

Summary
The development of T cell tolerance in the thymus requires the presentation of host proteins by multiple antigen-presenting-cell (APC) types. However, the importance of transferring host antigens from transcription factor AIRE-dependent medullary thymic epithelial cells (mTECs) to bone marrow (BM) APCs is unknown. We report that antigen was primarily transferred from mTECs to CD8α+ dendritic cells (DCs) and showed that CD36, a scavenger receptor selectively expressed on CD8α+ DCs, mediated the transfer of cell-surface, but not cytoplasmic, antigens. The absence of CD8α+ DCs or CD36 altered thymic T cell selection, as evidenced by TCR repertoire analysis and the loss of allo-tolerance in murine allogeneic BM transplantation (allo-BMT) studies. Decreases in these DCs and CD36 expression in peripheral blood of human allo-BMT patients correlated with graft-vs-host disease. Our findings suggest that CD36 facilitates transfer of mTEC-derived cell-surface antigen on CD8α+ DCs to promote tolerance to host antigens during homeostasis and allo-BMT.
Keywords: CD36, scavenger receptor, apoptotic cell clearance, efferocytosis, antigen transfer, thymic dendritic cells, regulatory T cells, medullary thymic epithelial cells, tolerance
ETOC paragraph
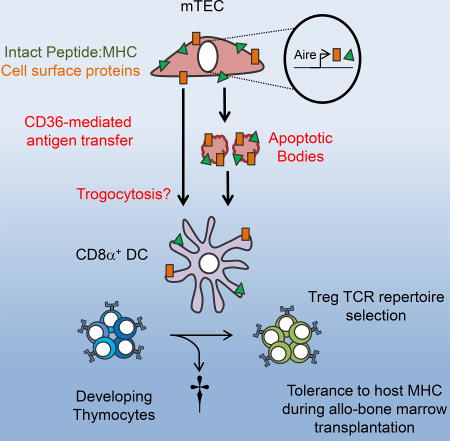
How cooperative antigen presentation between medullary thymic epithelial cells (mTECs) and dendritic cells (DCs) occurs remains unknown. Perry et al. show that CD36, a scavenger receptor expressed on CD8α+ DCs, mediates acquisition and presentation of cell-surface antigens from mTECs for T-cell receptor repertoire development and allo-tolerance during bone marrow transplantation.
Introduction
During T cell development, T cell receptor (TCR) gene segments are rearranged to generate a diverse TCR repertoire necessary for immunity to invading pathogens (Klein et al., 2014; Vrisekoop et al., 2014; Weissler and Caton, 2014). An unintended consequence of this diversity is the recognition of self-antigen, which can result in autoimmunity. For T cells, two fundamental processes promote tolerance to self in the thymus (referred to as central tolerance) prior to their release into the periphery: (1) negative selection, whereby autoreactive T cells are eliminated; and (2) generation of CD4+ forkhead box P3 (Foxp3)+ regulatory T (Treg) cells (Bluestone et al., 2015; Klein et al., 2014; Li and Zheng, 2015; Richards et al., 2016). Both processes are driven by TCR recognition of self-antigens presented by antigen presenting cells (APCs). Although this can occur throughout the thymus, it commonly occurs in the thymic medulla and involves both medullary thymic epithelial cells (mTECs) and bone marrow (BM)-derived dendritic cells (DCs), B cells, and macrophages (Chan and Anderson, 2015; Klein et al., 2014; Oh and Shin, 2015; Perry and Hsieh, 2016). mTECs can directly present peripheral tissue antigens (PTAs) generated via the transcription factor AIRE via non-conventional mechanisms to tolerize CD4+ T cells (Aschenbrenner et al., 2007; Cowan et al., 2013; Hinterberger et al., 2010; Perry et al., 2014). It has become increasingly clear, however, that thymic tolerance to PTAs can occur via antigen transfer from mTEC cells to BM-derived APCs (Gallegos and Bevan, 2004; Hubert et al., 2011; Koble and Kyewski, 2009; Lin et al., 2016; Millet et al., 2008; Perry et al., 2014).
Recently, we proposed that CD8α+ DCs are the primary BM APC recipient of antigen transfer from mTECs (Perry et al., 2014). Our conclusion was based on an assessment of eight TCRs for their ability to induce Treg cell development in basic leucine zipper transcriptional factor ATF-like 3 (Batf3)−/− mice, which are deficient in CD8α+ DCs. These data are consistent with a recent study using a different TCR that recognizes an AIRE-dependent mTEC antigen and yet involves antigen presentation by non-migratory BM APCs which include CD8α+ DCs (Lin et al., 2016), as well as reports showing that CD8α+ DCs home to mTEC-rich regions of the medulla (Atibalentja et al., 2011; Baba et al., 2009; Klein et al., 2014; Lei et al., 2011) and uniquely acquire antigen from mTEC (Ardouin et al., 2016). However, a recent report argued that BATF3-dependent CD8α+ DCs are not involved in thymic Treg cell selection based on analysis of two Treg cell TCRs and of TCR repertoires using a different fixed-TCRβ chain (Leventhal et al., 2016). Thus, the importance of BATF3-dependent CD8α+ DCs in thymic selection, their role in antigen transfer from mTECs, and the mechanisms by which this occurs, remain unclear.
Here, we quantified the importance of CD8α+ DCs in shaping the conventional T cell (Tconv) and Treg TCR repertoire, and their involvement in facilitating Treg cell generation to AIRE-dependent self-antigens. We also showed that CD36, a class B scavenger receptor preferentially expressed on CD8α+ DCs, is important for the transfer of a model cell-surface, but not cytoplasmic, antigen from mTEC cells. This mechanism of antigen transfer was critical for the development of thymic allo-tolerance during bone marrow transplantation (BMT) in mice and may be important for the prevention of Graft-versus-Host Disease (GVHD) after human allo-BMT. Thus, these data suggest a model by which CD8α+ DCs utilize CD36 to cross-present cell-surface antigens from mTECs to mediate thymic T cell tolerance.
Results
BATF3-dependent BM APCs facilitate thymic Treg TCR repertoire development
The role of BATF3-dependent CD8α+ DCs on thymic tolerance is controversial, with studies suggesting they are either irrelevant (Leventhal et al., 2016) or important (Perry et al., 2014) for thymic Treg cell selection. To address this, we assessed the impact of Batf3-deficiency globally on the TCR repertoire. Because of the great diversity of the normal TCR repertoire, we utilized a fixed TCRβ model as previously described (Perry et al., 2014), generating mice expressing a TCRβ transgene deficient in CD8α+ DCs (Batf3−/− TCRβ transgenic Tcra+/− Foxp3Thy1.1). These mice were used as BM donors into congenic Ly5.1 hosts. We sequenced TCRα chains from CD4 single-positive (SP, CD4+ CD8−) Foxp3+ (Treg) and mature CD62Lhi CD24lo CD4SP Foxp3− (Tconv) cells (Figure S1A) (Chai et al., 2017), whose frequencies were not affected by Batf3-deficiency (Figure 1A and 1B). We found differences within the Tconv and Treg TCR repertoires using unbiased factor analysis which showed minimal inter-mouse variability while primarily clustering around Batf3-deficient and -sufficient groups (Figure 1C). This was partly due to a number of TCRs enriched in Batf3-deficient mice, suggestive of CD8α+ DC mediated negative selection (Figure 1C, top plots, data points found below reference line) as shown in our previous study (Perry et al., 2014). In total, BATF3-dependent DCs negatively selected ~2% of the unique Tconv and Treg cell TCR repertoires (Figure 1D and Figure S1B).
Figure 1. BATF3-dependent BM APCs facilitate development of the thymic Treg cell TCR repertoire.

(A,B) Summary of Foxp3+ CD4+ (Treg), Foxp3− CD4+ CD8− (CD4SP) and CD8+ CD4− (CD8SP) thymocyte frequencies (A) or HSAlo CD62hi and HSAhi CD62lo CD4+ frequencies (B) from TCliβ transgenic Batf3+/+ or Batf3−/− → Ly5.1 BM chimeras (9–12 mice in 3 independent experiments). Data are presented as mean + SEM.
(C) Top panels: The average TCR frequency in the Tconv (Foxp3− HSAlo CD62hi) and Treg (Foxp3+) cell subset in Batf3+/+ or Batf3−/− → Ly5.1 BM chimeras are shown. Red dots indicate TCRs that show p < .05 MWU and 5-fold change between Batf3−/− and Batf3+/+. Bottom panels: Unsupervised clustering analysis of Tconv and Treg cell TCR repertoires from Batf3 BM chimeras.
(D,E) The percentage of unique TCRs or total sequences dependent on Batf3 for negative selection (D) or Treg cell selection (E). Each dot represents a single rarefied sampling of data sets based on the smallest number of sequences as described in (C).
(F,G) Venn diagram of absolute number of TCRs involved in negative selection (F) or Treg cell selection (G). All data are representative of at least two independent experiments with at least 4 mice per condition.
See also Figure S1 and S2.
By contrast to BATF3-dependent negative selection, we observed a greater requirement for CD8α+ DCs in Treg cell selection (Figure 1C, top plot, red dots above reference line). TCRs that we previously identified as BATF3-dependent in vivo (Perry et al., 2014) were also decreased in our Batf3-deficient Treg TCR data set (Figure S1C). We observed that CD8α+ DCs were required for selection of approximately 12% of the unique Treg cell TCR repertoire (Figure 1E and Figure S1D). However, these conclusions were different from a previous report showing no role for CD8α+ DCs in thymic Treg cell generation using Batf3−/− mice (Leventhal et al., 2016). Analysis of their data set (Leventhal et al., 2016) using our approaches revealed that 5–10% of their Treg TCR repertoire is BATF3-dependent (Figure S1E), suggesting that the differences in interpretation may be due to the statistical methods used. Thus, we propose that both TCR repertoire studies support a role for BATF3-dependent CD8α+ DCs in thymic Treg cell selection.
AIRE-dependent antigens are transferred to and presented by CD8α+ DCs
AIRE plays an important role in T cell tolerance, in part via the induction of PTAs in mTECs. AIRE-dependent antigens can be presented autologously on mTECs, and may also be transferred to CD8α+ DCs (Ardouin et al., 2016; Hubert et al., 2011; Koble and Kyewski, 2009; Perry et al., 2014). Although we had previously assessed the AIRE-dependent repertoire using analysis of TRAV14 TCRs, we generated new data using multiplex PCR to permit direct comparison with the BATF3 data set. Within the range of previous results, we found that AIRE was involved in the deletion of both Tconv (~4%) and Treg (~3%) cells as well as Treg cell selection (~10%) (Figures S2A–S2D).
We then assessed the overlap of TCRs that are dependent on CD8α+ DCs versus those dependent on AIRE-induced self-antigens in mTECs. While we observed relatively few Tconv TCRs that were co-dependent on BATF3 and AIRE for negative selection, a much greater fraction of Treg TCRs were co-dependent, consistent with AIRE-dependent antigen transfer from mTECs to CD8α+ DCs (Figures 1F–1G and Figures S2E–S2F). In fact, ~40% of AIRE-dependent Treg TCRs were co-dependent on CD8α+ DCs (Figure 1G). In summary, the cross-comparison of BATF3- and AIRE-dependent TCRs supported our previous proposal that a substantial fraction of Aire-dependent antigens is transferred to and presented by CD8α+ DCs.
T cell selection by CD8α+ DCs prevents tissue-specific inflammation
The TCR repertoire data demonstrated that CD8α+ DCs were involved in deletion and Treg cell selection. To address whether these TCRs that escape thymic tolerance were capable of inducing inflammation, we transferred mature thymic CD4+ T cells, with both Tconv and Treg cells, into T cell-deficient TCRb−/−Tcrd−/− mice and assessed tissue inflammation using both Positron Emission Tomography (PET) and histology (Figures 2A–C). We observed a number of tissues, particularly the lungs, with increased inflammation in mice receiving cells from either Aire- or Batf3-deficient mice (Figures 2A–2C and Figure S3A). In addition to polyclonal T cells, we asked whether pathology could be induced by Tconv cells expressing the BATF3- and AIRE-codependent Treg TCR G25 (Perry et al., 2014). Transgenic mice were generated bearing the thymic Treg TCR G25 and a reporter of Foxp3 (Foxp3IRES-GFP). Cells from naïve G25 transgenic Foxp3IRES-GFP Rag1−/− mice induced weight loss (Figure 2E) and lung inflammation in TCRb−/− Tcrd−/− mice reminiscent of polyclonal T cell transfers from Batf3- or Aire-deficient mice. Thus, these data supported the notion that BATF3, like AIRE, contributes to the induction of thymic T cell tolerance.
Figure 2. T cell selection by CD8α+ DCs prevents tissue-specific inflammation.

(A–C) (A) Experimental design: TCRb−/−Tcrd−/− mice were injected with a mixture of sorted thymic Tconv (106) and Treg (5×104) cells from wild-type (WT), Aire−/−, or Batf3−/− mice. (B) Mice were imaged 2.5 weeks post transfer by FDG PET (top row) and hematoxylin and eosin (H&E) histology (bottom row) of the lung. Yellow arrows indicate regions of increased radiolabeled glucose uptake. Images are representative of two independent experiments with 2–3 mice per condition. (C) Quantification of radiolabeled glucose uptake in lung, spleen, and muscle. Data is represented as change from WT (counts KO ÷ WT). Each dot represents data from an individual mouse; bar indicates mean value.
(D) Weight loss was assessed in TCRb−/−Tcrd−/− mice injected with 106 G25 TCR transgenic or polyclonal thymic Tconv cells. Weights (mean ± SEM) were measured weekly, and analyzed using repeated measures ANOVA with Sidak’s multiple comparisons correction.
(E) 1×105 Naïve G25 TCR transgenic CD4+ T cells were sorted, cell trace violet (CTV) labeled, and injected into Batf3+/− or Batf3−/− mice. Proliferation of and CD44 expression by CD45.2 marked G25 cells was assessed by flow cytometry of the spleens and pooled lymph nodes (LN) 4 days after transfer. Each dot represents data from an individual recipient from two independent experiments. *p < .05, **p < .01, ***p < .001; Student’s t-test.
See also Figure S3.
Batf3-deficient mice are not reported to develop spontaneous autoimmunity (Hildner et al., 2008). We hypothesized that although harboring defective central tolerance, the delayed or absence of autoimmunity observed in these mice may be due to a defect in antigen presentation in the periphery. Consistent with this hypothesis, Batf3-deficiency abrogates the development of Type 1 Diabetes in the Non-Obese Diabetes (NOD) model (Ferris et al., 2014). To further test this possibility, we asked whether peripheral CD8α+ DCs were required to activate cells expressing thymic BATF3-dependent Treg TCRs in vivo. We assessed the proliferation of naïve G25 TCR transgenic cells (Figure 2F), and CD44 upregulation in 7 other TCRs (Perry et al., 2014) retrovirally transduced into the human CLIP antigen-specific TCliαβ TCR transgenic Rag1−/− CD4+ cells (Hsieh et al., 2006) (Figure S3B). This analysis of eight TCRs supported the hypothesis that BATF3-dependent CD8α+ DCs are involved in presenting a distinct array of antigens in both the thymus as well as the periphery.
CD36 is involved in transfer of cell-surface antigens from mTECs
We next sought to determine the mechanism(s) by which CD8α+ DCs acquire antigens from mTECs, hypothesizing that CD8α+ DCs uniquely express receptors that facilitate antigen transfer from mTECs. Query of the Immunological Genome Project (ImmGen) (Heng et al., 2008) database revealed one potential candidate, CD36, expressed at a much higher level on thymic and splenic CD8α+ DCs compared with other DC subsets. CD36 is reported to recognize phosphatidylserine (PS) (Rigotti et al., 1995), and may facilitate the acquisition and cross-presentation of antigens derived from apoptotic cells in some studies (Albert et al., 1998) though not others (Belz et al., 2002; Schulz et al., 2002). CD36 on CD8α+ DCs may thereby facilitate antigen transfer as apoptosis is part of the normal mTEC life cycle (Gray et al., 2007).
We first confirmed that CD36 was uniquely expressed on the CD8α+ subset of thymic DCs (Figure 3A). Similar to Batf3 deficiency (Figures 1A and 1B), we did not observe effects of CD36 deficiency on overall CD4SP, Tconv, or Treg cell frequencies (Figures S4A and S4B). We then tested whether CD36 was involved in the transfer of cell-surface antigens using BM chimeras into Balb/c hosts that express the MHCII molecule Eα. As C57BL/6 mice do not express Eα, the preferential generation of Eα:I-Ab complexes on CD8α+ vs. SIRPα+ DCs (Ardouin et al., 2016; Perry et al., 2014), as detected using the peptide-in-groove antibody Y-Ae, occurs via antigen transfer from mTECs (Humblet et al., 1994) (Figure 3B). This was markedly diminished in CD8α+ DCs from Cd36−/− BM (Figure 3B). CD36 expression was concomitant with Eα presentation as well as high MHCII and CD80 levels (Figure S4C). However, the decreased Eα presentation observed in Cd36−/− BM chimeras was not a result of lower MHCII levels (Figure 3C). Thus, CD36 is required for the transfer of cell-surface antigens from mTECs to CD8α+ DCs.
Figure 3. CD36 is involved in transfer of cell-surface antigens from mTECs.

(A) Expression of CD36 on DCs from WT thymi by flow cytometry (see Methods). Plots are representative of two separate experiments with at least 2 mice per condition.
(B) Transfer of Eα to DCs in vivo was assessed in BM chimeras generated from either Cd36+/− or Cd36−/− donors into irradiated H-2d hosts. CD8α+ and SIRPα+ DC subsets were analyzed for Y-Ae expression 4 weeks after transplantation. Gates were drawn according to H-2b syngeneic BM chimera controls. Plots summarize data from two experiments with 2 mice per condition (mean + SEM).
(C) Expression of I-Ab and CD80 was assessed on thymic DCs from Cd36+/− and Cd36−/− mice by flow cytometry. Plots are representative of two separate experiments with at least 2 mice per condition.
(D) Transfer of a cytoplasmic antigen, GFP, was assessed in BM chimeras generated from a 1:1 mix of Cd36+/+ Ly5.1+ and Cd36−/− donors into mice expressing a BAC transgene in which Aire drives GFP expression (Adig). CD8α+ and SIRPα+ DC subsets were analyzed for GFP expression 4 weeks after transplantation, using gates from BM chimeras using Adig-negative hosts. Plots summarize data from one experiment with four Adig+ mice (mean + SEM). ***p < .001; Student’s t-test.
We also asked whether CD36 was involved in mediating antigen transfer of cytoplasmic GFP antigen from mTECs using the Adig BAC transgene, in which GFP is expressed via the Aire promoter (Gardner et al., 2008). Analysis of Cd36+/+ BM donor APC subsets into Adig+ hosts confirmed our previous conclusion that CD8α+ DCs preferentially acquired GFP from host mTECs (Figure 3D), but did not do so in a CD36-dependent manner. Additionally, we tested a second intracellular source of antigen using the mitochondrial-localized GFP reporter mouse (mito-GFP). Similar to cytosolic-localized GFP, TEC-derived, mito-GFP was transferred to CD8α+ DCs in a CD36-independent manner (Figure S4D). Thus, these studies of model antigens demonstrated that CD36 is important for cell-surface, but not cytoplasmic- or mitochondrial-derived, antigen transfer from mTECs to CD8α+ DCs
CD36 facilitates Tconv and Treg cell TCR repertoire development
Though the effect of Cd36-deficiency on the transfer of mTEC Eα was substantial, it remained possible that this was a unique interaction between CD36 and Eα. We therefore tested whether Cd36-deficiency affected the thymic TCR repertoire as above. We found no major shifts in the TCR repertoire (Figure 4A, top panels, points on the diagonal). An overall normal T cell selection process is consistent with the observation that thymocytes rarely express CD36 (ImmGen, Figure S4E). However, the TCR repertoire analysis revealed a requirement for CD36 for negative selection of a subset of the Tconv (~3%) and Treg (~5%) cell TCR repertoire and the selection of ~7% of the Treg cell TCR repertoire (Figures 4A–4C and Figure S4F). In addition, we corroborated our sequencing data with in vivo Treg cell development studies using retroviral transduction of Cd36+/+ Rag1−/− thymocytes with CD36-dependent (R35, R117) and -independent (G25, G41) Treg cell TCRs (Figures 4D and 4E), which we previously found were BATF3- and AIRE-codependent (Perry et al., 2014). Four Treg TCRs that are not dependent on BATF3 or AIRE were also not affected by CD36 absence in vivo as expected (Figure S4G). Taken together, these data demonstrated that CD36 exerts T cell-extrinsic effects on the Treg and Tconv cell TCR repertoire.
Figure 4. CD36 facilitates Tconv and Treg cell TCR repertoire development.

(A) Top panels: Plotted are the average frequency of Tconv and Treg TCRs in Cd36+/− or CD36−/− → Ly5.1 BM chimeras. Red dots indicate TCRs that show p < .05 MWU and 5-fold change. Bottom panels: Unsupervised clustering analysis of Tconv and Treg cell TCR repertoires from Cd36+/− and Cd36−/− BM chimeras (n of 9–10 mice in two independent experiments).
(B,C) The percentage of unique TCRs or total sequences in the filtered data set that are dependent on CD36 for negative selection (B) or Treg cell selection (C), based on MWU and fold-change criteria in (A) as per Figure 1D.
(D) Frequency of Treg cell TCRs in Cd36+/− and Cd36−/− BM chimeras previously identified to be BATF3 and AIRE-codependent (G25, G41, R117, R35) from in vivo developmental studies.
(E) Induction of Foxp3 in Cd36+/+ Rag1−/− thymocytes retrovirally transduced with TCRs and intrathymically injected into Cd36+/− and Cd36−/− hosts. Shown are representative flow cytometry plots gated on retrovirally transduced CD4SP cells and summary plots. Each dot represents data from a single recipient. Each TCR was analyzed in at least 2 independent experiments (1–3 mice per genotype).
(F) Venn diagram representations of Tconv and Treg cell TCRs dependent on BATF3, AIRE, and CD36. Values represent absolute number of TCRs, and Venn diagram sections are sized proportional to number. All sequencing data are representative of two independent experiments with 5 mice per condition. **p < .01, ***p < .001; Student’s t-test.
See also Figure S4.
By cross-referencing our data sets from Cd36 and Batf3-deficient conditions, we observed that many BATF3-dependent TCRs were not affected by CD36, suggesting that CD36 does not globally affect CD8α+ DC antigen presentation (Figure 4F). However, the majority of our CD36-dependent TCRs were BATF3-dependent (Figure 4F), suggesting that the effect of CD36 on the TCR repertoire is primarily due to its expression on CD8α+ DCs and not macrophages.
We then asked whether CD36 was important for antigen transfer, which we infer based on TCRs that are codependent on AIRE and BATF3. Of the few Tconv cell TCRs codependent on BATF3 and AIRE for negative selection, 50% were also dependent on CD36 (Figure 4F, left diagram). Similarly, approximately 60% of the Treg cell TCRs codependent on BATF3 and AIRE also required CD36 for Treg cell selection (Figure 4F, right diagram). Though it remains unknown how prevalent antigen presentation via transfer of cell-surface or cytoplasmic antigens is in vivo, inspection of AIRE-dependent tissue-restricted transcripts revealed that ~25% have a trans-membrane domain by GO (207/804 genes; >50 counts, 10× enriched vs Aire−/− (Sansom et al., 2014)). This is similar to the percentage of TCRs that we observe to be BATF3-, CD36-and AIRE-dependent (27/105 AIRE-dependent TCR). Thus, these data suggested that CD36 contributes substantially to cooperative antigen presentation of AIRE-dependent antigens by BATF3-dependent CD8α+ DCs for the development of the Treg and Tconv cell TCR repertoires.
CD36 acquires cell-surface antigen via scavenging of apoptotic bodies
We next sought to determine the process by which CD36 facilitated antigen acquisition from mTECs in vitro. As we are not aware of an Aire+ H-2d-expressing mTEC line, we used the TA3 hybridoma line as an imperfect surrogate (Glimcher et al., 1983). To assess antigen transfer, cell trace violet (CTV)-labeled BM-derived CD24+ DCs (BMDCs), which are of a similar developmental lineage as thymic CD8α+ DCs (Naik et al., 2005), were co-cultured with apoptotic GFP+ TA3 cells for 6 hours (Figure S5A). Transfer and presentation of TA3-derived Eα on I-Ab in DCs was CD36-dependent (Figure 5A), consistent with our in vivo data (Figure 3B), To demonstrate that our findings were not confounded by the use of BM-derived DCs, we isolated thymic DCs from Cd36-deficient and sufficient mice, followed by co-culture with apoptotic TA3 cells. As observed with CD24+ BMDCs, the absence of CD36 resulted in significantly decreased presentation of Eα (Figure S5B). To rule out that our observations are due to an underlying developmental phenotype, we isolated and cultured Cd36-sufficient thymic CD11c+ DCs with apoptotic TA3 cells in the presence or absence of a CD36 blocking antibody. Similar to our findings with both CD24+ BMDCs and CD8α+ thymic DCs, we observed significant reduction in Eα presentation by thymic CD8α+ DCs (Figure S5C). GFP was not detected with DCs by flow cytometry (Figure S5A), which suggested that the apoptotic bodies were not stuck to DCs in this in vitro system, although GFP transfer appeared to occur in vivo (Figure 3D).
Figure 5. CD36 acquires cell-surface antigen via scavenging of apoptotic bodies.

(A) DC acquisition and presentation of Eα using the Y-Ae antibody was assessed on Cd36+/− and Cd36−/− BM-derived CD24+ DCs co-cultured with apoptotic H-2d-bearing GFP+ TA3 cells for 6h at 0 °C and 37°C. FACS plots are representative o f three independent experiments with at least two independent replicates per condition.
(B) Acquisition of intact peptide/MHC from TA3 cells was determined by flow cytometry of I-Ad expression on CD24+ DCs as per (A). FACS plots are representative of two independent experiments with at least two independent replicates per condition.
(C) TA3 cells were pre-treated with an inhibitor of apoptosis, 50µM z-VAD-FMK (zVAD), 1h prior to co-culture as in (A). For transwell (0.4 µm) experiments, CD24+ DCs and apoptotic TA3 cells were cultured in the bottom and top chambers, respectively. FACS plots are representative of two independent experiments with at least two independent replicates per condition.
(D) Antigen transfer of Eα on DCs was assessed by Y-Ae expression in BM chimeras generated by mixing 1:1 P2ry14+/+ Ly5.1+ and P2ry14−/− BM donors injected into lethally irradiated Balb/c. Thymi were harvested 4 weeks post injection for analysis of CD8α+ and SIRPα+ DC subsets as per Figure 3B. Data are representative of two experiments with 3–4 mice per condition. All data are summarized as mean + SEM. ***p < .001, one-way ANOVA with Tukey’s post hoc test.
See also Figure S5.
CD36-mediated acquisition of I-Eα could be via endocytosis, and/or directly to the DC cell membrane via trogocytosis or related processes followed by MHC internalization. Consistent with the latter, we observed diminished I-Ad expression on Cd36-deficient CD24+ DCs after co-culture with apoptotic GFP+ TA3 cells (Figure 5B). We also asked whether I-Ad could be picked up in this manner by CD8α+ DCs in vivo. However, it appeared that the process of enzymatic digestion at 37°C for DC isola tion resulted in cell-surface MHC transfer (Figure S5D), rendering the in vivo analysis uninterpretable. By contrast, the 30 minute digestion with APCs of different MHC haplotypes did not generate Eα:I-Ab complexes detectable by Y-Ae (Figure S5D), implying that generation of Y-Ae on CD8α+ DCs in our BM chimeras (Figure 3B) must have occurred in vivo. In summary, our in vitro data suggested that CD36 may be involved in the transfer of intact cell surface MHC molecules to be displayed on CD8α+ DCs.
CD36 has several known ligands, including PS and thrombospondin on apoptotic bodies or exosomes that could be involved in antigen transfer from mTECs. To assess the role of apoptotic bodies, we pre-treated GFP+ TA3 cells with the pan-caspase inhibitor z-VAD-FMK prior to co-culture with CTV+ CD24+ DCs. Inhibition of apoptosis resulted in significantly diminished acquisition and presentation of Eα, which was further decreased in conjunction with Cd36-deficiency (Figure 5C). To assess the role of exosomes, we co-cultured GFP+ TA3 cells and CTV+ CD24+ DCs separated by a 0.4 µm membrane sufficient to allow access to exosomes but restrict passage of larger bodies (e.g., apoptotic bodies). This reduced the acquisition and presentation of Eα, arguing against a role for exosomes (Figure 5C). However, it remains possible that TA3 generates less exosomes than mTECs, or that the concentration of exosomes generated was insufficient to overcome the different physical characteristics of the culture system. While these data cannot exclude a role for exosomes in vivo, these in vitro data supported the published notion that CD36 is involved in the recognition of apoptotic bodies.
We next sought to test if apoptosis was required for Eα acquisition and presentation in vivo. To do so, we performed C57BL/6→Balb/c BM chimeras but treated mice with either the pan-caspase inhibitor z-VAD-FMK or vehicle for three days prior to harvest. We found that CD8α+ DCs from mice treated with z-VAD-FMK presented significantly less Eα than vehicle-treated mice (Figure S5E). Along with “eat me” signals, the phagocytosis of apoptotic cells also requires a signal to home to a dying cell (“find me”), including purine nucleosides and nucleotides, such as ATP (Poon et al., 2014). We asked whether there were purinergic receptors that were differentially expressed by thymic CD8α+ DCs. Analysis of ImmGen revealed that P2Y14 (gene P2ry14; previously known as GPR105;), an UDP-glucose-responsive metabotropic receptor (Abbracchio et al., 2003; Lee et al., 2003), is highly expressed by both thymic and splenic CD8α+ DCs but not SIRPα+ DCs. Transfer of P2Y14-deficient BM into Balb/c mice resulted in a significant decrease in Eα presentation by CD8α+, but not SIRPα+, DCs (Figure 5D). Additionally, we treated C57BL/6→Balb/c BM chimeric mice with either the purinergic receptor inhibitor Suramin or vehicle for three days prior to harvest. We found that CD8α+ DCs from mice treated with Suramin presented significantly less Eα than vehicle-treated mice (Figure S5F). Taken together, our in vitro and in vivo data supported the notion that CD8α+ DCs use CD36 to acquire cell-surface antigens from apoptotic mTECs.
CD36-dependent antigen transfer is necessary for direct allo-tolerance development
Based on previous reports (Dolan et al., 2006; Li et al., 2012; Mazzini et al., 2014; Qu et al., 2009), as well as our in vitro observation that MHCII can be transferred directly to the cell surface of CD8α+ DCs, we asked whether this might be involved in the development of thymic tolerance in vivo during allo-mismatched BMT. To test this, we generated fully-MHC mismatched allogeneic BM chimeras using C57BL/6 (B6) donors and Balb/c hosts. To assess whether thymic allo-tolerance was achieved, mature CD25− Tconv thymocytes from these mice were used to induce acute Graft-versus-Host disease (GVHD) in the context of a new BM transplant (Figure 6A). As expected, mice receiving thymic Tconv cells from Batf3+/− B6 → Balb/c BM chimeras showed minimal evidence of acute GVHD compared with those receiving Tconv cells from B6 → B6 autologous BM chimeras that have never been exposed to H-2d (Figure 6B). The process of BM chimera generation did not appear to affect the ability of thymocytes to induce acute GVHD, as Tconv cells from syngeneic B6 BM chimeras were equivalent to those from normal B6 mice (Figure S6A). Thus, these data confirmed that thymic T cell tolerance to host MHC occurs during allogeneic BM transplantation.
Figure 6. CD36-dependent antigen transfer is necessary for direct allo-tolerance in the thymus in mouse BMT.

(A) Experimental design: Bone marrow (BM) from H-2bBatf3+/−Batf3−/−, or Cd36−/− mice was transplanted into Balb/c H-2d hosts. As a positive control, Batf3+/− H-2b BM was transplanted into Ly5.1+ H-2b hosts.
(B) Acute in vivo GVHD was assessed using 106 sorted thymic CD4+ Tconv (CD25−) cells from the indicated BM chimera co-injected with T cell-depleted H-2b BM into H-2d hosts as per (A). Weight change, clinical score, and the fraction of mice reaching 20% weight loss requiring euthanasia was measured. Summary plots are of 2–4 independent experiments with 2–4 mice per condition (mean ± SEM).
(C) In vitro direct MLR was assessed by culturing 2.5×104 CTV labelled Tconv cells obtained as per (A) with irradiated H-2d splenocytes (2.5×104 cells) for 5 days. Proliferation of Tconv cells was assessed by CTV dilution using flow cytometry. Plots are representative of 2–3 independent experiments. Each dot in the summary plot represents the average of 2 technical replicates from an individual mouse; bar indicates group mean. ***p < .001, one-way ANOVA with Tukey’s post hoc test.
See also Figure S6.
By contrast, loss of thymic tolerance to host antigens occurred with deficiency in either BATF3 or CD36 in the donor B6 background BM. Thymic Tconv cells from the respective BM chimeras induced acute GVHD as assessed by weight loss, clinical score, and time to euthanasia (Figure 6B). Tconv cells from Cd36−/− → Balb/c chimeras were marginally less potent than Batf3−/− → Balb/c chimeras, consistent with the incomplete block of MHC transfer with Cd36 deficiency (Figure 3B). We did not find evidence for a cell-intrinsic T cell hyperreactivity imparted by Batf3-deficiency, as T cells from non-chimeric Batf3-deficient and -sufficient mice showed similar induction of GVHD (Figure S6B). Moreover, Cd36-deficient and -sufficient thymocytes isolated from mixed BM chimeras with B6 hosts showed no difference in their ability to induce GVHD (Figure S6C), suggesting that expression of CD36 on T cells is not critical for this immune response. Thus, these data suggested that CD36-dependent MHC transfer to CD8α+ DCs is required for thymic allo-tolerance.
Allo-recognition can occur via direct recognition of allogeneic MHC by the TCR, or indirectly via recognition of allogeneic peptides presented on self-MHC (e.g. Eα peptide on I-Ab). To test direct allo-recognition, we cultured sorted thymic Tconv cells in the presence of irradiated Balb/c splenocytes (Figure 6A). Tconv cells from Batf3+/− → Balb/c chimeras showed minimal proliferation, consistent with the in vivo results suggesting that these cells were allotolerant (Figure 6C). By contrast, we observed a clear population of activated/proliferated (CD25+ CTVdim) Tconv cells from Batf3−/− → Balb/c or Cd36−/− → Balb/c chimeras (Figure 6C), consistent with direct allorecognition. The frequency of activated/proliferated cells (CD25+ CTVdim) from Batf3- or Cd36-deficient donors was lower than Tconv cells from non-tolerant conditions using autologous B6 hosts (Figure 6C), suggesting that partial allo-tolerance did occur to host MHC presented on cTECs and mTECs. A deficiency in tolerance to direct allo-recognition was also consistent with the early time course of GVHD in vivo (Figure 6B), as donor APCs would not have seeded the periphery in large numbers. Additionally, it was unlikely that the observed phenotype is due to killing of BM by donor T cells because mice lethally irradiated that received no BM did not exhibit any of the clinical features observed in our GVHD model (data not shown).
Our finding that both CD8α+ DCs and CD36 were required for allo-tolerance development in mice predicted that patients who receive partial HLA-mismatch BMT may depend on donor BM-derived CD141+ DCs (the human equivalent of CD8α+ DCs) and CD36 to develop thymic allo-tolerance and prevent chronic GVHD (cGHVD). To address this, we performed a blinded analysis of peripheral blood mononuclear cell samples collected 30 days post-transplantation from a cohort of allo-BMT recipients, some of whom developed cGVHD (Figure S7A). We observed that patients who develop cGVHD had significantly fewer CD141+ DCs which expressed lower levels of CD36 (Figure 7A and Figure S7B), regardless of demographic or clinical characteristics (χ2 = 18.7, df(2), p<.001). We observed no significant differences in white blood cell count, neutrophil count, neutrophil frequency (Figure 7A), nor in blood monocyte populations, plasmacytoid DCs, or CD1a+ DCs (Figure S7C). Thus, the development of cGVHD was associated with decreased abundance of CD141+ DCs and CD36 expression. In summary, these data supported a model in which the efficiency of CD8α+ (CD141+) DC reconstitution after allogeneic BMT was associated with the induction of tolerance to direct host MHC recognition via CD36-dependent transfer of thymic CD8α+ DCs (Figure 7B).
Figure 7. Reduction of CD141+ DCs in peripheral blood predicts progression to chronic GVHD in human BMT patients.

(A) Analysis of human PBMCs from allo-BMT recipients 30 days post-transplantation (see also Figure S7). Representative FACS plots of CD141+ DCs in patients who develop cGVHD or remain cGVHD-free. Percentage of CD141+ cells and CD36 MFI (median fluorescence intensity) are summarized to the right. For CD36 MFI, Open circles indicate a sample with fewer than 50 gated cells. White blood cell (WBC) count, absolute neutrophil count (ANC), and neutrophil percentage was obtained from a clinical complete blood count analysis from the same blood draw. Each individual point represents a single patient sample. Red dots indicate patients who self-reported cGVHD symptoms at least 30 days after this peripheral blood draw. *p<.05, ***p<.001.
(B) Working model of CD36-mediated antigen transfer to CD8α+ DCs (CD141+ DCs in humans). Apoptotic mTEC cells are sensed via P2Y14 on CD8α+ DCs. AIRE-dependent cell-surface self-antigens are then captured via a CD36-dependent mechanism that results in transfer and direct display of proteins such as MHC on CD8α+ DCs, as well as internalization and presentation of peptides on MHC synthesized by CD8α+ DCs. Both of these mechanisms may be involved in tolerance to self- and allo-antigen.
See also Figure S7.
Discussion
Although the process of self-antigen transfer from mTECs to BM APCs has been recognized for over 2 decades, its impact on thymic T cell development and the mechanism(s) by which this process occurs has not been described. We made the following observations: First, antigen transfer to CD8α+ DCs made a significant contribution to AIRE-dependent thymic tolerance based on TCR repertoire analysis in a fixed TCRβ model. Second, antigen transfer from AIRE-expressing mTECs to CD8α+ DCs occurred via at least two distinct routes that segregate between cell-surface and cytoplasmic antigens. Third, cell-surface antigen transfer was dependent on CD36, a scavenger receptor preferentially expressed on the CD8α+ subset of DCs. Fourth, CD36-mediated antigen transfer resulted in the initial display of mTEC cell-surface proteins on DCs. Finally, this process is required for allo-tolerance in murine BMT, and, in initial studies, may also be relevant for humans. Thus, our data demonstrated that antigen transfer can occur via multiple pathways, and suggested that CD36-mediated uptake of cell-surface proteins by CD8α+ DCs plays an important role in tolerance to mTEC-derived antigens.
Previous studies suggest that thymic CD8α+ DCs are important in thymic tolerance (Lei, 2011; Lin et al., 2016; Perry et al., 2014). Here, we have quantified the effect of Batf3-deficiency on the CD4SP TCR repertoire in a fixed TCRβ model. The analysis of a fully polyclonal repertoire introduces the experimental complexity of pairing a and b chains for sequencing, as well as increased mouse to mouse variability due to the much greater repertoire diversity. With this restriction of the repertoire in mind, our results demonstrated that BATF3-dependent DCs affect both negative selection and Treg cell selection. Cross-correlation with the effect of AIRE on the TCR repertoire led to an estimate that CD8α+ DCs contributes to only ~6% of AIRE-dependent negative selection of Tconv cells, versus ~20 and ~40% for AIRE-dependent deletion and Treg cell selection, respectively. Thus, our data showed that CD8α+ DCs affect thymic T cell selection and play an important role via cooperative antigen presentation with mTECs.
The mechanism utilized by CD8α+ DCs to acquire cell-surface antigens from mTECs appears to be the recognition of apoptotic bodies by CD36. CD36 is expressed on thymic CD8α+ DCs but not SIRPα+ or pDCs, and is reported to bind PS present on apoptotic bodies. We found using TCR repertoire analysis that CD36 was involved in both negative selection and Treg cell differentiation with a strong bias toward co-dependence with BATF3 and AIRE, implying that CD36 plays a role in antigen transfer to CD8α+ DCs. In addition, absence of CD36 resulted in the loss of CD8α+ DC acquisition and presentation of membrane-bound I-Eα antigen from mTECs in vivo. Finally, deficiency in the purinergic receptor P2Y14, which recognizes UDP potentially released by apoptotic cells, decreased CD8α+ DC presentation of cell-surface-bound antigen acquired from mTECs. Taken together, these data suggested a model in which mTECs mature, express AIRE, and ultimately undergo apoptosis to generate bodies that are recognized by CD36 on CD8α+ DCs.
The observation that MHC is directly transferred to the CD8α+ DC cell surface brings up the possibility that mTEC-loaded peptide:MHC complexes directly mediate negative selection or Treg cell differentiation (Koble and Kyewski, 2009). Alternatively, mTEC-derived cell surface antigens can be internalized, degraded, and loaded onto MHCII as detected by the peptide-in-groove antibody Y-Ae. This mechanism could explain the observation of antigen transfer using the RIP-mOVA transgene (Koble and Kyewski, 2009), which expresses a transmembrane protein that encodes the OVA peptide. Understanding which cell-surface proteins are transferred from mTECs has unfortunately been limited by the possibility of cross-contamination during our thymic DC preparation protocol. Future studies are required to determine the relative importance of CD8α+ presentation of intact peptide/MHC transferred from mTECs versus internalization and presentation of mTEC-associated cell-surface antigens.
Our finding in mice that CD8α+ DCs, partially through CD36, mediated allo-tolerance development, led us to question if this phenomenon may also be relevant to human allo-BMT in which HLA-mismatching can enhance graft versus leukemia effects. Quantification of CD36 and CD141+ DCs, the human CD8α+ DC-equivalent, in the peripheral blood of allo-BMT recipients 30 days after transplant revealed that patients who develop chronic GVHD also had fewer CD141+ DCs and lower CD36 expression. While this finding was correlative, it was consistent with our murine studies that antigen transfer from recipient thymic stroma to donor CD8α+ DCs is important for allograft tolerance. Prospective studies of both donor and recipient blood cell populations are needed to further delineate the clinical utility of our finding.
In summary, these data suggested that antigen transfer from mTECs is mediated to a large part by CD8α+ DCs via CD36 and is demonstrably important to thymic T cell selection and the shaping of the TCR repertoire. We have also determined that CD36 is required for direct transfer of MHC and presumably other cell-surface proteins from mTECs onto the cell surface of CD8α+ DCs. One intriguing hypothesis is that defects in this pathway may lead to loss of tolerance to cell-surface proteins, potentially contributing to antibody mediated diseases such as Graves autoimmune thyroiditis that target cell-surface receptors. In a similar vein, defects in the transfer of cell-surface antigens may contribute to the loss of allo-tolerance during BMT, resulting in chronic Graft-versus-Host disease.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for reagents should be directed to the Lead Contact, Chyi-Song Hsieh (chsieh@wustl.edu)
Experimental Model and Subject Details
Mice
Animal breeding and experiments were performed in a specific pathogen-free animal facility using protocols approved by the Washington University Animal Studies Committee. All mice were on a C57BL/6 genetic background unless otherwise indicated. TClib mice (Wong et al., 2007), Batf3−/− (Hildner et al., 2008), Cd36−/− (Febbraio et al., 1999), Aire-G6pc2/GFP (Adig, Gardner, et al., 2008), and P2ry14−/− (Cho et al., 2014) have all been described previously. Aire− /− (Stock# 004743), Rag1−/− (Stock# 002216), Tcra−/− (Stock# 002116), TCRb−/−Tcrd−/− (Stock# 002121) and Foxp3IRES-GFP (Stock# 006772) mice were purchased from The Jackson Laboratory. Male Balb/c mice (Stock# 028) were ordered from Charles River and acclimated for one week prior to use. G25 TCR transgenic mice were generated by cloning the cDNA for G25 TCRα chain into the VA-hCD2 expression vector, which was then co-injected with the TCli TCRβ chain in the pTβ vector (Bautista et al., 2009). G25 transgenic mice did not show any overt signs of autoimmunity, even on a Rag1−/− background. Expression of TCRβ was comparable between transgenic and WT cells from the thymus or spleen (data not shown). G25 TCR transgenic mice were bred to Foxp3IRES-GFP Rag1−/− mice. Animals were typically 6–10 weeks old at the time experiments were performed and consisted of males and females.
Human Samples
Peripheral blood samples were banked pre and post hematopoietic stem cell transplantation on an IRB approved institutional tissue repository protocol after recipients provided informed consent. Blood samples were collected in EDTA blood tubes at days 30 after transplant. Peripheral blood mononuclear cells were isolated by Ficoll gradient and control rate frozen and stored under liquid nitrogen until use. White blood cell count, absolute neutrophil count, and neutrophil frequency were obtained via clinical complete blood count analysis from the same blood draw. Samples were de-identified and assigned a unique patient number, then clinically annotated for chronic GVHD retrospectively using clinical records. Patient population characteristics, including age, gender, reason for bone marrow transplantation, donor type, HLA match grade, GVHD symptoms, time of GVHD onset, and days since last follow-up can be found in Figure S7.
Method Details
Reagents, antibodies and flow cytometry
Fluorescently conjugated monoclonal antibodies were purchased from Biolegend, eBioscience, and Becton Dickenson. Y-Ae anti-I-Ab:Eα (52–68) peptide was obtained from eBioscience. Samples were analyzed using a FACSAria or FACSCanto (Becton Dickinson) and data were processed with FlowJo X (Treestar).
Thymic dendritic cell isolation and staining
Thymus was mechanically separated with scissors and digested with Liberase TL (125 µg/µl, Roche) and DNase I (50 µg/µl, New England Biolabs) in DMEM for 30 min at 37°C. Cells were stained with 2.4G2, B220, MHCII, CD11c, CD11b, CD8α, SIRPα, CD24 and experiment-specific antibodies as indicated. DC subsets were identified as follows: CD8α+ DCs (CD11chi MHCIIhi B220− CD11b− SIRPα− CD24+ CD8α+), SIRPα+ DCs (CD11chi MHCIIhi B220− CD11b+ SIRPα+ CD24− CD8α−), and pDCs (CD11c+ B220+).
Assessment of thymic Treg cell selection in vivo
As described previously (Bautista et al., 2009), TCRa chains of interest were cloned into the MigR1-TCRa-P2A-TCliβ retroviral vector. Foxp3IRES-GFP Rag1−/− thymocytes were transduced with TCRs in vitro, injected intrathymically into sublethally (600 rad) irradiated mice, and analyzed approximately 2.5 weeks later. Thymocytes were transduced with TCR vector containing no reporter, an IRES-Thy1.1 or an IRES-huCD2 prior to injection.
Bone marrow chimeras
BM was obtained by flushing donor humerus, tibia, and femur. BM was then RBC lysed and T cell-depleted by labeling cells with biotinylated anti-CD4 and anti-CD8 and anti-biotin microbeads, followed by magnetic cell separation using an AutoMACS (Miltenyi Biotech). 5×106 cells were injected into either 950 rad lethally irradiated C57BL/6 host mice or 750 rad lethally irradiated Balb/c host mice. Mice were maintained on antibiotic water one day prior and one week after transplantation. Over 99% chimerism was confirmed using Ly5.1 WT donors.
In vitro phagocytosis assay
BM from Cd36+/− and Cd36−/− mice was cultured in 100ng/ml of Flt3L (Peprotech) for 11 days. CD24+ DCs were sorted, CTV labeled, and cultured with apoptotic H2d-expressing GFP+ TA3 cells for 6h. Induction of apoptosis was induced with 150mJ UV-C using a Stratalinker UV Crosslinker. CTV+ cells were then analyzed by FACS for Y-Ae and I-Ad expression. GFP+ CTV− cells were excluded from analysis as non-engulfed cells. In some experiments, TA3 cells were pre-treated with 50uM z-VAD-FMK (Cayman Chemicals) for 1h prior to induction of apoptosis; or TA3 cells were cultured on the top layer of 0.4µm Corning HTS 96 well transwell plates separated from the DCs on the bottom layer. In other experiments, thymi from C57BL/6 or Cd36−/− mice were harvested and pooled, followed by positive selection of CD11c+ cells via MACS column. CD11c+ cells were cultured with apoptotic GFP+ TA3 cells for 6h with either the antibody that functionally blocks CD36 (JC63:1, Cayman Chemicals) or vehicle. All experiments were carried out at 37°C, as well as on ice (0°C), which allows binding but inhibits phagocytosis.
Imaging studies
Mice were withheld food 12–24 h prior to imaging. On the day of imaging, mice were anesthetized with 1.5–2% isoflurane in oxygen inside of a Plexiglas chamber and subsequently injected with fludeoxyglucose (FDG, 100 µl/20g mouse). Mice were imaged using microCT to obtain anatomical scans, and then undergo a 10 min transmission scan followed by a 10 min emission scan. All imaging was performed via a Siemens Inveon-MM microCT/PET imager. Co-registration and analysis was performed using Siemens Inveon Research Workplace software.
Naïve T cell transfer studies
Foxp3− CD44lo CD62Lhi G25 TCR transgenic Rag1−/− cells were sorted and retro-orbital injected into indicated mice. In some experiments, cells were first labeled with Cell Trace Violet (Thermo Fisher). For histological studies, 6–8-week-old littermate male TCRβδ−/− mice were used. In peripheral activation studies, 6–8-week-old littermate Batf3+/− and Batf3−/− mice were used. 105 naïve T cells were injected per mouse. For peripheral activation studies, pooled lymph nodes and spleens were harvested 4 days after transfer.
Allogeneic bone marrow chimeras, mixed lymphocyte reaction, and acute GVHD
Initial BM chimeras were performed as described above. Eight weeks post-transplant, thymic CD62Lhi CD24loCD4SP Tconv cells were sorted. For in vitro experiments, sorted cells were CTV labeled and co-cultured with irradiated (2000 rad) splenocytes from Balb/c mice. Samples were analyzed for CD25 expression and CTV dilution 5 days post co-culture. Donor Tconv cells were distinguished from Balb/c splenocytes by H-2Kb (AF6-88.5) and H-2Kd (SF1–1.1) expression. For in vivo experiments, 106 sorted Tconv cells were injected into lethally-irradiated Balb/c mice concurrently receiving allogenic 5×106 T cell-depleted C57BL/6 BM. In some experiments, Tconv cells came from adult mice instead of from BM chimeras. Mice were assessed for weight loss beginning 4 days post injection. Mice were sacrificed after ≥20% loss of initial body weight. Additionally, mice were scored for disease severity using a standard 10 point scale that assesses weight loss, posture, mobility, fur texture, and skin integrity (Cooke et al., 1996).
Quantification and Statistical Analysis
Data processing and statistical analysis
For TCR analysis, only TRAVs with summed reads greater than 1% in the WT multiplex TCR data were analyzed. This accounted for more than 95% of the total TRAV repertoire. TRAV_CDR3 species frequencies were then multiplied by a correction factor determined by the ratio of template switch TRAV frequency (Mamedov et al., 2013) to multiplex TRAV frequency using data obtained from 2 biological replicates each with 2 technical replicates sequenced both ways. These frequencies were used to determine the number of a particular TRAV_CDR3 clonotype (clonotypes determined via IMGT, (Giudicelli et al., 2006)). All data were rarefied to the smallest sample size. Rarefaction was performed 10 times. Student’s t-tests, one-way ANOVAs, or two-way repeated measures ANOVAs with Tukey’s post hoc test was used for between-subjects analyses unless otherwise noted.
Data and Software Availability
Graphpad Prism v6, IBM SPSS v21, and R v3.2.3 were used for used for graphical and statistical analysis and are available commercially or via open source sharing (R v3.2.3). The R package DESeq2 (v1.10.3) was used for differential TCR expression analysis; and vegan (v2.3–5) for dimensional analysis, rarefaction, and visualization. TCR data is availability via the European Nucleotide Archive (ENA) under the accession #PRJEB25804. All code is available upon request.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse Y-Ae anti-I-Ab:Eα (52–68) peptide | eBioscience | Cat#13-5741-81 |
| anti-mouse CD16/CD32 (Clone# 2.4G2) | BioXCell | Cat#BE0307 |
| Anti-mouse B220 (Clone# RA3-6B2) – AF700 | Biolegend | Cat#103231 |
| Anti-mouse I-Ab (Clone# AF6-120.1) – PerCP-Cy5.5 | Biolegend | Cat#116415 |
| Anti-mouse CD11c (Clone# N418) – PE-Cy7 | Biolegend | Cat#117317 |
| Anti-mouse CD11b (Clone# M1/70) – APC-Cy7 | Biolegend | Cat#101225 |
| Anti-mouse CD8α (Clone# 53–6.7) – PE or APC-Cy7 | Biolegend | Cat#100707 |
| Anti-mouse SIRPα (Clone# P84) – FITC | Biolegend | Cat#144005 |
| Anti-mouse CD24 (Clone# 30-F1) – PE or APC | Biolegend | Cat#138503 |
| Anti-mouse CD80 (Clone# 16-10A1) – Pacific Blue | Biolegend | Cat#104739 |
| Anti-mouse CD45.2 (Clone# 104) – BV605 | Biolegend | Cat#109841 |
| Anti-mouse CD45.1 (Clone# A20) – BV711 | Biolegend | Cat#110739 |
| Anti-human CD45 (Clone# 2D1) – AF700 | Biolegend | Cat#368513 |
| Anti-human CD3 (Clone# OKT3) – PE | eBioscience | Cat#12-0037-42 |
| Anti-human CD19 (Clone# SJ25C1) – PE | eBioscience | Cat#12-0198-42 |
| Anti-human CD56 (Clone# TULY56) – PE | eBioscience | Cat#12-0566-42 |
| Anti-human CD14 (Clone# G3D3) – BV605 | Biolegend | Cat#367125 |
| Anti-human CD11c (Clone# 3.9) – PerCP-Cy5.5 | Biolegend | Cat#301623 |
| Anti-human CD16 (Clone# 3G8) – APC-Cy7 | Biolegend | Cat#302017 |
| Anti-human HLA-DR (Clone# L243) – PE-Cy7 | eBioscience | Cat#25-9952-42 |
| Anti-human CD303 (Clone# 201A) – BV421 | Biolegend | Cat#354211 |
| Anti-human CD1a (Clone# HI149) – FITC | eBioscience | Cat#11-0019-42 |
| Anti-human CD141 (Clone# 1A4) – BV711 | BD Bioscience | Cat#563155 |
| Anti-human CD36 (Clone# 5–271) – APC | Biolegend | Cat#336207 |
| Anti-human CD2 (Clone# 299812) – APC | R&D Systems | Cat#FAB18561A |
| Anti-mouse Thy1.1 (Clone# OX-7) – PE-Cy7 | Biolegend | Cat#202517 |
| Anti-mouse CD4 (Clone# GK1.5) – PerCP-Cy5.5 | Biolegend | Cat#100433 |
| Anti-mouse CD62L (Clone# MEL-14) – BV421 | Biolegend | Cat#104435 |
| Anti-mouse CD44 (Clone# IM7) – APC | Biolegend | Cat#103011 |
| Anti-mouse TCRβ (Clone# H57-597) – PE | Biolegend | Cat#109207 |
| Anti-mouse CD25 (Clone# 3C7) – PE | Biolegend | Cat#101903 |
| Anti-mouse I-Ad (Clone# 39-10-8) – AF647 | Biolegend | Cat#115009 |
| Anti-mouse H-2Kb (AF6-88.5) – APC | Biolegend | Cat#116517 |
| Anti-mouse H-2Kd (SF1-1.1) – FITC | Biolegend | Cat#116605 |
| Anti-mouse CD36 (Clone# D-2712) – APC | BD Bioscience | Cat#562744 |
| Biological Samples | ||
| Peripheral Blood Mononuclear Cells | BM Transplant Patients | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Murine Flt3-Ligand | Peprotech | Cat#250-31L |
| Liberase TL | Roche | Cat#05401020001 |
| DNase I | New England Biolabs | Cat#M0303S |
| CD36 (Clone: JC63:1) | Cayman Chemicals | Cat#10009893 |
| z-VAD-FMK | Cayman Chemicals | Cat#187389-52-2 |
| Critical Commercial Assays | ||
| UV Crosslinker | Stratalinker | N/A |
| Cell Trace Violet | Thermo Fisher | Cat#C34557 |
| Deposited Data | ||
| Aire−/− mice TCR sequencing data | ENA | #PRJEB25804 |
| Batf3−/− mice TCR sequencing data | ENA | #PRJEB25804 |
| Cd36−/− mice TCR sequencing data | ENA | #PRJEB25804 |
| Experimental Models: Cell Lines | ||
| TA3 | Murphy Lab | Glimcher, et al., 1983 |
| Experimental Models: Organisms/Strains | ||
| TCliβ mice | Rudensky Lab | Wong et al., 2007 |
| Batf3−/− mice | Murphy Lab | Hildner et al., 2008 |
| Cd36−/− mice | Abumrad Lab | Febbraio et al., 1999 |
| Aire-G6pc2/GF (Adig) mice | Anderson Lab | Gardner, et al., 2008 |
| P2ry14−/− mice | GlaxoSmithKlein | Cho et al., 2014 |
| Aire−/− mice | Jackson Laboratories | Stock# 004743 |
| Rag1−/− mice | Jackson Laboratories | Stock# 002216 |
| Tcra−/− mice | Jackson Laboratories | Stock# 002116 |
| TCRb−/−Tcrd−/− mice | Jackson Laboratories | Stock# 002121 |
| Foxp3IRES-GFP mice | Jackson Laboratories | Stock# 006772 |
| Balb/c mice | Charles River | Stock# 028 |
| G25 TCR Transgenic mice | N/A | N/A |
| Recombinant DNA | ||
| MigR1-TCRα-P2A-TCliβ-IRES-Thy1.1 | N/A | Bautista et al., 2009 |
| MigR1-TCRα-P2A-TCliβ-IRES-huCD2 | N/A | N/A |
| G25-VA-hCD2 | N/A | Bautista et al., 2009 |
| TCli-pTβ | Rudensky Lab | Wong et al., 2007 |
| Software and Algorithms | ||
| Prism v6 | Graphpad | N/A |
| SPSS v21 | IBM | N/A |
| FlowJo X | TreeStar | N/A |
| R v3.2.3 | The R Project | N/A |
Highlights.
Batf3-dependent CD8α+ DCs mediate negative selection and Treg cell generation
CD8α+ DCs express CD36 which is involved in Aire-dependent T cell tolerance
CD36 facilitates transfer and display of mTEC surface antigens
CD36-mediated antigen transfer is required for allo-tolerance
Acknowledgments
We thank Jinquin Luo (Div. of Biostatistics, WUSTL) for consultation regarding statistical analysis and Nicole Santacruz for expert technical assistance. Experimental support was provided by the Transgenic Facility of the Rheumatic Diseases Core Center. The PET scan studies presented in this work were conducted in the MIR Pre-Clinical PET-CT Facility of the Washington University School of Medicine. C.S.H. is supported by NIH NIAID AI079187 and the Burroughs Wellcome Fund; J.S.A.P. is the recipient of an R01 Research Supplement to Promote Diversity in Health-Related Research, NIH NIAID AI079187-06S1. JFD is supported by the National Cancer Institute (R35 CA210084-01 and P50 CA171963-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
J.S.A.P. and C.S.H. conceived of the project. J.S.A.P., M. C., J. C., T. E., B.-C. L., N. A., J. F. DiP., M. A., and C.-S. H. designed the experiments; J.S.A.P., E.V.R.-G., Y.W.Z., W.P., M.C., J.C., and V. S. performed the experiments; and J.S.A.P. and C.S.H. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
References
- Abbracchio MP, Boeynaems J-M, Barnard EA, Boyer JL, Kennedy C, Miras-Portugal MT, King BF, Gachet C, Jacobson KA, Weisman GA, Burnstock G. Characterization of the UDP-glucose receptor (renamed here the P2Y14 receptor) adds diversity to the P2Y receptor family. Trends in Pharmacological Sciences. 2003;24:52–55. doi: 10.1016/S0165-6147(02)00038-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert ML, Pearce SFA, Francisco LM, Sauter B, Roy P, Silverstein RL, Bhardwaj N. Immature Dendritic Cells Phagocytose Apoptotic Cells via αvβ5 and CD36, and Cross-present Antigens to Cytotoxic T Lymphocytes. The Journal of Experimental Medicine. 1998;188:1359–1368. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardouin L, Luche H, Chelbi R, Carpentier S, Shawket A, Montanana Sanchis F, Santa Maria C, Grenot P, Alexandre Y, Grégoire C, et al. Broad and Largely Concordant Molecular Changes Characterize Tolerogenic and Immunogenic Dendritic Cell Maturation in Thymus and Periphery. Immunity. 2016;45:305–318. doi: 10.1016/j.immuni.2016.07.019. [DOI] [PubMed] [Google Scholar]
- Aschenbrenner K, D'Cruz LM, Vollmann EH, Hinterberger M, Emmerich J, Swee LK, Rolink A, Klein L. Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat Immunol. 2007;8:351–358. doi: 10.1038/ni1444. [DOI] [PubMed] [Google Scholar]
- Atibalentja DF, Murphy KM, Unanue ER. Functional Redundancy between Thymic CD8α+ and Sirpα+ Conventional Dendritic Cells in Presentation of Blood-Derived Lysozyme by MHC Class II Proteins. The Journal of Immunology. 2011;186:1421–1431. doi: 10.4049/jimmunol.1002587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Nakamoto Y, Mukaida N. Crucial Contribution of Thymic Sirpα+ Conventional Dendritic Cells to Central Tolerance against Blood-Borne Antigens in a CCR2-Dependent Manner. The Journal of Immunology. 2009;183:3053–3063. doi: 10.4049/jimmunol.0900438. [DOI] [PubMed] [Google Scholar]
- Bautista JL, Lio C-WJ, Lathrop SK, Forbush K, Liang Y, Luo J, Rudensky AY, Hsieh C-S. Intraclonal competition limits the fate determination of regulatory T cells in the thymus. Nat Immunol. 2009;10:610–617. doi: 10.1038/ni.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belz GT, Vremec D, Febbraio M, Corcoran L, Shortman K, Carbone FR, Heath WR. CD36 Is Differentially Expressed by CD8+ Splenic Dendritic Cells But Is Not Required for Cross-Presentation In Vivo. The Journal of Immunology. 2002;168:6066–6070. doi: 10.4049/jimmunol.168.12.6066. [DOI] [PubMed] [Google Scholar]
- Bluestone JA, Bour-Jordan H, Cheng M, Anderson M. T cells in the control of organ-specific autoimmunity. The Journal of Clinical Investigation. 2015;125:2250–2260. doi: 10.1172/JCI78089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai JN, Peng Y, Rengarajan S, Solomon BD, Ai TL, Shen Z, Perry JSA, Knoop KA, Tanoue T, Narushima S, et al. Helicobacter species are potent drivers of colonic T cell responses in homeostasis and inflammation. Science Immunology. 2017:2. doi: 10.1126/sciimmunol.aal5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AY, Anderson MS. Central tolerance to self revealed by the autoimmune regulator. Annals of the New York Academy of Sciences. 2015;1356:80–89. doi: 10.1111/nyas.12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Yusuf R, Kook S, Attar E, Lee D, Park B, Cheng T, Scadden DT, Lee BC. Purinergic P2Y14 receptor modulates stress-induced hematopoietic stem/progenitor cell senescence. The Journal of Clinical Investigation. 2014;124:3159–3171. doi: 10.1172/JCI61636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke K, Kobzik L, Martin T, Brewer J, Delmonte JJ, Crawford J, Ferrara J. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–3239. [PubMed] [Google Scholar]
- Cowan JE, Parnell SM, Nakamura K, Caamano JH, Lane PJL, Jenkinson EJ, Jenkinson WE, Anderson G. The thymic medulla is required for Foxp3+ regulatory but not conventional CD4+ thymocyte development. The Journal of Experimental Medicine. 2013;210:675–681. doi: 10.1084/jem.20122070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan BP, Gibbs KD, Ostrand-Rosenberg S. Tumor-Specific CD4+ T Cells Are Activated by “Cross-Dressed” Dendritic Cells Presenting Peptide-MHC Class II Complexes Acquired from Cell-Based Cancer Vaccines. The Journal of Immunology. 2006;176:1447–1455. doi: 10.4049/jimmunol.176.3.1447. [DOI] [PubMed] [Google Scholar]
- Febbraio M, Abumrad NA, Hajjar DP, Sharma K, Cheng W, Pearce SFA, Silverstein RL. A Null Mutation in Murine CD36 Reveals an Important Role in Fatty Acid and Lipoprotein Metabolism. Journal of Biological Chemistry. 1999;274:19055–19062. doi: 10.1074/jbc.274.27.19055. [DOI] [PubMed] [Google Scholar]
- Ferris Stephen T, CarreroJavier A, Mohan James F, Calderon B, Murphy Kenneth M, Unanue Emil R. A Minor Subset of Batf3-Dependent Antigen-Presenting Cells in Islets of Langerhans Is Essential for the Development of Autoimmune Diabetes. Immunity. 2014;41:657–669. doi: 10.1016/j.immuni.2014.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos AM, Bevan MJ. Central Tolerance to Tissue-specific Antigens Mediated by Direct and Indirect Antigen Presentation. The Journal of Experimental Medicine. 2004;200:1039–1049. doi: 10.1084/jem.20041457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner JM, DeVoss JJ, Friedman RS, Wong DJ, Tan YX, Zhou X, Johannes KP, Su MA, Chang HY, Krummel MF, Anderson MS. Deletional Tolerance Mediated by Extrathymic Aire-Expressing Cells. Science. 2008;321:843–847. doi: 10.1126/science.1159407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giudicelli V, Duroux P, Ginestoux C, Folch G, Jabado-Michaloud J, Chaume D, Lefranc M-P. IMGT/LIGM-DB, the IMGT® comprehensive database of immunoglobulin and T cell receptor nucleotide sequences. Nucleic Acids Research. 2006;34:D781–D784. doi: 10.1093/nar/gkj088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimcher LH, Hamano T, Asofsky R, Sachs DH, Pierres M, Samelson LE, Sharrow SO, Paul WE. IA mutant functional antigen-presenting cell lines. The Journal of Immunology. 1983;130:2287–2294. [PubMed] [Google Scholar]
- Gray D, Abramson J, Benoist C, Mathis D. Proliferative arrest and rapid turnover of thymic epithelial cells expressing Aire. The Journal of Experimental Medicine. 2007;204:2521–2528. doi: 10.1084/jem.20070795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng TSP, Painter MW, Elpek K, Lukacs-Kornek V, Mauermann N, Turley SJ, Koller D, Kim FS, Wagers AJ, Asinovski N, et al. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol. 2008;9:1091–1094. doi: 10.1038/ni1008-1091. [DOI] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinterberger M, Aichinger M, da Costa OP, Voehringer D, Hoffmann R, Klein L. Autonomous role of medullary thymic epithelial cells in central CD4+ T cell tolerance. Nat Immunol. 2010;11:512–519. doi: 10.1038/ni.1874. [DOI] [PubMed] [Google Scholar]
- Hsieh C-S, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7:401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- Hubert F-X, Kinkel SA, Davey GM, Phipson B, Mueller SN, Liston A, Proietto AI, Cannon PZF, Forehan S, Smyth GK, et al. Aire regulates the transfer of antigen from mTECs to dendritic cells for induction of thymic tolerance. Blood. 2011;118:2462–2472. doi: 10.1182/blood-2010-06-286393. [DOI] [PubMed] [Google Scholar]
- Humblet C, Rudensky AY, Kyewski B. Presentation and intercellular transfer of self antigen within the thymic microenvironment: expression of the Eα peptide-l-Ab complex by isolated thymic stromal cells. International Immunology. 1994;6:1949–1958. doi: 10.1093/intimm/6.12.1949. [DOI] [PubMed] [Google Scholar]
- Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don't see) Nat Rev Immunol. 2014;14:377–391. doi: 10.1038/nri3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koble C, Kyewski B. The thymic medulla: a unique microenvironment for intercellular self-antigen transfer. The Journal of Experimental Medicine. 2009;206:1505–1513. doi: 10.1084/jem.20082449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B-C, Cheng T, Adams GB, Attar EC, Miura N, Lee SB, Saito Y, Olszak I, Dombkowski D, Olson DP, et al. P2Y-like receptor, GPR105 (P2Y14), identifies and mediates chemotaxis of bone-marrowhematopoietic stem cells. Genes & Development. 2003;17:1592–1604. doi: 10.1101/gad.1071503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y. Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J. Exp. Med. 2011;208:383–394. doi: 10.1084/jem.20102327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y, Ripen AM, Ishimaru N, Ohigashi I, Nagasawa T, Jeker LT, Bösl MR, Holländer GA, Hayashi Y, de Waal Malefyt R, et al. Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. The Journal of Experimental Medicine. 2011;208:383–394. doi: 10.1084/jem.20102327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventhal Daniel S, Gilmore Dana C, Berger Julian M, Nishi S, Lee V, Malchow S, Kline Douglas E, Kline J, Vander Griend Donald J, Huang H, et al. Dendritic Cells Coordinate the Development and Homeostasis of Organ-Specific Regulatory T Cells. Immunity. 2016;44:847–859. doi: 10.1016/j.immuni.2016.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Kim S, Herndon JM, Goedegebuure P, Belt BA, Satpathy AT, Fleming TP, Hansen TH, Murphy KM, Gillanders WE. Cross-dressed CD8α+/CD103+ dendritic cells prime CD8+ T cells following vaccination. Proceedings of the National Academy of Sciences. 2012;109:12716–12721. doi: 10.1073/pnas.1203468109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zheng Y. Regulatory T cell identity: formation and maintenance. Trends in Immunology. 2015;36:344–353. doi: 10.1016/j.it.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Yang L, Silva HM, Trzeciak A, Choi Y, Schwab SR, Dustin ML, Lafaille JJ. Increased generation of Foxp3+ regulatory T cells by manipulating antigen presentation in the thymus. Nat Commun. 2016:7. doi: 10.1038/ncomms10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamedov IZ, Britanova OV, Zvyagin IV, Turchaninova MA, Bolotin DA, Putintseva EV, Lebedev YB, Chudakov DM. Preparing unbiased T cell receptor and antibody cDNA libraries for the deep next generation sequencing profiling. Frontiers in Immunology. 2013:4. doi: 10.3389/fimmu.2013.00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzini E, Massimiliano L, Penna G, Rescigno M. Oral Tolerance Can Be Established via Gap Junction Transfer of Fed Antigens from CX3CR1+ Macrophages to CD103+ Dendritic Cells. Immunity. 2014;40:248–261. doi: 10.1016/j.immuni.2013.12.012. [DOI] [PubMed] [Google Scholar]
- Millet V, Naquet P, Guinamard RR. Intercellular MHC transfer between thymic epithelial and dendritic cells. Eur. J. Immunol. 2008;38:1257–1263. doi: 10.1002/eji.200737982. [DOI] [PubMed] [Google Scholar]
- Naik SH, Proietto AI, Wilson NS, Dakic A, Schnorrer P, Fuchsberger M, Lahoud MH, O’Keeffe M, Shao Q-x, Chen W-f, et al. Cutting Edge: Generation of Splenic CD8+ and CD8-Dendritic Cell Equivalents in Fms-Like Tyrosine Kinase 3 Ligand Bone Marrow Cultures. The Journal of Immunology. 2005;174:6592–6597. doi: 10.4049/jimmunol.174.11.6592. [DOI] [PubMed] [Google Scholar]
- Oh J, Shin J-S. The Role of Dendritic Cells in Central Tolerance. Immune Netw. 2015;15:111–120. doi: 10.4110/in.2015.15.3.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JSA, Hsieh C-S. Development of T-cell tolerance utilizes both cell-autonomous and cooperative presentation of self-antigen. Immunological Reviews. 2016;271:141–155. doi: 10.1111/imr.12403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry Justin SA, Lio C-Wang J, Kau Andrew L, Nutsch K, Yang Z, Gordon Jeffrey I, Murphy Kenneth M, Hsieh C-S. Distinct Contributions of Aire and Antigen-Presenting-Cell Subsets to the Generation of Self-Tolerance in the Thymus. Immunity. 2014;41:414–426. doi: 10.1016/j.immuni.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon IKH, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu C, Nguyen VA, Merad M, Randolph GJ. MHC Class I/Peptide Transfer between Dendritic Cells Overcomes Poor Cross-Presentation by Monocyte-Derived APCs That Engulf Dying Cells. The Journal of Immunology. 2009;182:3650–3659. doi: 10.4049/jimmunol.0801532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DM, Kyewski B, Feuerer M. Re-examining the Nature and Function of Self-Reactive T cells. Trends in Immunology. 2016;37:114–125. doi: 10.1016/j.it.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigotti A, Acton SL, Krieger M. The Class B Scavenger Receptors SR-BI and CD36 Are Receptors for Anionic Phospholipids. Journal of Biological Chemistry. 1995;270:16221–16224. doi: 10.1074/jbc.270.27.16221. [DOI] [PubMed] [Google Scholar]
- Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, Heger A, Ponting CP, Holländer GA. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Research. 2014;24:1918–1931. doi: 10.1101/gr.171645.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Pennington DJ, Hodivala-Dilke K, Febbraio M, Reise Sousa C. CD36 or αvβ3 and αvβ5 Integrins Are Not Essential for MHC Class I Cross-Presentation of Cell-Associated Antigen by CD8α+ Murine Dendritic Cells. The Journal of Immunology. 2002;168:6057–6065. doi: 10.4049/jimmunol.168.12.6057. [DOI] [PubMed] [Google Scholar]
- Vrisekoop N, Monteiro João P, Mandl Judith N, Germain Ronald N. Revisiting Thymic Positive Selection and the Mature T Cell Repertoire for Antigen. Immunity. 2014;41:181–190. doi: 10.1016/j.immuni.2014.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissler KA, Caton AJ. The role of T-cell receptor recognition of peptide:MHC complexes in the formation and activity of Foxp3+ regulatory T cells. Immunological Reviews. 2014;259:11–22. doi: 10.1111/imr.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J, Obst R, Correia-Neves M, Losyev G, Mathis D, Benoist C. Adaptation of TCR Repertoires to Self-Peptides in Regulatory and Nonregulatory CD4+ T Cells. The Journal of Immunology. 2007;178:7032–7041. doi: 10.4049/jimmunol.178.11.7032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.