

Key Clinical Message
Congenital erythrocytosis is a hereditary disorder due to an increase in red cell mass that can be caused by mutations in proteins involved in HIF‐α pathway, as PHD2. Hereby, we describe a new familial mutation in PHD2 gene. Considering an increased thrombotic potential, patients began antiplatelet aggregation therapy and phlebotomies.
Keywords: Congenital erythrocytosis, hypoxia, phd2, thrombosis
Background
Congenital erythrocytosis is a rare hereditary disorder of red cell production, characterized by an absolute increase in red cell mass with elevated hematocrit and hemoglobin levels. Contrary to polycythemias, it is not accompanied by increased numbers of white cells and platelets 8.
CE can be caused by the presence of high‐affinity hemoglobin variants or mutations involving erythropoietin receptor (EPOR), bisphosphoglycerate mutase (BPGM), and oxygen‐sensing pathway enzymes, namely prolyl hydroxylase domain (PHD) proteins, von Hippel‐Lindau tumor suppressor protein, and hypoxia‐inducible factor (HIF) 8.
Under conditions of normal oxygen tension, isoform 2 of PHD's (PHD2) is constitutively active and hydroxylates key prolines in oxygen‐dependent degradation domains of HIF‐subunit 2α (HIF‐α). After hydroxylation, it binds to VHL protein that forms an E3 ubiquitin ligase complex, allowing HIF‐α ubiquitination and subsequent degradation by a proteasome. However, under hypoxic conditions, after escaping to ubiquitin‐mediated degradation, HIF‐α heterodimerizes with aryl hydrocarbon nuclear translocator and this complex acts as a transcription factor which upregulates the expression of EPO gene (Fig. 1) 1, 2.
Figure 1.
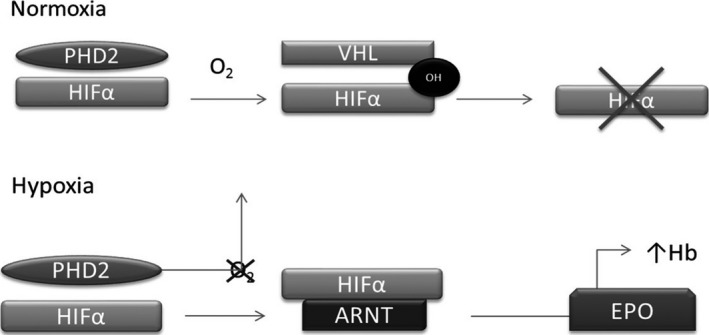
Control of erythropoietin (EPO) synthesis by the oxygen sensing pathway.
HIF‐α subunits are only stable under hypoxic conditions and are negatively regulated by PHD and VHL proteins. PHD or VHL loss of functions lead to an elevation in HIF‐α and, consequently, an abnormal upregulation of target genes, such as EPO 1.
After its release into the blood stream, EPO binds to its receptor (EPOR) on the surface of erythroid progenitor cells. Functioning as a dimeric molecule, the ligand‐occupied receptor activates the Jak2 protein tyrosine kinase which, in turn, phosphorylates tyrosine residues on the receptor and associated intracellular signaling molecules, promoting cellular proliferation, differentiation, and inhibition of apoptosis 2.
Online Mendelian Inheritance in Man classification divides genes involved in congenital erythrocytosis into four categories:
Mutations in EPOR, known as CE type 1, constitute a cause of primary erythrocytosis in 15% of cases. CE type 1 is associated with a hypersensitivity of erythroid progenitors to EPO, with subnormal serum levels, and prolonged activation of the JAK 7.
CE type 2 is caused by mutations in the VHL gene. This disorder has features of both primary and secondary erythrocytosis and is characterized by high sensitiveness of erythroid precursors to EPO and dysregulation of EPO, resulting in elevated EPO levels 7. Clinically important is the risk of peripheral and cerebrovascular thrombotic events and pulmonary hypertension that these patients presented in several studies.
Mutations in the PHD2 gene, EGLN1, are classified as CE type 3. In contrast to previous erythrocytosis types, all PHD2 mutations described to date are heterozygous with inappropriately normal EPO level 4, 7.
CE type 4 is a recently described category, characterized by mutations in the HIF2A gene, EPAS1. CE 4 is caused by gain‐of‐function mutations, affecting younger patients, with thrombotic events 7.
Objective: Report a new mutation in PHD2 gene (EGLN1) associated with CE.
Clinical Case
We describe a Portuguese family presenting with an isolated but sustained erythrocytosis, affecting three generations – grandfather, father (propositus), and son.
Propositus was referred to our hospital by general practitioner because of increased hemoglobin (Hb) of 182 g/L and hematocrit (Hct) of 58%, for at least 5 years. He had no medical history nor took any medication. He was a nonsmoker and presented intermittent headache and plethoric face. Examination revealed hypertension, no fever, and normal pulse and respiratory rate. Findings included a mildly ruddy complexion, lungs clear to auscultation bilaterally, and regular heart tones. There was neither hepatosplenomegaly nor other organomegalies. Extremities revealed no edema. Neurological examination was normal.
According to published guidelines, erythrocytosis was confirmed (Hb > 180 g/L and Htc>50%) with normal white blood cell and platelet count, without any other alterations, except an indirect hyperbilirubinemia. Arterial blood gas revealed an oxygen saturation of 99%. Hb electrophoresis did not reveal any abnormal Hb variant. Secondary causes of erythrocytosis were excluded as EPO level and partial oxygen pressure were normal. Urine examination, chest X‐ray, electrocardiogram, echocardiography, and pulmonary function tests were normal. Abdominal ultrasound showed a normal liver, spleen, kidneys, and suprarenal glands.
Bone marrow biopsy only showed an erythroid hyperplasia. JAK‐2 mutations were negative. HBB and EPOR gene sequencing were normal. We then proceeded to sequencing the genes included in EPO‐induced signaling pathway, and we detected a new mutation in exon 3 of EGLN1, c.1096T>C; p.Phe366Leu (F366L), in heterozygosity which provided the diagnosis of erythrocytosis secondary to mutant PHD2 gene (Fig. 2).
Figure 2.
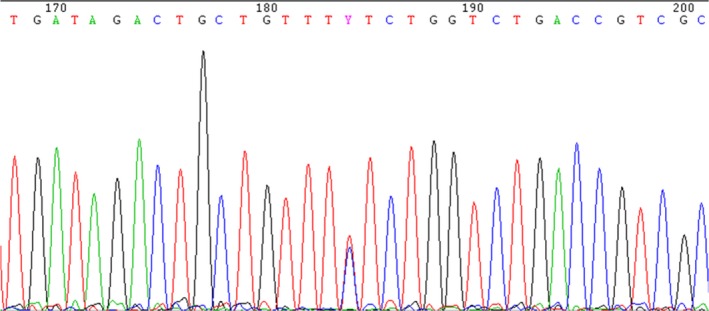
Sequencing of EGLN1 (exon 3). c.1096T>C; p.Phe366Leu (F366L) in silico: probably damaging (score 0.992).
Despite the lack of knowledge regarding its prothrombotic potential, once other mutations in PHD2 are known to be associated with a slightly increased thrombotic potential, our patients started antiplatelet aggregation therapy and phlebotomies on demand. Additionally, the patient was diagnosed with Gilbert's syndrome by a mutation in UGT1A1 gene promoter region [A(TA)7TAA] 5, 6.
After those findings, we conducted a mutation‐directed molecular study on propositus’ son which revealed the same exact mutation in heterozygosity. Of note, propositus’ father is now dead and both the propositus’ and his son are currently asymptomatic and under antiplatelet aggregation therapy and phlebotomies on demand.
Discussion
PHD2 plays an important role in the regulation of EPO production and subsequently in erythropoiesis.
A mutation in EGLN1 (PHD2 gene) has been detected in two generations of a Portuguese family with CE, and it cosegregates with the erythrocytosis phenotype. As far as we know, this is the first time this particular mutation is described. Also, according to the family studies and analysis in silico, this mutation is a pathogenic variant associated with the CE phenotype observed in this family 3.
Furthermore, more studies are required to better understand genotype–phenotype correlations and the most suitable therapeutic approach.
Conflict of Interest
The authors declare they have no conflict of interest.
Authorship
JB: Resident in Clinical Hematology, Centro Hospitalar de Tondela‐Viseu, EPE; principal author. CDR: Resident in Clinical Hematology, Centro Hospitalar de Tondela‐Viseu, EPE; co‐author. GF: Consultant in Clinical Hematology, Centro Hospitalar de Baixo Vouga; clinical reviewer. PR: Consultant in Clinical Hematology, Centro Hospitalar de Tondela‐Viseu. CC and MRA: Consultant in Clinical Hematology, Centro Hospitalar de Tondela‐Viseu. CB: PhD biology with specialization in Human Genetics, Congenital Anemia and Erythrocytosis Laboratory, Centro Hospitalar e Universitário de Coimbra; genetics reviewer. HMS: Director of Clinical Hematology Department, Centro Hospitalar de Tondela‐Viseu; consultant in Clinical Hematology, Centro Hospitalar de Tondela‐Viseu.
Acknowledgments
The authors would like to thank Molecular Hematology Unit of Centro Hospitalar e Universitário de Coimbra and Blood and Transfusion Medicine Service of Centro Hospitalar Tondela‐Viseu.
Clinical Case Reports 2018; 6(6): 1109–1111
References
- 1. Chowdhury, R. , Domene C., Schofield C. J., Pugh C. W., Cantrelle F. X., Landrieu I., et al. 2016. Structural basis for oxygen degradation domain selectivity of the HIF prolyl hydroxylases. Nat. Commun. 7:12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Percy, M. J. , and Lee F. S.. 2008. Familial erythrocytosis: molecular links to red blood cell control. Haematologica 93:963–967. [DOI] [PubMed] [Google Scholar]
- 3. Minervini, G. , Quaglia F., and Tosatto S. C.. 2016. Computational analysis of prolyl hydroxylase domain‐containing protein 2 (PHD2) mutations promoting polycythemia insurgence in humans. Sci. Rep. 6:18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gardie, B. , Percy M. J., Hoogewijs D., Chowdhury R., Bento C., Arsenault P. R., et al. 2014. The role of PHD2 mutations in the pathogenesis of erythrocytosis. Hypoxia 2:71–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilson, R. , Syed N., and Shah P.. 2016. Erythrocytosis due to PHD2 mutations: a review of clinical presentation, diagnosis, and genetics. Case Rep. Hematol. 2016:6373706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Albiero, E. , Ruggeri M., Fortuna S., Finotto S., Bernardi M., Madeo D., et al. 2012. Isolated erythrocytosis: study of 67 patients and identification of three novel germ‐line mutations in the prolyl hydroxylase domain protein 2 (PHD2) gene. Haematologica 97:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bento, C. , Percy M. J., Gardie B., Maia T. M., Wijk R., Perrotta S., et al. 2014. Genetic basis of congenital erythrocytosis: mutation update and online databases. Hum. Mut. 35:15–26. [DOI] [PubMed] [Google Scholar]
- 8. McMullin, M. 2009. Idiopathic erythrocytosis: a disappearing entity. Educ. Prog. Am. Soc. Hematol. 1:629–635. [DOI] [PubMed] [Google Scholar]