by UCP1 that uncouples the electron transport from ATP production 74, 75, 76. The differentiation of pre-adipocytes into mature insulin-sensitive adipocytes is stimulated by members of 2 transcription factor families, the CCAAT/enhancer binding proteins (C/EBPs) and PPARs (77). Adipocyte differentiation in WAT is driven by C/EBPα, C/EBPβ, and C/EBPδ that promote PPARγ expression, which is a master regulator of this process. Various KLFs regulate adipocyte metabolism and differentiation (Figure 4).
Figure 4.

KLF Isoforms That Control Adipocyte Differentiation
Summary of KLF isoforms and metabolic pathways involved in adipocyte biology. A detailed description of the pathways involved in included in the text. Figure was produced using Servier Medical Art (http://www.servier.com/). C/EBP = CCAAT/enhancer-binding protein; Dlk1 = delta-like noncanonical notch ligand 1; other abbreviations as in Figures 1 and 2.
In WAT, adipocyte differentiation is promoted by several members of the KLF family. KLF4 directly activates the C/EBPβ promoter, which at a later stage stimulates PPARγ expression (78). Interestingly, activation of C/EBPβ transcription is more robust if KLF4 interacts with the transcription factor Krox20, which itself can transactivate C/EBPβ transcription 78, 79. In addition to KLF4, KLF9 also induces the C/EBPβ promoter, and small interfering ribonucleic acid (RNA)–mediated knockdown reduces C/EBPβ expression, effectively suppressing adipocyte differentiation, but does not influence C/EBPδ expression (80). KLF9 physically interacts with C/EBPα (81), and KLF8 directly induces expression C/EBPα (82). Consistently, addition of KLF8 small interfering RNA in 3T3-L1 pre-adipocytes inhibits adipocyte differentiation, whereas KLF8 overexpression has the opposite effect. Protein–protein interactions occur between KLF5 and C/EBPβ as well as C/EBPδ (83). Thus, in WAT, KLF4, KLF5, KLF8, and KLF9 promote adipogenesis via stimulating C/EBPs.
A downstream target of C/EBPs, PPARγ, is also regulated by KLFs. KLF5 (83), KLF8 (82), KLF9 (81), KLF13 (84), and KLF15 (85) induce PPARγ expression. It has been shown that KLF5 regulates differentiation of embryonic fibroblasts to adipocytes by binding and activating the endogenous Pparg2 promoter (83). Adipocytes infected with retroviral vectors harboring a dominant-negative Klf5 construct exhibited reduced lipid accumulation compared with Klf5 overexpressing cells. White, but not brown, adipose tissue mass is reduced in Klf5+/– mice 3 days after birth, but that difference is no longer apparent in 4-week-old mice. Thus, although adipocyte KLF5 seems to drive adipose development, other KLF5-independent pathways that may be induced by the lipid-rich diet (milk) in the newborns may drive adipose development. KLF8 functions as a pro-adipogenic factor and directly induces expression of Pparg (82). KLF9 activates Pparg2 promoter (81). In addition, KLF15, which is markedly upregulated during differentiation of 3T3-L1 pre-adipocytes into mature adipocytes, also induces PPARγ expression (85).
The same PPARγ-inducing effect is conferred by Klf13, the expression of which is elevated early during porcine subcutaneous stromal vascular cell adipocyte differentiation (84). Knockdown of KLF13 results in decreased lipid accumulation and lower expression of adipogenesis-related genes, including Pparg. However, KLF13 does not seem to have the same role in mouse adipocyte differentiation. Klf13–/– mice do not have defective adipogenesis (86) and neither overexpression nor knockdown of KLF13 in the mouse 3T3-L1 pre-adipocyte cell line affected the expression of PPARγ and the adipocyte differentiation gene expression program (84). Thus, the role of KLF13 in causing adipose-specific changes in mice remains to be investigated, and a definite pro-adipogenic role for this family member has yet to be proven.
In WAT, KLF2 (78), KLF3 (87), and KLF7 (88) inhibit adipocyte differentiation. Mechanistically, KLF2 directly inhibits Pparg promoter, and adenovirus-mediated ectopic KLF2 expression inhibits Pparg expression, sterol regulatory element binding protein-1c (SREBP-1c), and C/EBPα, resulting in decreased intracellular lipid accumulation (89). However, KLF2 overexpression does not affect C/EBPβ or C/EBPδ expression. KLF3 directly represses the C/EBPα promoter inhibiting 3T3-L1 pre-adipocyte differentiation 87, 90. Similarly, KLF7 overexpression in pre-adipocytes inhibits adipocyte differentiation; however, the mechanism has not yet been studied (88).
KLF6 is another member of the KLF family that also affects adipogenesis but not in a C/EBP- or PPARγ-dependent manner. Silencing of KLF6 in pre-adipocytes markedly suppresses adipogenesis 91, 92. Mechanistically, lower Klf6 levels are associated with elevated expression of delta-like noncanonical notch ligand 1 (Dlk1), a transmembrane protein that inhibits differentiation of adipocytes. Accordingly, KLF6 overexpression inhibits Dlk1 expression in pre-adipocytes and 3T3 cells. The repressive effect of KLF6 on Dlk1 involves protein–protein interaction with HDAC3.
In BAT, KLF16 has been identified as a transcription factor inhibiting brown adipocyte differentiation. KLF16 expression is decreased during differentiation of primary murine brown pre-adipocytes into mature adipocytes. KLF16 directly represses Pparg expression. Consistently, overexpression of KLF16 inhibits adipogenesis in 3T3-L1 cells and in primary brown pre-adipocytes (93).
WAT contains thermogenic fat cells, the so-called inducible brown, brown-in-white, beige, or “brite” adipocytes, which are highly metabolically active and consume fat and carbohydrates via nonshivering thermogenesis 94, 95. The accumulation of beige adipocytes in WAT is referred to as browning of WAT. Due to the high metabolic activity of BAT, browning of WAT has been suggested as a therapeutic intervention for obesity in humans aiming to consume stored FAs by converting them to heat (96). KLF11 (97) and KLF15 (98) influence the browning of WAT, which is promoted by PPARγ and the glucocorticoid receptor. KLF11 is directly induced by PPARγ and serves as a PPARγ coregulator responsible for enhancing its effects (99). This interaction is required for the browning of human adipose tissue induced by the PPARγ agonist rosiglitazone, which is characterized by a stable gene program involved in increased oxidative capacity (97). Previously, KLF11 was identified as a direct activator of UCP1 expression, which is a central event in adipose tissue browning (98). Accordingly, knockdown of KLF11 results in reduced rosiglitazone-mediated UCP1 and beige-associated gene expression (97).
In another study, KLF15 was found to activate UCP1 synergistically with KLF11 during brown adipocyte differentiation through direct interaction with the Ucp1 promoter. KLF15, however, is not as essential as KLF11 for transcriptional activation of the Ucp1 gene and brown adipocyte differentiation (98). KLF15 mediates the “beiging effect” of PPARγ via the glucocorticoid receptor (100). Glucocorticoids are widely used as in vitro inducers of adipogenesis, exerting their effects through the glucocorticoid receptor (101). KLF15 is directly induced by the glucocorticoid receptor, and glucocorticoid response element sites have been located within the first intron of the Klf15 gene (100). c-Jun interrupts the glucocorticoid-mediated induction of KLF15 by occupying an intronic region near the glucocorticoid response element site of the Klf15 gene (102). Thus, the browning of WAT is promoted by KLF11 and KLF15.
Liver
The liver is a metabolically active and highly aerobic organ that is responsible for the synthesis and degradation of carbohydrates, proteins, and lipids. It sorts small molecules of digestion products and nutrients for further metabolism, storage, or distribution to extrahepatic tissues. The liver provides energy to other tissues mainly by exporting substrates that are critical for oxidization in peripheral tissues (i.e., FAs, glucose, ketone bodies).
Hepatic glucose metabolism plays a crucial role in maintaining normal concentrations of blood glucose. During food deprivation, when insulin levels are low and glucagon levels are high, the liver provides glucose, both by gluconeogenesis and glycogen breakdown. Gluconeogenesis is one of the most important functions of the liver. Maintaining normal plasma glucose concentrations is essential for energy homeostasis because glucose represents the primary energy source for many tissues, including the brain. Another pathway to deliver glucose to the circulation is by glycogenolysis, which involves hydrolysis of glycogen stores. After food intake, when insulin levels are high and glucagon levels are low, the liver removes glucose from the portal vein, where glucose is catabolized to pyruvate or used for glycogen synthesis in hepatocytes. Various cellular mechanisms that affect hepatic and systemic metabolism are regulated by KLFs (Figure 5).
Figure 5.

KLF Isoforms That Control Hepatic Metabolism
Summary of KLF isoforms and metabolic pathways involved in hepatocyte biology. A detailed description of the pathways involved in included in the text. Figure was produced using Servier Medical Art (http://www.servier.com/). CD = cluster of differentiation; FA = fatty acid; PEPCK = phosphoenolpyruvate carboxykinase; TGFβ = transforming growth factor beta; other abbreviations as in Figures 1 and 2.
In hepatic gluconeogenesis, the enzyme phosphoenolpyruvate carboxykinase (PEPCK) catalyzes the rate-limiting step. Changes in the level of Pepck messenger RNA or its activity are associated with the control of hepatic glucose output. Liver-specific Pepck knockout in mice inhibits gluconeogenesis (103), and total body Pepck knockout leads to severe hypoglycemia and death (104). KLF10 (105) and KLF11 (106) act as inhibitors of gluconeogenesis. KLF10 directly inhibits the Pepck gene (105). Transcriptome profiling in Klf10–/– mouse liver tissue revealed downregulation of glycolytic proteins and upregulation of gluconeogenic and lipogenic proteins. Compared with wild-type mice, Klf10–/– mice display a postprandial elevation of blood glucose in male mice and a postprandial elevation of blood TG levels in female mice. Interestingly, hepatic Klf10 displays a rhythmic expression pattern that is dependent on the clock protein, BMAL1 (105). Transcription of Klf10 is directly induced by the carbohydrate-responsive element-binding protein and is therefore increased by high glucose levels (107). KLF10 is further capable of inhibiting expression of Chrebp and its target genes in rat hepatocytes. Taken together, these data suggest a negative feedback loop between carbohydrate-responsive element-binding protein and KLF10 involved in the circadian regulation of hepatic gluconeogenesis. KLF11 regulates gluconeogenesis by directly inhibiting expression of Pepck (106). Adenovirus-mediated overexpression of KLF11 reduces PEPCK expression, lowers hepatic glucose output, and protects against hyperglycemia. In general, hepatic KLF11 is decreased after fasting of short duration but elevated after 24 h of fasting.
In contrast to KLF10 and KLF11, hepatic KLF15 acts as a nutritionally responsive positive regulator of gluconeogenesis, in part by directly inducing Pepck (108). KLF15 expression is greatest in the liver among various tissues (109). KLF15 expression follows the expression pattern of PEPCK and is elevated after food deprivation, or decreased in the fed state or after euglycemic insulin administration 67, 108.
Hepatic FA and lipid metabolism is also regulated by the same KLF family members affecting glucose metabolism: KLF10 (110), KLF11 (111), and KLF15 (112). During fasting, FAs are released from adipose tissue into circulation. When the flux of FAs toward the liver exceeds β-oxidation capacity or lipid export via lipoprotein secretion, TGs accumulate in the liver. FAs in the liver are continuously re-esterified into TGs. TGs from this pool can be used for hepatic very-low-density-lipoprotein–TG synthesis and secretion. Most of the plasma very-low-density-lipoprotein–TG pool is derived from plasma FA, re-esterified by the liver, and secreted into the plasma, whereas only a very small fraction of plasma very-low-density-lipoprotein–TG is derived from de novo FA synthesis in the liver (113).
Hepatic Klf10 expression is induced in primary mouse hepatocytes after treatment with a PPARα agonist, which promotes FA oxidation (110). Although a direct interaction between PPARα and the Klf10 promoter has not been established, coordinated action of these 2 transcription factors could be involved in the regulation of hepatic lipid metabolism. KLF11 activates PPARα and downstream lipid oxidation genes shown by adenovirus-mediated overexpression of KLF11, and it improves fatty liver phenotype in mice with diet-induced obesity (DIO) (111). Conversely, silencing of KLF11 in wild-type mice increases TG levels, caused by decreased FA oxidation, which can be accounted for by inhibition of PPARα expression. Furthermore, overexpression of hepatic KLF15 reduces expression of SREBP-1c leading to a decrease in plasma TG levels (112). KLF15 facilitates the switch from lipogenesis to gluconeogenesis during fasting by inhibiting expression of Srebp1c.
Another major role of the liver is to take up, and metabolize, dietary amino acids that are absorbed by the gastrointestinal tract and transported to the liver via the portal vein. Hepatic amino acid metabolism is influenced by KLF15. Klf15–/– mice demonstrate severe hypoglycemia after an overnight fast (67). Surprisingly, no decrease in Pepck expression is observed in Klf15/– mice; however, these mice exhibit reduced expression of amino acid–degrading enzymes in the liver and skeletal muscle after overnight fasting (67). Klf15 acts as a clock-controlled gene, and rhythmic expression of amino acid–metabolizing and urea cycle enzymes is driven by KLF15 in the liver and skeletal muscle (114). Therefore, KLF15 promotes protein catabolism in the liver and skeletal muscle, and coordinates these processes with nitrogen excretion.
The liver is an insulin-sensitive organ, and defective insulin signaling as well as development of insulin resistance in the liver can have major consequences on energy balance and metabolism. Hepatic insulin sensitivity is increased in the liver by KLF14, and overexpression of KLF14 in Hepa1-6 cells increases Akt phosphorylation and insulin-stimulated glucose uptake. KLF14 exhibits a synergistic effect with insulin administration leading to increased messenger RNA levels of sirtuin (SIRT)1, PGC-1α, and hepatocyte nuclear factor–4α (115). Insulin sensitivity improves with lower glucokinase levels, and hepatocyte Klf6 promotes glucokinase transcription that is associated with insulin resistance 116, 117.
Bile acid synthesis occurs exclusively in the liver via a series of enzymatic reactions that convert hydrophobic cholesterol into more water-soluble amphipathic compounds (118). Bile acids are potent “digestive surfactants” that promote absorption of lipids, including fat-soluble vitamins, by acting as emulsifiers. Bile acid synthesis is affected by KLF15-regulating factors produced by the ileum that are responsible for the circadian regulation of hepatic bile acid synthesis (119). Klf15–/– mice exhibit an interruption in the circadian expression of bile acid synthesis enzymes, which is not present in hepatic-specific Klf15–/– mice. KLF15 directly inhibits expression of fibroblast growth factor (Fgf) 15 in a circadian pattern, which is produced by the ileal epithelial cells and inhibits expression of bile acid–producing enzymes in the liver. Importantly, ileectomy restores expression of bile acid synthesis genes in Klf15–/– livers.
Pancreas
The pancreas plays multiple roles in systemic metabolism, including production of hormones responsible for regulating metabolic processes, and the production of enzymes necessary for the absorption of nutrients within the gastrointestinal tract. The pancreas may be separated into the exocrine and the endocrine pancreas. The exocrine pancreas consists of the pancreatic acini and ductal system, which secretes digestive enzymes and bicarbonate into the gastrointestinal tract. Alternatively, the endocrine pancreas is derived from the pancreatic islets of Langerhans, which contain multiple hormone-producing cell types that secrete their products into the circulation and regulate metabolism at distant tissues. These cell types include alpha cells that produce glucagon, beta cells that produce insulin, delta cells that produce somatostatin, PP cells that produce pancreatic polypeptide, and epsilon cells that produce ghrelin (120).
Because of the importance of the pancreas in the regulation of nutrient absorption and use, processes that disrupt pancreatic development often result in metabolic phenotypes. KLF10 regulates islet cell development by directly activating transcription of SEI-1, which is capable of inducing cell cycle inhibitor p21 (121). Klf10–/– mice surprisingly display reduced number of islets of Langerhans with reduction in the overall islet mass and area. These changes are associated with impaired glucose tolerance and impaired insulin secretion. The exact mechanism by which KLF10 regulates islets of Langerhans development and β-cell mass, as well as whether KLF10 participates in the production of pancreas-derived hormones, remains to be investigated.
Pancreatic β cells play a significant role in systemic metabolism and glucose homeostasis by synthesizing and secreting insulin in response to increases in blood glucose concentration. KLF11 is predominantly expressed within the pancreas, where it regulates β-cell function in response to glucose (122). KLF11 coordinates with coactivator p300 and promotes expression of the insulin (INS) gene 122, 123 and of the pancreatic and duodenal homeobox (Pdx)-1 gene (124), a master regulator of pancreatic development and β-cell function (125). Phenotypic characterization of Klf11–/– mice revealed defects in glucose homeostasis, resulting in part from β-cell dysfunction. These mice demonstrate impaired insulin production but surprisingly maintain lower blood glucose levels resulting from increased peripheral insulin sensitivity 123, 126.
KLF7 seems to oppose KLF11 in regulating insulin production. In the insulin-secreting HIT-T15 cell line, overexpression of KLF7 impairs glucose-stimulated insulin secretion that is accompanied by reduced expression of PDX-1 (127). The in vivo effects of KLF7 on pancreatic development and β-cell function, and potential interactions between KLF7 and KLF11 on the Insulin and Pdx-1 promoters, remain to be investigated.
Erythrocytes
An important component of metabolism is the sufficient delivery of oxygen to tissues, which serves as the terminal electron acceptor for oxidative phosphorylation and ATP synthesis in mitochondria. Erythrocytes carrying hemoglobin, the iron-containing molecular carrier for oxygen, have a central role in oxygen delivery. Although KLFs are not known to have a functional role in mature adult erythrocytes, which lack nuclei, multiple KLFs have a critical role in regulating erythrocyte progenitor cells.
In adults, erythropoiesis (i.e., the process of generating new erythrocytes from progenitor cells) occurs primarily in the bone marrow in response to erythropoietin, which is produced during hypoxia within the kidney. KLF1, also known as erythroid KLF, is a master regulator of human erythroid cell production. Klf1 is expressed in bone marrow and the spleen (128), where it controls the final transition from fetal γ-globin to adult β-globin expression (129) and cell cycle progression during erythroid differentiation 130, 131. Furthermore, KLF1 coordinates the gene expression of transmembrane proteins, cytoskeletal proteins, red blood cell antigens, and proteins involved in cell division, heme production, and iron procurement (132). In humans, monoallelic Klf1 mutations result in the persistence of fetal hemoglobin and a mild phenotype of beta-thalassemia (133), and homozygous loss of KLF1 results in severe nonspherocytic hemolytic anemia and very high levels of fetal hemoglobin into childhood (132).
In contrast to adult erythropoiesis, which occurs in the bone marrow, embryonic erythropoiesis occurs in the yolk sac, and fetal erythropoiesis in the spleen and liver (134). Although Klf1–/– mice are normal during the embryonic stage of erythrocyte development, these mice die around embryonic day 14 due to severe anemia and defective erythropoiesis in the liver 135, 136, 137. KLF1 directly activates transcription of the macrophage-expressed DNase II-alpha (Dnase2α) gene, an essential player in the digestion of extruded nuclei during erythrocyte maturation (138). Residual blood cells retain their nuclei and contain reduced hemoglobin. This phenotype likely results from reduced Dnase2a activity in macrophages (138) and defective β-globin synthesis in erythrocyte precursors (135).
KLF3, along with KLF8, silences embryonic globin expression during development (139). The expression pattern of KLF3 in bone marrow and the spleen overlaps with Kfl1 (128), and KLF1 and KLF3 bind similar DNA sequences (140). KLF1 typically activates and KLF3 represses transcription, suggesting opposing roles for these 2 KLF family members (141). Consistent with this observation, KLF3 represses the expression of a subset of KLF1 target genes that are crucial for normal erythroid maturation (142). In this network, Klf3 and Klf8 are activated by KLF1, and KLF3 feeds back as a repressor for Klf8 141, 143. Importantly, Klf3/Klf8 double knockout mice die in utero and are characterized by de-repression of fetal globin expression, which cannot be accounted for by lack of KLF3 or KLF8 alone (139).
KLF2 is an important regulator of embryonic, but not adult, erythrocyte development. Klf2-/- mice die in utero, resulting from intraembryonic hemorrhage and anemia, and display significant reductions in embryonic globin levels (144). KLF2 regulates the embryonic ε-globin gene but not the adult β-globin gene, suggesting that KLF2 primarily exerts its effects on embryonic erythrocyte generation. KLF2 also seems to affect the stability and morphology of primitive erythroid cells, suggesting that KLF2 regulates embryonic erythrocyte function similarly to how KLF1 regulates adult erythrocyte development.
Role of KLFs in Diseases That Are Associated With Metabolic Abnormalities
Heart failure
Heart failure is a clinical syndrome in which the heart is unable to effectively pump enough blood to meet the metabolic requirements of the organism. As the end stage of many diseases of the cardiovascular system, including acute coronary syndrome, various cardiomyopathies, and hypertension, the prevalence of heart failure has been increasing in the developing world as a result of the aging population and increased survival after myocardial infarction (145). Therefore, strategies aimed at the primary prevention of heart failure, as well as management of existing heart failure, are highly sought.
Metabolic dysfunction is a characteristic of advanced heart failure that occurs due to ischemia, pathological hypertrophy, and various forms of dilated cardiomyopathy 55, 146, 147, 148. These defects include reduced capacity for respiration, reduced FA oxidation, and increased carbohydrate oxidation. Modulation of cardiac FA oxidation has been proposed as a potential therapeutic strategy to improve cardiac function in patients with heart failure. Changes in metabolic function and substrate use likely contribute to cardiac disease progression as evidenced by animal models of cardiac disease (58). For example, cardiomyocyte-specific deletion of PPARα aggravates pressure overload hypertrophy (149). Alternatively, treatment with PPARα agonist protects against pressure overload hypertrophy 150, 151 and post–myocardial infarction remodeling (152).
Multiple members of the KLF family protect against heart failure progression in part by regulating metabolic gene expression. Cardiomyocyte KLF4 is induced in cultured cardiomyocytes after hypertrophic stimuli and seems to protect against pathologic hypertrophy (Figure 6) (153). Alternatively, cardiomyocyte-specific Klf4–/– mice cope significantly worse with introduced pressure overload, resulting in 75% mortality 2 weeks’ post-operatively. The protective effect of KLF4 is related to the interaction between KLF4 and the PGC-1α axis (Figure 2), as Klf4–/– mice have defects in the electron transport chain, associated with a lower mitochondrial respiration rate (56).
Figure 6.

KLF Isoforms Associated With the Development of Cardiac Hypertrophy and Dilated Cardiomyopathy and Fibrosis
Green indicates protective effect, and red indicates aggravating effect. The (+) sign indicates Krüppel-like factor (KLF) isoforms that promote the phenotype described next to the organs. The (-) sign indicates alleviation of the phenotype. Figures were produced using Servier Medical Art (http://www.servier.com/).
Because KLF5 is a positive regulator of PPARα and FA oxidation–related gene expression (59), one could anticipate a protective role for KLF5 against pressure overload hypertrophy. Surprisingly, KLF5 haploinsufficiency seems to be protective (154). Tissue-specific animal models found that the protective effect of KLF5 haploinsufficiency results from reduced expression of fibroblast KLF5 but not cardiomyocyte-specific KLF5 ablation. Changes in cardiomyocyte KLF5 in rodent models of heart failure remain to be investigated, and cardiomyocyte KLF5 activation may represent an approach to increase FA oxidation–related gene expression and to improve function of the diseased heart.
Human hearts with advanced heart failure exhibit decreased expression of Klf15 (60), which likely reduces expression of FA oxidation–related genes through impaired PPARα-mediated transcriptional regulation. Consistently, Klf15–/– mice that are subjected to pressure overload develop eccentric cardiac hypertrophy, whereas Klf15 overexpression protects against cardiac remodeling (155). Cardiac KLF15 also regulates the catabolism of BCAAs, which is postulated to protect against pressure overload–induced heart failure (156) and ischemia-reperfusion injury (157). Mechanistically, elevated levels of BCAAs inhibit glucose metabolism by suppressing glucose uptake and inhibiting mitochondrial respiration 156, 157. Importantly, KLF15 is a key inducer of BCAA catabolic genes, including BCAT, the BCKD complex subunits, and PP2Cm, which are reduced in KLF15 knockout hearts (156). Taken together, these observations suggest that reduced KLF15 may not only be involved in the fetal metabolic profile of failing hearts but also in BCAA catabolic deficiency, which contributes to mitochondrial dysfunction. Therefore, restoring expression of KLF15 in heart failure may be the target of future therapies designed to manage cardiac function in advanced heart failure.
KLF6 exacerbates cardiac fibrosis as demonstrated by reduced fibrosis and preserved cardiac function in Klf6+/– mice after angiotensin II infusion (158). This outcome seems to result from cardiomyocyte KLF6 because cardiomyocyte-specific, but not fibroblast-specific, Klf6–/– mice display the same protection from fibrosis. Cardiomyocyte KLF6 is induced in cardiomyocytes early after angiotensin stimulation and suppresses expression of thrombospondin 4, which is secreted by cardiomyocytes to regulate fibroblast activity (158). Therefore, cardiomyocyte KLF6 regulates factors responsible for intercellular communication, fibroblast activation, and fibrosis.
Atherosclerosis
Atherosclerosis, or hardening of the blood vessels, is a leading cause of death in the developed world that results from coronary artery disease and stroke. There are numerous risk factors for atherosclerosis, including various factors of metabolic origin, such as high blood glucose levels, dyslipidemia, and diet (159). Atherosclerosis results from a chronic inflammatory process of the vascular system and involves dysregulation of many components, including circulating lipoproteins, endothelial cells, immune cells, and vascular smooth muscle cells. Atherosclerotic lesions begin as a fatty streak on the artery’s intimal layer resulting from early endothelial dysfunction. Eventually, these lesions progress through multiple stages involving lipoprotein infiltration and oxidation, foam cell formation, plaque development, and smooth muscle cell migration to lead to mature atherosclerotic lesions (160). Members of the KLF family have been shown to interact with these components of atherosclerosis pathogenesis to provide a protective or exacerbating effect.
To clear cholesterol and TG-rich lipoprotein particles from the blood, both apolipoprotein E (APOE) and low-density lipoprotein receptors are necessary. APOE–/– and Ldlr–/– mice are widely used to study atherosclerosis. Klf2+/– mice crossed with APOE–/– mice display augmented atherosclerotic lesion formation due to a 40% enhanced lipid uptake in macrophages. Overexpression of KLF2 in a mouse macrophage cell line markedly inhibited lipid uptake (161). Along the same lines, myeloid-specific Klf2–/– mice crossed with Ldlr–/– mice exhibit a substantial increase in atherosclerosis (162). Thus, KLF2 acts as an atheroprotective factor in experimental atherosclerosis.
KLF4 affects the development of atherosclerosis through its expression in endothelial cells, myeloid cells, and vascular smooth muscle cells. Studies on mice with endothelial-specific overexpression or knockout of Klf4 on an APOE–/– background show that KLF4 is a protective factor against atherosclerosis (163). This outcome results from the antiadhesive and antithrombotic effects of endothelial KLF4. Mechanistically, endothelial KLF4 reduces atherosclerosis development by competing with nuclear factor–κB for coactivator p300. Recently, endothelial microRNA-103 was shown to reduce Klf4 expression and promote atherosclerosis development (164). Myeloid KLF4 also plays an important role in atherosclerosis development and foam cell generation. In macrophages, KLF4 is significantly downregulated by pro-inflammatory cytokines and oxidized lipids. Myeloid-specific Klf4–/– on an APOE–/– background demonstrates increased susceptibility to atherosclerotic lesion development likely resulting from enhanced pro-inflammatory gene expression (163). Therefore, both endothelial and myeloid KLF4 seem to protect against atherosclerosis development in part by conferring anti-inflammatory effects. Alternatively, vascular smooth muscle KLF4 promotes atherosclerosis development in part by promoting proinflammatory gene expression in smooth muscle cells and reducing fibrous cap thickness. Smooth muscle cell–specific conditional Klf4–/– mice exhibit a 50% reduction in atherosclerotic lesion size and increased plaque stability, suggesting that smooth muscle cell KLF4 is critical to the development of atherosclerotic lesions (165).
Klf14 is a ubiquitously expressed gene, demonstrating monoallelic maternal expression in humans and in mice (166). Genome-wide association studies have correlated single-nucleotide polymorphisms located near Klf14 to the development of atherosclerosis. The single-nucleotide polymorphism rs4731702 was associated with increased levels of circulating high-density lipoprotein and decreased risk of atherosclerotic cardiovascular disease 167, 168. Importantly, these associations are only significant when the variant is located on the maternally inherited allele, suggesting that the observed disease associations are driven by KLF14 (169). Mechanistically, KLF14 likely exerts its effect by regulating the production of lipoproteins in the liver. Hepatic KLF14 directly activates apolipoprotein A-I, a major component of the protective high-density lipoprotein. Overexpression of KLF14 in mice fed a high-fat diet increased plasma high-density lipoprotein cholesterol levels and cholesterol efflux (170). KLF14 was identified as a pharmacological agent to treat atherosclerosis. Administration of perhexiline, which was identified as an activator of endogenous KLF14, increases high-density lipoprotein cholesterol plasma levels and cholesterol efflux in vivo and protects against atherosclerosis development in APOE–/– mice.
Obesity
Increased weight and obesity are characterized by an abnormal excessively high amount of body fat in relation to lean body mass driven by energetic imbalance, in which caloric intake is greater than the calories expended through the body’s metabolic rate and physical activity. Several members of the KLF family affect energy homeostasis by affecting lipid and glucose metabolism, as well as adipocyte differentiation, and hence play a role in obesity development. KLF4 78, 79, KLF5 (83), KLF6 91, 92, KLF8 (82), KLF9 80, 81, KLF13 (84), and KLF15 (85) promote adipocyte differentiation and adipose tissue expansion, whereas KLF2 (89), KLF3 (87), KLF7 (88), and KLF16 (93) have the opposite effect (Figure 4). Moreover, KLF3 (87), KLF11 (126), KLF14 (115), and KLF15 171, 172 promote weight gain.
Despite KLF3 being a negative regulator of adipocyte differentiation, Klf3–/– mice surprisingly exhibit reduced white adipose tissue mass and adipocyte size, and are resistant to DIO and glucose intolerance (87). Differences in body weight in Klf3–/– mice do not arise from altered food intake or energy expenditure but may result from increased levels of the insulin-sensitizing hormone adipolin (FAM132A/CTRP12/C1qdc2) (90). Adipolin functions as an anti-inflammatory adipokine that improves glucose tolerance 173, 174. Moreover, KLF3 represses the expression of Lgals3, which encodes galectin-3, a protein suggested to protect from obesity and other metabolic disorders (175). Thus, increased galectin levels in Klf3–/– mice might account for the lean phenotype as galectin-3–/– mice demonstrate defective glucose metabolism, and age dependently increase white adipose tissue mass 175, 176. Thus, although reduced KLF3 expression is clearly associated with less weight gain in mice, the underlying mechanism and physiological relevance in humans remain undetermined.
In mouse models of obesity, hepatic KLF11 is decreased (111). In Klf11–/– mice fed a normal diet, body weight and energy expenditure are not affected. However, loss of Klf11 provides a protective effect against high-fat DIO and hypercholesterolemia, and also results in an increased metabolic rate in female mice only fed a high-fat diet (126). The observed reduction of KLF11 in obesity may denote a negative feedback loop that aims to block uncontrolled expansion of adipose tissue.
Association studies conducted through MuTHER (the Multiple Tissue Human Expression Resource Consortium) have identified KLF14 as a master regulator of gene expression in adipose tissue (177). KLF14 exerts its effect as a disease risk allele through the transregulation of genes associated with metabolic traits. Furthermore, KLF14 is decreased in the liver, adipose, and muscle of mice fed a high-fat diet and in diabetic db/db mice (115). More recently, a putative transregulatory network for KLF14 has been identified, providing information about regulators of Klf14 expression and protein–protein interactions (178). Taken together, these studies provide a basis for mechanistic studies characterizing the role of KLF14 in metabolic disease.
Despite the prominent role of KLF15 in promoting adipocyte differentiation, Klf15 expression is decreased in WAT and BAT of wild-type mice fed a high-fat diet, as well as in db/db mice (171). This finding is consistent with reports of reduced Klf15 expression in the visceral adipose tissue of obese individuals 171, 172. The relationship between reduced KLF15 expression and increased adiposity remains to be investigated. Screening for KLF15 targets in 3T3-L1 adipocytes by ChIP-chip integrated with expression microarray data has revealed genes likely regulated by KLF15. This study found that adrenomedullin, which has been implicated in metabolic complications of obesity, is directly repressed by KLF15 (179).
In the gastrointestinal tract, KLF4 directly activates transcription of the human ghrelin promoter and thereby increases appetite (180). Ghrelin, a peptide hormone expressed predominantly in the stomach, influences metabolic processes, including adipogenesis, growth hormone secretion, food intake, energy balance, and insulin secretion (181). KLF4 expression is induced by butyrate, a short-chain FA found in the gut, and promotes ghrelin expression in cultured human gastric cancer cells (180). These studies suggest that KLF4 may coordinate ghrelin expression within the gut environment, and higher expression levels are accompanied by increased appetite.
Genome-wide association studies identified a single-nucleotide polymorphism (rs7568369) in the promoter of Klf7, which was associated with protection against obesity in the Danish population (182). In this regard, increased expression of Klf7 together with toll-like receptor 4 correlates with obesity-induced inflammation in visceral adipose tissue studied in Wistar rats fed a high-fat diet and also in humans (183). Another study of East Asian subjects identified a single-nucleotide polymorphism (rs11142387) in the Klf9 locus that was associated with increased body mass index, which is in accordance with the pro-adipogenic role of KLF9 (184). Notably, no association has been detected within the European population. These association studies are in alignment with the roles of KLF7 and KLF9 as inhibitory and promoting factors in adipocyte differentiation, respectively.
Fatty liver disease
Obesity is frequently accompanied by ectopic lipid accumulation, primarily in the liver, skeletal muscle, and heart. Excess accumulation of lipids in the hepatocytes leads to hepatic steatosis, which may result from inhibition of β-oxidation, impaired very-low-density-lipoprotein secretion, and pathways involved in the synthesis of FAs. Hepatic KLF2 (185), KLF5 (186), KLF6 187, 188, 189, 190, 191, and KLF9 (188) increase hepatic lipid accumulation. Conversely, KLF10 (105), KLF11 (111), and KLF15 (112) inhibit hepatic steatosis (Figure 5).
Hepatic KLF2 messenger RNA and protein levels are significantly increased in wild-type mice fed a high-fat diet and leptin-deficient B6.V-Lepob/J (ob/ob) mice, which is a model of obesity and steatohepatitis. Adenoviral KLF2 overexpression induces hepatic TG accumulation in wild-type mice, whereas KLF2 silencing ameliorates hepatosteatosis in ob/ob mice. Because PPARγ promotes hepatic steatosis 192, 193, the KLF2-driven mechanism that mediates hepatic lipid accumulation must be different from the adipose tissue, where KLF2 inhibits PPARγ expression (77). The mechanism for hepatic lipid accumulation may, at least in part, derive from KLF2 induction of cluster of differentiation 36 (CD36), a membrane protein that facilitates FA uptake 185, 194. The KLF2-mediated hepatosteatosis development is severely diminished in cluster of differentiation 36–deficient mice (185).
Although KLF2 has opposite roles in the liver and the adipose tissue, KLF5 seems to have similar effects in both organs. Hepatic KLF5 is a positive regulator of Pparg2 expression and genes involved in FA uptake and TG synthesis (186), and it promotes hepatic lipid accumulation. The regulation of KLF5 activity in the liver is controlled by a ubiquitin-dependent degradation mechanism that is mediated by F box and WD repeat domain containing 7 (FBW7). Adenovirus-mediated knockdown of hepatic Fbw7 results in hepatosteatosis due to reduced KLF5 degradation, and increased expression of Pparg2 and genes involved in FA uptake and TG synthesis. Thus, KLF5 is a positive regulator of TG accumulation in the liver via activation of PPARγ expression.
KLF6 is another regulator of hepatic PPARγ and promotes hepatic steatosis. Klf6+/– and hepatocyte-specific Klf6–/– mice are protected from diet-induced steatohepatitis and exhibit improved insulin sensitivity. Klf6-ablated hepatocytes show a reduction in PPARα protein but not messenger RNA levels, suggesting alternative pathways that reduce lipid uptake or promote hepatic FA oxidation (187). One possibility involves regulation of PPARγ by KLF6. KLF6 directly induces Pparg gene expression, which promotes TG accumulation (188). In vitro experiments in palmitic acid–treated HepG2 cells mimicking conditions similar to that of a fatty liver showed increased KLF6 expression and TG accumulation. Contrary, silencing of KLF6 represses Pparg levels and reduces TG accumulation. KLF9 also drives the expression of PPARγ and leads to hepatic steatosis in mice with DIO. Similarly, palmitic acid–treated HepG2 cells exhibit increased KLF9 levels and augmented TG accumulation. KLF9 directly regulates the Pparg promoter. Silencing KLF9 represses Pparg expression and reduces TG accumulation in vitro.
Opposite to KLF2, KLF5, KLF6, and KLF9, which promote hepatic steatosis, KLF10 and KLF11 seem to play a protective role. KLF10 is involved in the regulation of hepatic glucose and lipid metabolism. High glucose levels induce KLF10 expression in primary hepatocytes (107), as does PPARα activation (110), which promotes FA oxidation, implicating KLF10 as an inhibitor of hepatic steatosis. Hepatocyte KLF10 inhibits the expression of Chrebp (107), which increases lipogenesis (195), and Pepck (105), which catalyzes gluconeogenesis. KLF11 activates lipid oxidation genes via induction of PPARα and improves fatty liver phenotype in DIO mice (111). KLF11 is also important for the inhibition of gluconeogenesis as it directly inhibits the expression of Pepck and protects against hyperglycemia (106).
KLF15 regulates carbohydrate, lipid, and protein homeostasis in the liver. KLF15 is a positive regulator of gluconeogenesis by increasing Pepck expression (108) and substrate availability (67). Activation of KLF15 during fasting also prevents hepatic lipogenesis by inhibiting SREBP-1c (112) and leads to lower plasma TG levels (112). Inhibition of KLF15 is protective against high-fat-diet–induced hepatic steatosis via increased expression of FA oxidation genes (196). Metformin, which inhibits hepatic gluconeogenesis and is a pharmacological agent for type 2 diabetes mellitus (T2DM), promotes degradation of KLF15 protein (197) and alleviates hepatic steatosis (198).
Hepatic chronic inflammation induces fibrosis, which eventually progresses to cirrhosis and hepatocellular carcinoma. Members of the KLF family have been implicated in liver fibrosis, an excessive accumulation of scar tissue that results from inflammation and liver cell death that occurs in most types of chronic liver diseases. KLF2 199, 200, 201 ameliorates liver fibrosis, whereas KLF6 189, 191 promotes it. Furthermore, KLF10 (202) is associated with liver disease. Although KLF10 has a protective role in hepatic steatosis, which might imply prevention of steatohepatitis and liver fibrosis, its role in the progression or protection from fibrosis remains to be elucidated.
KLF2 modulates inflammation or collagen production in the liver that may be accounted for by its expression in nonhepatocytes. In cirrhotic livers of Wistar rats, KLF2 expression is increased in hepatic endothelial cells compared with healthy controls, which is accompanied by the induction of vasoprotective target genes. Hepatic KLF2 expression is increased in rodents treated with simvastatin, a cholesterol-lowering statin drug that has been proposed to have antifibrotic effects due to KLF5 reduction of collagen production by hepatic stellate cells (199). KLF2 upregulates the antioxidant transcription factor nuclear factor erythroid 2-related factor 2 200, 201. Therefore, hepatic KLF2 upregulation in cirrhosis ameliorates liver fibrosis, counteracts endothelial dysfunction, and reduces oxidative stress, improving portal hypertension (201).
In the spectrum of nonalcoholic fatty liver disease, activation of fibrosis is a critical event for the progression from hepatic steatosis to nonalcoholic steatohepatitis and cirrhosis. The fibrosis program is primarily regulated by transforming growth factor beta (TGFβ). Increased hepatic KLF6 expression directly activates the promoters of Tgfb1 and types I and II TGFβ receptors (189) and may play a crucial role in the progression from steatosis to steatohepatitis with progressive fibrosis 189, 191. Thus, KLF6 activation may constitute a significant risk factor for steatohepatitis via progression of both lipid accumulation and fibrosis. Accordingly, a functional polymorphism in the Klf6 gene is associated with advanced forms of nonalcoholic fatty liver disease. KLF6-IVS1-27G>A promotes alternative splicing of KLF6 and delays progression of nonalcoholic fatty liver disease (190). In addition, KLF10 is increased in the livers of mice with diet-induced nonalcoholic steatohepatitis.
Increased KLF10 expression has been associated with increased activity of TGFβ signaling (202) that accounts for the hepatic fibrosis which follows nonalcoholic steatohepatitis. It remains unclear whether KLF10 is a component of an adaptive process that aims to reduce hepatic glucose or lipid levels when these are increased beyond physiological levels or if it participates in maladaptive mechanisms that lead to hepatic fibrosis.
Diabetes mellitus
Diabetes is a metabolic disease that involves dysfunction of several organs, and KLF biology is altered in most of them during disease progression (Figure 7). Type 1 diabetes mellitus is a chronic autoimmune disease, which is driven by the lack of insulin production from the pancreas that leads to hyperglycemia. T2DM is associated with obesity, which is driven by excessive adipocyte differentiation and adipose tissue expansion. Obesity accompanied by ectopic lipid accumulation compromises intracellular insulin signaling and aggravates insulin resistance that leads to hyperglycemia. Diabetes is linked to cardiovascular diseases, which represent the most prevalent cause of mortality and morbidity in the diabetic population (203). Patients with diabetes commonly experience obesity, hypertension, and dyslipidemia, which are cardiovascular risk factors often seen in conjunction with an increased risk for cardiac events such as coronary artery disease, stroke, and diabetic cardiomyopathy. Several studies have identified involvement of multiple KLF isoforms in the regulation of metabolic pathways in cardiomyocytes (Figure 2), skeletal muscle cells (Figure 3), adipocytes (Figure 4), and hepatocytes (Figure 5). Here we discuss KLFs that are directly involved in complications associated with diabetes.
Figure 7.

KLF Isoforms That Have Been Linked Either Directly or Indirectly With Complications That Are Associated With Diabetes
Green indicates protective effect, and red indicates aggravating effect. The (+) sign indicates the Krüppel-like factor (KLF) isoforms that promote the phenotype described next to the organs. The (-) sign indicates alleviation of the phenotype. Figures were produced using Servier Medical Art (http://www.servier.com/).
Maturity-onset diabetes of the young (MODY) is a monogenic form of diabetes, inherited in an autosomal dominant mode and characterized by a defect in the pancreatic β-cell function. Two KLF isoforms have been linked to pancreatic function: KLF11 is a positive regulator (122), and KLF7 acts as an inhibitor of insulin production (127). KLF11 is expressed ubiquitously but is enriched in the pancreas, where it acts as a glucose-inducible positive regulator of the INS gene (122). KLF11 loss-of-function mutations are associated with MODY7 and human neonatal diabetes mellitus 124, 204. The Q62R mutation is a gain-of-function variant that represses activity of KLF11 (122). Individuals with the Q62R variant demonstrate decreased insulin levels after glucose tolerance testing, and normoglycemic carriers have increased insulin sensitivity. Alternatively, homozygous mutation of the KLF11-binding site of the INS promoter results in neonatal diabetes mellitus, manifesting as intrauterine growth delay, impaired insulin synthesis, and a requirement for insulin supplementation at birth (123). Furthermore, hepatic KLF11 is decreased in mouse models of diabetes (111). Genome-wide expression profiling of liver tissue from Klf11-ablated mice demonstrates enrichment for dysregulated genes controlled by PPARγ (126). Pancreatic KLF11 also directly induces Pdx-1, a master regulator of the development of the pancreas (124).
T2DM, the most common form of diabetes (90% of all cases), results from impaired insulin secretion, insulin resistance, or a combination of both. T2DM is linked to the epidemic of obesity. Thus, KLF family members promoting adipogenesis (KLF4 78, 79, KLF5 [83], KLF6 91, 92, KLF8 [82], KLF9 80, 81, KLF13 [84], and KLF15 [85]), as well as inhibiting (KLF2 [89], KLF3 [87], KLF7 [88], and KLF16 [93]) adipocyte differentiation, may influence T2DM development. In addition, KLF7 overexpression decreases levels of adiponectin and leptin, adipocyte-derived hormones that regulate insulin sensitivity and appetite, respectively (127). Interestingly, a Klf7 single-nucleotide polymorphism (rs2302870) detected in the Japanese population is linked to T2DM susceptibility (88).
Increased hepatic gluconeogenesis is conferred a primary role in T2DM. Maintenance of normal glucose homeostasis in the liver is essential, and dysregulation of hepatic gluconeogenic processes contributes to imbalances linked to T2DM. High plasma glucose levels may be corrected by inhibition of hepatic glucose production and increased peripheral glucose use. Antidiabetic drugs, such as biguanides (205) and glucagon-like peptide-1 modulators (206), target gluconeogenesis and lower hepatic glucose production. Biguanides (e.g., metformin) repress hepatic glucose production and consequently reduce fasting plasma glucose levels and glycosylated hemoglobin A (205). Metformin is the most commonly prescribed antidiabetic medication worldwide (207). Hepatic gluconeogenesis is affected by KLF10 (105) and KLF11 (106), both inhibiting Pepck, and KLF15 (108) inducing Pepck. Liver-specific knockdown of KLF15 reduces expression of gluconeogenesis and amino acid metabolism–related genes, and treats hyperglycemia in diabetic mice (197). Interestingly, there seems to be a crossroad between metformin and KLF15, as metformin promotes ubiquitination and decreases KLF15 protein levels, as well as the expression of KLF15 targets in vitro and in db/db mice. This effect of metformin is reversed by adenovirus-mediated KLF15 restoration. Klf15–/– mice are protected from hepatic insulin resistance induced by a high-fat diet (196). Although metformin administration has been shown to improve hepatic steatosis (198), the contribution of metformin-induced KLF15 degradation on hepatic insulin sensitivity has not been evaluated.
Peripheral insulin sensitivity reflects the ability of the body, particularly muscle and adipose tissue, to absorb glucose. Insulin sensitivity in peripheral tissues is affected by KLF11 (122), KLF14 (115), and KLF15 (171). In adipose and muscle tissue, KLF11 reduces insulin sensitivity, and Klf11–/– mice maintain lower blood glucose levels despite impaired insulin production 123, 126. KLF14 regulates glucose uptake in skeletal muscle by increasing insulin sensitivity and GLUT4 translocation. Accordingly, KLF14 expression is inhibited in the muscle of mice fed a high-fat diet and in db/db mice, which is a model of T2DM (115). KLF14 activates PI3K/Akt signaling cascade and increases insulin sensitivity in the liver of models of T2DM, such as mice fed a high-fat diet and the db/db mice (115). In cultured endothelial cells, KLF14 activates sphingosine kinase-1 (SK1), which regulates ceramide synthesis and signaling. Klf14 expression is induced after FGF2 stimulation, and induction of Klf14 is required for FGF-mediated SK1 expression (209). Because recent evidence has implicated SK1 in the regulation of adipose inflammation and insulin resistance, dysregulation of SK1 may account for some of the KLF14-mediated metabolic phenotypes (208). Adipocyte-specific overexpression of KLF15 induces insulin resistance within the adipose tissue. Moreover, those mice are protected against weight gain induced by a high-fat diet (171). Adipose KLF15 directly represses expression of stearoyl–coenzyme A desaturase 1 (SCD1). SCD1 is important for TG synthesis and lipid accumulation (210). Thus, KLF15-mediated inhibition of SCD1 may be responsible for the insulin-resistant state of the transgenic mice (171). Despite insulin resistance, mice overexpressing adipose KLF15 maintain tighter control of glucose levels after glucose challenge tests, resulting from increased synthesis of insulin by the pancreas. Importantly, double transgenic mice overexpressing adipose KLF15 and SCD1 normalize insulin production by the endocrine pancreas, suggesting SCD1 downregulation is necessary for adipose–pancreas crosstalk.
Diabetic cardiomyopathy defines the structural and functional abnormalities of the myocardium of patients with diabetes without coronary artery disease or hypertension. Diabetic cardiomyopathy is associated with an early decrease followed by upregulation in cardiac Ppara expression 211, 212, 213. In diabetes mellitus, altered cardiac Klf5 expression runs in parallel to changes of Ppara levels due to direct binding of KLF5 to the Ppara promoter and subsequent activation of PPARα expression (59). Nevertheless, cardiomyocyte Ppara downregulation may account for the lack of protection from hypertrophy in these mice, as shown by increased cardiac hypertrophy in Ppara–/– mice that underwent chronic pressure overload (149).
Kidney disease also occurs with diabetes and may account for increased mortality due to cardiovascular disease (214). KLF2 expression is reduced in glomerular endothelial cells. In streptozotocin-induced diabetic nephropathy, endothelial cell–specific Klf2–/– worsens glomerular endothelial cell injury (215). Furthermore, decreased KLF4 expression in a human podocyte cell line, and in mice with podocyte-specific Klf4–/–, is connected to proteinuria, a sign of kidney disease that can result from diabetes, high blood pressure, and diseases that cause inflammation in the kidney (216).
Conclusions
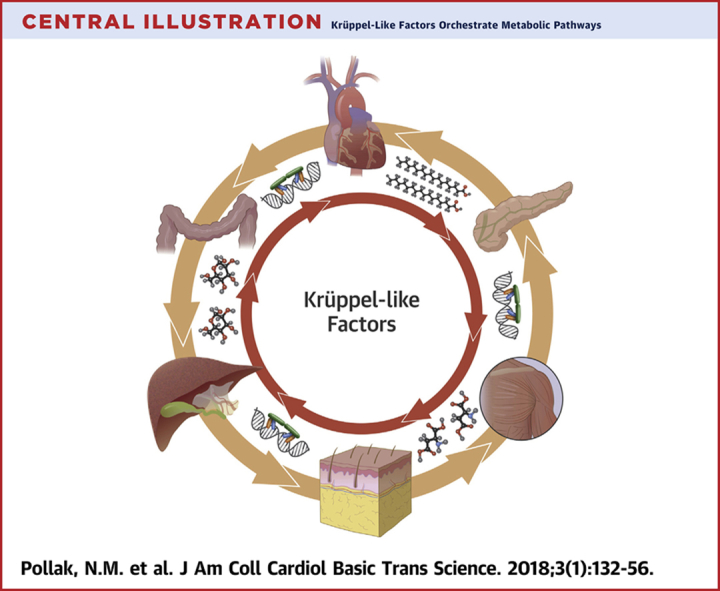
KLFs are transcriptional factors involved in the regulation of metabolic pathways in various tissues (Central Illustration). Altered expression or post-translational modifications of KLFs modulate metabolism at the cellular or systemic level. KLFs may therefore be associated with several components of metabolism-related diseases, such as the energetic balance, nutrients handling, inflammation, and responses to extracellular signals. The large variety of existing KLF isoforms, the various effects that one isoform may exert in different tissues, and the potential redundancy in the DNA regions that different isoforms can occupy set certain limitations that require careful design in potential translational applications. However, the rapid expansion of chemical substances that may act as either activators or inhibitors for KLFs in combination with other tools that mediate post-transcriptional or post-translational control constitute a promising potential application for KLF-mediated interventions in metabolic complications.
Central Illustration.

Krüppel-Like Factors Orchestrate Metabolic Pathways
Footnotes
This work was supported by a National Heart, Lung, and Blood Institute “Pathway to Independence” R00 award (HL112853) and HL130218. The authors have reported that they have no relationships relevant to the contents of this paper to disclose. Drs. Pollak and Hoffman contributed equally to this work.
All authors attest they are in compliance with human studies committees and animal welfare regulations of the authors' institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
References
- 1.McConnell B.B., Yang V.W. Mammalian Krüppel-like factors in health and diseases. Physiol Rev. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehta T.S., Lu H., Wang X. A unique sequence in the N-terminal regulatory region controls the nuclear localization of KLF8 by cooperating with the C-terminal zinc-fingers. Cell Res. 2009;19:1098–1109. doi: 10.1038/cr.2009.64. [DOI] [PubMed] [Google Scholar]
- 3.Pandya A.Y., Talley L.I., Frost A.R. Nuclear localization of KLF4 is associated with an aggressive phenotype in early-stage breast cancer. Clin Cancer Res. 2004;10:2709–2719. doi: 10.1158/1078-0432.ccr-03-0484. [DOI] [PubMed] [Google Scholar]
- 4.Quadrini K.J., Bieker J.J. Kruppel-like zinc fingers bind to nuclear import proteins and are required for efficient nuclear localization of erythroid Kruppel-like factor. J Biol Chem. 2002;277:32243–32252. doi: 10.1074/jbc.M205677200. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez E., Aburjania N., Priedigkeit N.M., DiFeo A., Martignetti J.A. Nucleo-cytoplasmic localization domains regulate Kruppel-like factor 6 (KLF6) protein stability and tumor suppressor function. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shields J.M., Yang V.W. Two potent nuclear localization signals in the gut-enriched Kruppel like factor define a subfamily of closely related Kruppel proteins. J Biol Chem. 1997;272:18504–18507. doi: 10.1074/jbc.272.29.18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans P.M., Zhang W., Chen X., Yang J., Bhakat K.K., Liu C. Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J Biol Chem. 2007;282:33994–34002. doi: 10.1074/jbc.M701847200. [DOI] [PubMed] [Google Scholar]
- 8.Li D., Yea S., Dolios G. Regulation of Kruppel-like factor 6 tumor suppressor activity by acetylation. Cancer Res. 2005;65:9216–9225. doi: 10.1158/0008-5472.CAN-05-1040. [DOI] [PubMed] [Google Scholar]
- 9.Miyamoto S., Suzuki T., Muto S. Positive and negative regulation of the cardiovascular transcription factor KLF5 by p300 and the oncogenic regulator SET through interaction and acetylation on the DNA-binding domain. Mol Cell Biol. 2003;23:8528–8541. doi: 10.1128/MCB.23.23.8528-8541.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song C.Z., Keller K., Chen Y., Stamatoyannopoulos G. Functional interplay between CBP and PCAF in acetylation and regulation of transcription factor KLF13 activity. J Mol Biol. 2003;329:207–215. doi: 10.1016/s0022-2836(03)00429-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W., Bieker J.J. Acetylation and modulation of erythroid Kruppel-like factor (EKLF) activity by interaction with histone acetyltransferases. Proc Natl Acad Sci U S A. 1998;95:9855–9860. doi: 10.1073/pnas.95.17.9855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sengupta T., Chen K., Milot E., Bieker J.J. Acetylation of EKLF is essential for epigenetic modification and transcriptional activation of the beta-globin locus. Mol Cell Biol. 2008;28:6160–6170. doi: 10.1128/MCB.00919-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geiman D.E., Ton-That H., Johnson J.M., Yang V.W. Transactivation and growth suppression by the gut-enriched Kruppel-like factor (Kruppel-like factor 4) are dependent on acidic amino acid residues and protein-protein interaction. Nucleic Acids Res. 2000;28:1106–1113. doi: 10.1093/nar/28.5.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He M., Zheng B., Zhang Y. KLF4 mediates the link between TGF-beta1-induced gene transcription and H3 acetylation in vascular smooth muscle cells. FASEB J. 2015;29:4059–4070. doi: 10.1096/fj.15-272658. [DOI] [PubMed] [Google Scholar]
- 15.Papadakis K.A., Krempski J., Svingen P. Kruppel-like factor KLF10 deficiency predisposes to colitis through colonic macrophage dysregulation. Am J Physiol Gastrointest Liver Physiol. 2015;309:G900–G909. doi: 10.1152/ajpgi.00309.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenzweig J.M., Glenn J.D., Calabresi P.A., Whartenby K.A. KLF4 modulates expression of IL-6 in dendritic cells via both promoter activation and epigenetic modification. J Biol Chem. 2013;288:23868–23874. doi: 10.1074/jbc.M113.479576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seo S., Lomberk G., Mathison A. Kruppel-like factor 11 differentially couples to histone acetyltransferase and histone methyltransferase chromatin remodeling pathways to transcriptionally regulate dopamine D2 receptor in neuronal cells. J Biol Chem. 2012;287:12723–12735. doi: 10.1074/jbc.M112.351395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsumura T., Suzuki T., Aizawa K. The deacetylase HDAC1 negatively regulates the cardiovascular transcription factor Kruppel-like factor 5 through direct interaction. J Biol Chem. 2005;280:12123–12129. doi: 10.1074/jbc.M410578200. [DOI] [PubMed] [Google Scholar]
- 19.Chen X., Bieker J.J. Stage-specific repression by the EKLF transcriptional activator. Mol Cell Biol. 2004;24:10416–10424. doi: 10.1128/MCB.24.23.10416-10424.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng Y., Tabbaa Z.M., Khan Z. Epigenetic regulation of uterine biology by transcription factor KLF11 via posttranslational histone deacetylation of cytochrome p450 metabolic enzymes. Endocrinology. 2014;155:4507–4520. doi: 10.1210/en.2014-1139. [DOI] [PubMed] [Google Scholar]
- 21.Salmon M., Gomez D., Greene E., Shankman L., Owens G.K. Cooperative binding of KLF4, pELK-1, and HDAC2 to a G/C repressor element in the SM22alpha promoter mediates transcriptional silencing during SMC phenotypic switching in vivo. Circ Res. 2012;111:685–696. doi: 10.1161/CIRCRESAHA.112.269811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J.S., Moncrieffe M.C., Kaczynski J., Ellenrieder V., Prendergast F.G., Urrutia R. A conserved alpha-helical motif mediates the interaction of Sp1-like transcriptional repressors with the corepressor mSin3A. Mol Cell Biol. 2001;21:5041–5049. doi: 10.1128/MCB.21.15.5041-5049.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuierer M., Hilger-Eversheim K., Dobner T. Induction of AP-2alpha expression by adenoviral infection involves inactivation of the AP-2rep transcriptional corepressor CtBP1. J Biol Chem. 2001;276:27944–27949. doi: 10.1074/jbc.M100070200. [DOI] [PubMed] [Google Scholar]
- 24.Turner J., Crossley M. Cloning and characterization of mCtBP2, a co-repressor that associates with basic Kruppel-like factor and other mammalian transcriptional regulators. EMBO J. 1998;17:5129–5140. doi: 10.1093/emboj/17.17.5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Vliet J., Turner J., Crossley M. Human Kruppel-like factor 8: a CACCC-box binding protein that associates with CtBP and represses transcription. Nucleic Acids Res. 2000;28:1955–1962. doi: 10.1093/nar/28.9.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi Y., Sawada J., Sui G. Coordinated histone modifications mediated by a CtBP corepressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- 27.Ueda J., Tachibana M., Ikura T., Shinkai Y. Zinc finger protein Wiz links G9a/GLP histone methyltransferases to the co-repressor molecule CtBP. J Biol Chem. 2006;281:20120–20128. doi: 10.1074/jbc.M603087200. [DOI] [PubMed] [Google Scholar]
- 28.Sewalt R.G., Gunster M.J., van der Vlag J., Satijn D.P., Otte A.P. C-terminal binding protein is a transcriptional repressor that interacts with a specific class of vertebrate polycomb proteins. Mol Cell Biol. 1999;19:777–787. doi: 10.1128/mcb.19.1.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koipally J., Georgopoulos K. Ikaros interactions with CtBP reveal a repression mechanism that is independent of histone deacetylase activity. J Biol Chem. 2000;275:19594–19602. doi: 10.1074/jbc.M000254200. [DOI] [PubMed] [Google Scholar]
- 30.Dewi V., Kwok A., Lee S. Phosphorylation of Kruppel-like factor 3 (KLF3/BKLF) and C-terminal binding protein 2 (CtBP2) by homeodomain-interacting protein kinase 2 (HIPK2) modulates KLF3 DNA binding and activity. J Biol Chem. 2015;290:8591–8605. doi: 10.1074/jbc.M115.638338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Daftary G.S., Lomberk G.A., Buttar N.S. Detailed structural-functional analysis of the Krüppel-like factor 16 (KLF16) transcription factor reveals novel mechanisms for silencing Sp/KLF sites involved in metabolism and endocrinology. J Biol Chem. 2012;287:7010–7025. doi: 10.1074/jbc.M111.266007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Slavin D.A., Koritschoner N.P., Prieto C.C., Lopez-Diaz F.J., Chatton B., Bocco J.L. A new role for the Kruppel-like transcription factor KLF6 as an inhibitor of c-Jun proto-oncoprotein function. Oncogene. 2004;23:8196–8205. doi: 10.1038/sj.onc.1208020. [DOI] [PubMed] [Google Scholar]
- 33.Ouyang L., Chen X., Bieker J.J. Regulation of erythroid Kruppel-like factor (EKLF) transcriptional activity by phosphorylation of a protein kinase casein kinase II site within its interaction domain. J Biol Chem. 1998;273:23019–23025. doi: 10.1074/jbc.273.36.23019. [DOI] [PubMed] [Google Scholar]
- 34.Huang B., Ahn Y.T., McPherson L., Clayberger C., Krensky A.M. Interaction of PRP4 with Kruppel-like factor 13 regulates CCL5 transcription. J Immunol. 2007;178:7081–7087. doi: 10.4049/jimmunol.178.11.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X.H., Zheng B., Han M., Miao S.B., Wen J.K. Synthetic retinoid Am80 inhibits interaction of KLF5 with RAR alpha through inducing KLF5 dephosphorylation mediated by the PI3K/Akt signaling in vascular smooth muscle cells. FEBS Lett. 2009;583:1231–1236. doi: 10.1016/j.febslet.2009.03.016. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Z., Teng C.T. Phosphorylation of Kruppel-like factor 5 (KLF5/IKLF) at the CBP interaction region enhances its transactivation function. Nucleic Acids Res. 2003;31:2196–2208. doi: 10.1093/nar/gkg310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellenrieder V., Buck A., Harth A. KLF11 mediates a critical mechanism in TGF-beta signaling that is inactivated by Erk-MAPK in pancreatic cancer cells. Gastroenterology. 2004;127:607–620. doi: 10.1053/j.gastro.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 38.Banck M.S., Beaven S.W., Narla G., Walsh M.J., Friedman S.L., Beutler A.S. KLF6 degradation after apoptotic DNA damage. FEBS Lett. 2006;580:6981–6986. doi: 10.1016/j.febslet.2006.10.077. [DOI] [PubMed] [Google Scholar]
- 39.Chen C., Sun X., Ran Q. Ubiquitin-proteasome degradation of KLF5 transcription factor in cancer and untransformed epithelial cells. Oncogene. 2005;24:3319–3327. doi: 10.1038/sj.onc.1208497. [DOI] [PubMed] [Google Scholar]
- 40.Chen Z.Y., Wang X., Zhou Y., Offner G., Tseng C.C. Destabilization of Kruppel-like factor 4 protein in response to serum stimulation involves the ubiquitin-proteasome pathway. Cancer Res. 2005;65:10394–10400. doi: 10.1158/0008-5472.CAN-05-2059. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X., Srinivasan S.V., Lingrel J.B. WWP1-dependent ubiquitination and degradation of the lung Kruppel-like factor, KLF2. Biochem Biophys Res Commun. 2004;316:139–148. doi: 10.1016/j.bbrc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 42.Carrano A.C., Dillin A., Hunter T. A Kruppel-like factor downstream of the E3 ligase WWP1 mediates dietary-restriction-induced longevity in Caenorhabditis elegans. Nat Commun. 2014;5:3772. doi: 10.1038/ncomms4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chang H.S., Lin C.H., Yang C.H., Liang Y.J., Yu W.C. The human papillomavirus-16 (HPV-16) oncoprotein E7 conjugates with and mediates the role of the transforming growth factor beta inducible early gene 1 (TIEG1) in apoptosis. Int J Biochem Cell Biol. 2010;42:1831–1839. doi: 10.1016/j.biocel.2010.07.019. [DOI] [PubMed] [Google Scholar]
- 44.Kim J., Kwak H.J., Cha J.Y., Jeong Y.S., Rhee S.D., Cheon H.G. The role of prolyl hydroxylase domain protein (PHD) during rosiglitazone-induced adipocyte differentiation. J Biol Chem. 2014;289:2755–2764. doi: 10.1074/jbc.M113.493650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tian X., Dai S., Sun J. F-box protein FBXO22 mediates polyubiquitination and degradation of KLF4 to promote hepatocellular carcinoma progression. Oncotarget. 2015;6:22767–22775. doi: 10.18632/oncotarget.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venuprasad K., Huang H., Harada Y. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat Immunol. 2008;9:245–253. doi: 10.1038/niXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Du J.X., Bialkowska A.B., McConnell B.B., Yang V.W. SUMOylation regulates nuclear localization of Kruppel-like factor 5. J Biol Chem. 2008;283:31991–32002. doi: 10.1074/jbc.M803612200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tahmasebi S., Ghorbani M., Savage P. Sumoylation of Kruppel-like factor 4 inhibits pluripotency induction but promotes adipocyte differentiation. J Biol Chem. 2013;288:12791–12804. doi: 10.1074/jbc.M113.465443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siatecka M., Soni S., Planutis A., Bieker J.J. Transcriptional activity of erythroid Kruppel-like factor (EKLF/KLF1) modulated by PIAS3 (protein inhibitor of activated STAT3) J Biol Chem. 2015;290:9929–9940. doi: 10.1074/jbc.M114.610246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kawai-Kowase K., Kumar M.S., Hoofnagle M.H., Yoshida T., Owens G.K. PIAS1 activates the expression of smooth muscle cell differentiation marker genes by interacting with serum response factor and class I basic helix-loop-helix proteins. Mol Cell Biol. 2005;25:8009–8023. doi: 10.1128/MCB.25.18.8009-8023.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oishi Y., Manabe I., Tobe K. SUMOylation of Kruppel-like transcription factor 5 acts as a molecular switch in transcriptional programs of lipid metabolism involving PPARdelta. Nat Med. 2008;14:656–666. doi: 10.1038/nm1756. [DOI] [PubMed] [Google Scholar]
- 52.Lopaschuk G.D., Ussher J.R., Folmes C.D., Jaswal J.S., Stanley W.C. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 53.Noh H.L., Okajima K., Molkentin J.D., Homma S., Goldberg I.J. Acute lipoprotein lipase deletion in adult mice leads to dyslipidemia and cardiac dysfunction. Am J Physiol Endocrinol Metab. 2006;291:E755–E760. doi: 10.1152/ajpendo.00111.2006. [DOI] [PubMed] [Google Scholar]
- 54.Levak-Frank S., Hofmann W., Weinstock P.H. Induced mutant mouse lines that express lipoprotein lipase in cardiac muscle, but not in skeletal muscle and adipose tissue, have normal plasma triglyceride and high-density lipoprotein-cholesterol levels. Proc Natl Acad Sci U S A. 1999;96:3165–3170. doi: 10.1073/pnas.96.6.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schilling J., Kelly D.P. The PGC-1 cascade as a therapeutic target for heart failure. J Mol Cell Cardiol. 2011;51:578–583. doi: 10.1016/j.yjmcc.2010.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liao X., Zhang R., Lu Y. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest. 2015;125:3461–3476. doi: 10.1172/JCI79964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sunaga H., Matsui H., Anjo S. Elongation of long-chain fatty acid family member 6 (Elovl6)-driven fatty acid metabolism regulates vascular smooth muscle cell phenotype through AMP-activated protein kinase/Kruppel-like factor 4 (AMPK/KLF4) signaling. J Am Heart Assoc. 2016;5 doi: 10.1161/JAHA.116.004014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pol C.J., Lieu M., Drosatos K. PPARs: protectors or opponents of myocardial function? PPAR Res. 2015;2015:835985. doi: 10.1155/2015/835985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drosatos K., Pollak N.M., Pol C.J. Cardiac myocyte KLF5 regulates Ppara expression and cardiac function. Circ Res. 2016;118:241–253. doi: 10.1161/CIRCRESAHA.115.306383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prosdocimo D.A., Anand P., Liao X. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. J Biol Chem. 2014;289:5914–5924. doi: 10.1074/jbc.M113.531384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prosdocimo D.A., John J.E., Zhang L. KLF15 and PPARalpha cooperate to regulate cardiomyocyte lipid gene expression and oxidation. PPAR Res. 2015;2015:201625. doi: 10.1155/2015/201625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang L., Prosdocimo D.A., Bai X. KLF15 establishes the landscape of diurnal expression in the heart. Cell Reports. 2015;13:2368–2375. doi: 10.1016/j.celrep.2015.11.038. [DOI] [PubMed] [Google Scholar]
- 63.Hargreaves M. Skeletal muscle metabolism during exercise in humans. Clin Exp Pharmacol Physiol. 2000;27:225–228. doi: 10.1046/j.1440-1681.2000.03225.x. [DOI] [PubMed] [Google Scholar]
- 64.Schiaffino S., Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev. 2011;91:1447–1531. doi: 10.1152/physrev.00031.2010. [DOI] [PubMed] [Google Scholar]
- 65.Ehrenborg E., Krook A. Regulation of skeletal muscle physiology and metabolism by peroxisome proliferator-activated receptor delta. Pharmacol Rev. 2009;61:373–393. doi: 10.1124/pr.109.001560. [DOI] [PubMed] [Google Scholar]
- 66.Haldar S.M., Jeyaraj D., Anand P. Kruppel-like factor 15 regulates skeletal muscle lipid flux and exercise adaptation. Proc Natl Acad Sci U S A. 2012;109:6739–6744. doi: 10.1073/pnas.1121060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gray S., Wang B., Orihuela Y. Regulation of gluconeogenesis by Kruppel-like factor 15. Cell Metabol. 2007;5:305–312. doi: 10.1016/j.cmet.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yamamoto J., Ikeda Y., Iguchi H. A Kruppel-like factor KLF15 contributes fasting induced transcriptional activation of mitochondrial acetyl-CoA synthetase gene AceCS2. J Biol Chem. 2004;279:16954–16962. doi: 10.1074/jbc.M312079200. [DOI] [PubMed] [Google Scholar]
- 69.Gray S., Feinberg M.W., Hull S. The Kruppel-like factor KLF15 regulates the insulin sensitive glucose transporter GLUT4. J Biol Chem. 2002;277:34322–34328. doi: 10.1074/jbc.M201304200. [DOI] [PubMed] [Google Scholar]
- 70.Felig P., Owen O., Wahren J., Cahill G.F.J. Amino acid metabolism during prolonged starvation. J Clin Invest. 1969;48:584–594. doi: 10.1172/JCI106017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Felig P., Pozefsky T., Marliss E., Cahill G.F.J. Alanine: key role in gluconeogenesis. Science. 1970;167:1003–1004. doi: 10.1126/science.167.3920.1003. [DOI] [PubMed] [Google Scholar]
- 72.Kim S., Moustaid-Moussa N. Secretory, endocrine and autocrine/paracrine function of the adipocyte. J Nutr. 2000;130:3110S–3115S. doi: 10.1093/jn/130.12.3110S. [DOI] [PubMed] [Google Scholar]
- 73.MacDougald O.A., Burant C.F. The rapidly expanding family of adipokines. Cell Metab. 2007;6:159–161. doi: 10.1016/j.cmet.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 74.Cinti S. The adipose organ. Prostaglandins Leukot Essent Fatty Acids. 2005;73:9–15. doi: 10.1016/j.plefa.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 75.Rial E., Zardoya R. Oxidative stress, thermogenesis and evolution of uncoupling proteins. J Biol. 2009;8:58. doi: 10.1186/jbiol155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim S.H., Plutzky J. Brown fat and browning for the treatment of obesity and related metabolic disorders. Diabetes Metab J. 2016;40:12–21. doi: 10.4093/dmj.2016.40.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosen E.D., Walkey C.J., Puigserver P., Spiegelman B.M. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- 78.Birsoy K., Chen Z., Friedman J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab. 2008;7:339–347. doi: 10.1016/j.cmet.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen Z., Torrens J.I., Anand A., Spiegelman B.M., Friedman J.M. Krox20 stimulates adipogenesis via C/EBPbeta-dependent and -independent mechanisms. Cell Metab. 2005;1:93–106. doi: 10.1016/j.cmet.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 80.Kimura H., Fujimori K. Activation of early phase of adipogenesis through Kruppel-like factor KLF9-mediated, enhanced expression of CCAAT/enhancer-binding protein beta in 3T3L1 cells. Gene. 2014;534:169–176. doi: 10.1016/j.gene.2013.10.065. [DOI] [PubMed] [Google Scholar]
- 81.Pei H., Yao Y., Yang Y., Liao K., Wu J.R. Kruppel-like factor KLF9 regulates PPARgamma transactivation at the middle stage of adipogenesis. Cell Death Differ. 2011;18:315–327. doi: 10.1038/cdd.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee H., Kim H.J., Lee Y.J. Kruppel-like factor KLF8 plays a critical role in adipocyte differentiation. PLoS One. 2012;7 doi: 10.1371/journal.pone.0052474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oishi Y., Manabe I., Tobe K. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab. 2005;1:27–39. doi: 10.1016/j.cmet.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 84.Jiang S., Wei H., Song T. KLF13 promotes porcine adipocyte differentiation through PPARgamma activation. Cell Biosci. 2015;5:28. doi: 10.1186/s13578-015-0016-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mori T., Sakaue H., Iguchi H. Role of Krüppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem. 2005;280:12867–12875. doi: 10.1074/jbc.M410515200. [DOI] [PubMed] [Google Scholar]
- 86.Gordon A.R., Outram S.V., Keramatipour M. Splenomegaly and modified erythropoiesis in KLF13-/- mice. J Biol Chem. 2008;283:11897–11904. doi: 10.1074/jbc.M709569200. [DOI] [PubMed] [Google Scholar]
- 87.Sue N., Jack B.H., Eaton S.A. Targeted disruption of the basic Kruppel-like factor gene (Klf3) reveals a role in adipogenesis. Mol Cell Biol. 2008;28:3967–3978. doi: 10.1128/MCB.01942-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kanazawa A., Kawamura Y., Sekine A. Single nucleotide polymorphisms in the gene encoding Kruppel-like factor 7 are associated with type 2 diabetes. Diabetologia. 2005;48:1315–1322. doi: 10.1007/s00125-005-1797-0. [DOI] [PubMed] [Google Scholar]
- 89.Banerjee S.S., Feinberg M.W., Watanabe M. The Kruppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-gamma expression and adipogenesis. J Biol Chem. 2003;278:2581–2584. doi: 10.1074/jbc.M210859200. [DOI] [PubMed] [Google Scholar]
- 90.Bell-Anderson K.S., Funnell A.P., Williams H. Loss of Kruppel-like factor 3 (KLF3/BKLF) leads to upregulation of the insulin-sensitizing factor adipolin (FAM132A/CTRP12/C1qdc2) Diabetes. 2013;62:2728–2737. doi: 10.2337/db12-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Inuzuka H., Nanbu-Wakao R., Masuho Y., Muramatsu M., Tojo H., Wakao H. Differential regulation of immediate early gene expression in preadipocyte cells through multiple signaling pathways. Biochem Biophys Res Commun. 1999;265:664–668. doi: 10.1006/bbrc.1999.1734. [DOI] [PubMed] [Google Scholar]
- 92.Li D., Yea S., Li S. Kruppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J Biol Chem. 2005;280:26941–26952. doi: 10.1074/jbc.M500463200. [DOI] [PubMed] [Google Scholar]
- 93.Jang M.K., Lee S., Jung M.H. RNA-Seq analysis reveals a negative role of KLF16 in adipogenesis. PLoS One. 2016;11 doi: 10.1371/journal.pone.0162238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Petrovic N., Walden T.B., Shabalina I.G., Timmons J.A., Cannon B., Nedergaard J. Chronic peroxisome proliferator-activated receptor gamma (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J Biol Chem. 2010;285:7153–7164. doi: 10.1074/jbc.M109.053942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vegiopoulos A., Muller-Decker K., Strzoda D. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science. 2010;328:1158–1161. doi: 10.1126/science.1186034. [DOI] [PubMed] [Google Scholar]
- 96.Kajimura S., Spiegelman B.M., Seale P. Brown and beige fat: physiological roles beyond heat generation. Cell Metab. 2015;22:546–559. doi: 10.1016/j.cmet.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Loft A., Forss I., Siersbaek M.S. Browning of human adipocytes requires KLF11 and reprogramming of PPARgamma superenhancers. Genes Dev. 2015;29:7–22. doi: 10.1101/gad.250829.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yamamoto K., Sakaguchi M., Medina R.J. Transcriptional regulation of a brown adipocyte-specific gene, UCP1, by KLF11 and KLF15. Biochem Biophys Res Commun. 2010;400:175–180. doi: 10.1016/j.bbrc.2010.08.039. [DOI] [PubMed] [Google Scholar]
- 99.Yin K.J., Fan Y., Hamblin M. KLF11 mediates PPARγ cerebrovascular protection in ischaemic stroke. Brain. 2013;136:1274–1287. doi: 10.1093/brain/awt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Asada M., Rauch A., Shimizu H. DNA binding-dependent glucocorticoid receptor activity promotes adipogenesis via Krüppel-like factor 15 gene expression. Lab Invest. 2011;91:203–215. doi: 10.1038/labinvest.2010.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lee M.J., Pramyothin P., Karastergiou K., Fried S.K. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta. 2014;1842:473–481. doi: 10.1016/j.bbadis.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee da S., Choi H., Han B.S. c-Jun regulates adipocyte differentiation via the KLF15mediated mode. Biochem Biophys Res Commun. 2016;469:552–558. doi: 10.1016/j.bbrc.2015.12.035. [DOI] [PubMed] [Google Scholar]
- 103.She P., Burgess S.C., Shiota M. Mechanisms by which liver-specific PEPCK knockout mice preserve euglycemia during starvation. Diabetes. 2003;52:1649–1654. doi: 10.2337/diabetes.52.7.1649. [DOI] [PubMed] [Google Scholar]
- 104.Hakimi P., Johnson M.T., Yang J. Phosphoenolpyruvate carboxykinase and the critical role of cataplerosis in the control of hepatic metabolism. Nutr Metab (Lond) 2005;2:33. doi: 10.1186/1743-7075-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Guillaumond F., Grechez-Cassiau A., Subramaniam M. Krüppel-like factor KLF10 is a link between the circadian clock and metabolism in liver. Mol Cell Biol. 2010;30:3059–3070. doi: 10.1128/MCB.01141-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang H., Chen Q., Jiao T. Involvement of KLF11 in hepatic glucose metabolism in mice via suppressing of PEPCK-C expression. PLoS One. 2014;9 doi: 10.1371/journal.pone.0089552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Iizuka K., Takeda J., Horikawa Y. Krüppel-like factor-10 is directly regulated by carbohydrate response element-binding protein in rat primary hepatocytes. Biochem Biophys Res Commun. 2011;412:638–643. doi: 10.1016/j.bbrc.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 108.Teshigawara K., Ogawa W., Mori T. Role of Krüppel-like factor 15 in PEPCK gene expression in the liver. Biochem Biophys Res Commun. 2005;327:920–926. doi: 10.1016/j.bbrc.2004.12.096. [DOI] [PubMed] [Google Scholar]
- 109.Uchida S., Tanaka Y., Ito H. Transcriptional regulation of the CLC-K1 promoter by myc-associated zinc finger protein and kidney-enriched Krüppel-like factor, a novel zinc finger repressor. Mol Cell Biol. 2000;20:7319–7331. doi: 10.1128/mcb.20.19.7319-7331.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rakhshandehroo M., Hooiveld G., Muller M., Kersten S. Comparative analysis of gene regulation by the transcription factor PPARalpha between mouse and human. PLoS One. 2009;4 doi: 10.1371/journal.pone.0006796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhang H., Chen Q., Yang M. Mouse KLF11 regulates hepatic lipid metabolism. J Hepatol. 2013;58:763–770. doi: 10.1016/j.jhep.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 112.Takeuchi Y., Yahagi N., Aita Y. KLF15 enables rapid switching between lipogenesis and gluconeogenesis during fasting. Cell Reports. 2016;16:2373–2386. doi: 10.1016/j.celrep.2016.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zechner R., Strauss J.G., Haemmerle G., Lass A., Zimmermann R. Lipolysis: pathway under construction. Curr Opin Lipidol. 2005;16:333–340. doi: 10.1097/01.mol.0000169354.20395.1c. [DOI] [PubMed] [Google Scholar]
- 114.Jeyaraj D., Scheer F.A., Ripperger J.A. Klf15 orchestrates circadian nitrogen homeostasis. Cell Metab. 2012;15:311–323. doi: 10.1016/j.cmet.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang M., Ren Y., Lin Z. Krüppel-like factor 14 increases insulin sensitivity through activation of PI3K/Akt signal pathway. Cell Signal. 2015;27:2201–2208. doi: 10.1016/j.cellsig.2015.07.019. [DOI] [PubMed] [Google Scholar]
- 116.Ferre T., Riu E., Franckhauser S., Agudo J., Bosch F. Long-term overexpression of glucokinase in the liver of transgenic mice leads to insulin resistance. Diabetologia. 2003;46:1662–1668. doi: 10.1007/s00125-003-1244-z. [DOI] [PubMed] [Google Scholar]
- 117.Bechmann L.P., Gastaldelli A., Vetter D. Glucokinase links Kruppel-like factor 6 to the regulation of hepatic insulin sensitivity in nonalcoholic fatty liver disease. Hepatology. 2012;55:1083–1093. doi: 10.1002/hep.24793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lefebvre P., Cariou B., Lien F., Kuipers F., Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 119.Han S., Zhang R., Jain R. Circadian control of bile acid synthesis by a KLF15-Fgf15 axis. Nat Commun. 2015;6:7231. doi: 10.1038/ncomms8231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Puri S., Folias A.E., Hebrok M. Plasticity and dedifferentiation within the pancreas: development, homeostasis, and disease. Cell Stem Cell. 2015;16:18–31. doi: 10.1016/j.stem.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu M.J., Wu W.C., Chang H.W. KLF10 affects pancreatic function via the SEI-1/p21Cip1 pathway. Int J Biochem Cell Biol. 2015;60:53–59. doi: 10.1016/j.biocel.2014.12.021. [DOI] [PubMed] [Google Scholar]
- 122.Neve B., Fernandez-Zapico M.E., Ashkenazi-Katalan V. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U S A. 2005;102:4807–4812. doi: 10.1073/pnas.0409177102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bonnefond A., Lomberk G., Buttar N. Disruption of a novel Kruppel-like transcription factor p300-regulated pathway for insulin biosynthesis revealed by studies of the c.-331 INS mutation found in neonatal diabetes mellitus. J Biol Chem. 2011;286:28414–28424. doi: 10.1074/jbc.M110.215822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fernandez-Zapico M.E., van Velkinburgh J.C., Gutierrez-Aguilar R. MODY7 gene, KLF11, is a novel p300-dependent regulator of Pdx-1 (MODY4) transcription in pancreatic islet beta cells. J Biol Chem. 2009;284:36482–36490. doi: 10.1074/jbc.M109.028852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.McKinnon C., Docherty K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia. 2001;44:1203–1214. doi: 10.1007/s001250100628. [DOI] [PubMed] [Google Scholar]
- 126.Mathison A., Escande C., Calvo E. Phenotypic characterization of mice carrying homozygous deletion of KLF11, a gene in which mutations cause human neonatal and MODY VII diabetes. Endocrinology. 2015;156:3581–3595. doi: 10.1210/en.2015-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kawamura Y., Tanaka Y., Kawamori R., Maeda S. Overexpression of Kruppel-like factor 7 regulates adipocytokine gene expressions in human adipocytes and inhibits glucose induced insulin secretion in pancreatic beta-cell line. Mol Endocrinol. 2006;20:844–856. doi: 10.1210/me.2005-0138. [DOI] [PubMed] [Google Scholar]
- 128.Miller I.J., Bieker J.J. A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol Cell Biol. 1993;13:2776–2786. doi: 10.1128/mcb.13.5.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhou D., Liu K., Sun C.W., Pawlik K.M., Townes T.M. KLF1 regulates BCL11A expression and gamma- to beta-globin gene switching. Nat Genet. 2010;42:742–744. doi: 10.1038/ng.637. [DOI] [PubMed] [Google Scholar]
- 130.Pilon A.M., Arcasoy M.O., Dressman H.K. Failure of terminal erythroid differentiation in EKLF-deficient mice is associated with cell cycle perturbation and reduced expression of E2F2. Mol Cell Biol. 2008;28:7394–7401. doi: 10.1128/MCB.01087-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tallack M.R., Keys J.R., Humbert P.O., Perkins A.C. EKLF/KLF1 controls cell cycle entry via direct regulation of E2f2. J Biol Chem. 2009;284:20966–20974. doi: 10.1074/jbc.M109.006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Magor G.W., Tallack M.R., Gillinder K.R. KLF1-null neonates display hydrops fetalis and a deranged erythroid transcriptome. Blood. 2015;125:2405–2417. doi: 10.1182/blood-2014-08-590968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Perseu L., Satta S., Moi P. KLF1 gene mutations cause borderline HbA(2) Blood. 2011;118:4454–4458. doi: 10.1182/blood-2011-04-345736. [DOI] [PubMed] [Google Scholar]
- 134.Schechter A. Hemoglobin research and the origins of molecular medicine. Blood. 2008;112:3927–3938. doi: 10.1182/blood-2008-04-078188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nuez B., Michalovich D., Bygrave A., Ploemacher R., Grosveld F. Defective haematopoiesis in fetal liver resulting from inactivation of the EKLF gene. Nature. 1995;375:316–318. doi: 10.1038/375316a0. [DOI] [PubMed] [Google Scholar]
- 136.Perkins A.C., Sharpe A.H., Orkin S.H. Lethal beta-thalassaemia in mice lacking the erythroid CACCC-transcription factor EKLF. Nature. 1995;375:318–322. doi: 10.1038/375318a0. [DOI] [PubMed] [Google Scholar]
- 137.Borg J., Papadopoulos P., Georgitsi M. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat Genet. 2010;42:801–805. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Porcu S., Manchinu M.F., Marongiu M.F. Klf1 affects DNase II-alpha expression in the central macrophage of a fetal liver erythroblastic island: a non-cell-autonomous role in definitive erythropoiesis. Mol Cell Biol. 2011;31:4144–4154. doi: 10.1128/MCB.05532-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Funnell A.P., Mak K.S., Twine N.A. Generation of mice deficient in both KLF3/BKLF and KLF8 reveals a genetic interaction and a role for these factors in embryonic globin gene silencing. Mol Cell Biol. 2013;33:2976–2987. doi: 10.1128/MCB.00074-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Crossley M., Whitelaw E., Perkins A., Williams G., Fujiwara Y., Orkin S.H. Isolation and characterization of the cDNA encoding BKLF/TEF-2, a major CACCC-box-binding protein in erythroid cells and selected other cells. Mol Cell Biol. 1996;16:1695–1705. doi: 10.1128/mcb.16.4.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Funnell A.P., Maloney C.A., Thompson L.J. Erythroid Kruppel-like factor directly activates the basic Kruppel-like factor gene in erythroid cells. Mol Cell Biol. 2007;27:2777–2790. doi: 10.1128/MCB.01658-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Funnell A.P., Norton L.J., Mak K.S. The CACCC-binding protein KLF3/BKLF represses a subset of KLF1/EKLF target genes and is required for proper erythroid maturation in vivo. Mol Cell Biol. 2012;32:3281–3292. doi: 10.1128/MCB.00173-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Eaton S.A., Funnell A.P., Sue N., Nicholas H., Pearson R.C., Crossley M. A network of Kruppel like factors (Klfs). Klf8 is repressed by Klf3 and activated by Klf1 in vivo. J Biol Chem. 2008;283:26937–26947. doi: 10.1074/jbc.M804831200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Basu P., Morris P.E., Haar J.L. KLF2 is essential for primitive erythropoiesis and regulates the human and murine embryonic beta-like globin genes in vivo. Blood. 2005;106:2566–2571. doi: 10.1182/blood-2005-02-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Roger V.L. Epidemiology of heart failure. Circ Res. 2013;113:646–659. doi: 10.1161/CIRCRESAHA.113.300268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lopaschuk G., Ussher J., Folmes C., Jaswal J., Stanley W. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207–258. doi: 10.1152/physrev.00015.2009. [DOI] [PubMed] [Google Scholar]
- 147.Neubauer S. The failing heart—an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 148.Stanley W., Recchia F., Lopaschuk G. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 149.Smeets P.J., Teunissen B.E., Willemsen P.H. Cardiac hypertrophy is enhanced in PPAR alpha-/- mice in response to chronic pressure overload. Cardiovasc Res. 2008;78:79–89. doi: 10.1093/cvr/cvn001. [DOI] [PubMed] [Google Scholar]
- 150.Lebrasseur N.K., Duhaney T.A., De Silva D.S. Effects of fenofibrate on cardiac remodeling in aldosterone-induced hypertension. Hypertension. 2007;50:489–496. doi: 10.1161/HYPERTENSIONAHA.107.092403. [DOI] [PubMed] [Google Scholar]
- 151.Zou J., Le K., Xu S. Fenofibrate ameliorates cardiac hypertrophy by activation of peroxisome proliferator-activated receptor-alpha partly via preventing p65-NFkappaB binding to NFATc4. Mol Cell Endocrinol. 2013;370:103–112. doi: 10.1016/j.mce.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 152.Linz W., Wohlfart P., Baader M. The peroxisome proliferator-activated receptor-alpha (PPAR-alpha) agonist, AVE8134, attenuates the progression of heart failure and increases survival in rats. Acta Pharmacologica Sinica. 2009;30:935–946. doi: 10.1038/aps.2009.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Liao X., Haldar S.M., Lu Y. Kruppel-like factor 4 regulates pressure-induced cardiac hypertrophy. J Mol Cell Cardiol. 2010;49:334–338. doi: 10.1016/j.yjmcc.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Takeda N., Manabe I., Uchino Y. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest. 2010;120:254–265. doi: 10.1172/JCI40295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fisch S., Gray S., Heymans S. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2007;104:7074–7079. doi: 10.1073/pnas.0701981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sun H., Olson K.C., Gao C. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation. 2016;133:2038–2049. doi: 10.1161/CIRCULATIONAHA.115.020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li T., Zhang Z., Kolwicz S.C., Jr. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. 2017;25:374–385. doi: 10.1016/j.cmet.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sawaki D., Hou L., Tomida S. Modulation of cardiac fibrosis by Kruppel-like factor 6 through transcriptional control of thrombospondin 4 in cardiomyocytes. Cardiovasc Res. 2015;107:420–430. doi: 10.1093/cvr/cvv155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Benjamin E.J., Blaha M.J., Chiuve S.E. Heart disease and stroke statistics—2017 update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gisterå A., Hansson G.K. The immunology of atherosclerosis. Nat Rev Nephrol. 2017;13:368–380. doi: 10.1038/nrneph.2017.51. [DOI] [PubMed] [Google Scholar]
- 161.Atkins G.B., Wang Y., Mahabeleshwar G.H. Hemizygous deficiency of Kruppel-like factor 2 augments experimental atherosclerosis. Circ Res. 2008;103:690–693. doi: 10.1161/CIRCRESAHA.108.184663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lingrel J.B., Pilcher-Roberts R., Basford J.E. Myeloid-specific Kruppel-like factor 2 inactivation increases macrophage and neutrophil adhesion and promotes atherosclerosis. Circ Res. 2012;110:1294–1302. doi: 10.1161/CIRCRESAHA.112.267310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhou G., Hamik A., Nayak L. Endothelial Kruppel-like factor 4 protects against atherothrombosis in mice. J Clin Invest. 2012;122:4727–4731. doi: 10.1172/JCI66056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Hartmann P., Zhou Z., Natarelli L. Endothelial Dicer promotes atherosclerosis and vascular inflammation by miRNA-103-mediated suppression of KLF4. Nat Commun. 2016;7:10521. doi: 10.1038/ncomms10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shankman L.S., Gomez D., Cherepanova O.A. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015;21:628–637. doi: 10.1038/nm.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Parker-Katiraee L., Carson A.R., Yamada T. Identification of the Imprinted KLF14 Transcription Factor Undergoing Human-Specific Accelerated Evolution. PLoS Genetics. 2005 doi: 10.1371/journal.pgen.0030065. preprint:e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen X., Li S., Yang Y. Genome-wide association study validation identifies novel loci for atherosclerotic cardiovascular disease. J Thromb Haemost. 2012;10:1508–1514. doi: 10.1111/j.1538-7836.2012.04815.x. [DOI] [PubMed] [Google Scholar]
- 168.Teslovich T.M., Musunuru K., Smith A.V. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kong A., Steinthorsdottir V., Masson G. Parental origin of sequence variants associated with complex diseases. Nature. 2009;462:868–874. doi: 10.1038/nature08625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Guo Y., Fan Y., Zhang J. Perhexiline activates KLF14 and reduces atherosclerosis by modulating ApoA-I production. J Clin Invest. 2015;125:11. doi: 10.1172/JCI79048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Nagare T., Sakaue H., Matsumoto M. Overexpression of KLF15 transcription factor in adipocytes of mice results in down-regulation of SCD1 protein expression in adipocytes and consequent enhancement of glucose-induced insulin secretion. J Biol Chem. 2011;286:37458–37469. doi: 10.1074/jbc.M111.242651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang C., Ha X., Li W. Correlation of A2bAR and KLF4/KLF15 with obesity dyslipidemia induced inflammation in Uygur population. Mediators Inflamm. 2016;2016:7015620. doi: 10.1155/2016/7015620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Enomoto T., Ohashi K., Shibata R. Adipolin/C1qdc2/CTRP12 protein functions as an adipokine that improves glucose metabolism. J Biol Chem. 2011;286:34552–34558. doi: 10.1074/jbc.M111.277319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wei Z., Peterson J.M., Lei X. C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J Biol Chem. 2012;287:10301–10315. doi: 10.1074/jbc.M111.303651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Knights A.J., Yik J.J., Mat Jusoh H. Kruppel-like factor 3 (KLF3/BKLF) is required for widespread repression of the inflammatory modulator galectin-3 (Lgals3) J Biol Chem. 2016;291:16048–16058. doi: 10.1074/jbc.M116.715748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Pang J., Rhodes D.H., Pini M. Increased adiposity, dysregulated glucose metabolism and systemic inflammation in Galectin-3 KO mice. PLoS One. 2013;8 doi: 10.1371/journal.pone.0057915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Small K.S., Hedman A.K., Grundberg E. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nat Genet. 2011;43:561–564. doi: 10.1038/ng.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Fokeng M.G., Atogho-Tiedeu B., Sobngwi E., Mbanya J., Fon Mbacham W. The Krüppel-like factor 14 (KLF14), master gene of multiple metabolic phenotypes: Putative TransRegulator Network. Transl Biomed. 2016;7 [Google Scholar]
- 179.Nagare T., Sakaue H., Takashima M. The Krüppel-like factor KLF15 inhibits transcription of the adrenomedullin gene in adipocytes. Biochem Biophys Res Commun. 2009;379:98–103. doi: 10.1016/j.bbrc.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 180.Lee H.J., Kang Y.M., Moon C.S. KLF4 positively regulates human ghrelin expression. Biochem J. 2009;420:403–411. doi: 10.1042/BJ20081850. [DOI] [PubMed] [Google Scholar]
- 181.Chen C.Y., Asakawa A., Fujimiya M., Lee S.D., Inui A. Ghrelin gene products and the regulation of food intake and gut motility. Pharmacol Rev. 2009;61:430–481. doi: 10.1124/pr.109.001958. [DOI] [PubMed] [Google Scholar]
- 182.Zobel D.P., Andreasen C.H., Burgdorf K.S. Variation in the gene encoding Kruppel-like factor 7 influences body fat: studies of 14 818 Danes. Eur J Endocrinol. 2009;160:603–609. doi: 10.1530/EJE-08-0688. [DOI] [PubMed] [Google Scholar]
- 183.Wang C., Ha X., Li W. Correlation of TLR4 and KLF7 in inflammation induced by obesity. Inflammation. 2017;40:42–51. doi: 10.1007/s10753-016-0450-z. [DOI] [PubMed] [Google Scholar]
- 184.Okada Y., Kubo M., Ohmiya H. Common variants at CDKAL1 and KLF9 are associated with body mass index in east Asian populations. Nat Genet. 2012;44:302–306. doi: 10.1038/ng.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen J.L., Lu X.J., Zou K.L., Ye K. Kruppel-like factor 2 promotes liver steatosis through upregulation of CD36. J Lipid Res. 2014;55:32–40. doi: 10.1194/jlr.M039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kumadaki S., Karasawa T., Matsuzaka T. Inhibition of ubiquitin ligase F-box and WD repeat domain-containing 7alpha (Fbw7alpha) causes hepatosteatosis through Kruppellike factor 5 (KLF5)/peroxisome proliferator-activated receptor gamma2 (PPARgamma2) pathway but not SREBP-1c protein in mice. J Biol Chem. 2011;286:40835–40846. doi: 10.1074/jbc.M111.235283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bechmann L.P., Vetter D., Ishida J. Post-transcriptional activation of PPAR alpha by KLF6 in hepatic steatosis. J Hepatol. 2013;58:1000–1006. doi: 10.1016/j.jhep.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Escalona-Nandez I., Guerrero-Escalera D., Estanes-Hernandez A., Ortiz-Ortega V., Tovar A.R., Perez-Monter C. The activation of peroxisome proliferator-activated receptor gamma is regulated by Kruppel-like transcription factors 6 & 9 under steatotic conditions. Biochem Biophys Res Commun. 2015;458:751–756. doi: 10.1016/j.bbrc.2015.01.145. [DOI] [PubMed] [Google Scholar]
- 189.Kim Y., Ratziu V., Choi S.G. Transcriptional activation of transforming growth factor beta1 and its receptors by the Kruppel-like factor Zf9/core promoter-binding protein and Sp1. Potential mechanisms for autocrine fibrogenesis in response to injury. J Biol Chem. 1998;273:33750–33758. doi: 10.1074/jbc.273.50.33750. [DOI] [PubMed] [Google Scholar]
- 190.Miele L., Beale G., Patman G. The Kruppel-like factor 6 genotype is associated with fibrosis in nonalcoholic fatty liver disease. Gastroenterology. 2008;135:282–291.e1. doi: 10.1053/j.gastro.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Starkel P., Sempoux C., Leclercq I. Oxidative stress, KLF6 and transforming growth factor-beta up-regulation differentiate non-alcoholic steatohepatitis progressing to fibrosis from uncomplicated steatosis in rats. J Hepatol. 2003;39:538–546. doi: 10.1016/s0168-8278(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 192.Inoue M., Ohtake T., Motomura W. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun. 2005;336:215–222. doi: 10.1016/j.bbrc.2005.08.070. [DOI] [PubMed] [Google Scholar]
- 193.Gavrilova O., Haluzik M., Matsusue K. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- 194.Abumrad N.A., Goldberg I.J. CD36 actions in the heart: lipids, calcium, inflammation, repair and more? Biochim Biophys Acta. 2016;1860:1442–1449. doi: 10.1016/j.bbalip.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Ishii S., Iizuka K., Miller B.C., Uyeda K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc Natl Acad Sci U S A. 2004;101:15597–15602. doi: 10.1073/pnas.0405238101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Jung D.Y., Chalasani U., Pan N. KLF15 is a molecular link between endoplasmic reticulum stress and insulin resistance. PLoS One. 2013;8 doi: 10.1371/journal.pone.0077851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Takashima M., Ogawa W., Hayashi K. Role of KLF15 in regulation of hepatic gluconeogenesis and metformin action. Diabetes. 2010;59:1608–1615. doi: 10.2337/db09-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Woo S.L., Xu H., Li H. Metformin ameliorates hepatic steatosis and inflammation without altering adipose phenotype in diet-induced obesity. PLoS One. 2014;9 doi: 10.1371/journal.pone.0091111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Gracia-Sancho J., Russo L., Garcia-Caldero H., Garcia-Pagan J.C., Garcia-Cardena G., Bosch J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut. 2011;60:517–524. doi: 10.1136/gut.2010.220913. [DOI] [PubMed] [Google Scholar]
- 200.Habeos I.G., Ziros P.G., Chartoumpekis D., Psyrogiannis A., Kyriazopoulou V., Papavassiliou A.G. Simvastatin activates Keap1/Nrf2 signaling in rat liver. J Mol Med (Berl) 2008;86:1279–1285. doi: 10.1007/s00109-008-0393-4. [DOI] [PubMed] [Google Scholar]
- 201.Marrone G., Maeso-Diaz R., Garcia-Cardena G. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: behind the molecular mechanisms of statins. Gut. 2015;64:1434–1443. doi: 10.1136/gutjnl-2014-308338. [DOI] [PubMed] [Google Scholar]
- 202.Kim J.K., Lee K.S., Chang H.Y., Lee W.K., Lee J.I. Progression of diet induced nonalcoholic steatohepatitis is accompanied by increased expression of Kruppel-like-factor 10 in mice. J Translational Med. 2014;12:186–197. doi: 10.1186/1479-5876-12-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Matheus A.S., Tannus L.R., Cobas R.A., Palma C.C., Negrato C.A., Gomes M.B. Impact of diabetes on cardiovascular disease: an update. Int J Hypertens. 2013;2013:653789. doi: 10.1155/2013/653789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lomberk G., Grzenda A., Mathison A. Krüppel-like factor 11 regulates the expression of metabolic genes via an evolutionarily conserved protein interaction domain functionally disrupted in maturity onset diabetes of the young. J Biol Chem. 2013;288:17745–17758. doi: 10.1074/jbc.M112.434670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Cusi K., Consoli A., DeFronzo R.A. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1996;81:4059–4067. doi: 10.1210/jcem.81.11.8923861. [DOI] [PubMed] [Google Scholar]
- 206.Balas B., Baig M.R., Watson C. The dipeptidyl peptidase IV inhibitor vildagliptin suppresses endogenous glucose production and enhances islet function after single-dose administration in type 2 diabetic patients. J Clin Endocrinol Metab. 2007;92:1249–1255. doi: 10.1210/jc.2006-1882. [DOI] [PubMed] [Google Scholar]
- 207.DeFronzo R.A., Ferrannini E., Groop L. Type 2 diabetes mellitus. Nat Rev Dis Primers. 2015;1:15019. doi: 10.1038/nrdp.2015.19. [DOI] [PubMed] [Google Scholar]
- 208.Wang J., Badeanlou L., Bielawski J., Ciaraldi T.P., Samad F. Sphingosine kinase 1 regulates adipose proinflammatory responses and insulin resistance. Am J Physiol Endocrinol Metab. 2014;306:756–768. doi: 10.1152/ajpendo.00549.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.de Assuncao T.M., Lomberk G., Cao S. New role for Krüppel-like factor 14 as a transcriptional activator involved in the generation of signaling lipids. J Biol Chem. 2014;289:15798–15809. doi: 10.1074/jbc.M113.544346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sampath H., Ntambi J.M. The role of stearoyl-CoA desaturase in obesity, insulin resistance, and inflammation. Ann N Y Acad Sci. 2011;1243:47–53. doi: 10.1111/j.1749-6632.2011.06303.x. [DOI] [PubMed] [Google Scholar]
- 211.Buchanan J., Mazumder P.K., Hu P. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349. doi: 10.1210/en.2005-0938. [DOI] [PubMed] [Google Scholar]
- 212.Bugger H., Chen D., Riehle C. Tissue-specific remodeling of the mitochondrial proteome in type 1 diabetic akita mice. Diabetes. 2009;58:1986–1997. doi: 10.2337/db09-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Finck B.N., Lehman J.J., Leone T.C. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. doi: 10.1172/JCI14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Webster A.C., Nagler E.V., Morton R.L., Masson P. Chronic kidney disease. Lancet. 2017;389:1238–1252. doi: 10.1016/S0140-6736(16)32064-5. [DOI] [PubMed] [Google Scholar]
- 215.Zhong F., Chen H., Wei C. Reduced Kruppel-like factor 2 expression may aggravate the endothelial injury of diabetic nephropathy. Kidney Int. 2015;87:382–395. doi: 10.1038/ki.2014.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Hayashi K., Sasamura H., Nakamura M. KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J Clin Invest. 2014;124:2523–2537. doi: 10.1172/JCI69557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lee J.Y., Skon C.N., Lee Y.J. The transcription factor KLF2 restrains CD4(+) T follicular helper cell differentiation. Immunity. 2015;42:252–264. doi: 10.1016/j.immuni.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kuo C.T., Veselits M.L., Barton K.P., Lu M.M., Clendenin C., Leiden J.M. The LKLF transcription factor is required for normal tunica media formation and blood vessel stabilization during murine embryogenesis. Genes Dev. 1997;11:2996–3006. doi: 10.1101/gad.11.22.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kuo C.T., Veselits M.L., Leiden J.M. LKLF: a transcriptional regulator of single-positive T cell quiescence and survival. Science. 1997;277:1986–1990. doi: 10.1126/science.277.5334.1986. [DOI] [PubMed] [Google Scholar]
- 220.Winkelmann R., Sandrock L., Porstner M. B cell homeostasis and plasma cell homing controlled by Kruppel-like factor 2. Proc Natl Acad Sci U S A. 2011;108:710–715. doi: 10.1073/pnas.1012858108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Hart G.T., Wang X., Hogquist K.A., Jameson S.C. Kruppel-like factor 2 (KLF2) regulates B cell reactivity, subset differentiation, and trafficking molecule expression. Proc Natl Acad Sci U S A. 2011;108:716–721. doi: 10.1073/pnas.1013168108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lee J.S., Yu Q., Shin J.T. Klf2 is an essential regulator of vascular hemodynamic forces in vivo. Dev Cell. 2006;11:845–857. doi: 10.1016/j.devcel.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 223.Zhou Z., Tang A.T., Wong W.Y. Cerebral cavernous malformations arise from endothelial gain of MEKK3-KLF2/4 signalling. Nature. 2016;532:122–126. doi: 10.1038/nature17178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Sebzda E., Zou Z., Lee J.S., Wang T., Kahn M.L. Transcription factor KLF2 regulates the migration of naive T cells by restricting chemokine receptor expression patterns. Nat Immunol. 2008;9:292–300. doi: 10.1038/ni1565. [DOI] [PubMed] [Google Scholar]
- 225.Weinreich M.A., Takada K., Skon C., Reiner S.L., Jameson S.C., Hogquist K.A. KLF2 transcription-factor deficiency in T cells results in unrestrained cytokine production and upregulation of bystander chemokine receptors. Immunity. 2009;31:122–130. doi: 10.1016/j.immuni.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Carlson C.M., Endrizzi B.T., Wu J. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature. 2006;442:299–302. doi: 10.1038/nature04882. [DOI] [PubMed] [Google Scholar]
- 227.Kelsey L., Flenniken A.M., Qu D. ENU-induced mutation in the DNA-binding domain of KLF3 reveals important roles for KLF3 in cardiovascular development and function in mice. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Jaubert J., Cheng J., Segre J.A. Ectopic expression of Kruppel like factor 4 (Klf4) accelerates formation of the epidermal permeability barrier. Development. 2003;130:2767–2777. doi: 10.1242/dev.00477. [DOI] [PubMed] [Google Scholar]
- 229.Segre J.A., Bauer C., Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat Genet. 1999;22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 230.Shindo T., Manabe I., Fukushima Y. Kruppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat Med. 2002;8:856–863. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- 231.Wan H., Luo F., Wert S.E. Kruppel-like factor 5 is required for perinatal lung morphogenesis and function. Development. 2008;135:2563–2572. doi: 10.1242/dev.021964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.McConnell B.B., Kim S.S., Yu K. Kruppel-like factor 5 is important for maintenance of crypt architecture and barrier function in mouse intestine. Gastroenterology. 2011;141:1302–1313. doi: 10.1053/j.gastro.2011.06.086. 1313.e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shahrin N.H., Diakiw S., Dent L.A., Brown A.L., D'Andrea R.J. Conditional knockout mice demonstrate function of Klf5 as a myeloid transcription factor. Blood. 2016;128:55–59. [Google Scholar]
- 234.Matsumoto N., Kubo A., Liu H. Developmental regulation of yolk sac hematopoiesis by Kruppel-like factor 6. Blood. 2006;107:1357–1365. doi: 10.1182/blood-2005-05-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Laub F., Lei L., Sumiyoshi H. Transcription factor KLF7 is important for neuronal morphogenesis in selected regions of the nervous system. Mol Cell Biol. 2005;25:5699711. doi: 10.1128/MCB.25.13.5699-5711.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Blackmore M.G., Wang Z., Lerch J.K. Kruppel-like factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proc Natl Acad Sci U S A. 2012;109:7517–7522. doi: 10.1073/pnas.1120684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Morita M., Kobayashi A., Yamashita T. Functional analysis of basic transcription element binding protein by gene targeting technology. Mol Cell Biol. 2003;23:2489–2500. doi: 10.1128/MCB.23.7.2489-2500.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]