

SUMMARY
Despite its success in several clinical trials, cancer immunotherapy remains limited by the rarity of targetable tumor-specific antigens, tumor-mediated immune suppression, and toxicity triggered by systemic delivery of potent immunomodulators. Here, we present a proof-of-concept immunomodulatory gene circuit platform that enables tumor-specific expression of immunostimulators, which could potentially overcome these limitations. Our design comprised de novo synthetic cancer-specific promoters and, to enhance specificity, an RNA-based AND gate that generates combinatorial immunomodulatory outputs only when both promoters are mutually active. These outputs included an immunogenic cell-surface protein, a cytokine, a chemokine, and a checkpoint inhibitor antibody. The circuits triggered selective T cell-mediated killing of cancer cells, but not of normal cells, in vitro. In in vivo efficacy assays, lentiviral circuit delivery mediated significant tumor reduction and prolonged mouse survival. Our design could be adapted to drive additional immunomodulators, sense other cancers, and potentially treat other diseases that require precise immunological programming.
In Brief
An immunomodulatory gene circuit platform that enables tumor-specific expression of immunostimulators that permits selective T cell-mediated killing of cancer cells, but not of normal cells, is developed. This platform shows prolonged survival in a mouse cancer model and has the potential to be adapted to express a range of other immune regulators and to treat other cancer types.
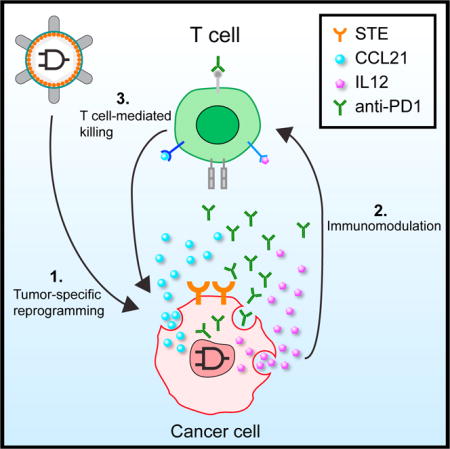
INTRODUCTION
The potential of cancer immunotherapy has been demonstrated in several clinical trials, yet this approach is still limited by several major challenges, including the lack of targetable tumor-specific antigens and tumor-mediated immunosuppression. For example, chimeric antigen receptor (CAR)-T cells and bispecific antibodies should ideally target cell-surface antigens that are exclusively present on tumor cells, since targeting normal tissues can result in severe side effects (Morgan et al., 2010). However, finding highly tumor-specific surface antigens is difficult, which limits the range of targetable tumors (Klebanoff et al., 2016). In addition, even when ideal targetable tumor antigens are available, tumor-mediated immunosuppression can prevent successful immunotherapy by disrupting important immunological functions that are necessary for effective anti-tumor immune responses (Rabinovich et al., 2007).
Immunostimulatory factors can be utilized to overcome these challenges, but off-target activity often results in severe toxicity. For example, surface T cell engagers (STEs) are artificial immunogenic cell-surface proteins that bind the non-variable regions of the T cell receptor complex (Liao et al., 2000, 2003). Consequently, STE-expressing cells are designated for T cell-mediated killing regardless of T cell receptor antigen specificity (see Figure 1B for our STE design based on membrane-anchored anti-CD3 single-chain variable fragment [scFv]). Tumor-specific STE expression can substitute for targetable tumor antigens but must be constrained to cancer cells to avoid damage to healthy tissues (Liao et al., 2003). Other immunomodulators, such as chemokines, cytokines, and immune checkpoint inhibitors, can assist in overcoming tumor-mediated immunosuppression but have caused severe side effects in clinical trials (Lasek et al., 2014; Leonard et al., 1997). Furthermore, individual immunomodulators are frequently insufficient on their own to yield strong anti-tumor efficacy (Mahoney et al., 2015). Combinatorial immunotherapy can provide significantly stronger efficacy but also increase the risk and severity of adverse effects (Boutros et al., 2016). Tumor-localized release of immunomodulators could potentially decrease these systemic side effects and improve the therapeutic efficacy, but is challenging. Thus, although various strategies are being explored to overcome this problem (Neri and Sondel, 2016; Shukla and Steinmetz, 2016; Tugues et al., 2015), further development is required.
Figure 1. Immunomodulatory Synthetic Gene Circuits Are Programmed to Be Selectively Activated in Cancer Cells.
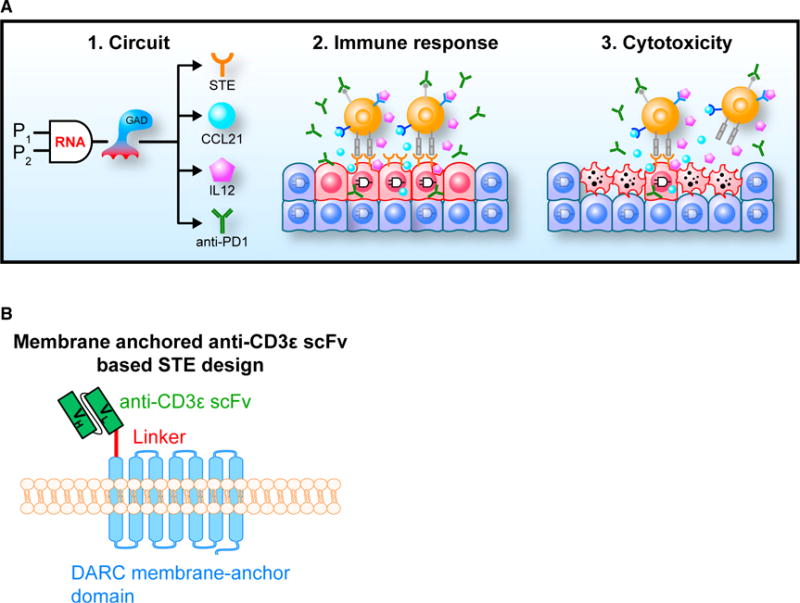
(A) Panel 1: Immunomodulatory synthetic gene circuits are designed to integrate the activity of two tumor-specific synthetic promoters (P1 and P2) with an RNA-based AND gate mechanism and generate combinatorial immunomodulator outputs only when both input promoters are mutually active. When activated, the AND gate expresses a synthetic transcription factor (GAD: a fusion protein consisting of the GAL4 DNA binding domain and VP16 transcription activating domain), which drives the co-expression of combinatorial immunomodulators, including surface T cell engagers (STEs, which are anti-CD3ε scFvs displayed on the cell surface) and secreted CCL21, IL12, and an anti-PD1 antibody. Panel 2: The circuits are triggered to express immunomodulators in cancer cells (red), but not normal cells (blue). Black-line circuit diagrams indicate circuits are active and gray-line circuit diagrams indicate circuits are inactive. Panel 3: Cancer-specific expression of combinatorial immunomodulators triggers effective T cell-mediated killing of the cancer cells.
(B) Schematic drawing of STE displayed on cell surfaces.
Synthetic biology enables the creation of powerful genetic biological tools for studying, diagnosing, and treating disease (Bacchus et al., 2013; Kotula et al., 2014; Pardee et al., 2016; Schukur et al., 2015; Xie et al., 2016). For example, synthetic tumor-targeting gene circuits have previously been designed to target tumor cells with enhanced specificity but have not leveraged the anti-tumor potential of the immune system, which may limit their efficacy (Liu et al., 2014; Morel et al., 2016; Nissim and Bar-Ziv, 2010; Xie et al., 2011). We hypothesized that gene circuits could be designed to achieve tumor-specific production of multiple immunomodulators and thus overcome major obstacles in current immunotherapies (Figure 1A).
Here, we demonstrate the potential of gene circuits for cancer immunotherapy. Tumor-targeting gene circuits often require promoters with high cell-state specificity to function as cancer sensors, which can be hard to find (Selvakumaran et al., 2001; Yun et al., 2008). Moreover, native mammalian promoters can span several kb and are therefore difficult to fit into the limited encoding capacity of many viral vectors. Thus, we constructed compact synthetic promoters de novo that exhibited enhanced specificity for human cancer cells compared with previously described endogenous cancer-specific promoters.
Discriminating cancer cells based on the integration of multiple cancer markers significantly enhances targeting specificity (Liu et al., 2014; Morel et al., 2016; Nissim and Bar-Ziv, 2010; Roybal et al., 2016; Xie et al., 2011). Thus, to enhance the circuit selectivity over a single synthetic promoter, we designed a synthetic RNA-based circuit that integrates the activity of two synthetic ovarian-cancer-specific promoters and generates output proteins only when both promoters are decidedly active, analogous to a Boolean AND gate. Specifically, input promoter 1 (P1) regulates an auto-inhibitory mRNA that encodes the output protein, while input promoter 2 (P2) regulates an RNA molecule that relieves the auto-inhibition and thus enables output production. This RNA-based design has the potential to reduce the off-target immunogenic signature of the circuit by minimizing the expression of foreign proteins in normal cells. Immunomodulatory outputs used in this study include a cell-surface antigen (STE), a chemokine that promotes T cell trafficking to tumor sites (CCL21), a cytokine that enhances T cell activation and function (IL12), and an immune checkpoint inhibitor (anti-PD1 antibody), hereafter referred to as the SCIP combination (Lasek et al., 2014; Liao et al., 2000, 2003; Lin et al., 2014; Postow et al., 2015).
We characterized our circuits with in vitro specificity assays using normal and cancer cell lines and in vivo efficacy assays using a disseminated ovarian cancer model, which recapitulates advanced human ovarian cancer (Lengyel et al., 2014). In addition, we demonstrated that our circuit can be readily modified to target breast cancer cells by replacing the synthetic input promoters. Our circuit design is modular and enables facile construction, testing, and optimization of arbitrary promoter inputs, circuit components, and outputs. The outputs can be any genetically encodable element, including intracellular, secreted, and cell-surface proteins. This flexibility will enable circuits to be optimized using advanced tumor models and patient-derived tumors, which will be important for translation into clinical use. Thus, our programmable immunomodulatory gene circuits have broad potential utility for cancer immunotherapy and studying tumor immunology.
RESULTS
RNA-Based AND Gate Circuit
We first designed and optimized the RNA-based AND gate. We engineered two genetic modules such that each was regulated by a separate promoter (Figures 2A and 2B). Module 1 consists of P1, which regulates the expression of an RNA that encodes two exons of an output protein mKate2 (mK-Ex1, mK-Ex2), separated by a synthetic intronic microRNA (miRNA) (miR1), as well as perfect match miR1 binding sites (BS[Pe]) downstream to the second exon. Following splicing, the exons are assembled into a mature output mRNA that contains BS(Pe)s in the 3′ untranslated region (UTR), while the intron is processed by the cell to produce miR1. Consequently, when only P1 is active, miR1 binds the BS(Pe) sites in the output mRNA and targets it for degradation. Therefore, module 1 constitutes an auto-inhibitory loop for output protein production (Figure 2A). Module 2 consists of P2, which regulates a miRNA sponge that consists of multiple bulged complementary miR1 binding sites (BS[B]) and thus acts as a miR1 shunt that relieves the auto-inhibitory nature of module 1 when it is expressed (Figures 2A and 2B). In this system, the output protein is expressed at a high level only when the promoters regulating both modules are mutually active. However, when only one of the promoters is active or when none of the promoters are active, output protein expression is low.
Figure 2. The Basic RNA-Only Single-Output AND Gate Design.
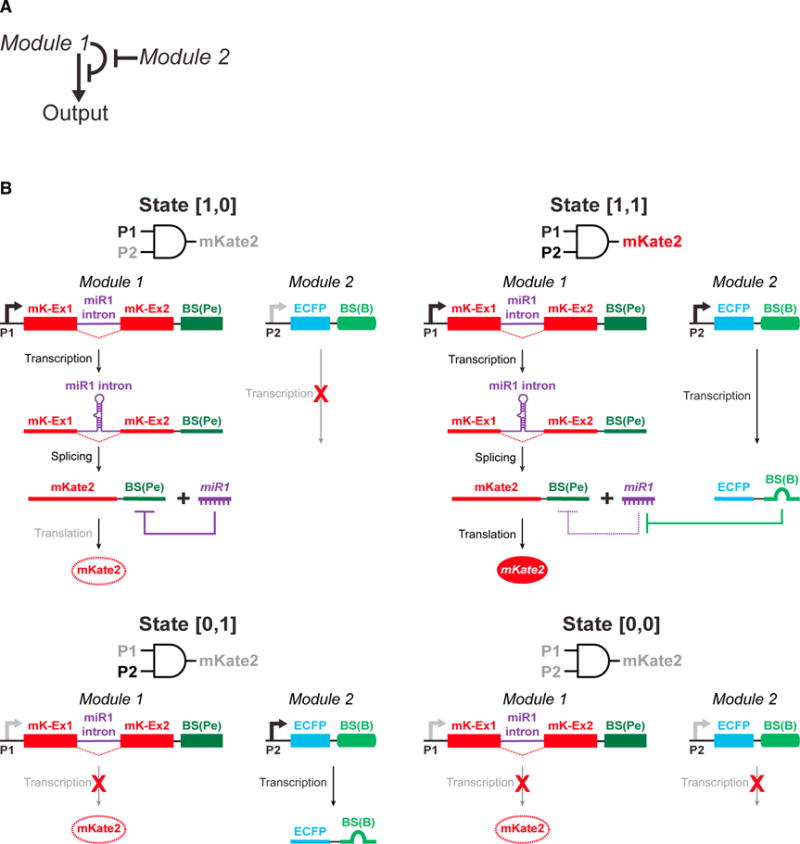
(A) Module 1 of the AND gate is designed as an auto-inhibitory loop such that it represses its own output. Module 2 is designed to inhibit the auto-inhibition of module 1. Module 1 and module 2 are regulated by cancer-specific promoters P1 and P2, respectively. The output from module 1 is expressed at a high level only when both P1 and P2 are active, which enhances the tumor specificity of the circuit.
(B) All four possible input states and their respective output states are shown for the RNA-only single-output AND gate. Input states are defined within the square brackets by whether module 1 and module 2 are active, where 0 means inactive and 1 means active. In state [1,0], P1 is active and the mKate2 transcript is expressed (mK-Ex1 and mK-Ex2 denote mKate2 exon1 and mKate2 exon2, respectively). However, this transcript encodes miR1 within the mKate2 gene that inhibits the mKate2 transcript by targeting BS(Pe)s in the 3′ UTR. Thus, the mKate2 levels are minimal. In state [0,1], P1 is inactive, so the output protein mKate2 is not expressed. In state [0,0], neither P1 nor P2 are active, and thus, the output protein mKate2 is not expressed. In state [1,1], the P2 promoter expresses a sponge for miR1 that is based on BS(B)s. This enables sequestration of miR1 away from inhibiting the mKate2 transcript expressed by P1, thus allowing for mKate2 expression.
We systematically optimized various aspects of the miRNA-based mechanism (Figures 3, S1, S2, S3, and STAR Methods) (Auyeung et al., 2013; Ebert and Sharp, 2010; Fellmann et al., 2013; Suzuki et al., 2015). In module 2, we characterized how the miRNA binding site (BS) sequence, expression level, and sponge architecture determines the efficiency of the miR1 sponge (Figures 3, S1, and S2). We defined four different states for this circuit: in state [0,0], both module 1 and module 2 are inactive; in state [0,1], module 1 is inactive, while module 2 is active; in state [1,0], module 1 is active, while module 2 is inactive; and in state [1,1], both module 1 and module 2 are active (Figure 2B). The ON-OFF ratio for the AND gate circuit was defined as the mKate2 output level in the presence versus the absence of the sponge (state [1,1] versus state [1,0] in Figure 2B). Ultimately, we chose Sponge 7: H2A1p-EC1-10BZRANB1-EC2-5B (Figure S2A) as the final “optimized sponge” design because it yielded the highest ON-OFF ratio for the AND gate circuit (~6-fold, Figure S2A).
Figure 3. Flowchart of Circuit Optimization Strategies for the RNA-Based AND Gate.
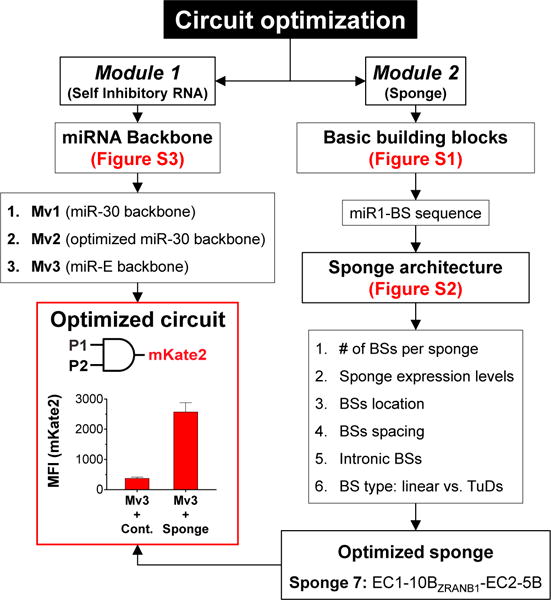
Multiple versions of the miR1 production backbone in module 1, as well as the miR1 BS sequences and sponge architecture in module 2, were systematically tested. The optimized circuit utilized the miR-E backbone (Mv3, Table S2) in module 1 to generate miR1 and a sponge architecture “Sponge 7” in module 2 that encoded ten optimized BS(B)s within an enhanced cyan fluorescent protein (ECFP) gene (EC1-10BZRANB1-EC2) and five additional BS(B)s in the downstream 3′ UTR (Figure S2A). The mKate2 output levels for the optimized circuit with a control sponge with no miR1 BSs (“Mv3 + Cont.”, representing the [1,0] state in Figure 2B), and the optimized circuit with the optimized sponge (“Mv3 + Sponge”, representing the [1,1] state in Figure 2B) are shown. Error bars represent SEM, n = 3 biological replicates. TuD = tough-decoy architecture.
See also Figures S1, S2, and S3; Table S2; and STAR Methods.
In module 1, we optimized the synthetic intronic pri-miRNA backbone in the self-inhibitory mKate2 transcript for enhanced miR1 production efficiency (Greber et al., 2008; Nissim et al., 2014; Xie et al., 2011). The absolute mKate2 signal in the OFF (no sponge, state [1,0]) and ON states (optimized sponge, state [1,1]) were both affected by the pri-miRNA designs we tested (Mv1-Mv3, Figure S3). Mv1 exhibited an OFF-ON shift from ~1400 to ~8400 mean fluorescence intensity (MFI) (5.8-fold activation ratio), Mv2 from ~170 to ~500 MFI (3-fold activation), and Mv3 from ~380 to ~2600 MFI (6.8-fold activation) (Figures S3D and S3E). We selected the Mv3 architecture for further experimentation because it had low mKate2 level in the OFF state, substantial absolute mKate2 levels in the ON state, and the highest fold activation. However, we note that different designs could be more suitable for other targeting scenarios. For example, when using effector outputs with low potency, Mv1 could be chosen to achieve high absolute levels of expression in the ON state. Conversely, Mv2 could be suitable for expressing extremely potent outputs for which even low background output activity could be detrimental.
Synthetic Cancer-Specific Promoters
To generate synthetic ovarian-cancer-specific promoters, we identified transcription factors (TFs) that are overexpressed in ovarian cancer cells by comparing gene expression in primary human ovarian tumors versus healthy controls, and the respective binding motifs of these TFs, using publically available databases (STAR Methods). In each synthetic promoter S(TF)p, multiple binding motifs for a single cancer-specific TF were encoded in tandem upstream to a synthetic minimal promoter derived from the major adenoviral late promoter (Figure 4A). This design approach resulted in compact promoters with evenly distributed and dense TF-BSs corresponding to a small number of TF families. For example, analysis of the 1-kb upstream of the transcriptional start site (TSS) of the native tumor-specific promoter H2A1p (Rogakou et al., 1998) revealed ~82 possible BSs for 40 different TF families, with a mean of ~8 TF-BSs per 100 bp. In comparison, the synthetic S(E2F1)p promoter, comprised of E2F1 BSs, had only 44 possible BSs for 7 different TF families, with a mean of ~16 TF-BSs per 100 bp (Table S1 and STAR Methods).
Figure 4. Synthetic Tumor-Specific Promoters Exhibited High Tumor Specificity.
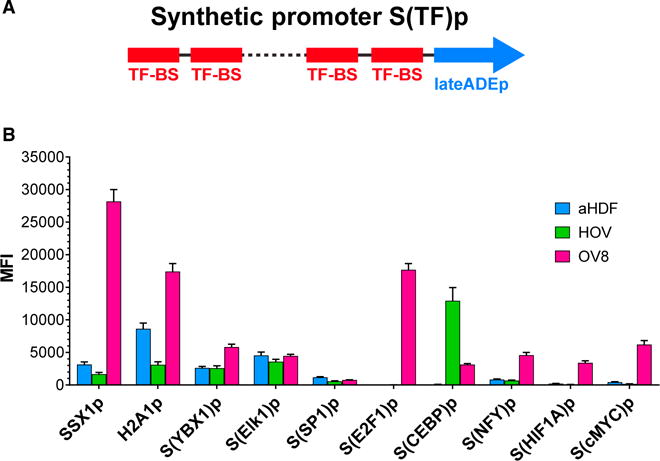
(A) Design of synthetic tumor-specific promoters, S(TF)p. Multiple BSs for a TF that is overexpressed in cancer (TF-BS) were cloned in tandem upstream of a minimal late adenovirus promoter (lateADEp).
(B) Synthetic tumor-specific promoters regulating mKate2 exhibited enhanced tumor specificity. Representative native tumor-specific promoters, SSX1p and H2A1p, are shown for comparison. Parentheses indicate the TF-BSs used to construct each synthetic tumor-specific promoter. aHDF and HOV are normal primary cells. OV8 is a human ovarian cancer cell line. Error bars represent SEM, n = 3 biological replicates.
See also Table S1.
We assayed the specificity and activity of natural and artificial promoters in human OVCAR8 (OV8) ovarian cancer cells and normal primary cells, including primary adult human dermal fibroblasts (aHDF) and primary human ovarian epithelium (HOV). Several synthetic promoters, including S(E2F1)p and S(cMYC) p (comprised of E2F1 and cMYC BSs, respectively), were highly active in OV8 ovarian cancer cells, but not in normal cells. These synthetic promoters exhibited greater activation ratios compared with known native tumor-specific promoters such as SSX1p (Gure et al., 1997) and H2A1p (Rogakou et al., 1998) (Figure 4B for selected promoters).
For example, SSX1p exhibited 9-fold activation in OV8 compared to aHDF and 17-fold activation compared to HOV (Figure 4B). H2A1p had 2-fold activation in OV8 compared to aHDF and 5.6-fold compared to HOV. The synthetic promoter S(E2F1)p had 224-fold induction in OV8 versus aHDF and 570-fold activation in OV8 compared to HOV, with absolute maximal expression levels comparable to the native promoters. S(cMYC)p exhibited 15-fold induction in OV8 versus aHDF and 36-fold activation in OV8 compared to HOV. Since the maximal activity of S(cMYC)p was lower than that of S(E2F1)p in OV8 cells, we selected S(cMYC)p to drive expression of module 1 and S(E2F1)p to drive the expression of module 2. This was done so that the sponge could be expressed at a greater level than the miR1-encoding construct, which is important for achieving a high activation ratio with our circuit (Figure S2A).
Multi-output AND Gate for Combinatorial Immunomodulation
To facilitate the expression of combinatorial immunomodulators from our circuit, we replaced mKate2 in module 1 with a synthetic transcription factor (GAD) that binds to its cognate synthetic promoter and activates the expression of downstream genes (Figure 5A). This GAD was previously made by fusing the yeast GAL4 DNA-binding domain to the viral VP16 transcriptional activation domain. Specifically, we used the synthetic promoter S(cMYC)p to express module 1 (S(cMYC)p-GADEX1-[miR1]cons-GADEX2-11Pe). Module 1 encoded a self-inhibitory RNA transcript with miR1 as an intron within the GAD coding sequence followed by 11 tandem BS(Pe)s in the 3′ UTR. In the module 2 vector, the synthetic promoter S(E2F1)p regulated the expression of the optimized sponge architecture (Sponge 7: EC1-10BZRANB1-EC2-5B). Finally, we encoded an mKate2 output under the regulation of an artificial promoter targeted by GAD that contains GAL4-BSs upstream to a synthetic minimal late adenoviral promoter (GALp), namely module 3 (Figure 5A). We hypothesized that increasing the number of GAL4-BSs in the GALp would increase output expression levels. We therefore built 3 different versions of GALp, either with 5, 8, or 14 tandem GAL4-BSs (G5p, G8p, and G14p, respectively). We further surmised that output levels in the OFF state could be lowered by adding miR1-BS(Pe) to the mKate2 output, thus allowing miR1 to reduce both GAD and mKate2 levels directly when the sponge is not active (Figure 5A). Thus, we built three additional versions of the output vector in which a single BS(Pe) was encoded in the 3′ UTR of the output mKate2 transcript (G5-Pe, G8-Pe and G14-Pe, respectively). This design architecture enables convenient tuning of each output’s expression levels based on the promoter and whether one Pe was incorporated into the output transcript.
Figure 5. Multi-output AND Gate Architecture and Performance.
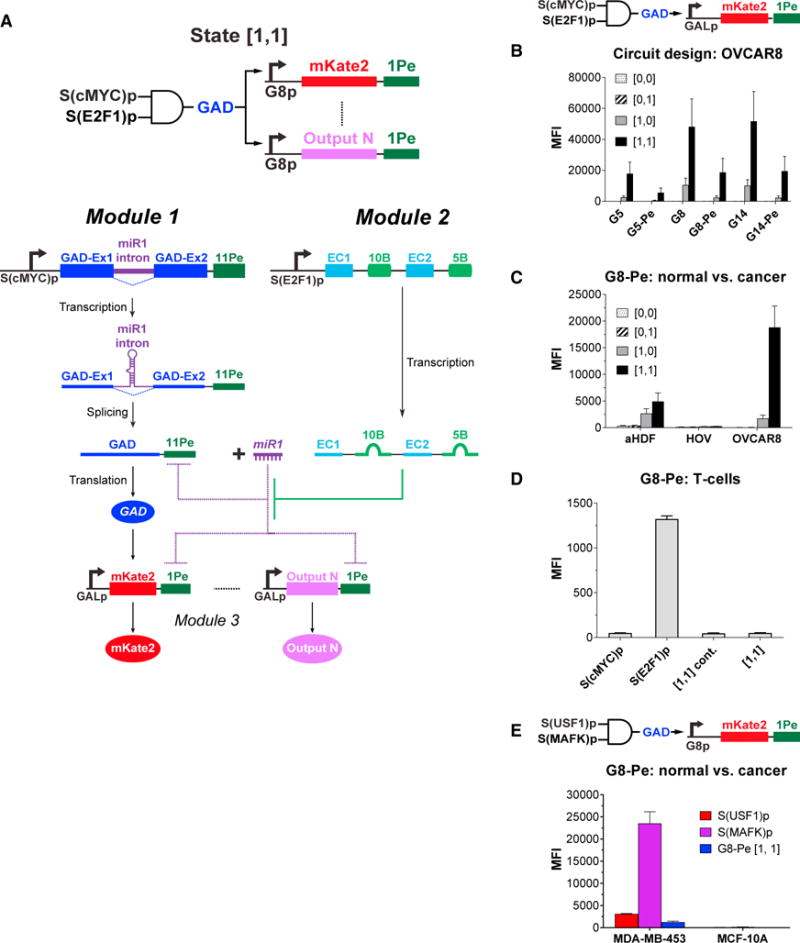
(A) The incorporation of a synthetic transcription factor (GAD) as the output of module 1 means that it is only expressed at high levels when both P1 and P2 are high (state [1,1]). GAD enables facile tuning of downstream gene expression (mKate2 ⋯ Output N).
(B) mKate2 expression can be modulated by modifying the number of GAD BSs in the synthetic GALp expressing mKate2 (G5, G8, G14 for 5, 8, 14 GAD BSs in GALp) and the presence of miRNA BSs at the mKate2 3′ UTR (G5-Pe, G8-Pe, G14-Pe indicate transcripts with a BS[Pe]). Note the circuit diagram depicts the version with a perfect miR1 BS in the mKate2 transcript. State [0,0] indicates cells containing the negative control modules, module 1con and module 2con (see STAR Methods for details). State [0,1] indicates cells containing module 1con and module 2. State [1,0] indicates cells containing module 1 and module 2con. State [1,1] indicates cells containing module 1 and module 2. All cell states tested also contained their respective module 3.
(C) The G8-Pe AND gate architecture triggered high mKate2 expression in human ovarian cancer cells (OV8) but not in normal cells in state [1,1].
(D) Our circuit design prevents potential off-target effects in primary human T cells. The S(E2F1)p promoter was active in T cells but S(cMYC)p was inactive in T cells. The G8-Pe circuit triggered minimal mKate2 output in T cells, thus minimizing the potential off-target effects that would have been observed if the S(E2F1)p was used to directly drive mKate2 expression.
(E) Our circuit design can be readily adapted to distinguish a different tumor type from its normal counterparts. By using two different promoters, the G8-Pe circuit triggered a strong output in the breast cancer cell line MDA-MB-453 but minimal output in the non-tumorigenic breast epithelial cell line MCF-10A.
Error bars represent SEM, n = 3 biological replicates for all experiments, except n = 6 biological replicates for OV8 group in (C).
See also Figure S4A.
We first characterized the multi-output AND-gate architectures by modeling scenarios in which neither (state [0,0]), only one (states [0,1] or [1,0]), or both (state [1,1]) cancer-specific promoters are active in OV8 cells (Figure 5B and STAR Methods). The G8-Pe circuit exhibited negligible mKate2 levels in states [0,0] and [0,1] and high 8-fold mKate2 activation in state [1,1] over the minimal mKate2 expression in state [1,0] and was thus chosen for further use.
We next compared mKate2 expression from the G8-Pe circuit delivered via lentivirus in OV8 cells versus normal cells, including primary aHDF, HOV, and human T cells. The circuit exhibited higher mKate2 expression in OV8 versus aHDF and HOV in the state [1,1] circuit configuration (4-fold and 71-fold, respectively; Figure 5C). We next determined the activity of the S(cMYC)p and S(E2F1)p promoters individually, as well as the activity of the G8-Pe circuit with S(cMYC)p and S(E2F1)p promoters as inputs, in primary human T cells. We observed that S(E2F1)p was significantly activated in primary human T cells. However, since S(cMYC)p was not active in these cells, the integrated output generated by the G8-Pe circuit was 30-fold lower compared to the activity of S(E2F1)p on its own (Figures 5D and S4A). Furthermore, we tested the specificity of this design using additional cell lines, including non-tumorigenic human ovarian fibroblasts (HOFs), human ovarian microvasculature epithelial cells (HOMECs), colonic epithelial cells (CCD-841-CoN), and mammary epithelial cells (MCF-10A and MCF-12A), as well as inducible pluripotent stem cells (iPSCs). The circuit achieved high specificity for OV8 cells, with 10-fold to 800-fold higher circuit output in OV8 cells versus these additional cell lines (Figure S4B). These data demonstrate the stringency of our circuit and the potential of our circuit design to enhance therapeutic specificity by reducing the probability of false-positive targeting compared to targeting with a single promoter.
Finally, to demonstrate the modularity of our design, we examined whether the G8-Pe circuit could be adapted to target breast cancer cells by simple modification of the input promoters (Figure 5E). For this purpose, we created two additional synthetic promoters, S(USF1)p and S(MAFK)p, that were designed to specifically target the MDA-MB-453 breast cancer cell line, but not the MCF-10A non-tumorigenic mammary epithelial cell line. We compared the mKate2 outputs generated by each promoter individually versus the G8-Pe circuit in both cell lines. This new design generated a high output only in MDA-MB-453 cells, demonstrating the versatility of our circuit architecture and its ability to be adapted to a variety of different therapeutic scenarios and applications.
STEs Trigger T Cell-Mediated Cancer-Cell Killing and IFN-γ Secretion
Next, we sought to encode immunotherapies as the outputs of our synthetic cancer-specific circuits. An STE was constructed by fusing an scFv targeting human CD3ε with an inert membrane anchor derived from the Duffy antigen/receptor for chemokines (DARC) of erythrocytes (von Nickisch-Rosenegk et al., 2012) (Figure 1B). We replaced mKate2 with STE as the output of the G8-Pe circuit and validated the therapeutic specificity of this circuit in vitro. We observed 20- to 118-fold higher STE expression on circuit-transduced OV8 cells than on aHDF or HOV cells (Figure 6A). We also observed robust T cell-mediated killing of circuit-transduced OV8 cells, which was significantly higher than in aHDF or HOV cells (Figures 6B and S4C–S4E). There was minimal T cell-mediated killing of non-STE-expressing cells (Figures S4C–S4E), which could have been caused by our experimental conditions, including major histocompatibility complex (MHC) mismatch between T cells and target cells, as well as T cell-mediated killing with non-STE-based mechanisms. In addition to direct tumor killing, robust interferon (IFN)-γ secretion by T cells is critical for strong therapeutic efficacy. We observed strong and specific IFN-γ secretion by T cells only when they were co-cultured with circuit-transduced OV8 cells (Figures 6C and S4F), which was 20- to 28-fold higher than when T cells were co-cultured with circuit-transduced aHDF or HOV cells. We also observed minimal OV8 killing and IFN-γ secretion by T cells when module 2 was removed from the G8-Pe circuit (state [1,0]) to model the potential scenario in which one of the promoters exhibits some off-target activity (Figures 6A–6C and S4C–S4F).
Figure 6. Synthetic Circuits Triggered Human Ovarian Cancer-Specific Expression of STE, T Cell-Mediated Lysis, and IFN-γ Secretion on Tumor Cells.
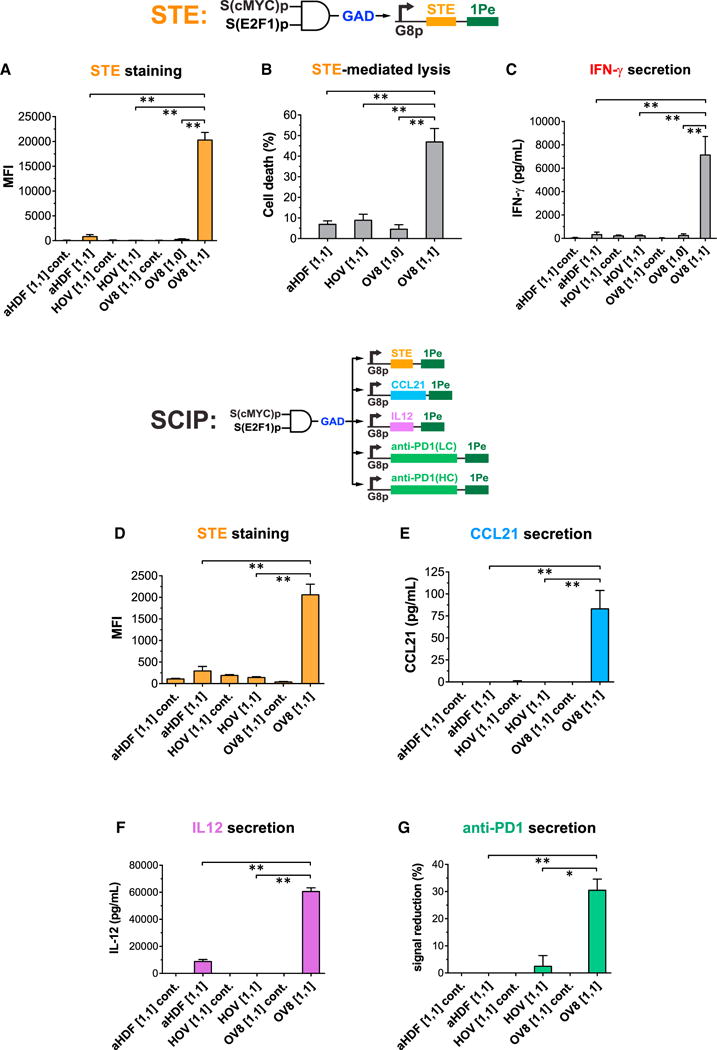
(A–C) The circuit in Figure 5A was engineered to display STE on cell surfaces as the output. STE was encoded in module 3. State “[1,1] cont.” indicates cells containing module 1 and module 2 and a negative control module 3con that expresses the non-specific transcription factor rtTA3 as the output. State [1,1] indicates cells containing module 1, module 2, and module 3. State [1,0] indicates cells containing module 1, a negative control module 2con in which S(E2F1)p expresses ECFP without miR1-BS(B), and module 3. Circuit-transduced tumor cells but not normal cells expressed high levels of STE in state [1,1], which triggered robust T cell killing and IFN-γ secretion. Student’s t test was performed to compare the [1,1] states between normal cells and OV8 cells, and the [1,1] state versus the [1,0] state in OV8 cells.
(D–G) Synthetic circuits triggered human ovarian cancer-specific expression of combinatorial immunomodulators. The circuit in Figure 5A was extended to express multiple immunomodulatory outputs (surface-displayed STE and secreted CCL21, IL12, and anti-PD1 Ab) specifically in OV8 cells but not normal primary cells. State [1,1] cont. indicates cells containing module 1 and module 2 and a negative control module 3con that expresses the non-specific TF rtTA3 as the output. State [1,1] indicates cells containing module 1, module 2, and module 3. Each immunomodulatory gene was expressed from its own G8p promoter encoded on a lentivirus, except for the anti-PD1 Ab, which was split into two lentiviruses encoding LC and HC respectively. Cell lines were co-infected with these lentiviral constructs. Student’s t test was performed to compare output levels in the [1,1] states between normal cells and OV8 cells.
Error bars represent SEM, n = 3 biological replicates (*p < 0.05; **p < 0.005).
See also Figure S4.
We also performed a control experiment in which STE expression was regulated by the human ubiquitin C (hubC) promoter, which is constitutively active in OV8, aHDF, and HOV cells. We examined the sensitivity of these cells to STE-mediated killing and the cells’ ability to trigger IFN-γ production by T cells (Figures S4G–S4I). We found that while STE expression levels in OV8 cells were ~3-fold higher than in aHDF and HOV cells, STE-mediated killing was comparable in aHDF and OV8 and ~2-fold higher in HOV. In addition, all three STE-expressing cell lines triggered comparably strong IFN-γ production by T cells. These results highlight the stringency of our AND gate, which mediates significantly higher OV8 specificity over these other cell lines.
We next examined whether STE expression could trigger T cell-mediated anti-tumor activity in vivo. OV8 cells were trans-duced with lentiviruses encoding a doxcycline-inducible STE expression system and then injected intraperitoneally (i.p.) into immunodeficient NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice on day 0. Since NSG mice lack T cells, human T cells were also periodically injected i.p. Specifically, STE expression was induced at day 14, followed by human T cell injections at days 16 and 31. In the absence of T cells, tumor growth was similar between the STE-induced and non-induced groups. STE expression led to a 6-fold decrease in tumor burden in mice treated with T cells compared to non-treated mice (Figures S4J–S4L), demonstrating that the anti-tumor activity of STEs was dependent on T cells. However, consistent with previous studies (Liao et al., 2003), STE expression on its own was unable to completely abrogate tumor growth, even when expressed in high levels in nearly all the tumor cells (Figure S4L).
Tumor-Specific Expression of Multiple Immunotherapeutic Outputs Triggers Effective T Cell-Dependent Tumor Killing
Since combination therapy has shown enhanced therapeutic efficacy over monotherapies (Lasek et al., 2014; Mahoney et al., 2015; Tugues et al., 2015) and STE expression alone was insufficient to achieve strong anti-tumor effects in vivo, we engineered our optimized cancer-specific gene circuit to generate multiple immunomodulators as outputs. To demonstrate the capability of the circuit to generate various types of commonly used immu-nomodulators, we implemented the SCIP combination.
First, we transduced OV8 cells with lentiviruses encoding the G8-Pe AND gate and the SCIP combination. We validated the selective co-production of STE, CCL21, IL12, and an anti-PD1 antibody (Ab) from OV8 cells, but not from aHDF or HOV cells, in vitro (Figure 6D–6G). Next, we validated the functionality of our SCIP circuit with an in vivo model of human ovarian cancer. We engineered the G8-Pe circuit to produce a control output, STE only, or the SCIP combination in OV8 cells. We then established i.p. disseminated ovarian cancers by injecting these OV8 cells into the peritoneal cavity of NSG mice. This experimental setup was used to model the functionality of the circuit throughout the course of the treatment and its effects on circuit efficacy. For example, the activity of our input promoters could be altered in response to environmental signals or due to tumor heterogeneity, resulting in circuit inactivation and reduced efficacy.
In the absence of T cells, all tumors grew robustly (Figure S5A). When administered with T cells i.p., tumor sizes in the control output and STE-only groups were significantly larger than the STE+CCL21+IL12+anti-PD1 Ab (SCIP) combination, in which tumors were undetectable in most mice (Figures 7A, S5A, and S5B). Significantly prolonged mice survival was also observed in the SCIP group (Figure 7B). Thus, combinatorial immunotherapies expressed from within tumor cells can mediate strong anti-tumor effects.
Figure 7. Combination Immunotherapies Triggered by Cancer-Specific Circuits Significantly Reduced Tumor Burden and Increased Survival in an Intraperitoneally Disseminated Human Ovarian Cancer Model in Mice.
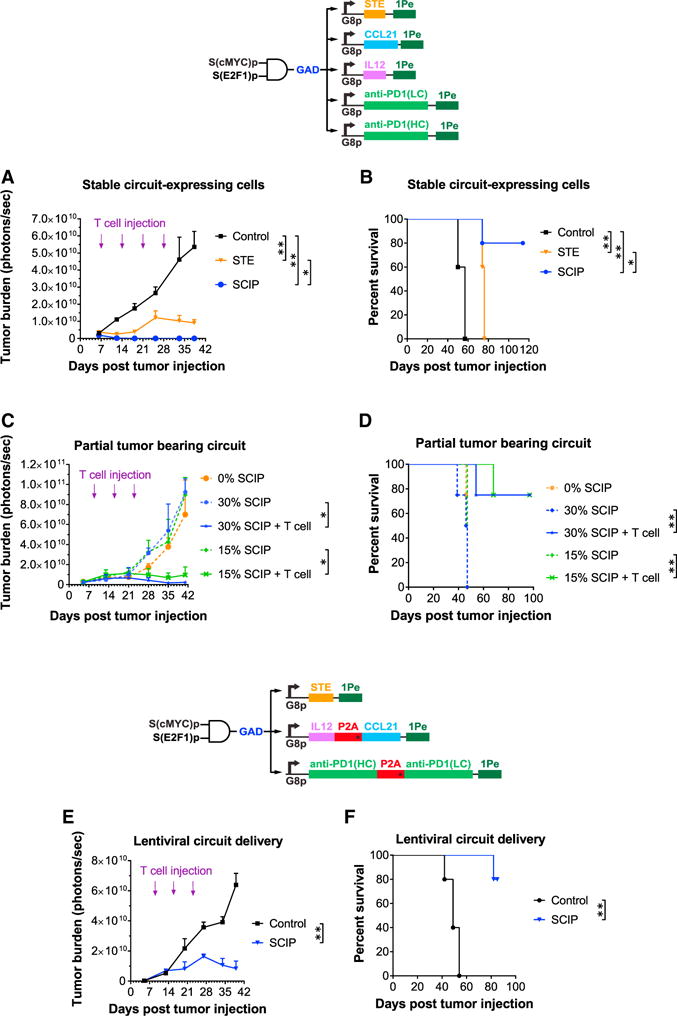
(A) OV8 cells were transduced with constructs encoding a control output rtTA3 (Control), STE-only (STE), or combination immunomodulators (SCIP). All groups were implanted into NSG mice and injected periodically with human T cells. Student’s t test was performed to compare tumor burden between groups at day 37.
(B) Kaplan-Meier survival curves of various groups. A log-rank (Mantel-Cox) test was performed to compare survival between groups.
(C) Robust therapeutic efficacy was achieved even when only 15% or 30% of tumor cells were transduced with the SCIP. Student’s t test was performed to compare tumor burden between groups at day 41.
(D) Kaplan-Meier survival curves of various groups with different percentages of the overall tumor cells transduced with the SCIP circuit. A log-rank (Mantel-Cox) test was performed to compare survival between groups.
(E) Lentiviral delivery of the SCIP-expressing circuit significantly reduced ovarian cancer burden. NSG mice were injected with OV8 cells into the peritoneal space at day 0. Lentiviruses encoding circuit module 1 and module 2 and a control output rtTA3 (Control), or combination immunomodulators (SCIP) were then injected i.p. on day 7. These mice were also injected i.p. periodically (days 9, 16, and 23) with human T cells. Student’s t test was performed to compare tumor burden between groups at day 39.
(F) Kaplan-Meier survival curves of various groups. A log-rank (Mantel-Cox) test was performed to compare survival between groups.
Error bars represent SEM, n = 5 biological replicates for all experiments, except n = 4 biological replicates for groups in (C–D).
(*p < 0.05; **p < 0.005).
See also Figure S5, S6, and S7.
Next, we sought to confirm that the SCIP-expressing circuit could mediate robust efficacy even when it was only introduced into a small fraction of tumor cells. This experiment was used to model a common problem with tumor-targeting therapies in which the therapeutic circuit is activated only in a fraction of the tumor. For example, this phenomenon can be mediated by modulation of gene expression, cellular heterogeneity within tumors, or partial circuit delivery. We mixed wild-type OV8 cells with OV8 cells containing the SCIP-expressing circuit in defined ratios. Even when only 15% or 30% of the overall tumor was composed of SCIP-expressing OV8 cells, significant tumor reduction and survival was still achieved, but only in the presence of T cells (Figures 7C, 7D, S6A, and S6B). These data suggest that immunomodulatory gene circuits may be able to mediate strong anti-tumor effects despite incomplete delivery in future applications.
Finally, we directly tested the efficacy of our SCIP circuit within in vivo delivery settings. To minimize the total number of viruses required to encode the entire SCIP circuit and thus simplify the delivery process, we combined the CCL21 and IL12 output modules into a single lentiviral construct by encoding both outputs, separated by a P2A peptide (Szymczak et al., 2004), under the regulation of a single G8 output promoter. We similarly combined the anti-PD1 Ab heavy-chain (HC) and light-chain (LC) modules into a single lentivirus. Mice with i.p. disseminated OV8 tumors were injected i.p. with either a control lentivirus mix that contained module 1, module 2, and a negative control output or a therapeutic virus mix that contained module 1, module 2, and the SCIP outputs. Human T cells were also periodically injected i.p. Under these settings, we observed significantly reduced tu-mor burden and enhanced survival in mice treated with the SCIP circuit versus the control unit (Figures 7E, 7F, S7A, and S7B). These data indicate that in vivo delivery of our immunomodula-tory gene circuits has the potential to trigger anti-tumor effects, even when the components of our gene circuit are encoded on multiple vectors.
DISCUSSION
In this study, we present a proof-of-concept demonstration that versatile genetic circuits can be engineered for highly specific production of combinatorial immunomodulators from within cancer cells. These circuits were delivered in vivo to trigger robust anti-tumor responses and increased survival of treated mice. Our modular design strategy enables the facile optimization required for future clinical translation and targeting of additional tumor types.
However, several issues must be addressed to further advance this approach toward therapeutic applications. Design and testing of circuits will likely need to be performed in additional models, such as patient-derived tumor cells and a panel of normal tissues. The potential off-target activity of circuit components, such as miR1 and the miRNA sponge, should also be examined, for example, via transcriptomics. In vitro assays only allow for testing of short-term specificity, and thus, future efforts should also examine long-term specificity and side effects within in vivo settings.
Furthermore, the NSG mice supplemented with human T cells that we utilized in this study have several drawbacks that limit our ability to fully evaluate safety and efficacy. In this model, tumor cells are of human origin, but normal tissues are of mouse origin, thus making it challenging to characterize targeting specificity for human applications. Second, NSG mice injected with human T cells eventually develop life-threatening graft-versus-host disease (Covassin et al., 2011), making it difficult to evaluate long-term safety concerns. Third, these mice only contain a partial human immune system, established by human T cell injections, and may not be sufficient to determine the exact optimal combination of immunomodulatory outputs required for efficacy in humans. Therefore, future work will be needed to optimize and characterize specific immunomodulatory combinations in additional animal models, including humanized mice, syngeneic mouse models with intact immune systems, and patient-derived xenograft models, which can account for tumor heterogeneity.
Improved delivery strategies will be needed to convey our immunomodulatory gene circuits into tumors, such as non-viral carriers or oncolytic viruses. Yet, given that only 15% of tumor cells had to express the circuit to achieve robust anti-tumor responses in our model (Figures 7C, 7D, and S6) and that our circuit could mediate anti-tumor effects when delivered in vivo by multiple lentiviruses (Figures 7E, 7F, and S7), we hypothesize that delivery efficiencies will not need to be perfect to achieve a therapeutic effect. However, we note that delivery requirements will likely vary in other tumor models.
Furthermore, it will be useful to test whether our approach can be used in combination with other therapeutic strategies. For example, the efficacy of engineered T cells may be enhanced with tumor-specific secretion of immunomodulators optimized to attract these cells to the tumor site and activate them while simultaneously disrupting the immunosuppressive microenvironment. Additionally, recent studies have shown that many tumors express immunogenic neoantigens that can be targeted by the host immune system (Schumacher and Schreiber, 2015). We speculate that using our strategy to trigger tumor killing in combination with neoantigen-based immunotherapies could be a fruitful approach to investigate. For example, our circuits could trigger robust killing of cancer cells and release such neoantigens. Furthermore, secreted immunomodulators could enhance neoantigen presentation by antigen-presenting cells and further improve anti-tumor immune responses.
Finally, since our approach enables tumor-specific secretion of defined combinations of immunomodulators, it could be a useful framework to study the immunological mechanisms that underlie tumor biology. Moreover, synthetic gene circuits have been shown to have the potential to treat immunological disorders by sensing extracellular disease markers (Schukur et al., 2015). Similarly, our approach for sensing internal cell states with multiple artificial promoters could be adapted to treat other complex diseases that require highly specific and multifactorial programming of immunological functions with simple modifications to the synthetic promoter inputs and immunomodulatory outputs.
In summary, modulating the immune system is a promising approach to treating complex diseases. However, multiple disease-mediated mechanisms can limit the effectiveness of mono-therapies (Mahoney et al., 2015). Combinatorial immunomodulators can be used to overcome this difficulty, but given the broad effects of the immune system on human physiology, it will be important to constrain the activity of immunomodulators to specific disease compartments to avoid undesirable side effects and maximize efficacy. Thus, we envision that synthetic gene circuit immunotherapies that are highly specific, tunable, and amenable to combinatorial effector expression will provide powerful strategies for engineering the immune system to understand and treat disease.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE anti-HA tag clone GG8-1F3.3.1 | Miltenyi Biotec | Cat# 130-092-257; RRID: AB_871939 |
| PE anti-His tag clone GG11-8F3.5.1 | Miltenyi Biotec | Cat# 130-092-691; RRID: AB_1103227 |
| Purified anti-human CD3 clone OKT3 | BioLegend | Cat# 317302; RRID: AB_571927 |
| Brilliant Violet 421 anti-human CD279 (PD-1) clone EH12.2H7 | BioLegend | Cat# 329920; RRID: AB_10960742 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human IL-2 protein | NCI BRB Preclinical Repository | https://ncifrederick.cancer.gov/research/brb/ |
| Fugene HD – Transfection Reagent | Promega | Cat #E2311 |
| VivoGlo Luciferin, In Vivo Grade | Promega | Cat #P1043 |
| Critical Commercial Assays | ||
| CytoTox 96® Non-Radioactive Cytotoxicity Assay Kit | Promega | Cat #G1780 |
| Human IFN-γ DuoSet ELISA | R&D systems | Cat #DY285 |
| Human CCL21/6Ckine DuoSet ELISA | R&D systems | Cat #DY366 |
| Human IL-12 p70 DuoSet ELISA | R&D systems | Cat #DY1270 |
| Cell Culture Reagents | ||
| DMEM | Life Technologies | Cat #10569-044 |
| Fetal bovine serum | VWR | Cat #95042-108 |
| Non-Essential Amino Acids | Hyclone | Cat #16777-186 |
| Pen/Strep | Life Technologies | Cat #15140-122 |
| RPMI-1640 | Invitrogen | Cat #11875119 |
| HEPES | Life Technologies | Cat #15630080 |
| Sodium Pyruvate | Life Technologies | Cat #11360-070 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat #M3148-25ML |
| Ovarian Epithelial Cell Medium | ScienCell Research Laboratories | Cat #7311 |
| Pen/Strep | ScienCell Research Laboratories | Cat #0503 |
| Ovarian Epithelial Cell Growth Supplement | ScienCell Research Laboratories | Cat #7352 |
| Poly-L-lysine | Sigma-Aldrich | Cat #P4707-50ML |
| Fibroblast Medium | ScienCell Research Laboratories | Cat #2301 |
| Fibroblast Growth Supplement | ScienCell Research Laboratories | Cat #2352 |
| Endothelial Cell Medium | ScienCell Research Laboratories | Cat #1001 |
| Endothelial Cell Growth Supplement | ScienCell Research Laboratories | Cat #1052 |
| Fibronectin | Sigma-Aldrich | Cat #F1141-2MG |
| MEGM BulletKit | Lonza | Cat #CC-3151 & CC-4136 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat #CRL-3216 |
| Adult Human Dermal Fibroblast | ATCC | Cat #PCS-201-012 |
| MDA-MB-453 | ATCC | Cat #HTB-131 |
| MCF-10A | ATCC | Cat #CRL-10317 |
| MCF-12A | ATCC | Cat #CRL-10782 |
| CCD-841-CoN | ATCC | Cat #CRL-1790 |
| OV8-Luc | Laboratory of Sangeeta N. Bhatia (MIT) | N/A |
| Primary human ovarian epithelial (HOV) | ScienCell Research Laboratories | Cat #7310 |
| Primary human ovarian fibroblasts (HOF) | ScienCell Research Laboratories | Cat #7330 |
| Primary human ovarian microvascular endothelial cells (HOMEC) | ScienCell Research Laboratories | Cat #7300 |
| iPSC | Laboratory of Ron Weiss (MIT) | N/A |
| Experimental Models: Organisms/Strains | ||
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) (female 8-13weeks) | Jackson Laboratories | Stock #005557 |
| Recombinant DNA | ||
| Please see “Plasmids by figures.docx” and “All sequences.docx” | This paper | N/A |
| Software and Algorithms | ||
| FlowJo software v7.6.1 | TreeStar | N/A |
| Prism Version 7 | GraphPad | N/A |
| Living Image v4.5.2 | Perkin Elmer | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Timothy Lu (timlu@mit.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Source and Culture of Primary Human T cells
Human PBMCs were obtained from leukoreduction collar (Brigham and Women’s hospital Crimson Core Laboratory, Boston, MA) with gradient centrifugation. To culture and expand human T cells, human PBMCs were stimulated with 40 ng/mL of anti-human CD3 antibody (clone: OKT3; BioLegend, San Diego, CA;Catalog #317304) for 3 days in RPMI 1640 (Invitrogen, Carlsbad, CA; Catalog #11875119), supplemented with 10% heat-inactivated FBS, 10 mM HEPES (Life Technologies Catalog #15630080), 0.1 mM nonessential amino acids, 1 mM sodium pyruvate (Life Technologies Catalog #11360-070), 1% Pen/Strep, 50 mM 2-Mercaptoethanol (Sigma-Aldrich Catalog #M3148-25ML), and 50 IU/mL recombinant human IL-2 (NCI, Frederick, MD).
Animal Model Details
Animal studies were conducted at the Koch Institute Animal Facility under a protocol approved by MIT’s Committee on Animal Care. Female NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; Stock No: 005557) were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were used in experiments at the age of 8-13 weeks old.
Cell Culture and Cell Lines
HEK293T, adult Human Dermal Fibroblast (aHDF), MDA-MB-453, MCF-10A, MCF-12A, and CCD-841-CoN cells were obtained from the American Type Culture Collection, Rockville, MD (HEK293T, Catalog #CRL-3216; aHDF, Catalog #PCS-201-012; MDA-MB-453, Catalog #HTB-131; MCF-10A, Catalog #CRL-10317; MCF-12A, Catalog #CRL-10782; CCD-841-CoN, Catalog #CRL-1790). Ovarian carcinoma cell line 8 (OV8) engineered to stably express firefly luciferase (OV8-Luc) was a gift from Sangeeta N. Bhatia (Massachu-setts Institute of Technology, Cambridge, MA). Primary human ovarian epithelial cells (HOV), primary human ovarian fibroblasts (HOF), and primary human ovarian microvasculature endothelial cells (HOMEC) were obtained from ScienCell Research Laboratories, Carlsbad, CA (HOV, Catalog #7310; HOF, Catalog #7330; HOMEC, Catalog #7300). Inducible pluripotent stem cell (iPSC) was a gift from R. Weiss (Massachusetts Institute of Technology). HEK293T, OV8-Luc, aHDF, and MDA-MB-453 cells were cultured in DMEM (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; VWR, Radnor, PA; Catalog #95042-108), 1% Non-Essential Amino Acids (MEM/NEAA; Hyclone; Catalog #16777-186), and 1% Pen/Strep (Life Technologies Catalog #15140-122) at 37 C with 5% CO2. HOV cells were cultured in Ovarian Epithelial Cell Medium (ScienCell Research Laboratories Cat-alog #7311) supplemented with Pen/Strep (ScienCell Research Laboratories Catalog #0503), Ovarian Epithelial Cell Growth Supplement (ScienCell Research Laboratories Catalog #7352) and were grown on a poly-L-lysine (Sigma-Aldrich, St. Louis, MO; Catalog #P4707-50ML) coated-plate. HOF cells were cultured in Fibroblast Medium (ScienCell Research Laboratories Catalog #2301) supplemented with Pen/Strep (ScienCell Research Laboratories Catalog #0503), Fetal Bovine Serum (ScienCell Research Laboratories Catalog #0010), Fibroblast Growth Supplement (ScienCell Re-search Laboratories Catalog #2352) and were grown on a poly-L-lysine (Sigma-Aldrich Catalog #P4707-50ML) coated-plate. HOMEC cells were cultured in Endothelial Cell Medium (ScienCell Research Laboratories Catalog #1001) supplemented with Pen/Strep (ScienCell Research Laboratories Catalog #0503), Fetal Bovine Serum (ScienCell Research Laboratories Catalog #0025), Endothelial Cell Growth Supplement (ScienCell Research Laboratories Catalog #1052) and were grown on a Fibronectin (Sigma-Aldrich Catalog #F1141-2MG) coated-plate. MCF-10A and MCF-12A cells were cultured in MEGM BulletKit (Lonza, Walkersville, MD; Catalog #CC-3151 and CC-4136). All cell lines were banked directly after being purchased from vendors and used at low passage numbers. Cell lines were tested negative for mycoplasma contamination with IMPACT PCR by the Division of Comparative Medicine Diagnostic Lab at MIT.
METHOD DETAILS
Plasmid Construction
The various circuit modules, synthetic promoters, and immunomodulatory constructs were built using conventional restriction enzyme cloning and Gibson assembly. Plasmids compositions used in each experiment are detailed in supplemental file “Plasmids by figures.docx.” All DNA sequences used in this study are detailed in GenBank format in the single text file “All sequences.docx.” Both files can be downloaded at Mendeley database: https://data.mendeley.com/datasets/8jsp7jzmt3/draft?a=56eab839-e2b7-47d1-9a6f-ef4d6d114f97
Additionally, individual GenBank and SnapGene sequence files for the plasmids in this study can be downloaded from a public Dropbox folder using the link below: https://www.dropbox.com/sh/wbq2y3lx87yzppe/AAAvFM7g6b1K35Ay1zSreoGDa?dl=0
Transfection for AND Gate Optimization
To optimize the AND gate circuit in Figures 2, 3, S1, S2, and S3, we tested different combinations of two plasmids: module 1 variants expressed an auto-inhibitory mKate2 RNA transcript regulated by synthetic miRNA (miR1) and module 2 variants expressed sponges for miR1 within an ECFP transcript, where ECFP enables us to assess sponge expression. In brief, 12 μL of FuGENE HD transfection reagent (Promega, Madison, WI; Catalog #E2311) mixed with 100 μL of OptiMEM medium (Life Technologies Catalog #31985) was added to the mixture of two plasmids (1 mg each). During 20 minutes incubation of FuGENE HD/DNA complexes at room temperature, HEK293T suspension cells were prepared and diluted to 1.2 × 106 cells/mL in culture medium. 0.5 mL of diluted cells (0.6 × 106 cells) were added to each FuGENE HD/DNA complex tube, mixed well, and after 5 min incubation at room temperature transferred to a designated well in 6-well plate containing 2 mL cell culture medium, followed by incubation at 37°C with 5% CO2. Transfected cells were prepared for FACS analysis at 48-hour post-transfection.
Lentivirus Production and Transduction
Lentiviruses were produced in HEK293T cells using co-transfection in 6-well plate format. In brief, 12 μL of FuGENE HD was mixed with 100 μL of OptiMEM medium and was added to a mixture of 3 plasmids: 0.5 μg of pCMV-VSV-G vector, 0.5 μg of lentiviral packaging psPAX2 vector, and 1 μg of lentiviral expression vector. During 20 minutes incubation of FuGENE HD/DNA complexes at room temperature, HEK293T suspension cells were prepared and diluted to 3.6 × 106 cells/mL in cell culture medium. 0.5 mL of diluted cells (1.8 × 106 cells) were added to each FuGENE HD/DNA complex tube, mixed well, and incubated for 5 min at room temperature before being added to a designated well in 6-well plate containing 1 mL cell culture medium, followed by incubation at 37°C with 5% CO2. Media of transfected cells were replaced with 2.5 mL fresh culture media 18 hours post transfection. Supernatant containing newly produced viruses was collected at 48-hour post-transfection, and filtered through a 0.45 mm syringe filter (Pall Corporation, Ann Arbor, MI; Catalog #4614).
For testing of synthetic promoters (Figure 4A and 4B), 1:9 dilutions of filtered viral supernatants were prepared and 3 mL of each individual virus was used to infect 2.5 × 105 OV8-Luc, aHDF or HOV cells in the presence of 8 μg/mL polybrene (Sigma) overnight. For optimizing and testing the GAD-expressing gate (Figure 5A–5E), we transduced three viruses encoding module 1, module 2, and module 3. Each virus was diluted 1:3 and we used 3 mL of the pooled viruses to infect 2.5 × 105 target cells in the presence of 8 μg/mL polybrene (Sigma) overnight. Cell culture medium was replaced the next day after infection and cells were cultured for at least one week prior to FACS analysis or cytotoxicity assay. For creating cell lines bearing circuits expressing multiple outputs (Figures 6D–6G and 7), two rounds of lentiviral infection were performed. The viruses for the first round consisted of module 1, module 2, and an output construct expressing STE. The viruses for the second round consisted of output constructs expressing all other outputs.
Flow Cytometry
For characterizing fluorescent protein expression, cells were resuspended with DMEM and analyzed by a LSRII Fortessa cytometer (BD Biosciences, San Jose, CA). To characterize STE display and expression level on cell surfaces, various cell lines were stained with phycoerythrin labeled anti-HA tag Ab (Miltenyi Biotec; catalog #130-092-257), or anti-His tag Ab (Miltenyi Biotec; catalog #130-092-691). Data analysis was performed by FlowJo software (TreeStar Inc, Ashland, OR).
T Cell-Mediated Cell Lysis In Vitro
T cell-mediated cell lysis was measured by LDH release assay with CytoTox 96® Non-Radioactive Cytotoxicity Assay Kit (Promega, Madison, WI; catalog #G1780). Human T cells were incubated with circuit-expressing target cells at a effector to target ratio (E:T ratio) of 20:1 (6×105:3×104) in duplicate wells in 96 well flat-bottom plates. T cells and target cells were also plated alone to determine spontaneous LDH release from T cells (“effector spontaneous”) and from target cells (“target spontaneous”). A set of target cells were also plated and lysed with lysis buffer for determining maximal LDH release (“target maximum”). 8.5 hours later, cell-free culture supernatant was harvested and LDH release was determined following the manufacturer’s protocol. T cell-mediated lysis was calculated as: percentage of T cell-mediated lysis = [(experimental O.D. value - effector spontaneous O.D. value - target spontaneous O.D. value) / (target maximum O.D. value - target spontaneous O.D. value)] × 100. Media O.D. values were subtracted from all samples before calculation. STE mediated lysis was calculated as: T cell mediated lysis of STE expressing cells - T cell mediated lysis of non-STE expressing cells. The results of each experiment represent three biological replicates.
IFN-γ Production by T Cells
Human T cells were co-cultured with tumor cells at an E:T ratio = 20:1 (6×105:3×104) in duplicate wells in 96 well flat-bottom plates. Cell-free medium was collected after 24h and IFN-γ concentration determined by Human IFN-γ DuoSet ELISA (R&D systems, Minneapolis, MN; catalog #DY285).
Multiple-Output Circuit Triggers STE, CCL21, IL12, and Anti-PD1 Ab Production Specifically by OV8 Cells
To validate STE, CCL21, IL12, and anti-PD1 Ab production by OV8 cells but not by normal cells (Figure 6D–6G), 2×105 OV8-Luc, HOV, or aHDF cells transduced with the SCIP-expressing circuit were plated in 6-well plates and cell-free medium was collected after 72h. Displayed STE on cell surfaces was measured via staining with phycoerythrin labeled anti-HA tag Ab (Miltenyi Biotec) or phycoery-thrin labeled anti-His tag Ab (Miltenyi Biotec) and quantified by flow cytometry analysis. CCL21 concentration was determined by Human CCL21/6Ckine DuoSet ELISA (R&D systems; catalog #DY366). IL12 concentration was determined by Human IL12 p70 DuoSet ELISA (R&D systems; catalog #DY1270), and anti-PD1 Ab production was validated by a competitive staining approach. The competitive staining was done by incubating cell-free medium (circuit-conditioned or control medium) with activated human T cells for 30 minutes, then followed by staining with Brilliant Violet 421 anti-human CD279 (PD-1) Ab [clone: EH12.2H7; BioLegend; catalog #329920]. The percentage signal reduction was calculated by the following equation: (1 - PD1 staining MFI of T cells incubated with circuit-conditioned medium / PD1 staining MFI of T cells incubated with control medium)* 100%.
In Vivo Experiments Validating Circuit Therapeutic Efficacy
For in vivo experiments, animals were randomly assigned to each experimental group. No specific inclusion/exclusion criteria were used for animal studies. Investigators were not blinded to animal groups. Four to five mice were used in each experimental group. To validate STE therapeutic efficacy on ovarian cancer, OV8-Luc cells expressing doxycycline (Dox)-inducible STE (5×105 cells) were injected intraperitoneally (i.p.) into NSG mice on day 0, and STE expression were induced by Dox [0.2mg/mL Dox + 2% Sucrose (Sigma-Aldrich; catalog #S3929-1KG) in drinking water] at day 14. Activated human T cells (107 cells) were injected at day 16 and 31. For measuring tumor burden, mice were injected with 3mg of VivoGlo Luciferin, In Vivo Grade (Promega; catalog #P1043) 10 minutes prior to bioluminescence imaging (IVIS spectrum optical imaging system, Xenogen, Alameda, CA).
To measure the efficacy of different therapeutic output combinations on ovarian cancer, OV8-Luc cells (5×105 cells) engineered to express various outputs were injected i.p. into NSG mice on day 0. Activated human T cells (107 cells) were injected at day 7, 14, 21 and 28. Tumor burden was monitored by IVIS imaging using the protocol described above. To measure survival, mice were euthanized when they developed signs of distress, such as ruffled fur, poor body posture, distended abdomen, and jaundice.
To identify the minimal percentage of tumor cells needed to be transduced with the SCIP-expressing circuit to mediate robust therapeutic efficacy, OV8-Luc cells engineered with the SCIP-expressing circuit were mixed with wild-type OV8-Luc cells at various ratios (15% to 85%; 30% to 70%). 5×105 mixed OV8-Luc cells were injected i.p. into NSG mice on day 0. Activated human T cells (107 cells) were injected at day 9, 16, and 23. Tumor burden was monitored by IVIS imaging using the protocol described above. We used the criteria described above to measure survival.
To deliver the SCIP circuit with lentivirus in vivo, lentiviruses for each circuit component (module 1, module 2, STE, CCL21+IL12, and anti-PD1 Ab, see Figure 7E for more details) were produced as described above. For the SCIP therapy group, 10 mL of viruses for each of the five circuit components were collected and concentrated 15-fold using Amicon 100K MW cutoff ultrafiltration columns (Millipore, catalog #UFC910024). For the control group, 10 mL of viruses for module 1, 10 mL of viruses for module 2, and 30 mL of viruses encoding the control output (rtTA3) was collected and concentrated. After concentration, viruses were pooled and a p24 ELISA kit (Advanced Bioscience Laboratories, catalog #5421) was used to determine the viral titer. The pooled viruses had titers of ~7×1010 viral particles/mL. Polybrene was added to the viruses to reach a final concentration of 16 μg/mL. 600 μL of viral supernatant was injected into mice intraperitoneally.
Sponge Optimization in Module 2
We first optimized the miR1 binding-sites (miR1-BSs) in module 2 for maximal sponging efficiency. We designed 14 variants of miR1-BSs with different mismatches, insertions, and deletions compared to perfect-match miR1-BSs (Figures S1B–S1D). We measured the sponging efficiency of each variant by comparing mKate2 levels generated by module 1 in the presence of the module 2 sponge encoding 5 identical tandem miR1-BSs in the 3′ UTR of ECFP versus no miR1-BSs (hereafter reported as “fold ON-OFF induction”). As an indication of miR1 binding to the sponge, we also expected to observe a decrease in ECFP expression in constructs where the 3′ UTR of ECFP contained miR1-BSs versus no miR1-BSs. We used two highly active native promoters in HEK293T cells, SSX1p (Gure et al., 1997) and H2A1p (Rogakou et al., 1998), to express module 1 and module 2, respectively. In HEK293T cells, H2A1p, which expresses the sponge in module 2, is > 3-fold stronger than SSX1p, which expresses module 1 (Figure S1A). Thus, the module 2 sponge transcript concentrations were expected to be in excess compared to the module 1 transcript, which ensured enhanced sponging efficiency. When we tested a sponge transcript with perfect match to miR1 generated by module 1, we found a 80% decrease in the levels of ECFP, even though only a 33% decrease was expected due to the 1:3 ratio of miR1:sponge transcript concentrations. This phenomenon may reflect recycling of miRNA molecules following target transcript inhibition, which is especially efficient for perfect–match binding sites (Baccarini et al., 2011) (Figure S1D).
We calculated the binding efficiency of miR1 to its corresponding binding sites in the sponge (ΔΔG) with a previously described model (Kertesz et al., 2007), but we found that ΔΔG did not predict sponging efficiency (Figure S1B). Specifically, calculations for microRNA binding efficiencies to target sites were performed at: https://genie.weizmann.ac.il/pubs/mir07/mir07_prediction.html. For example, binding sites with perfect complementary to miR1 (Pe) had the strongest binding efficiency (ΔΔG = 26), but limited mKate2 activation (Figure S1C, 1.7-fold ON-OFF induction), which could be a result of efficient sponge transcript degradation due to perfect miRNA complementarity (Ebert and Sharp, 2010). A ΔΔG = −15 was calculated for several binding sites that contained various mismatches to miR1 that resulted in bulged complementarity to miR1 (Bv2, Bv4, Bv6 and Bv15). Yet, the measured ON-OFF induction for these binding sites ranged from 1.25-fold (Bv15) to 2.62-fold (Bv2), which covered almost the entire dynamic range of mKate2 activation. miR1 binding site version Bv2 contained mismatches to nucleotides 9–12 of miR1 and exhibited the best sponging efficiency (2.6-fold mKate2 ON-OFF induction), likely because mismatches at these positions protect the sponge from Ago2-mediated degradation while allowing efficient miR1 binding by preserving seed sequence complementarity (Ebert and Sharp, 2010). We therefore used Bv2 for the sponge in further optimization of the circuit. Hereafter, we use the abbreviation ‘#B’ to refer to the number of tandem Bv2 binding sites separated by 5 bp spacers (for example, ‘5B’ refers to 5 Bv2 binding sites). Finally, we also observed that ECFP levels could not predict mKate2 activation. These results highlight the complexity of miRNA-mediated regulation, in which the complementarity of the miRNA to its cognate binding sites determines not only the inhibitory pathway to which the target mRNA is designated (mRNA cleavage, destabilization or translation inhibition), but also the rate of unloading of miRNAs from Ago2, which is enhanced by high complementarity of the miRNA to its target (De et al., 2013). While bulged BSs generally have better efficiency in shunting miRNAs compared to perfect match BSs, we believe that deriving optimized rules on how to introduce effective bulges will require additional studies.
We then tested how the number, spacing, location and, architecture of miR1 Bv2 binding sites in the sponge transcript, together with sponge expression levels, affect sponging efficiency. We observed 2.9 and 3.6-fold mKate2 ON-OFF induction when 5B versus 10B binding sites were encoded in the 3′ UTR of the module 2 sponge transcript expressed by H2A1p, respectively (Figure S2A, Sponge 1: H2A1p-ECFP-5B versus Sponge 4: H2A1p-ECFP-10B). Switching the promoter in the sponge encoding the 5B binding sites from H2A1p to CMVp, a 4.3-fold stronger promoter compared to H2A1p (Figure S1A), resulted in a 6-fold ON-OFF induction in mKate2 levels (Figure S2A, Sponge 3: CMVp-ECFP-5B). Therefore, increasing the sponge transcript concentration efficiently increases miR1 shunting, suggesting that the promoter selected to express module 2 should be stronger than the one selected to express module 1. Switching the location of the 10B from the 3′ UTR to the 5′ UTR of ECFP in module 2 (Sponge 4: H2A1p-ECFP-10B versus Sponge 5: H2A1p-10B-ECFP) reduced the sponging efficacy from 3.6-fold mKate2 ON-OFF induction to 2.4-fold (Figure S2A), which suggests that the miR1-BS(B) were more accessible when encoded in the 3′ UTR of the mRNA rather than in the 5′ UTR. Interestingly, increasing the spacing between miR1-BSs located in the 3′ UTR of the sponge transcript from 5 bp to 100 bp (Sponge 2: H2A1p-ECFP-5B/100bp) only mildly reduced the sponging efficiency from 2.9 to 2.3-fold mKate2 ON-OFF induction. Additionally, encoding 24 miR1-BSs in the loop of an RNA hairpin with previously described tough-decoy (TuD) architecture (2 miR1-BSs per loop, 12 loops overall, Sponge 9: H2A1p-ECFP-12TuD (Ebert and Sharp, 2010; Haraguchi et al., 2009) provided only 3.8-fold mKate2 ON-OFF induction (Figure S2A).
Finally, to increase the molar ratio of sponges produced from a given promoter, we designed a module 2 variant where 10B were encoded as an intron within an ECFP coding sequence in addition to 5B encoded in the 3′UTR of the transcript (Figure S2A). In this architecture, the donor and acceptor consensus sequences that flank the 10 intronic miR1-BS(B) were derived from a previously described synthetic intron (Sponge 6: H2A1p-EC1-10BCons-EC2-5B) (Greber et al., 2008). Based on ECFP expression, this sponge was correctly spliced, thus producing ECFP mRNA that contained 5B and an intron with 10B. This sponge demonstrated 4-fold mKate2 ON-OFF induction, slightly better than Sponge 4: H2A1p-ECFP-10B.
We designed an additional architecture in which the intronic consensus sequences that flank the 10B were derived from the ZRANB1 gene (Memczak et al., 2013) (Sponge 7: H2A1p-EC1-10BZRANB1-EC2-5B). This design was supposed to generate a stable circular 10B exon and EC1/EC2-5B introns, thus disrupting ECFP expression. This architecture demonstrated enhanced sponging efficacy with 6-fold mKate2 activation (Figures S2A and S2B) and did not produce ECFP signal. To test whether that the lack of ECFP signal was caused by the expected improper splicing, resulting in an impaired ECFP transcript, or because the transcript was not spliced at all, we generated an additional sponge in which the ZRANB1 splicing sequences were deleted (Sponge 8: H2A1p-EC1-10B-EC2-5B). Sponge 8 demonstrated almost identical sponging efficacy as Sponge 7 (Figure S2A), which supports the hypothesis that Sponge 7 did not splice. Since Sponge 7 and Sponge 8, which did not splice, demonstrated higher sponging efficiency than Sponge 6, which did splice, we concluded that intronically encoded miRNA sponges did not significantly contribute to sponging activity. Thus, Sponge 7: H2A1p-EC1-10BZRANB1-EC2-5B architecture was chosen as the final ‘optimized sponge’ design for our circuit.
miRNA Backbone Optimization in Module 1
We hypothesized that our circuit performance could be optimized by enhancing miR1 production from module 1. We therefore modified the pri-miRNA stems for efficient pri-miRNA processing (Figure S3A and Table S2) (Auyeung et al., 2013; Fellmann et al., 2013; Suzuki et al., 2015). Previous studies have described multiple features of pri-miRNA stems that promote Drosha-mediated pri-miRNA processing, including a UG motif at the base of the pri-miRNA hairpin, a narrow range of tolerable pri-miRNA stem lengths, a mismatched motif in the basal stem region, and a CNNC motif 16-18bp downstream to the Drosha processing site that is required for efficient SRp20-dependent processing of the pri-miRNAs (Auyeung et al., 2013; Fellmann et al., 2013). Our miR1 design Mv1 was based on a previously described intronically encoded synthetic miRNA (Greber et al., 2008; Nissim et al., 2014; Xie et al., 2011) derived from a miR-30 backbone (Fellmann et al., 2013). The EcoRI restriction site at position 231-236 was generated by mutating the original sequence of endogenous miR-30 from GACTTC to GAATTC (where position 1 is the first intronic nucleotide following the splicing of module 1). This point mutation disrupted an essential CNNC motif in the miRNA backbone and resulted in reduced pri-miRNA processing efficiency. The XhoI restriction site at position 128–133 was generated by mutating the original sequence of the endogenous miR-30 from CTAAAG to CTCGAG, which could further impair the pri-miRNA secondary structure.
We generated our Mv2 design by adding a CNNC motif (CTTCAAGGGGCTA) downstream to base pair 231 of Mv1 (Insert 1) and an additional insert (AAGGTATAT) downstream to base pair 133 of Mv1 (Insert 2) to improve the secondary pri-miRNA structure that was altered by the formation of the XhoI restriction site (Fellmann et al., 2013) (Figure S3A and Table S2). To generate our Mv3 design, we added an additional insert (GACTTC) downstream to base pair 111 of Mv1 (Insert 3) to improve the secondary pri-miRNA structure that was altered by the formation of the EcoRI restriction site (Fellmann et al., 2013) (Figure S3A and Table S2). This resulted in a pri-miRNA backbone similar to the optimized miR-E backbone, which is superior to the miR-30 backbone (Fellmann et al., 2013).
We compared miR1 production from each backbone by characterizing their efficiency at inhibiting mKate2 levels in the module 1 transcript, in which they were encoded, and ECFP levels in a module 2 mRNA containing 5 perfect match miR1-BSs (5Pe) (Figures S3B–S3E). As reference, we used a control construct with no miR1-BSs (Figure S3B). Pri-miRNA Mv3 mediated a 93% decrease in ECFP expression in the module 2 construct with the 5Pe sponge compared to the control construct, which was superior to Mv1 (80% inhibition) and Mv2 (71% inhibition), demonstrating that pri-miRNA design can tune the efficiency of miR1 production. These data suggest that that miR1 production was more potent in Mv3 compared to Mv2 and Mv1.
We also validated the effects of the miR1 backbone on the efficiency of mKate2 production by measuring mKate2 activation levels in the presence of the optimized sponge in module 2 (Figures S3D and S3E). The absolute mKate2 signal in the OFF and ON states (e.g., mKate2 levels measured with control versus optimal sponge) were both affected by the pri-miRNA design: Mv1 exhibited an OFF-ON shift from ~1400 to ~8400 Mean Fluorescence Intensity (MFI) (5.8-fold activation ratio), Mv2 from ~170 to ~500 MFI (3-fold activation) and Mv3 from ~380 to ~2600 MFI (6.8-fold activation). Based on the ECFP repression data from comparing the 5Pe sponge to the control construct, we expected Mv3 to be more efficient at inhibiting mKate2 than Mv1 and Mv2 in the presence of the 5Pe sponge. However, Mv2 exhibited potent inhibition of mKate2 levels in both the OFF and ON states. This suggests that our modifications to the pri-miRNA design may not only affect the efficiency of miR1 production, but also other regulatory mechanisms. We hypothesize that these pri-miRNA modifications could modify the efficiency of existing splicing sites or may have added alternative splicing sites in the miRNA intron that could impair mKate2 production by alternative splicing. This hypothesis is supported by the fact that the pri-miRNAs in designs Mv2 and Mv3 include a potential splicing donor sequence AAGGT introduced by Insert 2, and that a potential donor site CAGGT in Mv1 and Mv2 was modified to CAGGG by Insert 3 in Mv3 (Table S2).
As noted in the main text, we selected the Mv3 architecture for further experimentation since it had low mKate2 level in OFF state and exhibited the highest fold activation by the sponge in ON state.
Finally, the miRNA duplex sequence can be modified to promote asymmetric guide strand selectivity that favors miR1 production. The miR1 pri-miRNA is processed into a miRNA duplex capable producing both miR1 encoded by the duplex 3p arm and an alternative miRNA encoded by the duplex 5p arm (Table S2). The ratio of 3p (miR1):5p production is determined by the superposed patterns of the 5′ end nucleotide identity and the thermodynamic stability of two miRNA duplex termini (Suzuki et al., 2015). Quantitative thermodynamic and sequence analysis of the miR1 duplex has shown that the 3p arm has 5′-uridine and a thermodynamically unstable 5′ end, whereas 5p arm has 5′-guanine and a thermodynamically stable 5′ end (Suzuki et al., 2015). This indicates that the miR1 duplex is optimized to produce mostly the 3p arm (e.g., miR1); thus, it was therefore not modified. Nevertheless, we note that asymmetric guide strand selectivity is an important consideration in designing synthetic miRNAs.
Bioinformatics Framework for Synthetic Promoters Design
We identified genes that were differentially expressed in human ovarian cancer cells compared to healthy tissues from the publicly available The Cancer Genome Atlas (TCGA) database (Bell et al., 2011) using R (version 3.0.2). We followed standard procedures to calculate differentially expressed genes between cancer cases compared to controls using t test. The p values were corrected for multiple testing using the Bonferroni correction. We then selected genes that were previously identified as transcription factors (TFs) from the literature (Vaquerizas et al., 2009; Wingender et al., 2013). From these genes, we selected the ones that were hypomethylated in cancer. For that purpose, we used the publicly available database of DNA Methylation and gene expression in Human Cancer MethHC (Huang et al., 2015). This database provides the most hyper- and hypomethylated genes for different cancers. We selected TF genes with the most hypomethylated promoter regions using default settings. TF expression in OV8 cells was confirmed using the CCLE database (Barretina et al., 2012). We then determined the binding motifs for ovarian cancer-enriched TFs using GREAT, MEME, JASPAR and MOTIFMAP (Bailey et al., 2009; Daily et al., 2011; McLean et al., 2010; Sandelin et al., 2004) and assembled them into our synthetic promoters (Figures 4A and 4B).
Analysis of Natural and Synthetic Promoters
We analyzed promoter sequences using MatInspector software to predict transcription factor binding sites (TF-BSs) (Cartharius et al., 2005). We ran our analysis on two natural promoters (SSX1p, H2A1p) and two synthetic promoters (S(E2F1)p, S(cMYC)p) using core similarity 0.9 and matrix similarity optimized + 0.05. We examined individual TF-BSs as well as TF-BS families (defined as TFs with highly similar TF-BS – for example, the binding sites for MYC, USF and Max were grouped as the family E-BOX). We found that the synthetic promoters had more predicted TF-BSs per 100bp. For example, S(cMYC)p contained 35.8 TF-BS matches per 100 bp while H2A1p and SSX1p contained ~7.9 TF-BS matches per 100bp. The synthetic promoters had matches to less TF-BS families (~6–7 different TF-BS families) compared with the natural promoters (~40–41 different TF-BS families). The synthetic promoters had more TF-BS matches for each of the different TF-BS families compared with the natural promoters. Furthermore, we identified the TF-BS family with the highest number of matches for each promoter, and found that the top TF-BS family for the synthetic promoters had more TF-BS matches than the natural promoters. For example, E2F TF-BS family was found to be 66% of the total predicted TF-BSs in S(E2F1)p and the E-BOX TF-BS family (corresponding to MYC, USF, MAX) were found to be 29% of the total predicted TF-BSs in S(cMYC)p. In contrast, the ZF5F (ZFP 161) TF-BS family was found to be only 11% of the total predicted TF-BSs in H2A1p while the MYBL (MYB) TF-BS family was found to be only 6% of the total predicted TF-BSs in SSX1p.
Multi-output AND Gate Optimization for Combinatorial Immunomodulation
We first characterized the output levels of four distinct circuit states in OV8 cells (Figure 5B). To do so, we encoded each module on separate lentiviruses and infected OV8 cells with the corresponding module 3 architecture and either: (A) a negative control module 1 that expresses the non-specific transcription factor rtTA3 (module 1con) and a negative control module 2 in which S(E2F1)p expresses ECFP without miR1-BS(B) (module 2con) to model the scenario when neither cancer-specific promoters are active [0,0]; (B) module 1con and module 2 to model the scenario when only S(E2F1)p is active [0,1]; (C) module 1 and module 2con to model the scenario when only S(cMYC)p is active [1,0]; (D) module 1 and module 2 to model the scenario when both promoters are active [1,1].
The circuits expressed minimal mKate2 fluorescence in states [0,0] and [0,1]. In state [1,0], there was a low background mKate2 level that increased from 5 to 8 GAL4-BSs in the synthetic GALp and was attenuated with the presence of miR1-BS(Pe) in module 3 (Figure 5B). Only when both input constructs were present (state [1,1]) did we observe a significantly increased mKate2 output. Furthermore, the mKate2 output level could be enhanced by increasing the number of GAL4-BSs in the module 3 synthetic promoter from 5 to 8 GAL4-BSs. Although adding perfect match miR1 binding sites in the 3′ UTR of the mKate2 transcript decreased the basal and maximal output levels, it also improved fold activation (G5-Pe, G8-Pe, and G14-Pe versus G5, G8, and G14, respectively). The G8-Pe circuit exhibited negligible mKate2 levels in states [0,0] and [0,1], and high 8-fold mKate2 activation in state [1,1] over the minimal mKate2 expression in state [1,0], and was thus chosen for further use.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was done by Prism software version 7.01 (GraphPad Software, Inc., La Jolla, CA).
DATA AND SOFTWARE AVAILABILITY
Plasmids compositions used in each experiment are detailed in supplemental file “Plasmids by figures.docx”. All DNA sequences used in this study are detailed in GenBank format in the single text file “All sequences.docx”. Both files can be downloaded at Men-deley Data: https://data.mendeley.com/datasets/8jsp7jzmt3/draft?a=56eab839-e2b7-47d1-9a6f-ef4d6d114f97.
Additionally, individual GenBank and SnapGene sequence files for the plasmids in this study can be downloaded from a public Dropbox folder using this link: https://www.dropbox.com/sh/wbq2y3lx87yzppe/AAAvFM7g6b1K35Ay1zSreoGDa?dl=0.
Supplementary Material
Highlights.
Synthetic RNA-based circuits enable tumor-specific immunomodulator expression
These circuits trigger tumor-specific killing by T cells in vitro
Circuit-mediated immunomodulation enacts effective anti-tumor responses in vivo
This approach can be adapted to target multiple cancer types
Acknowledgments
L.N. dedicates this work to Zmira Nissim. L.N. thanks Yaakov Nissim, Aviva Yitzhak, and Ovad Nissim for their invaluable support. We thank Ramez Daniel for discussion. We thank Mark Mimee and Karen Weisinger for editing the manuscript. We thank the staff of the Division of Comparative Medicine at Koch Institute for Integrative Cancer Research at Massachusetts Institute of Technology for helping with animal experiments. We thank the Swanson Biotechnology Center at Koch Institute for assisting with animal imaging. This work was supported by the National Institutes of Health (1P50GM098792, R01-GM034277, R01-CA133404), the Department of De-fense (W81XWH-16-1-0565, W81XWH-16-1-0452), the Defense Advanced Research Projects Agency, and the Koch Institute Frontier Research Program and in part by the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute. H.I.S. is supported by the Uehara Memorial Foundation Research Fellowship and the Osamu Hayaishi Memorial Scholarship for Study Abroad. L.N., M.-R.W., and T.K.L. have filed a patent application on this work. T.K.L. is a member of the scientific advisory board at Senti Biosci-ences and holds shares in the company.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two tables and can be found with this article online at https://doi.org/10.1016/j.cell.2017.09.049.
AUTHOR CONTRIBUTIONS
L.N., M.-R.W., and T.K.L. conceived and designed the study. L.N., M.-R.W., E.P., and A.B.-N. performed experiments and analyzed data. H.I.S. and P.A.S. performed the miRNA backbone optimization. D.S., C.W., and Y.T. performed computational analysis and bioinformatics framework for synthetic promoters design. L.N., M.-R.W., E.P., A.B.-N., H.I.S., D.S., C.W., Y.T., P.A.S., and T.K.L. wrote the paper. All authors discussed the results and reviewed the manuscript.
References
- Auyeung VC, Ulitsky I, McGeary SE, Bartel DP. Beyond secondary structure: primary-sequence determinants license pri-miRNA hairpins for processing. Cell. 2013;152:844–858. doi: 10.1016/j.cell.2013.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarini A, Chauhan H, Gardner TJ, Jayaprakash AD, Sachidanan-dam R, Brown BD. Kinetic analysis reveals the fate of a micro-RNA following target regulation in mammalian cells. Curr Biol. 2011;21:369–376. doi: 10.1016/j.cub.2011.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchus W, Aubel D, Fussenegger M. Biomedically relevant circuit-design strategies in mammalian synthetic biology. Mol Syst Biol. 2013;9:691. doi: 10.1038/msb.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell D, Berchuck A, Birrer M, Chien J, Cramer DW, Dao F, Dhir R, DiSaia P, Gabra H, Glenn P, et al. Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–615. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros C, Tarhini A, Routier E, Lambotte O, Ladurie FL, Carbonnel F, Izzeddine H, Marabelle A, Champiat S, Berdelou A, et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol. 2016;13:473–486. doi: 10.1038/nrclinonc.2016.58. [DOI] [PubMed] [Google Scholar]
- Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- Covassin L, Laning J, Abdi R, Langevin DL, Phillips NE, Shultz LD, Brehm MA. Human peripheral blood CD4 T cell-engrafted non-obese diabetic-scid IL2rg(null) H2-Ab1 (tm1Gru) Tg (human leucocyte antigen D-related 4) mice: a mouse model of human allogeneic graft-versus-host disease. Clin Exp Immunol. 2011;166:269–280. doi: 10.1111/j.1365-2249.2011.04462.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daily K, Patel VR, Rigor P, Xie X, Baldi P. MotifMap: integrative genome-wide maps of regulatory motif sites for model species. BMC Bio-informatics. 2011;12:495. doi: 10.1186/1471-2105-12-495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De N, Young L, Lau PW, Meisner NC, Morrissey DV, MacRae IJ. Highly complementary target RNAs promote release of guide RNAs from human Argonaute2. Mol Cell. 2013;50:344–355. doi: 10.1016/j.molcel.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MS, Sharp PA. MicroRNA sponges: progress and possibilities. RNA. 2010;16:2043–2050. doi: 10.1261/rna.2414110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. An optimized micro-RNA backbone for effective single-copy RNAi. Cell Rep. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Greber D, El-Baba MD, Fussenegger M. Intronically encoded siRNAs improve dynamic range of mammalian gene regulation systems and toggle switch. Nucleic Acids Res. 2008;36:e101. doi: 10.1093/nar/gkn443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gure AO, Türeci O, Sahin U, Tsang S, Scanlan MJ, Jäger E, Knuth A, Pfreundschuh M, Old LJ, Chen YT. SSX: a multigene family with several members transcribed in normal testis and human cancer. Int J Cancer. 1997;72:965–971. doi: 10.1002/(sici)1097-0215(19970917)72:6<965::aid-ijc8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Haraguchi T, Ozaki Y, Iba H. Vectors expressing efficient RNA decoys achieve the long-term suppression of specific microRNA activity in mammalian cells. Nucleic Acids Res. 2009;37:e43. doi: 10.1093/nar/gkp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang WY, Hsu SD, Huang HY, Sun YM, Chou CH, Weng SL, Huang HD. MethHC: a database of DNA methylation and gene expression in human cancer. Nucleic Acids Res. 2015;43:D856–D861. doi: 10.1093/nar/gku1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kertesz M, Iovino N, Unnerstall U, Gaul U, Segal E. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39:1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22:26–36. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotula JW, Kerns SJ, Shaket LA, Siraj L, Collins JJ, Way JC, Silver PA. Programmable bacteria detect and record an environmental signal in the mammalian gut. Proc Natl Acad Sci USA. 2014;111:4838–4843. doi: 10.1073/pnas.1321321111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasek W, Zagożdżon R, Jakobisiak M. Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immun-other. 2014;63:419–435. doi: 10.1007/s00262-014-1523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengyel E, Burdette JE, Kenny HA, Matei D, Pilrose J, Haluska P, Nephew KP, Hales DB, Stack MS. Epithelial ovarian cancer experimental models. Oncogene. 2014;33:3619–3633. doi: 10.1038/onc.2013.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, Sosman JA, Dutcher JP, Vogelzang NJ, Ryan JL. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. 1997;90:2541–2548. [PubMed] [Google Scholar]
- Liao KW, Lo YC, Roffler SR. Activation of lymphocytes by anti-CD3 single-chain antibody dimers expressed on the plasma membrane of tumor cells. Gene Ther. 2000;7:339–347. doi: 10.1038/sj.gt.3301080. [DOI] [PubMed] [Google Scholar]
- Liao KW, Chen BM, Liu TB, Tzou SC, Lin YM, Lin KF, Su CI, Roffler SR. Stable expression of chimeric anti-CD3 receptors on mammalian cells for stimulation of antitumor immunity. Cancer Gene Ther. 2003;10:779–790. doi: 10.1038/sj.cgt.7700637. [DOI] [PubMed] [Google Scholar]
- Lin Y, Sharma S, John MS. CCL21 Cancer Immunotherapy. Cancers (Basel) 2014;6:1098–1110. doi: 10.3390/cancers6021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zeng Y, Liu L, Zhuang C, Fu X, Huang W, Cai Z. Synthesizing AND gate genetic circuits based on CRISPR-Cas9 for identification of bladder cancer cells. Nat Commun. 2014;5:5393. doi: 10.1038/ncomms6393. [DOI] [PubMed] [Google Scholar]
- Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–584. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cisregulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- Morel M, Shtrahman R, Rotter V, Nissim L, Bar-Ziv RH. Cellular heterogeneity mediates inherent sensitivity-specificity tradeoff in cancer targeting by synthetic circuits. Proc Natl Acad Sci USA. 2016;113:8133–8138. doi: 10.1073/pnas.1604391113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recog-nizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol. 2016;40:96–102. doi: 10.1016/j.coi.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissim L, Bar-Ziv RH. A tunable dual-promoter integrator for targeting of cancer cells. Mol Syst Biol. 2010;6:444. doi: 10.1038/msb.2010.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissim L, Perli SD, Fridkin A, Perez-Pinera P, Lu TK. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol Cell. 2014;54:698–710. doi: 10.1016/j.molcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee K, Green AA, Takahashi MK, Braff D, Lambert G, Lee JW, Ferrante T, Ma D, Donghia N, Fan M, et al. Rapid, Low-Cost Detection of Zika Virus Using Programmable Biomolecular Components. Cell. 2016;165:1255–1266. doi: 10.1016/j.cell.2016.04.059. [DOI] [PubMed] [Google Scholar]
- Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosup-pressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016;164:770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schukur L, Geering B, Charpin-El Hamri G, Fussenegger M. Implantable synthetic cytokine converter cells with AND-gate logic treat experimental psoriasis. Sci Transl Med. 2015;7:318ra201. doi: 10.1126/scitranslmed.aac4964. [DOI] [PubMed] [Google Scholar]
- Schumacher TN, Schreiber RD. Neoantigens in cancer immu-notherapy. Science. 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Selvakumaran M, Bao R, Crijns AP, Connolly DC, Weinstein JK, Hamilton TC. Ovarian epithelial cell lineage-specific gene expression using the promoter of a retrovirus-like element. Cancer Res. 2001;61:1291–1295. [PubMed] [Google Scholar]
- Shukla S, Steinmetz NF. Emerging nanotechnologies for cancer immunotherapy. Exp Biol Med (Maywood) 2016;241:1116–1126. doi: 10.1177/1535370216647123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki HI, Katsura A, Yasuda T, Ueno T, Mano H, Sugimoto K, Miyazono K. Small-RNA asymmetry is directly driven by mammalian Argonautes. Nat Struct Mol Biol. 2015;22:512–521. doi: 10.1038/nsmb.3050. [DOI] [PubMed] [Google Scholar]
- Szymczak AL, Workman CJ, Wang Y, Vignali KM, Dilioglou S, Vanin EF, Vignali DA. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- Tugues S, Burkhard SH, Ohs I, Vrohlings M, Nussbaum K, Vom Berg J, Kulig P, Becher B. New insights into IL-12-mediated tumor suppression. Cell Death Differ. 2015;22:237–246. doi: 10.1038/cdd.2014.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- von Nickisch-Rosenegk M, Teschke T, Bier FF. Construction of an artificial cell membrane anchor using DARC as a fitting for artificial extracellular functionalities of eukaryotic cells. J Nanobiotechnology. 2012;10:1. doi: 10.1186/1477-3155-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E, Schoeps T, Dönitz J. TFClass: an expandable hierarchical classification of human transcription factors. Nucleic Acids Res. 2013;41:D165–D170. doi: 10.1093/nar/gks1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Wroblewska L, Prochazka L, Weiss R, Benenson Y. Multi-input RNAi-based logic circuit for identification of specific cancer cells. Science. 2011;333:1307–1311. doi: 10.1126/science.1205527. [DOI] [PubMed] [Google Scholar]
- Xie M, Ye H, Wang H, Charpin-El Hamri G, Lormeau C, Saxena P, Stel-ling J, Fussenegger M. β-cell-mimetic designer cells provide closed-loop glycemic control. Science. 2016;354:1296–1301. doi: 10.1126/science.aaf4006. [DOI] [PubMed] [Google Scholar]
- Yun HJ, Cho YH, Moon Y, Park YW, Yoon HK, Kim YJ, Cho SH, Lee YI, Kang BS, Kim WJ, et al. Transcriptional targeting of gene expression in breast cancer by the promoters of protein regulator of cyto-kinesis 1 and ribonuclease reductase 2. Exp Mol Med. 2008;40:345–353. doi: 10.3858/emm.2008.40.3.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.