

Abstract
The nitric oxide (NO) donor sodium nitroprusside (SNP) is known to reduce aqueous humor (AH) secretion in the isolated porcine eye. Previously, SNP was found to inhibit Na,K-ATPase activity in nonpigmented ciliary epithelium (NPE), AH-secreting cells, through a cGMP/protein kinase G (PKG)-mediated pathway. Here we show Src family kinase (SFK) activation in the Na,K-ATPase activity response to SNP. Ouabain-sensitive 86Rb uptake was reduced by >35% in cultured NPE cells exposed to SNP (100 μM) or exogenously added cGMP (8-Br-cGMP) (100 μM) and the SFK inhibitor PP2 (10 μM) prevented the response. Ouabain-sensitive ATP hydrolysis was reduced by ~40% in samples detected in material obtained from SNP- and 8-Br-cGMP-treated cells following homogenization, pointing to an intrinsic change of Na,K-ATPase activity. Tyrosine-10 phosphorylation of Na,K-ATPase α1 subunit was detected in SNP and L-arginine-treated cells and the response prevented by PP2. SNP elicited an increase in cell cGMP. Cells exposed to 8-Br-cGMP displayed SFK activation (phosphorylation) and inhibition of both ouabain-sensitive 86Rb uptake and Na,K-ATPase activity that was prevented by PP2. SFK activation, which also occurred in SNP-treated cells, was suppressed by inhibitors of soluble guanylate cyclase (ODQ; 10 μM) and PKG (KT5823; 1 μM). SNP and 8-Br-cGMP also increased phosphorylation of ERK1/2 and p38 MAPK and the response prevented by PP2. However, U0126 did not prevent SNP or 8-Br-cGMP-induced inhibition of Na,K-ATPase activity. Taken together, the results suggest that NO activates guanylate cyclase to cause a rise in cGMP and subsequent PKG-dependent SFK activation. Inhibition of Na,K-ATPase activity depends on SFK activation.
Controlling elevated intraocular pressure (IOP) is currently the only available remedy to prevent or delay vision loss and retinal ganglion cell death in persons with glaucoma. Reduction of aqueous humor (AH) secretion is one of the common strategies used to control IOP. AH is secreted through translocation of solutes and water across the ciliary body epithelium bilayer, the pigmented (PE) and nonpigmented (NPE) epithelium. The two epithelial layers contact each other at their apical surfaces where there are numerous gap junctions. The PE basolateral surface contacts the stroma of the ciliary process and the basolateral surface of the NPE contacts the AH that fills the posterior chamber of the eye. Solutes and water are taken up by the PE from the stromal fluid, pass through the gap junction to NPE and then enter the posterior chamber. Aqueous humor then flows through the pupil to the anterior chamber and exits via the trabecular meshwork located at the anterior chamber angle. Na,K-ATPase is the primary active transporter that establishes the ion gradients which drive AH formation. In the intact eye, Na, K-ATPase inhibition by ouabain reduces AH secretion by ~62% (Shahidullah et al., 2003). Na,K-ATPase is localized to the basolateral surface of both layers but expression is considerably more abundant in the NPE than the PE (Ghosh et al., 1990). Na,K-ATPase on the NPE plays key role in AH secretion.
Nitric oxide (NO) is an important signaling molecule responsible for numerous biological activities. It is widely believed that the biological effects of NO-donors, such as nitrovasodilators, are due to the release of NO which activates soluble guanylate cyclase (sGC), giving rise to an increase of intracellular cGMP (Feelisch and Noack, 1987). Using an arterially perfused in vitro intact eye preparation, we reported previously that sodium azide and sodium nitroprusside (SNP), two vasodilator drugs that act through the generation of NO, both reduce AH secretion and cause a reduction of IOP. This occurs in the bovine (Millar et al., 2001) and porcine eye (Shahidullah et al., 2005). As an ocular hypotensive agent, NO has an added advantage that it has been also shown to be neuroprotective at physiological concentrations (Kojima et al., 1996; Chuman et al., 2000; Mohanakumar et al., 2002; Nakazawa et al., 2002). The ability of NO to reduce AH formation is consistent with reports that NO has an inhibitory effect on fluid transport in other tissues including kidney (Ortiz and Garvin, 2002) and salivary gland (Lomniczi et al., 1998). In the in vitro pig eye model, we confirmed that the inhibitory effect of NO donors on AH secretion could be suppressed by ODQ, a specific inhibitor of soluble guanylate cyclase (sGC), suggesting that the effect involved the generation of cGMP (Shahidullah et al., 2005). Recently we have shown that NO donor SNP causes inhibition of Na,K-ATPase activity in freshly isolated porcine NPE cells (Shahidullah and Delamere, 2006). It is noteworthy that NO and NO donors have been shown to inhibit Na,K-ATPase in other secretory tissues including choroid plexus (Ellis et al., 2000, 2001), trachea (de Oliveira Elias et al., 1999) and kidney tubule (Guzman et al., 1995; Seven et al., 2005). Early reports indicated convincingly that the inhibitory effect of NO on Na,K-ATPase activity involves a cGMP- and protein kinase G (PKG)-mediated signaling mechanism (Shahidullah and Delamere, 2006). More recently, studies in the lens have shown that agonist-elicited changes in Na,K-ATPase activity are the result of tyrosine kinase activation (Tamiya et al., 2007; Mandal et al., 2011). Here we provide evidence that Src family tyrosine kinase (SFK) activation, elicited by cGMP/PKG signaling, is a required step in the mechanism responsible for the reduction in Na,K-ATPase activity caused by NO generation in porcine NPE cells.
Materials and Methods
Cells and reagents
Fresh porcine eyes, purchased from the University of Arizona Meat Science laboratory, were delivered on ice. The use of porcine tissue was approved by the University of Arizona Institutional Animal Care and Use Committee and conformed to the ARVO Resolution for the Use of Animals in Ophthalmic and Vision Research. Porcine NPE was established in primary culture as described earlier (Shahidullah et al., 2007) and grown in HEPES-buffered DMEM containing 10% fetal bovine serum. Prior to use, the cell monolayers were serum-starved for 2 h and then the medium was replaced with Krebs solution that contained (in mM) 119 NaCl, 4.7 KCl, 1.2 KH2PO4, 25 NaHCO3, 2.5 CaCl2, 1 MgCl2, and 5.5 glucose, equilibrated with 5% CO2 and adjusted to pH 7.4. Experiments were carried out in Krebs solution at 37°C in a humidified incubator, saturated with 95% air and 5% CO2.
HEPES-buffered DMEM, fetal bovine serum and newborn calf serum were purchased from Invitrogen (Carlsbad, CA). ATP, ouabain (octahydrate), sodium nitroprusside (SNP), gentamycin, Dowex 50 W × 4–400 mesh cation exchange resin (hydrogen form), 1H-[1,2,4]Oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio] butadiene (U0126), dimethyl Sulfoxide (DMSO), 3-Isobutyl-1-methylxanthine (IBMX) and all other chemicals to prepare the Krebs’ solution were purchased from Sigma (St. Louis, MO). The cGMP [125I] RIA Kit and 86Rb were purchased from Perkin Elmer (Waltham, MA). Alamethicin, 8-bromo-cGMP (sodium salt), 4-amino-3-(4-chlorophenyl)-1-(t-butyl)-1H-pyrazolo[3,4-d] pyrimidine, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) were purchased from Tocris Biosciences (Minneapolis, MN). Mouse monoclonal anti-Na,K-ATPase α1 antibody was purchased from Sigma. Rabbit polyclonal anti p44/42 MAP kinase antibody, mouse monoclonal anti phospho-p44/42 MAP kinase (Thr-202/Tyr-204) antibody, rabbit polyclonal anti phospho-Na,K-ATPase α1 (Ser 16) antibody, rabbit polyclonal anti phospho-Src-family (Tyr 416) antibody, rabbit polyclonal anti phospho-Na,K-ATPase α1 (Tyr 10) antibody, rabbit polyclonal anti-β-actin antibody, rabbit polyclonal anti-phospho-p38 MAPK (Tyr 180/Tyr 182) antibody were obtained from Cell Signaling Technology (Danvers, MA). Mouse monoclonal anti-β-actin antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Mouse monoclonal anti-p38 MAPK antibody was purchased from Thermo Scientific (Rockford, IL). Goat anti-rabbit IRDye 680 or goat anti-mouse IRDye 800 conjugated secondary antibodies were purchased from LI-COR Biosciences (Lincoln, NE).
Sample preparation and Na,K-ATPase activity assay
NPE cells were grown to confluence as monolayers on 24 mm polyester permeable culture inserts with a 0.4 μm pore size (Corning). The cells were preincubated with Krebs solution for 1 h then the Krebs solution was changed, exposing both surfaces of the culture insert to test compounds. Following treatment, the membrane with the cell monolayer was removed from each culture insert using a custom-made stainless steel cutter. Cells from two inserts were pooled as one sample for the Na,K-ATPase assay. The two inserts were arranged into a sandwich with the cells facing inside and the membrane sandwich was then cut into small pieces, placed in a 2.0 ml Eppendorf tube, frozen in liquid nitrogen and stored at −80°C until the Na,K-ATPase assay.
The culture insert membranes, which are brittle when frozen, were pulverised using a glass pestle that fits snugly in the 2.0 ml Eppendorf tube. The samples were subjected to two cycles of freezing in liquid nitrogen and pulverization, then 300 μl of ice cold 2×-strength ATPase assay buffer was added. Assay buffer composition was (in mM): L-Histidine, 80; NaCl 200; KCl, 10; MgCl2, 6.0; EGTA, 2.0 (pH 7.4) and a protease inhibitor mixture at manufacturer’s recommended concentration (1 tablet/7 ml of the ATPase buffer, Roche Diagnostics, Indianapolis, IN). The mixture was homogenized at ice-cold temperature for 1 min (4 strokes of 15 sec at 5 sec intervals) using Misonix S3000 sonicator at a 6 W power setting (Misonix, Farmingdale, NY). The homogenate was subjected to centrifugation at 13,000g for 30 min at 4°C to remove the polyester membrane fragments, cell nuclei and larger mitochondria. Protein in the supernatant was measured by bicinchoninic acid (BCA) assay (Smith et al., 1985; Pierce Biotechnology, Rockford, IL), using bovine serum albumin as a standard. The supernatant was used to measure Na,K-ATPase activity.
Na,K-ATPase activity was measured according to our previously published method (Shahidullah et al., 2012). Samples obtained from treated or control cells (80 μl) were placed in duplicate to 75 mm × 12 mm glass assay tubes and an additional 120 μl of 2× assay buffer was added to each tube. To improve access of ions and ATP to membrane vesicles, alamethicin solution in ethanol (5 μl) was added to give a final approximate concentration of 0.1 mg of alamethicin per mg protein (Xie et al., 1989). Half the tubes received ouabain, a highly specific Na,K-ATPase inhibitor (3) (final concentration 300 μM) and the remaining tubes received an equivalent volume (5 μl) of distilled water. An additional 150 μl of distilled water was added to each tube. The tubes were pre-incubated at 37°C for 5 min then ATP stock solution (40 μl) was added to each tube (final ATP concentration 2 mM) bringing the total assay mixture volume to 400 μl and the concentration of the 2 × -strength Na,K-ATPase buffer to single-strength. After 30 min of incubation at 37°C in the dark, the ATP hydrolysis reaction was stopped by the addition of 150 μl of 15% ice-cold trichloroacetic acid (TCA) and placing the tubes on ice for 20 min with occasional shaking.
ATP hydrolysis was determined as the amount of inorganic phosphate released in each reaction tube. To detect inorganic phosphate each tube was placed in a centrifuge at 3,000 rpm (2,680g) for 15 min at 4°C, then 400 μl of the supernatant was removed, placed in pre-marked glass tubes and mixed with 400 μl of 4.0% FeSO4 solution in ammonium molybdate (1.25 g of ammonium molybdate in 100 ml of 2.5 N sulfuric acid). Standard solutions containing NaH2PO4 equivalent to 0, 10, 62.5, 125, 250, and 500 nmol of PO4, were treated similarly. After 5 min at room temperature the tubes with the samples (not the standards) were placed in a centrifuge and spun at 3,000 rpm (2,680g) for 10 min to pellet additional precipitates. A 250 μl aliquot of each standard or sample was then transferred to each well of a 96-well plate and the absorbance was measured at 750 nm in a Perkin Elmer plate reader (Victor V3, Perkin Elmer, CT). Na,K-ATPase activity is calculated as the difference between ATP hydrolysis in the presence and in the absence of ouabain. Values are presented as nmoles ATP hydrolyzed per milligram protein per 30 min. Because Na,K-ATPase activity was variable between batches of cells, data for different experiments were not pooled.
Measurement of 86Rb uptake
NPE monolayers grown to confluence on 24 mm permeable culture inserts were preincubated in Krebs’ solution for 1 h at 37°C. Then the Krebs’ solution was changed and the cells were incubated in the presence or absence of test compounds added to both sides of the culture insert for a specified time (10 min in the case of SNP or 8-Br-cGMP) before the addition of 86RbCl (1 μCi/ml) for 5 min. Half the samples received ouabain (500 μM) along with the 86RbCl. Uptake of 86Rb was stopped by removing the culture inserts and washing them three times by quick submersion in ice-cold Krebs solution. The cell monolayer and permeable membrane was cut from each insert and placed in 7 ml of scintillation fluid in a scintillation vial. Radioactivity was measured in a scintillation counter. Results are expressed as cpm/insert and Na,K-ATPase-mediated 86Rb is the difference between uptake in the presence and absence of ouabain.
Cyclic GMP radioimmunoassay (RIA)
NPE monolayers grown to confluence on 35 mm dishes were preincubated with Krebs solution for 1 h then exposed to Krebs solution containing 100 μM SNP and some cells also received IBMX (1 mM). After incubation, the monolayers were washed briefly with ice-cold Krebs’ solution, 0.5 ml of 10% TCA was added and the cells were scraped and collected in 2.0 ml Eppendorf tubes, frozen in liquid nitrogen and stored at −80°C until assay.
Samples were subjected to three cycles of freeze-thaw then sonicated using a Misonix S3000 sonicator (6 W power setting, 4 strokes of 15 sec each with a 5 sec interval). After sonication, ~1500 cpm of 3H-cGMP was added to each tube to determine cGMP recovery efficiency. At a specific activity of 8.0 Ci/mmol and at a 50% counting efficiency, 1,500 cpm represented approximately 0.2 pmol (200 fmol) of cGMP. This was taken into account when calculating cGMP content of a sample. The samples were centrifuged 13,000g for 30 min and each supernatant was transferred to new Eppendorf tubes, designated an SN tube. Each pellet was washed with 200 μl of 10% TCA, briefly sonicated, centrifuged again at 13,000g for 30 min and the supernatant transferred to the SN tube. The supernatant samples were used for cGMP assay and pellet samples were used for protein measurement.
Cyclic GMP was extracted from the supernatant samples using ion exchange chromatography. Cation exchange columns (5 cm) were prepared using washed and pre-swollen (overnight) Dowex 50 × W 4–400 mesh cation exchange resin (hydrogen form) loaded into glass Pasteur pipettes (the narrow ends were plugged with cotton wool). The columns were washed 10 times (1 ml each) with double distilled water and three times (1 ml each) with 0.02 N HCl. Each supernatant sample (~0.7 ml) was applied to a chromatography column which was first washed with 0.5 ml of water and the eluted material discarded. The next four washes (1 ml each) were collected and pooled in glass tubes. The eluted samples were dried under a stream of air at 40°C. Dried samples were reconstituted in 400 μl of cGMP assay buffer (Na-acetate buffer). A 100 μl aliquot of each reconstituted sample was mixed with 7 ml of liquid scintillation fluid in a scintillation vial and counted using a beta counter to calculate recovery percentage. Recovery efficiency was calculated from the total and blank counts. Blank-count tubes contained only the assay buffer and the total-count tubes contained 1,500 cpm of 3H cGMP. The samples obtained from ion exchange chromatography were subjected to radio-immunoassay performed using a cGMP assay kit (Perkin Elmer NEX133001KT) following the acelylated cGMP assay procedure recommended by the manufacturer. Results were expressed as pmoles of cGMP/mg protein.
Western blot analysis
NPE monolayers cultured to confluence on 60 mm dishes were preincubated in control Krebs solution then exposed to Krebs solution containing specified test compounds. After a specified time the incubation medium was removed and the cells were lysed in RIPA buffer containing (in mM) 50 HEPES, 150 NaCl, 1.0 EDTA, 10 sodium pyrophosphate, 2.0 sodium orthovanadate, 10 sodium fluoride, and 1 phenylmethylsulfonyl fluoride (PMSF) with 10% glycerol, 1.0% Triton X-100, 1.0% sodium deoxycholate, and Complete Mini Protease Inhibitor Cocktail tablets (Roche Diagnostics; 1 tab/7 ml). The cell lysate was centrifuged at 14,000g for 30 min, the supernatant collected and protein concentration was measured with a BCA protein assay kit (Pierce Biotechnology). The supernatant was mixed with Laemmli buffer and the proteins were separated by electrophoresis on a 7.5% SDS-polyacrylamide minigel. Proteins were then transferred by electrophoresis to nitrocellulose membrane which was blocked overnight with Aqua Block blocking buffer (East Coast Bio, ME). The nitrocellulose membranes were incubated overnight at 4°C with the following primary antibodies: Mouse monoclonal anti-Na, K-ATPase α1 (1:450), rabbit polyclonal anti-p44/42 MAP kinase (1:1,000), mouse monoclonal anti phospho-p44/42 MAPK (Thr-202/Tyr-204) (1:2,000), rabbit polyclonal anti phosphor Ser16-Na, K-ATPase α1 (1:1,000), rabbit polyclonal anti-phospho Tyr-416 Src family kinase (1:1,000), rabbit polyclonal anti phospho-Na,K-ATPase α1 (Tyr-10) (1:1,000), rabbit polyclonal anti-β-actin (1:3,000), rabbit polyclonal anti-phospho-p38 MAPK (Tyr-180/Tyr-182) (1:1,000), mouse monoclonal anti-β-actin (1:10,000), mouse monoclonal anti-p38 MAPK (1:1,000). All antibodies were diluted in Aqua Block blocking buffer. After three washes in 30 mM Tris, 150 mM NaCl, 0.5% (v/v) Tween-20 (TTBS) at pH 7.4, each nitrocellulose membrane was incubated for 1 h at 37°C with an appropriate secondary antibody conjugated with IRDye 800 or 680; IRDye goat anti-mouse or anti-rabbit secondary antibody (1:20,000) (Licor Biosciences, Lincoln, NE). Protein bands were visualized and band density quantified by infrared laser scan detection (LI-COR Odyssey). By using secondary antibodies at two different infrared wavelengths, two proteins were quantified simultaneously (e.g., Na,K-ATPase α1 and phospho-Tyr-10-Na,K-ATPase α1) permitting calculation of a band density ratio.
Reactive oxygen species detection
Cells cultured to confluence on a 96-well plate were loaded with cell-permeable fluorogenic probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (100 μM) dissolved in serum-free culture medium for 60 min at 37°C. Then the cells were washed twice with Hank’s balanced salt solution containing calcium and magnesium (HBSS) and each well received 200 μl of DMEM containing SNP, SNAP, L-arginine or H2O2 at a specified concentration. After incubation at 37°C for 10 min the treatment medium was removed, the cells were washed twice with HBSS then lysed and generation of reactive oxygen species (ROS) was assayed using a Green Fluorescence ROS assay kit which measures relative concentration of ROS in the cytoplasm (OxiSelect Intracellular ROS Assay Kit, Cell Biolabs, Inc., Lawrenceville, GA). Fluorescence was determined at an excitation wave-length of 480 nm and an emission wave-length of 530 nm using a Varioskan Flash multimode reader (Thermo Scientific).
Statistical analysis
The results are expressed as the mean ±SE and comparison was made by one way analysis of variance followed by the Bonferroni post hoc multiple comparison test.
Results
Previous studies demonstrated that NO causes Na,K-ATPase inhibition. Although there is convincing evidence that the Na, K-ATPase response to NO donors is linked to PKG activation following an increase of cGMP, Na,K-ATPase responses to other types of stimulus are known to hinge upon Src family tyrosine kinase (SFK) activation. Here we carried out 86Rb uptake studies to examine the functional response of Na,K-ATPase to the NO donor SNP in intact NPE cells treated with the SFK inhibitor PP2. SNP (100 μM) reduced the rate of ouabain-sensitive 86Rb uptake by approximately ~60%. In the presence of 10 μM PP2, the effect of SNP on ouabain-sensitive 86Rb uptake was abolished (Fig. 1). Exposing intact cells to 100 μM 8-Br-cGMP also caused a decrease in the rate of ouabain-sensitive 86Rb uptake and, in similar fashion, PP2 eliminated the response (Fig. 2). Added alone, PP2 did not significantly alter the rate of ouabain-sensitive 86Rb uptake.
Fig. 1.
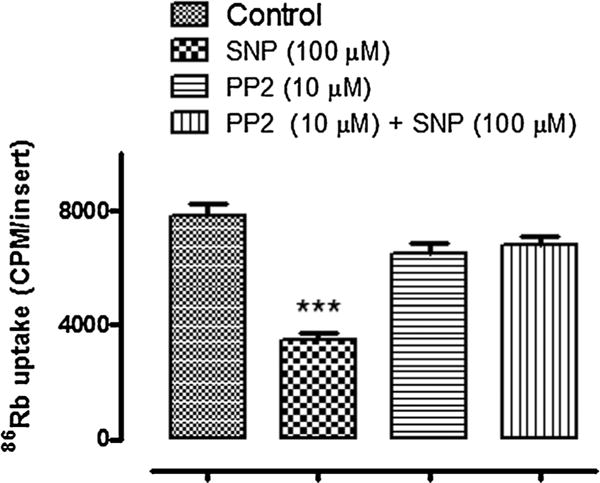
The influence of PP2 on the ouabain-sensitive 86Rb uptake response to SNP. Cells were cultured to confluence on permeable inserts and pre-incubated with PP2 for 40 min before being exposed to SNP (100 μM) in the continued presence of PP2 (10 μM). SNP was introduced for 5 min and then 86RbCl ± ouabain (500 μM) was added for a further 5 min in the continued presence of SNP. The values are the mean ± SEM of results from four cell monolayers. ***Indicates a significant difference from control, P < 0.001.
Fig. 2.
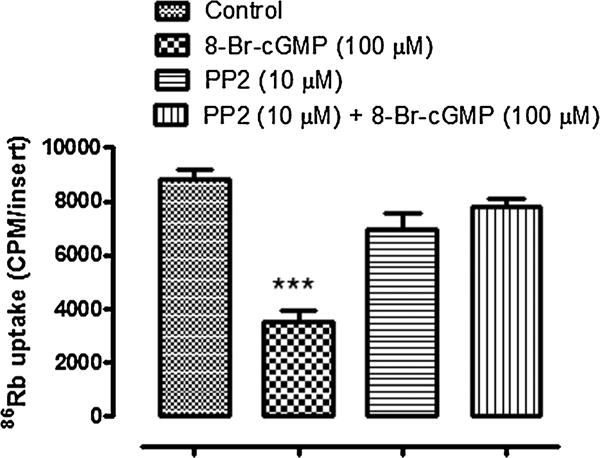
The influence of PP2 on the ouabain-sensitive 86Rb uptake response to 8-Br-cGMP. Cells were cultured to confluence on permeable inserts and pre-incubated with PP2 (10 μM) for 40 min before being exposed to 8-Br-cGMP (100 μM) in the continued presence of PP2. 8-Br-cGMP was introduced for 5 min and then 86RbCl ± ouabain (500 μM) was added for a further 5 min in the continued presence of 8-Br-cGMP. The values are the mean ± SEM of results from four cell monolayers. ***Indicates a significant difference from control, P < 0.001.
In separate experiments, intact cells were exposed to either SNP or 8-Br-cGMP for 10 min before being homogenized then used for measurements of ouabain-sensitive ATP hydrolysis. The rate of ouabain-sensitive ATP hydrolysis was reduced by ~40% in samples obtained from cells that had been exposed to either SNP or 8-Br-cGMP (Fig. 3). In contrast, no significant change of ouabain-sensitive ATP hydrolysis was observed in samples obtained from cells that had been exposed to either SNP or 8-Br-cGMP in the presence of 10 μM PP2 (Fig. 3).
Fig. 3.
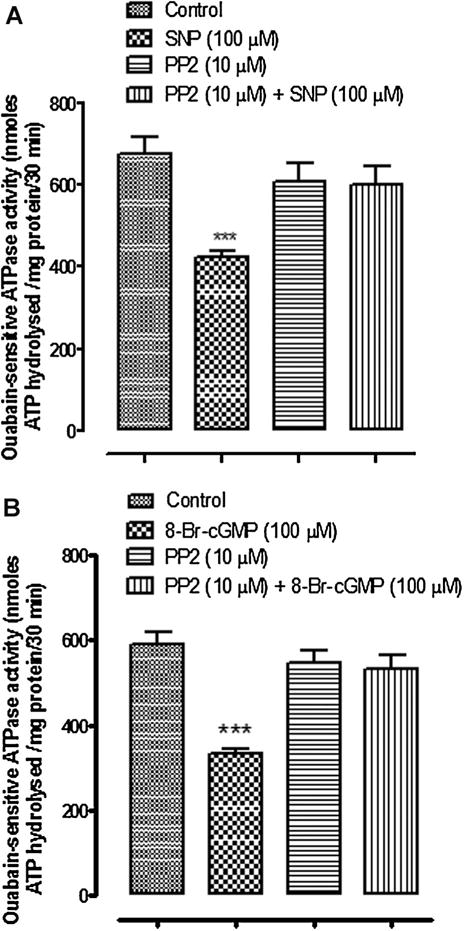
The influence of PP2 on Na,K-ATPase activity in cells exposed to SNP (Part A) or cGMP (Part B). Ouabain-sensitive ATP hydrolysis (Na,K-ATPase activity) was measured in samples obtained by homogenizing cells that had been pre-incubated with PP2 (10 μM) for 40 min before being exposed to either SNP (100 μM) or 8-Br-cGMP (100 μM) for 10 min in the continued presence of PP2. The values are the mean ± SEM of results from four to six samples, each pooled from two cell monolayers. ***Indicates a significant difference from control, P < 0.001.
The detection of reduced ouabain-sensitive ATP hydrolysis in material obtained from SNP-treated cells points to an intrinsic change of Na,K-ATPase activity that is maintained following homogenization, centrifugation and freezing. Western blot analysis of cells exposed to SNP revealed an increase in tyrosine phosphorylation of the Na,K-ATPase α1 catalytic subunit, detectable within 2 min that was maintained for 15 min (Fig. 4A). The response was abolished by PP2 (Fig. 4B). Similarly, phosphorylation of Na,K-ATPase α1 catalytic subunit was observed when cells were exposed to the endogenous substrate for nitric oxide synthase, L-arginine (Fig. 5A). L-Arginine response also was blocked by PP2 (Fig. 5). No Ser-16 phosphorylation of Na,K-ATPase α1 was observed in cells treated with SNP or L-arginine (data not shown).
Fig. 4.
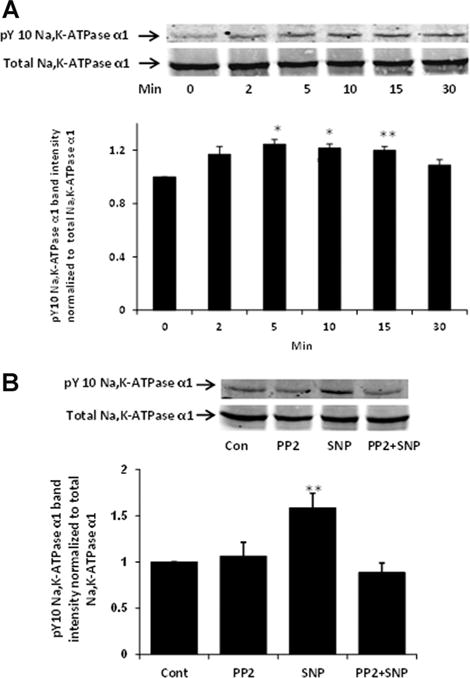
Western blots showing tyrosine phosphorylation of Na,K-ATPase α1 catalytic subunit in cells treated with SNP (100 μM) (Part A). A typical time course is presented alongside a bar graph that shows band density results pooled from three independent experiments (mean ± SEM). Part B shows the influence of PP2 on SNP-induced tyrosine phosphorylation of the Na,K-ATPase α1 subunit. Western blot analysis was performed on samples obtained by homogenizing cells that had been pre-incubated with PP2 (10 μM) for 40 min before being exposed to SNP (100 μM) for 10 min in the continued presence of PP2. A typical Western blot of pY-10-Na,K-ATPase-α1 bands (upper) and total Na,K-ATPase α1 subunit bands (lower) detected simultaneously in the same samples alongside a bar graph (lower) that shows pY-10-Na,K-ATPase-α1 band density normalized to total Na,K-ATPase α1 (mean ± SEM) of three independent samples, are presented. *P < 0.05, **P < 0.01, and ***P < 0.001 indicate significant differences from control.
Fig. 5.
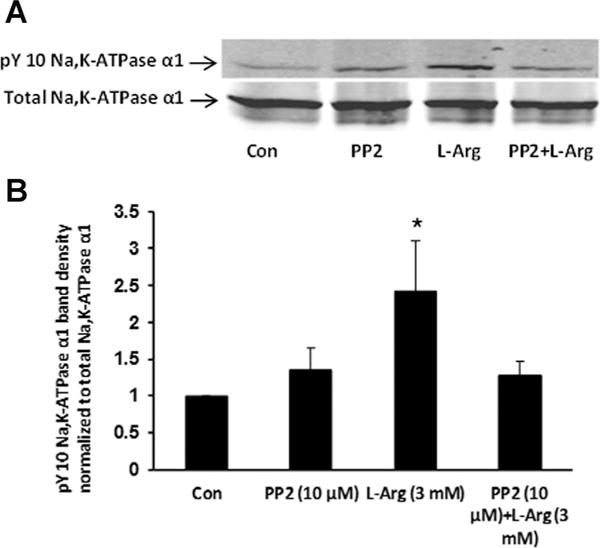
The influence of PP2 on L-arginine-induced tyrosine phosphorylation of the Na,K-ATPase α1 subunit. The samples were obtained from cells, cultured on permeable inserts, that had been pre-incubated with PP2 (10 μM) for 40 min before being exposed to L-arginine (3 mM) for 10 min in the continued presence of PP2. Part A shows a typical Western blot. Part B shows pY-10-Na,K-ATPase-α1 band density normalized to total Na,K-ATPase α1 (mean ± SEM) of three independent experiments. *P < 0.05 indicates a significant difference from control.
Increased SFK phosphorylation at Y-416, a modification that signifies SFK activation, was detectable after 2 min exposure of the intact cells to SNP and lasted up to 15 min (Fig. 6A). Increased SFK phosphorylation also was observed in cells treated with L-arginine (Fig. 6B). In both cases the SFK phosphorylation response was transient and occurred over similar time frame. The increase in SFK phosphorylation was not observed in material obtained from cells that were exposed to SNP in the presence of 10 μM PP2 (Fig. 7). The principal phospho-SFK band appeared at ~60 kDa but faint phospho-SFK bands at lower MW were also observed.
Fig. 6.
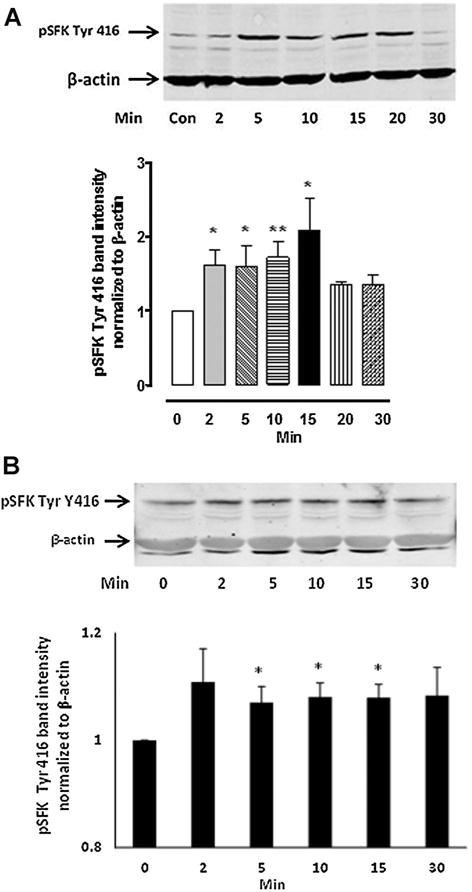
The time course of SFK phosphorylation detected by Western blot analysis in NPE cells treated with either 100 μM SNP (Part A) or 3 mM L-arginine (Part B). In each part, a typical Western blot is shown together with a bar graph (lower) of pSFK Tyr 416 band density normalized to β-actin (mean ± SEM), pooled from three independent experiments. *P < 0.05 and **P < 0.01 indicate significant differences from control.
Fig. 7.
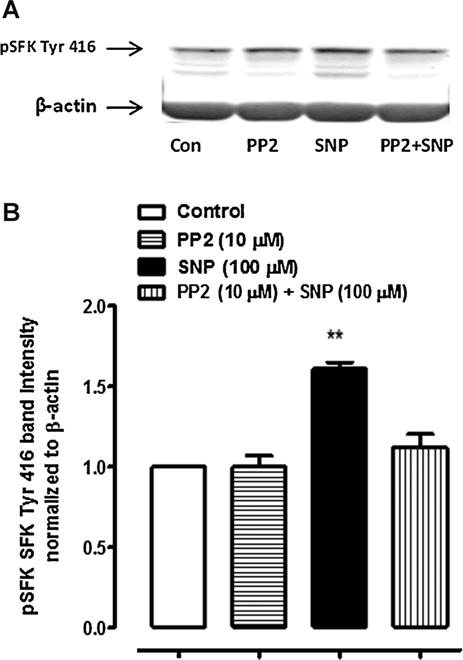
Western blots showing the inhibitory effect of PP2 on SFK phosphorylation in cells treated with 100 μM SNP. The samples were obtained from cells that had been pre-incubated with PP2 (10 μM) for 40 min before being exposed to either SNP (100 μM) for 10 min in the continued presence of PP2. A typical western blot is shown (Part A) together with a bar graph (Part B) of pSFK Tyr 416 band density normalized to β-actin (mean ± SEM), pooled from three independent experiments. **Indicates a significant difference from control, P < 0.01.
Studies were carried out to further examine the possible link between the cGMP and SFK activation. A significant increase of cGMP was observed in NPE cells exposed to SNP (Fig. 8). Elevated cGMP was detectable after 2 min exposure of the intact cells to SNP and reached a maximum at 10 min. The rise in cGMP was similar in the presence and absence of IBMX (1 mM), a phosphodiesterase inhibitor added to suppress cGMP breakdown (data not shown). If the cGMP rise contributes to the SFK response in SNP-treated cells, we reasoned that soluble guanylate cyclase inhibition would suppress the rise of cGMP and PKG activation that should prevent the SFK activation. This was the case. The increase in SFK phosphorylation caused by SNP was suppressed in the presence of the guanylate cyclase inhibitor, ODQ (10 μM; Fig. 9A) or the PKG inhibitor, KT5823 (1 μM; Fig. 9B). Moreover, SFK phosphorylation was found to be significantly increased in samples obtained from cells that had been exposed to 8-Br-cGMP and SFK phosphorylation response was eliminated when cells were exposed to 8-Br-cGMP in the presence of PP2 (Fig. 10).
Fig. 8.
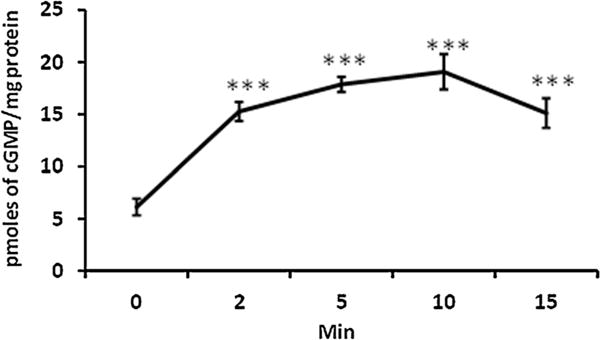
Time-dependent accumulation of cGMP measured in NPE cells exposed to SNP (100 μM) for 2–15 min. The results shown are mean ± SEM of six independent samples for each time point. ***Indicates a significant difference (P < 0.001) from the control (time zero) cGMP value.
Fig. 9.
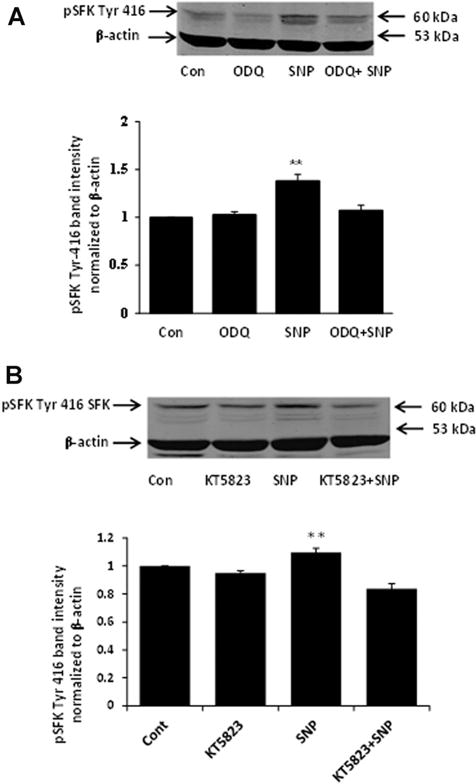
Western blots showing the inhibitory effect of ODQ (Part A) and KT5823 (Part B) on SFK phosphorylation in cells treated with 100 μM SNP. The samples were obtained from cells that had been pre-incubated with ODQ (10 μM) or KT5823 (1 μM) for 20 min before being exposed to SNP for 10 min in the continued presence of either ODQ or KT5823. Each part shows a typical Western blot (upper) together with a bar graph (lower) of pSFK Tyr 416 band density normalized to β-actin (mean ± SEM), pooled from three independent experiments. **Indicates a significant difference from control, P < 0.01.
Fig. 10.
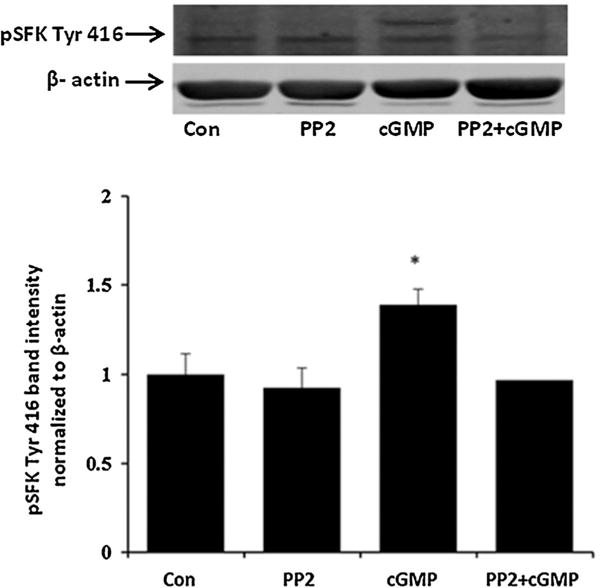
Western blots showing the inhibitory effect of PP2 on SFK phosphorylation in cells treated with 10 μM 8-pCPT-cGMP (cGMP). The samples were obtained from cells that had been pre-incubated with PP2 (10 μM) for 40 min before being exposed to 8-pCPT-cGMP (10 μM) for 10 min in the continued presence of PP2. A typical Western blot is presented (upper) alongside a bar graph (lower) that shows band density results pooled from three independent experiments (mean ± SEM). *Indicates a significant difference from control (P < 0.05).
The ability of PP2 to prevent the inhibition of Na,K-ATPase activity and ouabain sensitive 86Rb uptake in SNP-treated cells is consistent with a response that involves cGMP/PKG-dependent activation of SFK and SFK-dependent inhibition of Na,K-ATPase activity. However, it is noteworthy that SNP also was found to cause a marked transient increase in ERK1/2 and p38 MAPK phosphorylation (Fig. 11). Transient ERK1/2 and p38 MAPK phosphorylation also was observed when cells were exposed to SNAP or L-arginine (data not shown). SNP- induced ERK1/2 and p38 activation was prevented in cells pretreated with the SFK inhibitor PP2 (Fig. 12). Because ERK1/2 activation has been reported elsewhere to lead to reduction of Na,K-ATPase activity, cells were exposed to SNP in the presence of UO126 (10 μM), a potent ERK1/2 inhibitor. UO126 did not prevent the Na,K-ATPase response to SNP or 8-Br-cGMP (Fig. 13).
Fig. 11.
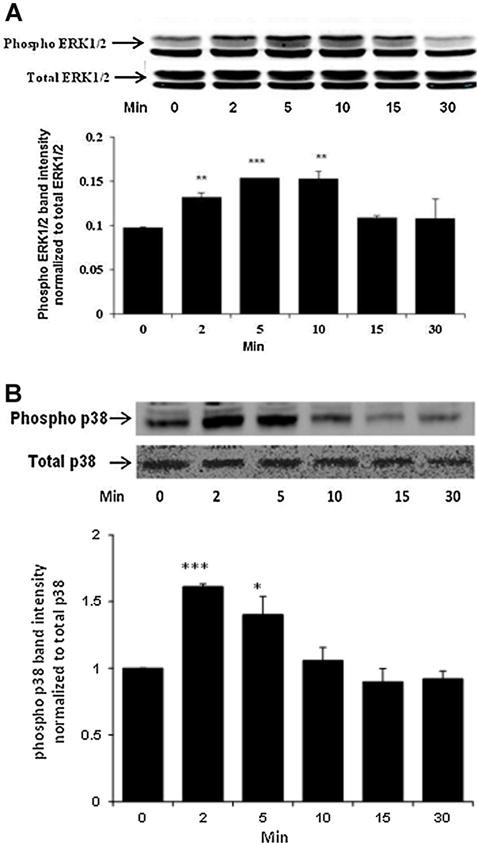
Western blot showing time-dependent phosphorylation of ERK1/2 (Part A) and p38 MAPK (Part B) in NPE cells treated with 100 μM SNP for 2–30 min. In each part, a typical Western blot for the time-dependent response (upper) together with a bar graph (lower) showing band density results (mean ± SEM), pooled from three independent experiments, are presented. *P < 0.05, **P < 0.01, and ***P < 0.001 indicate significant differences from control.
Fig. 12.
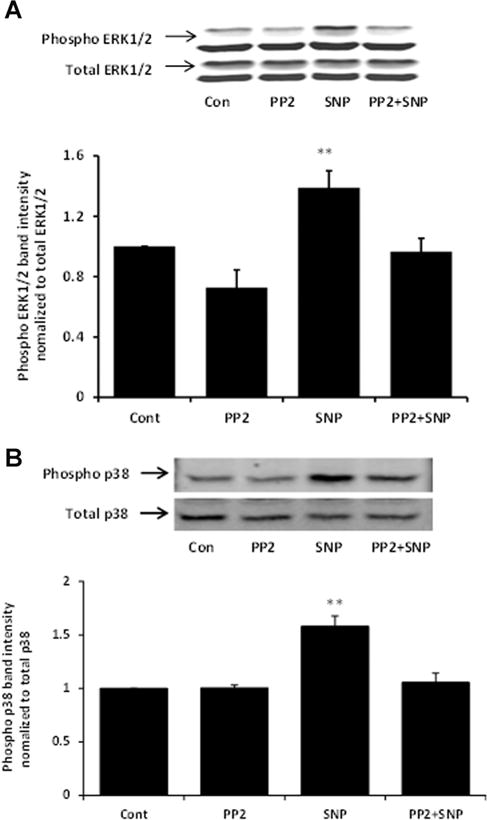
Western blots showing the inhibitory effect of PP2 on ERK1/2 (Part A) and p38 (Part B) phosphorylation in cells exposed to 100 μM SNP. The samples were obtained from cells that had been pre-incubated with PP2 (10 μM) for 40 min before being exposed to SNP for 10 min in the continued presence of PP2. In each part, a typical Western blot (upper) together with a bar graph (lower) showing the band density results (mean ±SEM), pooled from three independent experiments, are presented. **Indicates a significant difference from control, P < 0.01.
Fig. 13.
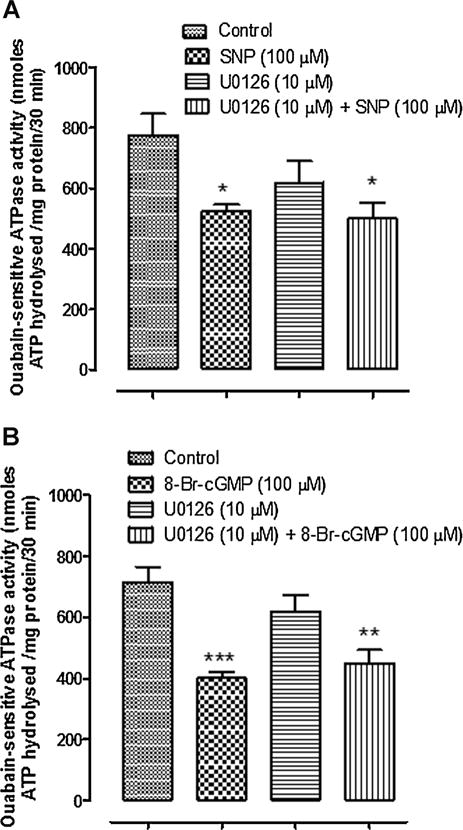
The effect of U0126 on Na,K-ATPase activity in cells exposed to SNP (Part A) or 8-Br-cGMP (Part B). Ouabain-sensitive ATP hydrolysis (Na,K-ATPase activity) was measured in samples obtained by homogenizing cells that had been exposed to SNP (100 μM) for 10 min in the presence or absence of the ERK1/2 inhibitor U0126 (10 μM). The values are the mean ± SEM of results from four to six samples, each pooled from two cell monolayers. *P < 0.05, **P < 0.01 and ***P < 0.001 indicate significant differences from control.
Since NO has the potential to cause generation of ROS we examined intracellular ROS in cells exposed to SNP, SNAP, or L-arginine. Increased ROS was not detectable. In contrast a significant ROS increase was observed in cells exposed to H2O2, a maneuver used here as a positive control (Fig. 14).
Fig. 14.
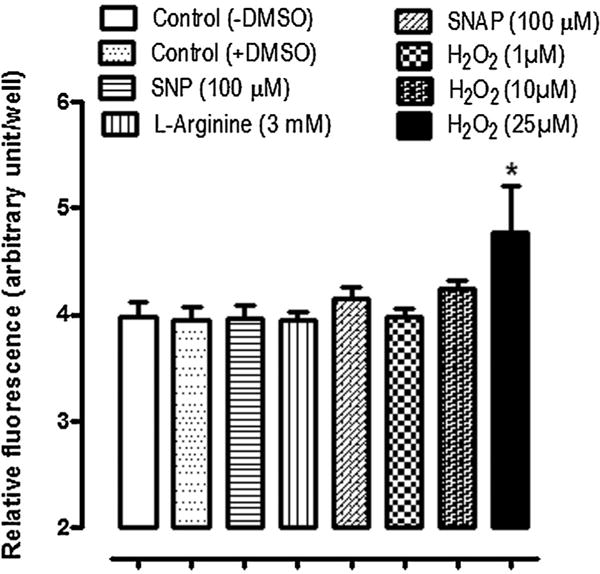
Estimation of cytoplasmic accumulation of reactive oxygen species (shown as relative fluorescence unit) in NPE cells treated with 100 μM SNP, 3 mM L-arginine, 100 μM SNAP or 1, 10, and 25 μM H2O2 for 10 min. One group of control cells received no vehicle and another group received DMSO. The values are the mean ± SEM of results from 12 independent samples for each concentration of the drug used. *P < 0.05 indicates a significant difference from control.
Discussion
NO donors SNP, SNAP and the NO synthase substrate L-arginine are used widely to study NO-activated pathways and previous experiments on NPE confirmed NO production on the basis of nitrite detection in the bathing medium. The results of the present study show NO causes SFK activation as judged by the ability of SNP and L-arginine to cause SFK activation by phosphorylating at residue Tyr 416 (Thomas and Brugge, 1997; Roskoski, 2004). Importantly, the SFK inhibitor PP2, which suppressed SFK phosphorylation, was found to prevent the inhibition of ouabain-sensitive 86Rb uptake caused by SNP. The findings are consistent with SFK activation being a required step in the chain of events that leads from NO generation to Na,K-ATPase inhibition.
Upon NO generation, downstream physiological events commonly are the result of a signaling pathway that involves cGMP and PKG (Feelisch and Noack, 1987). This is so for Na, K-ATPase inhibition that occurs when choroid plexus and NPE are exposed to SNP and L-arginine (Ellis et al., 2000; Shahidullah and Delamere, 2006). Consistent with this notion, direct measurement of cGMP in SNP-treated cells revealed an increase detectable at 2 min and sustained for 15 min. Moreover, exposure of NPE cells to the cell-permeable cGMP analog 8-Br-cGMP was found to inhibit ouabain-sensitive 86Rb uptake by more than 50%. In contrast, 86Rb uptake inhibition did not occur when cells were treated with 8-Br-cGMP in the presence of PP2, pointing to the likelihood that cGMP elicits Na,K-ATPase inhibition by a mechanism that involves SFK activation.
It is noteworthy that the inhibitory effect of SNP on Na,K-ATPase remained detectable as a reduction in ouabain-sensitive ATP hydrolysis measured following cell homogenization and centrifugation. This suggests NO causes an intrinsic modification of Na,K-ATPase function. When phosphorylation of the Na,K-ATPase α1 subunit in SNP-treated cells was examined by a Western blot approach, a measurable increase in tyrosine-10 but no change of Ser-16 phosphorylation was observed. It is notable that SNP-induced SFK phosphorylation and SFK-induced Na,K-ATPase α1 phosphorylation followed similar time course. L-Arginine also caused an SFK-activation-dependent increase in Na,K-ATPase α1 phosphorylation at Tyr 10. Although tyrosine-10 phosphorylation of Na,K-ATPase α1 subunit in SNP-treated NPE appears consistent with activation of tyrosine kinases such as SFKs, earlier studies in insulin-treated renal proximal tubule cells associated tyrosine-10 phosphorylation with an increase of Na,K-ATPase activity (Feraille et al., 1999). In the present study, tyrosine-10 phosphorylation was observed under conditions that caused Na,K-ATPase activity to decrease. We have no explanation for the discrepancy. However, in an earlier study it was shown that in synaptosomal membrane prepared from rat brain cortex, insulin inhibited Na,K-ATPase activity by 35–40% (Bojorge and de Lores Arnaiz, 1987).
The ability of PP2 to abolish the SNP effect on ouabain-sensitive 86Rb uptake and ATP hydrolysis points to a signaling mechanism that requires SFK activation. Transient SFK phosphorylation was detected within 2 min when cells were treated with SNP or L-arginine and the response was prevented by PP2. SFK phosphorylation in SNP-treated cells also was prevented by ODQ, a selective inhibitor of soluble guanylate cyclase, added to blunt the rise of cGMP. Similarly, the PKG inhibitor KT5823 suppressed SFK phosphorylation in SNP-treated cells. Moreover, 8-Br-cGMP was found to elicit an increase in SFK phosphorylation. Taken together, the results suggest that NO, generated in response to the NO donor, activates guanylate cyclase to cause a rise in cGMP, and subsequent PKG-dependent SFK activation. Activation of Src by NO-cGMP-PKG signaling pathway has previously been documented in several tissues, such as in osteoblasts (Rangaswami et al., 2010) and in fibroblasts (Monteiro et al., 2000).
Earlier studies with genistein demonstrated the requirement for tyrosine phosphorylation in the Na,K-ATPase inhibition caused by dopamine agonists in rabbit NPE (Nakai et al., 1999). Genistein also prevented the Na,K-ATPase stimulation in renal proximal tubule caused by insulin (Garvin and Sanders, 1991; Feraille et al., 1997). Several studies have linked modulation of Na,K-ATPase activity more specifically to SFK tyrosine kinase activation. In the epithelium of the porcine lens, activation of SFK following purinergic receptor activation was linked to an increase in Na,K-ATPase activity (Tamiya et al., 2007). Also in porcine lens, SFK activation was associated with the ability of endothelin-1 to cause a reduction of Na,K-ATPase activity (Mandal et al., 2011). In both instances, the SFK inhibitor PP2 prevented the Na,K-ATPase response. How SFK can be linked to either inhibition or activation of Na,K-ATPase, depending on the stimulus, is still under investigation and falls beyond the scope of the present study. There are several tyrosine kinases in the SFK family and the appearance of faint phospho-SFK bands in addition to the principal ~60 kDa band in SNP-treated cells suggests more than one SFK might be activated. Different patterns of SFK activation could elicit different downstream responses. The key finding here is that NO-mediated Na,K-ATPase inhibition, which we know to be cGMP-dependent, also depends on SFK activation.
The effects of NO are wide-ranging and an ERK1/2 activation response was observed in SNP-treated cells along with a p38 MAP kinase response. Typically p38 is activated subsequent to ERK1/2. Interestingly, PP2 prevented phosphorylation of both ERK1/2 and p38 indicating NO-induced activation of ERK1/2 and p38 MAPK is SFK-dependent. However, SNP-induced ERK1/2 and p38 activation did not play an obvious role in modulating Na,K-ATPase activity since a selective ERK1/2 inhibitor failed to prevent the SNP-induced reduction of Na,K-ATPase activity although the SFK inhibitor PP2 abolished both ERK1/2 and downstream p38 activation. This result is in contrast to the reports that ERK1/2 activation negatively modulates Na,K-ATPase in opossum kidney cells and lung alveolar epithelium (Khundmiri et al., 2008; Welch et al., 2010) and causes Ser phosphorylation of the Na,K-ATPase α1 subunit in vitro (Al-Khalili et al., 2004) and internalization of Na,K-ATPase α1 polypeptide (Khundmiri et al., 2004). The downstream consequences of ERK1/2 and p38 activation in SNP-treated NPE cells remain to be determined but SFK activation is clearly an early step in this part of the NO response as well as the Na,K-ATPase inhibition response.
NO donors have been reported to cause ROS generation (Xu et al., 2004) and we acknowledge the ability of ROS to activate a number of protein kinases, including Src tyrosine kinase (Giannoni et al., 2005), protein kinase C (Cosentino-Gomes et al., 2012), ERK1/2 and p38 MAPK (Son et al., 2011). However, this seems unlikely to explain the NO responses observed here. First, under the conditions of the present study no detectable increase of ROS was observed in cells exposed to SNP, SNAP, or L-arginine. Second, blockade of the sGC/cGMP/PKG pathway effectively prevented SFK, ERK and p38 activation.
Mechanistically, the results of these studies point to NO-dependent activation of guanylate cyclase that causes a rise in cGMP, and subsequent PKG-dependent SFK activation that causes tyrosine phosphorylation of the Na,K-ATPase α1 subunit and changes Na,K-ATPase activity. Na,K-ATPase activity establishes ion gradients that support the coordinated operation of ion transport mechanisms that enable the ciliary epithelium bilayer to secrete fluid into the eye (Civan, 1998; Civan and Macknight, 2004). By causing Na,K-ATPase inhibition, NO has the potential to suppresses the mechanism of aqueous humor formation. It should also be recognized that altered aqueous humor drainage from the eye could be influenced by NO. In vitro, NO has been found to cause a reduction in the volume of trabecular meshwork cells and Schlemm’s canal endothelium and these changes are paralleled by an increase in aqueous outflow facility (Ellis et al., 2009, 2010).
Acknowledgments
The authors are grateful to the University of Arizona Meat Science laboratory and Hatfield Quality Meat, Pennsylvania for the supply of porcine eyes.
Contract grant sponsor: National Institute of Health;
Contract grant number: EY006915.
Footnotes
The authors declared that they have no conflict of interest.
Literature Cited
- Al-Khalili L, Kotova O, Tsuchida H, Ehren I, Feraille E, Krook A, Chibalin AV. ERK1/2 mediates insulin stimulation of Na(+),K(+)-ATPase by phosphorylation of the alpha-subunit in human skeletal muscle cells. J Biol Chem. 2004;279:25211–25218. doi: 10.1074/jbc.M402152200. [DOI] [PubMed] [Google Scholar]
- Bojorge G, de Lores Arnaiz GR. Insulin modifies Na(+), K(+)-ATPase activity of synaptosomal membranes and whole homogenates prepared from rat cerebral cortex. Neurochem Int. 1987;11:11–16. doi: 10.1016/0197-0186(87)90142-2. [DOI] [PubMed] [Google Scholar]
- Chuman H, Chuman T, Nao-i N, Sawada A. The effect of L-arginine on intraocular pressure in the human eye. Curr Eye Res. 2000;20:511–516. [PubMed] [Google Scholar]
- Civan MM. The Eye’s Aqueous Humor: From secretion to glaucoma. San Diego, London, Boston, New York, Sydney, Tokyo, Toronto: Academic Press; 1998. Transport components of net secretion of the aqueous humour and their integrated regulation; pp. 1–24. [Google Scholar]
- Civan MM, Macknight AD. The ins and outs of aqueous humour secretion. Exp Eye Res. 2004;78:625–631. doi: 10.1016/j.exer.2003.09.021. [DOI] [PubMed] [Google Scholar]
- Cosentino-Gomes D, Rocco-Machado N, Meyer-Fernandes JR. Cell signaling through protein kinase C oxidation and activation. Int J Mol Sci. 2012;13:10697–10721. doi: 10.3390/ijms130910697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira Elias M, Tavares de Lima W, Vannuchi YB, Marcourakis T, da Silva ZL, Trezena AG, Scavone C. Nitric oxide modulates Na+, K+-ATPase activity through cyclic GMP pathway in proximal rat trachea. Eur J Pharmacol. 1999;367:307–314. doi: 10.1016/s0014-2999(98)00928-5. [DOI] [PubMed] [Google Scholar]
- Ellis DZ, Nathanson JA, Sweadner KJ. Carbachol inhibits Na(+)-K(+)-ATPase activity in choroid plexus via stimulation of the NO/cGMP pathway. Am J Physiol Cell Physiol. 2000;279:C1685–C1693. doi: 10.1152/ajpcell.2000.279.6.C1685. [DOI] [PubMed] [Google Scholar]
- Ellis DZ, Nathanson JA, Rabe J, Sweadner KJ. Carbachol and nitric oxide inhibition of Na,K-ATPase activity in bovine ciliary processes. Invest Ophthalmol Vis Sci. 2001;42:2625–2631. [PubMed] [Google Scholar]
- Ellis DZ, Dismuke WM, Chokshi BM. Characterization of soluble guanylate cyclase in NO-induced increases in aqueous humor outflow facility and in the trabecular meshwork. Invest Ophthalmol Vis Sci. 2009;50:1808–1813. doi: 10.1167/iovs.08-2750. [DOI] [PubMed] [Google Scholar]
- Ellis DZ, Sharif NA, Dismuke WM. Endogenous regulation of human Schlemm’s canal cell volume by nitric oxide signaling. Invest Ophthalmol Vis Sci. 2010;51:5817–5824. doi: 10.1167/iovs.09-5072. [DOI] [PubMed] [Google Scholar]
- Feelisch M, Noack EA. Correlation between nitric oxide formation during degradation of organic nitrates and activation of guanylate cyclase. Eur J Pharmacol. 1987;139:19–30. doi: 10.1016/0014-2999(87)90493-6. [DOI] [PubMed] [Google Scholar]
- Feraille E, Carranza ML, Rousselot M, Favre H. Modulation of Na+,K(+)-ATPase activity by a tyrosine phosphorylation process in rat proximal convoluted tubule. Am J Physiol. 1997;498:99–108. doi: 10.1113/jphysiol.1997.sp021844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feraille E, Carranza ML, Gonin S, Beguin P, Pedemonte C, Rousselot M, Caverzasio J, Geering K, Martin PY, Favre H. Insulin-induced stimulation of Na+,K(+)-ATPase activity in kidney proximal tubule cells depends on phosphorylation of the alpha-subunit at Tyr-10. Mol Biol Cell. 1999;10:2847–2859. doi: 10.1091/mbc.10.9.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvin J, Sanders K. Endothelin inhibits fluid and bicarbonate transport in part by reducing Na+/K+ ATPase activity in the rat proximal straight tubule. J Am Soc Nephrol. 1991;2:976–982. doi: 10.1681/ASN.V25976. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Freitag AC, Martin-Vasallo P, Coca-Prados M. Cellular distribution and differential gene expression of the three alpha subunit isoforms of the Na,K-ATPase in the ocular ciliary epithelium. J Biol Chem. 1990;265:2935–2940. [PubMed] [Google Scholar]
- Giannoni E, Buricchi F, Raugei G, Ramponi G, Chiarugi P. Intracellular reactive oxygen species activate Src tyrosine kinase during cell adhesion and anchorage-dependent cell growth. Mol Cell Biol. 2005;25:6391–6403. doi: 10.1128/MCB.25.15.6391-6403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman NJ, Fang MZ, Tang SS, Ingelfinger JR, Garg LC. Autocrine inhibition of Na+/K(+)-ATPase by nitric oxide in mouse proximal tubule epithelial cells. J Clin Invest. 1995;95:2083–2088. doi: 10.1172/JCI117895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khundmiri SJ, Bertorello AM, Delamere NA, Lederer ED. Clathrin-mediated endocytosis of Na+,K+-ATPase in response to parathyroid hormone requires ERK-dependent phosphorylation of Ser-11 within the alpha1-subunit. J Biol Chem. 2004;279:17418–17427. doi: 10.1074/jbc.M311715200. [DOI] [PubMed] [Google Scholar]
- Khundmiri SJ, Ameen M, Delamere NA, Lederer ED. PTH-mediated regulation of Na+-K+-ATPase requires Src kinase-dependent ERK phosphorylation. Am J Physiol Renal Physiol. 2008;295:F426–F437. doi: 10.1152/ajprenal.00516.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S, Sugiyama T, Shimizu K, Ishida O, Nakajima M, Azuma I, Goh Y. Effect of a nitric oxide donor on intraocular pressure. Nippon Ganka Gakkai Zasshi- Acta Societatis Ophthalmologicae Japonicae. 1996;100:181–186. [PubMed] [Google Scholar]
- Lomniczi A, Suburo AM, Elverdin JC, Mastronardi CA, Diaz S, Rettori V, McCann SM. Role of nitric oxide in salivary secretion. Neuroimmunomodulation. 1998;5:226–233. doi: 10.1159/000026342. [DOI] [PubMed] [Google Scholar]
- Mandal A, Shahidullah M, Beimgraben C, Delamere NA. The effect of endothelin-1 on Src-family tyrosine kinases and Na,K-ATPase activity in porcine lens epithelium. J Cell Physiol. 2011;226:2555–2561. doi: 10.1002/jcp.22602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JC, Shahidullah M, Wilson WS. Intraocular pressure and vascular effects of sodium azide in bovine perfused eye. J Ocul Pharmacol Ther. 2001;17:225–234. doi: 10.1089/108076801750295263. [DOI] [PubMed] [Google Scholar]
- Mohanakumar KP, Thomas B, Sharma SM, Muralikrishnan D, Chowdhury R, Chiueh CC. Nitric oxide: An antioxidant and neuroprotector. Ann NY Acad Sci. 2002;962:389–401. doi: 10.1111/j.1749-6632.2002.tb04083.x. [DOI] [PubMed] [Google Scholar]
- Monteiro HP, Gruia-Gray J, Peranovich TMS, Barbosa de Oliveira LC, Stern A. Nitric oxide stimulates tyrosine phosphorylation of focal adhesion kinase, SRC kinase, and mitogen-activated protein kinases in murine fibroblasts. Free Radic Biol Med. 2000;28:174–182. doi: 10.1016/s0891-5849(99)00233-6. [DOI] [PubMed] [Google Scholar]
- Nakai Y, Dean WL, Hou Y, Delamere NA. Genistein inhibits the regulation of active sodium-potassium transport by dopaminergic agonists in nonpigmented ciliary epithelium. Invest Ophthalmol Vis Sci. 1999;40:1460–1466. [PubMed] [Google Scholar]
- Nakazawa T, Tomita H, Yamaguchi K, Sato Y, Shimura M, Kuwahara S, Tamai M. Neuroprotective effect of nipradilol on axotomized rat retinal ganglion cells. Curr Eye Res. 2002;24:114–122. doi: 10.1076/ceyr.24.2.114.8162. [DOI] [PubMed] [Google Scholar]
- Ortiz PA, Garvin JL. Role of nitric oxide in the regulation of nephron transport. Am J Physiol Renal Fluid Electrolyte Physiol. 2002;282:F777–F784. doi: 10.1152/ajprenal.00334.2001. [DOI] [PubMed] [Google Scholar]
- Rangaswami H, Schwappacher R, Marathe N, Zhuang S, Casteel DE, Haas B, Chen Y, Pfeifer A, Kato H, Shattil S, Boss GR, Pilz RB. Cyclic GMP and protein kinase G control a Src-containing mechanosome in osteoblasts. Sci Signal. 2010;3:ra91. doi: 10.1126/scisignal.2001423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R., Jr Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun. 2004;324:1155–1164. doi: 10.1016/j.bbrc.2004.09.171. [DOI] [PubMed] [Google Scholar]
- Seven I, Turkozkan N, Cimen B. The effects of nitric oxide synthesis on the Na+,K(+)-ATPase activity in guinea pig kidney exposed to lipopolysaccharides. Mol Cell Biochem. 2005;271:107–112. doi: 10.1007/s11010-005-5616-1. [DOI] [PubMed] [Google Scholar]
- Shahidullah M, Delamere NA. NO donors inhibit Na,K-ATPase activity by a protein kinase G-dependent mechanism in the nonpigmented ciliary epithelium of the porcine eye. Br J Pharmacol. 2006;148:871–880. doi: 10.1038/sj.bjp.0706795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M, Wilson WS, Yap M, To CH. Effects of ion transport and channel-blocking drugs on aqueous humor formation in isolated bovine eye. Invest Ophthalmol Vis Sci. 2003;44:1185–1191. doi: 10.1167/iovs.02-0397. [DOI] [PubMed] [Google Scholar]
- Shahidullah M, Yap MK, To CH. cGMP, sodiun nitroprusside and sodium azide reduce aqueous humour formation in the isolated arterially perfused pig eye. Br J Pharmacol. 2005;144:1–9. doi: 10.1038/sj.bjp.0706156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M, Tamiya S, Delamere NA. Primary culture of porcine nonpigmented ciliary epithelium. Curr Eye Res. 2007;32:511–522. doi: 10.1080/02713680701434899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahidullah M, Mandal A, Beimgraben C, Delamere NA. Hyposmotic stress causes ATP release and stimulates Na, K-ATPase activity in porcine lens. J Cell Physiol. 2012;227:1428–1437. doi: 10.1002/jcp.22858. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Son Y, Cheong Y-K, Kim N-H, Chung H-T, Kang DG, Pae H-O. Mitogen-activated protein kinases and reactive oxygen species: How can ROS activate MAPK pathways? J Signal Transduct. 2011;2011:792639. doi: 10.1155/2011/792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamiya S, Okafor MC, Delamere NA. Purinergic agonists stimulate lens Na-K-ATPase-mediated transport via a Src tyrosine kinase-dependent pathway. Am J Physiol Cell Physiol. 2007;293:C790–C796. doi: 10.1152/ajpcell.00579.2006. [DOI] [PubMed] [Google Scholar]
- Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- Welch LC, Lecuona E, Briva A, Trejo HE, Dada LA, Sznajder JI. Extracellular signal-regulated kinase (ERK) participates in the hypercapnia-induced Na,K-ATPase downregulation. FEBS Lett. 2010;584:3985–3989. doi: 10.1016/j.febslet.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie ZJ, Wang YH, Ganjeizadeh M, McGee R, Jr, Askari A. Determination of total (Na+- K+)-ATPase activity of isolated or cultured cells. Analytical Biochemistry. 1989;183:215–219. doi: 10.1016/0003-2697(89)90470-3. [DOI] [PubMed] [Google Scholar]
- Xu Z, Ji X, Boysen PG. Exogenous nitric oxide generates ROS and induces cardioprotection: Involvement of PKG, mitochondrial KATP channels, and ERK. Am J Physiol Heart Circ Physiol. 2004;286:H1433–H1440. doi: 10.1152/ajpheart.00882.2003. [DOI] [PubMed] [Google Scholar]