

Abstract
Mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders (LSDs) caused by a deficiency of lysosomal enzymes, leading to a wide range of various clinical symptoms depending upon the type of MPS or its severity. Enzyme replacement therapy (ERT), hematopoietic stem cell transplantation (HSCT), substrate reduction therapy (SRT), and various surgical procedures are currently available for patients with MPS. However, there is no curative treatment for this group of disorders. Gene therapy should be a one-time permanent therapy, repairing the cause of enzyme deficiency. Preclinical studies of gene therapy for MPS have been developed over the past three decades. Currently, clinical trials of gene therapy for some types of MPS are ongoing in the United States, some European countries, and Australia.
Here, in this review, we summarize the development of gene therapy for MPS in preclinical and clinical trials.
Keywords: Mucopolysaccharidoses, Gene therapy, Adeno-associated virus, Glycosaminoglycans
1. Introduction
Mucopolysaccharidoses (MPS) are a group of lysosomal storage disorders (LSDs) caused by a deficiency of lysosomal enzymes and subsequent accumulation of undegraded glycosaminoglycans (GAGs) in multiple tissues and organs. Seven types of MPS are classified based on a deficiency of particular enzyme and show various symptoms including coarse face, central nervous system (CNS) impairment, hearing loss, respiratory compromise, valvular heart disease, hepatosplenomegaly, skeletal dysplasia, and gait abnormality [1]. If untreated, patients often die within a couple of decades.
Enzyme replacement therapy (ERT), hematopoietic stem cell transplantation (HSCT), substrate reduction therapy (SRT), and various surgical interventions including adenotonsillectomy, spinal cord decompression/fusion, hip reconstruction or replacement, osteotomy, 8-plate knee surgery, etc. are currently available for patients with MPS in clinical practice. Conventional intravenous ERT results in partial improvement in soft tissues and activity of daily living (ADL) of patients with MPS I, II, IVA, VI, or VII; however, ERT has very limited impact on avascular lesions in bone and cartilage or the CNS because the native enzyme does not cross the blood-brain barrier (BBB). HSCT is a standard of care for patients with MPS I under 2 years of age or without CNS impairment and provides therapeutic effect in patients with MPS II, IVA, VI, and VII especially if treated at an early stage [2–7]. Although ERT and HSCT have some advantages to slow progression of the disease for MPS, there are several limitations of ERT: i) requiring weekly injection, ii) short circulation of the drug in blood, iii) high cost, and iv) limited penetration to bone and CNS lesions [8–11]. HSCT is promising and far more cost-effective compared with ERT [12,13]; however, this cell-based therapy may not be applicable to all patients because of the limited availability of matched donors and the mortality risk of the procedure such as graft-versus-host disease (GVHD), infection disease, and additional complications [2–5,7,8,10,11]. Therefore, more effective and feasible therapy for MPS is urgently required. Gene therapy for MPS is not yet approved in clinical practice. However, clinical trials for some types of MPS are under way in the United States, some European countries, and Australia [14–19]. Phases I/II clinical trials for MPS I, II, IIIA, IIIB, and VI are ongoing or are scheduled (Table 1). Gene therapy is a promising approach for treating a genetic disorder such as MPS and has been under investigation for the last three decades. It should provide a one-time permanent treatment since the active enzyme is released from transduced cells consistently, as observed in HSCT. However, initial approaches using retroviral vectors failed because of short-term gene expression and adverse effects. The approval of gene therapy products in China [20], Russia [21], and Europe [22], has brought new hopes into the gene therapy field. Recently, the Food and Drug Administration (FDA) approved the first gene therapy available in the United States, for certain patients with a form of acute lymphoblastic leukemia [23].
Table 1.
Current statuses of gene therapy clinical trials for mucopolysaccharidoses.
| Product | Organization | Vector/transgene | Injection route | Status | Clinical endpoint | Ref | |
|---|---|---|---|---|---|---|---|
| MPS I | RGX-111 | REGENXBIO Inc. | AAV9/IDUA | ICS | IND active. Phase I sheduled in 2018 | NA | [106] |
| SB-318 | Sangamo Therapeutics | AAV6/ZFN, Zinc finger nuclease | IV | Phase I ongoing | Adverse events. Urine GAG levels. AAV6 clearance in plasma, saliva, urine, stool, semen | [14,105] | |
| MPS II | RGX-121 | REGENXBIO Inc. | AAV9/hIDS | ICS | IND submission | NA | [106] |
| SB-913 | Sangamo Therapeutics | AAV6/ZFN, Zinc finger nuclease | IV | Phase I ongoing | Adverse events. Urine DS/HS/total GAG levels. AAV2/6 clearance in plasma, saliva, stool, and semen | [15] | |
| MPS IIIA | SAF-301 | Lysogene | AAV10/hSGSH and | IC | Phase I/II completed | Adverse events. Neurocognitive/behavioral tests. Biological markers in blood, urine, and CSF | [16,17,98] |
| ABO-102 | Abeona Therapeutics | AAV9/hSGSH | IV | Phase I/II ongoing | Toxicity. CSF/blood leukocyte SGSH enzyme activity. Urine/CSF HS and total GAG levels. Liver volume (MRI). Leiter International Performance Scale. VABS. Mullen Scales of Early Learning. Sanfilippo Behavior Rating Scale | [18,107] | |
| EGT-101 | Esteve | AAV9/hSGSH | ICV | Phase I/II scheduled in 2018 | NA | [108] | |
| MPS IIIB | ABO-101 | Abeona Therapeutics | AAV9/hNAGLU | IV | Phase I/II ongoing | NA | [109] |
| AAV2/5-NAGLU | UniQure | AAV2/5/NAGLU | IC | Phase I/II completed | Tolerance. Neurocognitive progression. Brain growth. NAGLU enzymatic activity in CSF. Anti-NAGLU immune response for 30 months after surgery | [99] | |
| MPS VI | AAV2/8.TBG. hARSB | Fondazione Telethon, Italy | AAV8/hARSB | IV | Phase I/II ongoing | Kidney function. C3 and C4 proteins. Urine GAGs. Leukocyte ARSB levels. 6MWT and 3MSCT. FVC and FEV1 | [19] |
Abbreviations: 3MSCT, 3-minute stair climb test: 6MWT, 6-minute walk test: AAV, adeno-associated virus: ARSB, arylsulfatase B: CSF, cerebrospinal fluid: DS, dermatan sulfate: FEV1, forced expiratory volume in 1 s: FVC, forced viral capacity: GAG, glycosaminoglycan: hARSB, human Arylsulfatase B: hIDS, human iduronate 2-sulfatase: hNAGLU, human N-acetyl-alpha-glucosaminidase: HS, heparan sulfate: hSGSH, human N-sulphoglucosamine sulphohydrolase: IC, intracerebral: ICS, intracisternal: ICV, intracerebroventricular: IDUA, alpha-L-Iduronidase: IND, investigational new drug: IV, intravenous: MRI, magnetic resonance imaging: NAGLU, N-acetyl-alpha-glucosaminidase: SGSH, N-sulphoglucosamine sulphohydrolase: SUMF1, Sulfatase Modifying Factor 1: VABS, Vineland Adaptive Behavior Scales.
At present, the two main approaches to gene therapy are: 1) in vivo gene therapy, in which a virus carrying the therapeutic gene is injected intravenously or locally, and the gene is delivered into somatic cells of the patients; and 2) ex vivo gene therapy, in which the gene is transduced into somatic cells derived from the patient and then transplanted back into the recipient (Fig. 1). For in vivo gene therapy, the genes are mainly transfected by using different viral vector systems, including retroviral, lentiviral, adenoviral, and adeno-associated virus (AAV)-based vectors, as well as non-viral vectors. For ex vivo gene therapy, gammaretrovirus (hereafter retrovirus) and lentiviral vectors are mainly used to transduce the genes of the lysosomal enzyme into recipient’s cells such as bone marrow, myoblast, and fibroblast cells. These transduced cells are amplified in the laboratory and then transplanted into recipient’s body using standard procedures, as developed for bone marrow transplantation. Many preclinical studies of in vivo or ex vivo gene therapy for MPS have been reported using rodents and large animal models.
Fig. 1. Strategies of in vivo gene therapy and ex vivo gene therapy.

In vivo gene therapy on the left starts with placing the therapeutic gene into the viral genome. Viral vector is then injected into the body via IV, IT, IM, or IP to transduce target cells. Ex vivo gene therapy on the right starts with an extraction of stem cells from the body. The virus with the therapeutic gene will transduce the stem cells outside of the patient. Once the viral vector has succesfully transferred the gene to the cells, the cells are reintroduced into the body via IV, IT, or IM where the stem cells can differentiate, and the gene can be expressed.
Abbreviation: IV, intravenous: IT, intrathecal: IM, intramuscular: IP, intraparenchyma.
Recombinant AAV (rAAV) vectors are useful tools for gene therapy to treat MPS since the AAV vectors efficiently infect different cell types, persist as episomes, and have a low risk of insertional mutagenesis and genotoxicity [24]. Currently, rAAV is the most frequently used virus vector in ongoing clinical trials of gene therapy for MPS. Various AAV serotypes have been identified from human and non-human primate, and AAV tropism is dependent on its serotype since AAV entry is determined by interaction between the virus capsid proteins and its specific receptor on the cell surface. This unique feature of AAV serotypes has potential to deliver enzymes to specific organs or areas such as CNS by receptor-mediated transcytosis across the endothelium of BBB [25]. rAAV gene delivery to targeted tissue depends on the administration route, and intraparenchymal (IP), intra-cerebrospinal fluid (CSF) and intravenous (IV) injection are available for AAV-mediated gene therapy in clinical trials of patients with MPS (Table 1).
In this review, we summarize recent studies of gene therapy for MPS in preclinical studies using animal models and AAV-derived vectors, and the current situation of ongoing clinical trials.
2. Pre-clinical study of gene therapy for MPS
2.1. Types of virus vectors
Several viruses are suitable tools for gene transfer in pre-clinical studies and clinical practice since they have high transduction efficiencies in a broad range of human cells. Viral vectors are mainly originated from retroviruses, lentiviruses, adenoviruses, and AAVs. Retrovirus vectors can infect and integrate into host genomes in dividing targeted cells such as hematopoietic cells. The introduced gene is transmitted and expressed for a long time in daughter cells. However, the integration could potentially activate an oncogene or inactivate a tumor suppressor gene, leading to promotion of tumorigenesis in targeted cells [26,27]. Lentivirus vectors that are based on human immunodeficiency virus-1 (HIV-1) are more feasible and effective. Lentivirus vectors integrate into the host genome and provide stable gene expression. Unlike retrovirus vectors, lentivirus vectors can mediate stable gene transfer into not only dividing cells such as hematopoietic stem cells, but also non-dividing cells such as nerve cells [28,29]. Adenovirus vectors can also allow gene transfer into both dividing cells and non-dividing cells and show high expression levels of the gene. However, the expression is transient since this virus vector is not inserted into the host chromosome. This vector is not associated with a risk of germline infection and mutagenesis [30,31]. The disadvantages of adenovirus vectors include adaptive immune response and cellular toxicity, which limit their application for gene therapy. Some pre-clinical studies using adenovirus vectors have been reported, and IV administration of this virus vector showed a positive impact on multiple tissues including skeletal and CNS lesions in an MPS VII model mouse [32–34]. Currently, there are no ongoing clinical trials of adenovirus vector for gene therapy for MPS due to adverse effects.
Adeno-associated viruses are small, replication-defective, non-pathogenic and non-enveloped viruses. These viruses require a so-called helper virus such as adenovirus or herpesvirus to replicate. Since the AAV vector is infected in cells and persists as an episome, there is a low risk of insertional mutagenesis and genotoxicity [24], critical concerns for lentivirus and retrovirus vectors. However, a small percentage of rAAV can integrate into host chromosomal sequences, and insertional mutagenesis has resulted in human hepatocellular carcinoma after rAAV gene therapy [35–37]. The AAV vector can transduce a wide range of non-dividing and dividing cells. Notably, the AAV vector provides a long-term transgene expression when non-dividing cells are infected. Because the AAV vector has stability, long-term expression, and low immunogenicity, it is currently the most popular virus tool in gene therapy for MPS fields. However, the AAV vector has a limitation of delayed expression in transduced cells. It takes 1 or 2 weeks to achieve expression at peak since AAV is a single strand (ss) DNA and requires second strand synthesis or annealing for gene expression [38]. The primary limitation of rAAV vectors is that they can package a maximum of 4.7 kb of exogenous DNA. The efficiency of rAAV vector gene delivery can be significantly enhanced by creating self-complementary (sc) AAV vectors, which package both DNA strands as a single, inverted repeat molecule that folds into double-stranded DNA immediately after uncoating in the cell. This allows a rapid and more efficient (5–20 fold) transduction compared to conventional ssAAV vector because it bypasses the requirement for DNA synthesis to convert the single-stranded vector genome into active double-stranded DNA. However, the small packaging capacity of the scAAV remains a limitation for clinical application of gene therapy because the complementary strand halves the size of gene that can be inserted to about 2.3 kb [39,40].
2.2. AAV serotypes
AAV has significant advantages over other viral vectors in the application for gene therapy since AAV shows non-pathogenicity in humans and provides long-lasting transgene expression. Various AAV serotypes (AAV1 to AAV13) and more than one hundred variants of AAVs have been identified from human and non-human primates to date [41,42]. AAV serotypes are defined by capsid protein motifs, which are recognized by neutralizing antibodies. Cell surface glycans interact with exposed capsid regions of AAV and work as primary AAV receptor. AAV serotypes 1, 5, and 6 bind to α2,3/α2,6 N-linked sialic acid (SA) [43,44]. AAV serotypes 2, 3, and 6 bind to heparan sulfate proteoglycans (HSPG) [45,46]. AAV serotype 4 binds to α2,3/α2,6 O-linked SA [47], and AAV serotype 9 requires N-linked galactose residues [48,49]. Primary receptors for AAV8 and AAVrh 10 remain unknown, but the 37/67-kDa laminin receptor has been reported to act as a co-receptor for AAV8 entry [50]. This difference of interaction between each AAV serotype and its receptor is critical to determine tissue tropisms of each serotype. Understanding the tissue tropism of each AAV serotype in an animal model is required to select appropriate AAV serotype for specific gene therapy application before starting clinical trials. Zincarelli et al. examined transgene expression and biodistribution of AAV 1–9 in mice after IV injection of AAV containing the cytomegalovirus driven luciferase transgene and assessed primary and secondary viral targets [51]. Bioluminescence imaging studies categorized different groups based on transduction levels of AAVs: i) high expression groups - AAV 7 and 9, ii) moderate expression groups – AAV 1, 6, and 8, and iii) low expression groups – AAV 2, 3, 4, and 5. In addition, this study showed two classes of AAV kinetics including rapid-onset, AAV 1, 6, 7, 8, and 9 and slow-onset, AAV 2, 3, 4, and 5. Luciferase protein expression profile after administration of AAV 1–9 serotypes demonstrated that liver is the primary target of AAV serotypes 1, 2, 5, 6, 7, and 9. AAV serotypes 3 and 4 have low transduction efficiency in the liver; however, these serotypes have a specific tropism for the heart. This study also showed that AAV serotype 9 has the most robust tissue expression, and only AAV serotypes 8 and 9 provided luciferase protein expression in brain after IV administration.
2.3. Targeting strategy to CNS and bone
ERT has no impact in some types of MPS which are characterized by CNS disorders because the mature BBB efficiently blocks enzyme diffusion to CNS lesions. Recombinant AAV also has difficulty penetrating the BBB, so that expression of the active enzyme into CNS lesions is limited. Different delivery routes of rAAV vectors to CNS lesions have been studied. Intraparenchymal administration of rAAV has a robust impact locally on CNS lesions and is useful for neurological disorders, characterized by local lesions such as Parkinson’s disease. IP administration of rAAV reduced storage materials and improved behavioral deficits in mouse models of MPS I [52], MPS IIIA [53,54], MPS IIIB [55,56], and MPS VII [57–61]. A single-gene defect disease such as lysosomal diseases needs systemic, widespread gene transduction including the brain and spinal cord [62]. Thus, the IP administration is impractical in clinical practice, and development of less invasive delivery system of gene vectors is required for patients with MPS. Intrathecal (IT) or IV administration of rAAV9 is a less invasive systemic delivery method that efficiently transduces genes into multiple tissues, including the CNS. Delivery of various rAAV serotypes to CNS lesions via CSF administration in a large animal has been studied [63–65]. These studies show that rAAV9 has high transduction efficiency in widespread CNS lesions including the spinal cord. Zhang et al. showed that gene transfer efficiency using rAAV9 was superior to rAAV 1, 6, 7, and 8 and that rAAV9 resulted in widespread neural transduction of neonatal mice after IV injection [66]. Transduction efficiency of rAAV 2 and 5 into CNS lesions was considerably lower than other rAAVs.
Delivery of sufficient enzyme into avascular bone and cartilage lesions also remains an unmet challenge for gene therapy. We showed that the insertion of an aspartic acid octapeptide (D8) into the C-terminus of tissue-nonspecific alkaline phosphatase [67], the N-terminus of β-glucuronidase [68] and N-acetylgalactosamine-6-sulfatase (GALNS) [69] significantly increases delivery of the enzymes to bone. We applied the same principle to engineer an AAV2 vector to target gene delivery to the bone (Fig. 2). To increase the vector affinity for hydroxyapatite (HA), we inserted D8 immediately after the N-terminal region of the VP2 capsid protein. The modified vector was generated with physical titers and transduction efficiencies comparable to the unmodified vector. The bone-targeting vector had significantly higher HA affinity and vector genome copies in bone compared with the unmodified vector. The modified vector was released from HA, and its enzyme activity in bone two-weeks p.i. was 8.7-fold higher than in mice treated with unmodified vector [10,70]. Immunohistochemistry analysis of the in vivo transduction experiment showed that the modified AAV2 vector increased expression of the GALNS gene and its enzyme activity in the bone of MPS IVA knock-out mice. Therefore, bone-targeting gene therapy with a modified AAV could be a novel therapy for skeletal dysplasia in MPS.
Fig. 2. Mechanism of multiple-AAA targeting system. Viral capsid in the lower panel has multiple copies of D8 integrated into capsid proteins, showing the retargeting of gene vector to bone (hydroxyapatite in the mineral region) schematically.
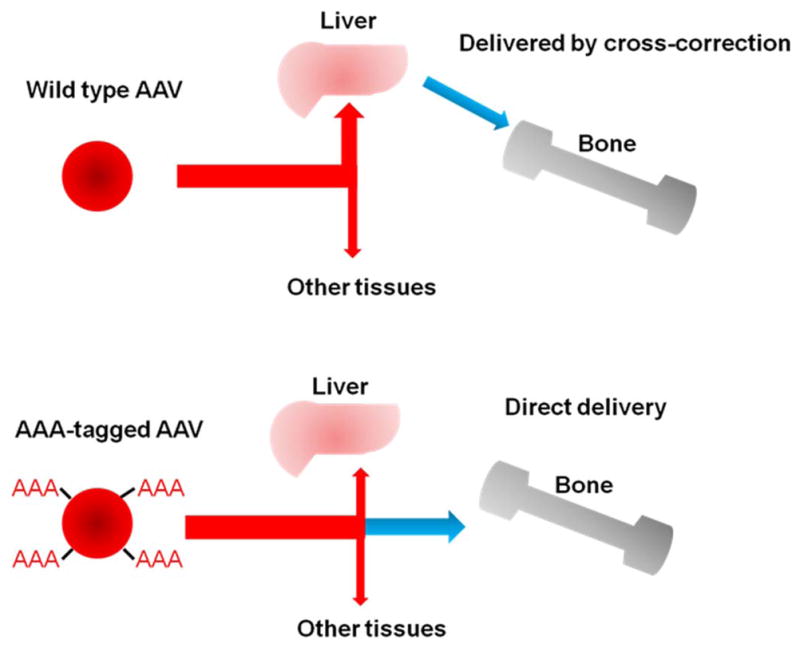
Adopted from Tomatsu et al. Mol Genet Metab. 2015;114(2):94–109.
2.4. AAV-mediated in vivo studies using animal models of MPS
AAV2 was the most common serotype studied during the early development of AAV vectors. However, rAAV2 shows poor transduction efficiency compared with other rAAVs. Recently, it has been shown that rAAV8 and 9 are more efficiently transduced into CNS lesions after IT or IV administration [51,64,66,71–75]. In this section, we summarized recent studies of rAAV-mediated gene therapy in animal models of each type of MPS. These studies led to the current ongoing and scheduled clinical trials of gene therapy in MPS.
Pre-existing immunity to AAV is a major problem for clinical translation, although the immune response against AAV8 or AAV9 is less common than for AAV2 serotype [76–79]. However, pre-existing neutralizing antibodies to AAV8 at low level provide a negative impact on gene transfer to liver and therapeutic efficacy in skeletal lesions in a MPS VI cat model [80]. Previous studies showed pre-existing neutralizing antibodies to AAV8 were undetectable in 58% patients of MPS VI investigated for gene therapy [81]. Almost half of the general human population has anti-AAV9 antibody at some level [79]. After a single systemic gene delivery of rAAV9 in non-human primates, pre-existing anti-AAV9 antibody at low level did not diminish vector transduction. However, high level pre-existing anti-AAV9 antibody leds to the reduction of vector transduction in somatic tissues, but had less impact on transduction in the CNS, indicating that CNS entry is less sensitive to pre-existing anti-AAV9 antibody [82]. These studies led to use of AAV antibody titer as a criterion of MPS patient eligibility to enter clinical trials of gene therapy.
2.4.1. MPS I
Hinderer et al. reported IT AAV9 gene therapy that improved CNS pathology in a feline model of MPS I [83]. In this study, MPS I cats were intrathecally administrated at 4–7 months of age with 1012 GC/kg AAV9 vector expressing normal feline α-L-iduronidase (IDUA) and followed for 6 months. After vector administration, IDUA activity was detectable in various parts of the brain and spinal cord, while no enzyme activity was detected in the same tissues of untreated MPS I cats. This AAV9 administration reduced the accumulation of ganglioside GM3 in the brain and also reduced accumulation of GAGs in the somatic tissues such as liver, spleen, heart, and lung. However, antibodies were produced against the expressed IDUA, potentially reducing efficacy of treatment. This group also reported that the IT administration of IDUA gene using an AAV9 vector improved the CNS pathology in a MPS I canine model of MPS I that was immunologically tolerant to human IDUA [84]. This study was conducted to investigate the efficacy of the AAV9 expressing human IDUA and to estimate the minimum efficacy dose for clinical trials of MPS I. After IT injection of different dose of the AAV9 vector (i.e., 1010, 1011, and 1012 GC/kg), the IDUA activity was dose-dependent elevated in the CSF and maintained above the enzyme activity in normal dogs at high dose (1012 GC/kg) during the 6-month study. Immunohistochemistry showed that the accumulation of GAG and GM3 was reduced in a dose-dependent manner and that there was also a decrease in the brain lesions of the MPS I canine model. Even the lowest dose used (1010 GC/kg), showed some efficacy, which is approximately equivalent to 6.2 × 1011 GC in adult humans [84]. Ellinwood et al. showed direct injection of AAV5 vector into the brain of two dog models of MPS, providing critical information on reproducibility, tolerance, appropriate vector type and dosage and optimal age for the AAV5 gene therapy in large animals to support clinical application [85]. Moreover, Vance et al. evaluated AAV8/9 chimeric capsid in in vitro study of human cornea. This study showed that AAV8/9 provided the efficient transduction and no significant apoptosis in the affected cells [86].
2.4.2. MPS II
Motas et al. reported that intracisternal (ICS) gene therapy corrected neurological and systemic symptoms in an MPS II model mouse, which had a deficiency of lysosomal enzyme iduronate-2-sulfatase (IDS) [87]. MPS II mice, with already progressive disease, were treated with 5 × 1010 vector genomes of an AAV9 vector encoding murine IDS. Four months after gene delivery, GAG accumulation in CNS lesions was significantly reduced in treated mice. ICS gene delivery of the AAV9 vector decreased GAG accumulation not only in CNS lesions but also in somatic tissues such as liver, spleen heart, kidney, and lung, indicating that circulating IDS cross-corrected non-transduced tissues. ICS AAV9 gene therapy also corrected behavioral deficits and prolonged survival of treated MPS II mice [87].
Hinderer et al. showed that ICV administration of an AAV9 vector carrying the human IDS gene improved the CNS pathology in 2 to 3-month-old MPS II mice [88]. After vector infusion, it was shown that the enzyme activity in brain lesions increased in a dose-dependent manner. Histological staining demonstrated that the accumulation of lysosomal storage materials such as and GM3 was decreased in the brain of the treated mice. Using a novel object recognition test, they also showed improvements in long-term memory in treated mice. GAG accumulation in the liver was also reduced, providing further evidence that gene therapy applied directly to the nervous system can cross-correct deficiencies in somatic tissues. Laoharawee et al. also evaluated the effectiveness of AAV9 encoding human IDS after intracerebroventricular injection in a MPS II mouse model [89]. Low level of IDS activity in CNS lesions (7–40% of wild-type) was observed; however, GAG accumulation was reduced in brain, and neurologic deficits were improved in treated mice. Thus, these studies show that intra-CSF administration of AAV9 should be useful for preventing neurologic deterioration in patients with MPS II.
2.4.3. MPS IIIA and IIIB
Several studies have shown that systemic delivery of rAAV8 or rAAV9 containing the deficient gene cause elevation of lysosomal enzyme activity in CNS lesions and correction of disease progression in mouse models of MPS IIIA [90–93] and MPS IIIB [82,94–96]. Fu et al. showed that a single IV administration of a scAAV9 vector encoding human N-sulfoglucosamine sulfohydrolase (hSGSH) (5e12 vg/kg) in an MPS IIIA model mouse significantly improved behavior performance and survival rate [93] Improvements were best when the vector was administered at an early stage of disease, but these phenotypes were also partially corrected when treatment was delayed until mice showed intermediate progression of disease. These results suggest that scAAV9-hSGSH vector may be clinically useful to reverse disease progression in patients with established MPS IIIA. This group also showed that IV administration of rAAV9 containing human α-N-acetylglucosaminidase (hNAGLU) corrected GAG accumulation in CNS and somatic tissues and provided long-term (> 18 months) neurological improvement in MPS IIIB model mouse [94]. An investigational new drug (IND)-enabling good laboratory practice (GLP)-compliant toxicology study demonstrated that injection of the rAAV9 did not provide adverse clinical events and chronic toxicity during a 6-month study [95]. Systemic rAAV8-mediated gene delivery also has been studied in MPS IIIA mouse models [90,92]. Liver-targeting AAV8 [90] and AAV8 carrying engineered enzyme expressing specific brain targeting peptide [92] both corrected brain pathology in MPS IIIA mice.
Direct injection of rAAV into multiple areas in brain lesions successfully increased enzyme activity, reduced storage materials and improved behavioral deficits in mouse models of MPS IIIA [53] and IIIB [85]. These preclinical studies provide the basis for ongoing or scheduled clinical trials of patients with MPS IIIA/B [97,98].
2.4.4. MPS VI
MPS VI is caused by a deficiency of arylsulfatase B (ARSB) and is characterized by hepatosplenomegaly, corneal clouding, heart valve disease, and skeletal abnormalities without CNS disorders. Intravascular high-dose administration of rAAV8 vector carrying ARSB with a liver-specific promoter into MPS VI cats and rats reduced GAG accumulation, resolved skeletal problems, and reduced heart valve thickness [99,100]. In the feline model, urinary GAG levels and skeletal abnormalities were reduced in a dose-dependent manner. Treatment was less effective for cats with pre-existing neutralized antibodies to AAV8, indicating that pre-existing immunity to AAV8 should be considered before starting gene therapy in clinical practice [80].
2.4.5. MPS VII
A single tail vein injection of rAAV9 vector expressing β-glucuronidase (GUSB) in an adult MPS VII model mouse led to systemic delivery of the gene vector in brain and other tissues. However, the AAV9 genome copies were lower in the CNS lesions than in other somatic tissues, and the treatment did not resolve storage lesions in different regions of the brain such as cortex, hippocampus, and striatum; and did not resolve the cognitive deficits. Survival rate was also not changed between treated and untreated MPS VII mice. Elevation of sialic acid in the brain of this disease model might inhibit transduction of the rAAV9 in CNS lesions [101]. Systemic administration of an epitope-modified AAV2 was superior to rAAV9 in resolving the CNS lesions of the MPS VII model [101].
Gurda et al. compared IV injection and IT injection via cisterna magna of GUSB in the rAAV9 vector on an MPS VII canine model [102]. Dogs treated with a single systemic administration of rAAV9 vector carrying GUSB at 3 days of age resulted in higher levels of GUSB activity in CSF, but GAG accumulation was not significantly reduced. However, IT injection at 3 weeks of age significantly reduced GAG accumulation in CNS lesions of treated MPS VII model, indicating that IT injection may be more effective in resolving CNS lesions than IV injection.
2.4.6. Genome editing using zinc finger nuclease
Genome editing with zinc finger nuclease (ZFN) is innovative genetic engineering tool to modify DNA sequences. ZFN generate double-strand breaks (DSB) at an appropriate position in the genome, and DSB is repaired through either non-homologous end joining or homologous recombination to restore the breakpoint. By using AAV8 vector delivery, ZFN-mediated site-specific insertion of a corrective gene at albumin locus provided robust expression of IDUA and IDS in a wild-type mouse [103]. Administration of AAV8 vector with albumin locus-targeting ZFN in hepatocyte induced enzyme expression of IDUA and IDS in liver and other tissues of MPS I and MPS II model mouse. This AAV8/ZFNs treatment reduced cellular vacuolation by histological analysis, and heparan and dermatan sulfate levels were also decreased in the brain. Cognitive deficits were prevented after the treatment in both model mice [104].
3. Clinical trials of gene therapy for MPS
Currently, clinical trials for some types of MPS are under way in the United States, some European countries, and Australia. Phase I/II clinical trials for MPS I, MPS II, MPS IIIA, MPS IIIB, and MPS VI are ongoing or are scheduled (Table 1). The AAV vector is most commonly used in clinical trials of gene therapy for MPS. The current status of gene therapy is described in this section.
3.1. MPS I
REGENXBIO has produced RGX-111, an AAV9 vector containing the gene IDUA for administered via IV infusion. An IND application is active, and plans to enroll patients for a dosing trial are set for the 1st half of 2018 [105].
SB-318 has been developed by Sangamo Therapeutics, and a phase I clinical trial is currently underway (NCT02702115) [14]. This gene therapy trial uses an AAV6 vector that contains a zinc finger nuclease designed to use gene editing to insert a correct copy of the IDUA gene into the albumin locus for expression in hepatocytes [104]. This should lead to long-term secretion of normal IDUA. Only adult (18+ years old) attenuated MPS I patients, such as Hurler-Scheie, Scheie, and Hurler post-HSCT are being recruited. This is an ascending dose safety study using high, medium, and low doses of the vector for IV infusion. The primary outcome in this study is the assessment of adverse effects by this gene therapy. Secondary outcomes are urinary GAG levels and clearance of the AAV6 in plasma, saliva, urine, stool, and semen [14].
3.2. MPS II
REGENXBIO has produced RGX-121, which consists of an AAV9 vector containing the human IDS gene. This is being administered by IT injection in preclinical testing. An IND application was planned for the 2nd half of 2017 [105].
Sangamo Therapeutics has developed SB-913 which, like SB-318, contains ZFN in AAV6 for IV infusion. A phase I clinical trial is underway (NCT03041324) [15]. This trial is recruiting adult (18+ years old) attenuated MPS II patients. As for the SB-318 trial, three different doses (high, medium, and low) are primarily being evaluated for adverse effects of this gene therapy. Secondary outcomes are urinary total GAG, heparan sulfate (HS), and dermatan sulfate (DS) levels and clearance of the AAV6 in plasma, saliva, urine, stool, and semen up to 36 months after treatment [15].
3.3. MPS IIIA
LYSOGENE developed SAF-301, which is AAV10 encoding hSGSH and SUMF1 for IC administration in phase I/II clinical trials (NCT01474343 and NCT02053064) [16,17]. LYSOGENE recruited 4 MPS IIIA patients between 18 months and 6 years of age into each trial. The primary outcome was the assessment of the tolerance and safety. Secondary outcomes were data of brain function using MRIs and neurocognitive tests, and levels of biological markers in blood, urine, and CSF [16]. IC administration of 7.2 × 1011 viral genomes/body of the AAV10 vector showed good tolerance and absence of adverse effect related to the product during one year of follow-up. Improvement of brain atrophy and behavior was also seen in some patients [98]. A long-term follow-up study for five years after the injection of SAF-301 was recently completed [17].
Abeona Therapeutics has produced ABO-102, which consists of scAAV9 encoding hSGSH with a U1a promoter administered for IV infusion. An open-label, dose-escalation phase I/II clinical trial is ongoing (NCT02716246) [18]. This trial is recruiting children and adult MPS IIIA patients 2 years of age or older. Patients are administered two different doses (5 × 1012 or 1 × 1013 vg/kg), and the primary outcome is an assessment of genotoxicity. Secondary outcomes are SGSH activity level in leukocyte and CSF, liver and spleen volume, cognitive ability, and urinary GAG levels [18]. After 30 days, cohort one showed an average reduction of CSF HS of 25.76%, while cohort 2 showed an average reduction of 60.71%. After 180 days, cohort 1 experienced an average reduction of 58.71% in CSF HS, and data for cohort 2 at 6 months is not available. Cohort 1 also showed an average 17.68% decrease in liver volume at day 30 and 31.26% at day 180. Cohort 2 has an average 14.81% decrease in liver volume at day 30. There are no reported serious adverse events (n = 6 through 1139 days follow-up) [106].
Esteve has developed EGT-101 that consists of AAV9 containing hSGSH and is proposed for ICV for treatment. They plan to enter phase I/II in the 1st quarter of 2018 [107].
3.4. MPS IIIB
Abeona Therapeutics have produced ABO-101, which contains AAV9 with hNAGLU for administration via IV infusion. The U.S. FDA has approved the IND for phase I/II clinical trials [108].
UniQure recently completed a phase I/II clinical trial with 4 children using rAAV2/5 containing NAGLU administered in 16 IP deposits, 4 in the cerebellum [99]. Tolerance to the virus, neurocognitive progression, brain volume, NAGLU enzymatic activity in CSF, and anti-NAGLU immune responses were measured over 2 years. 125 adverse events were recorded, and NAGLU activity 15–20% of that in unaffected children appeared in lumbar CSF.
3.5. MPS VI
Fondazione Telethon is developing a gene therapy that consists of an AAV8 encoding hARSB with liver-specific thyroxine-binding globulin (TBG) promoter in phase I/II clinical trial (NCT03173521) [19]. The trial currently recruits child and adult MPS VI patients from 4 to 65 years of age, who should have received ERT for at least 12 months before enrollment. Three different doses (2 × 1011, 6 × 1011, or 2 × 1012 gc/kg) are being evaluated for primary outcomes of assessments of liver and kidney function and urinary GAG levels. Secondary outcomes are ARSB activity in leukocyte, 6-minute walk test (6MWT), 3-minute stair climb test (3MSCT), forced vital capacity (FVC), and forced expiratory volume at 1 min (FEV1) [19].
4. Unmet challenge of viral vectors
The selection of a viral vector to deliver genetic material to human body results in some logistical issues. 1) The number of viral vectors available for therapeutic use is still limited. Maintaining therapeutic enzyme level is critically important for patients with MPS to prevent or improve clinical manifestations. However, there is limited information about the length of gene expression in MPS patients after AAV-mediated gene therapy. Murrey et al. showed that NAGLU activity was detected at 1.3–3 fold above endogenous levels in the brain after 6 months post-injection of single systemic administration of rAAV9 gene vector in non-human primates [82]. A recent clinical trial of MPS IIIB reported that CSF NAGLU activity was detected at 15–20% of that in unaffected children with MPS IIIB after 30 months treatment of IC rAAV5 gene therapy [99]. 2) Any viral vectors can lead to the possibility that the body will develop an immune response as a foreign body. Once an immune response and antibody for the viral vector are produced, the patient cannot be treated with the same viral vector effectively since it will be rejected by the body. If gene therapy fails for certain patients in a clinical trial, the same serotype of virus vector will not be used again in those patients in subsequent trials. 3) The probability of pre-existing immunity (antibody) against the viral vector increases with patient age, rendering the therapy ineffective. 4) There remains concern for genotoxicity despite the low percentage of rAAV integration [37]. Previous reports showed the incidence of hepatocellular carcinoma in mouse model of MPS VII after AAV-mediated gene therapy [35,36]. The AAV vector dose, its constructs selection including promoter/enhancer, and the timing of administration can be all critical factors that could lead to hepatocellular carcinoma [37,109]. 5) For CNS gene therapy, the appropriate administration method is not yet resolved. Several administrations including IV, IT, IC, or ICV injection are under investigation. 6) The price and commercialization of the approved therapies remain unclear.
Some shortcomings of viral vectors (such as genotoxicity and low transgenic expression) can be overcome through the use of different or hybrid vectors.
5. Conclusions
Currently, ERT and HSCT are available for patients with MPS. However, conventional ERT limits access to CNS and bone lesions while HSCT has some mortality risk. Gene therapy should be a one-time permanent treatment without an additional expensive cost and without high mortality and morbidity. AAV is the most popular virus in gene therapy for MPS, and AAV-mediated in vivo gene delivery is moving forward to clinical trials. However, there are still some problems to overcome such as vector selection, administration route, and immunogenicity as well as a restricted number of times that vectors can be administered to the body.
Acknowledgments
This work was supported by grants from National MPS Society, the Austrian MPS Society, and International Morquio Organization (Carol Ann Foundation). This work was also supported by Japanese MPS Family Society. R.W.M. and S.T. were supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of National Institutes of Health (NIH) under grant number P30GM114736. S.T. was supported by NIH grant 1R01HD065767. The content of the article has not been influenced by the sponsors. C.J.A-D. was supported by Fulbright Colombia.
Abbreviation
- 3MSCT
3-minute stair climb test
- 6MWT
6-minute walk test
- AAV
adeno-associated virus
- ADL
activity of daily living
- ARSB
arylsulfatase B
- BBB
blood-brain barrier
- CNS
central nervous system
- CSF
cerebrospinal fluid
- D8
aspartic acid octapeptide
- DS
dermatan sulfate
- DSB
double-strand breaks
- ERT
enzyme replacement therapy
- FDA
Food and Drug Administration
- FEV1
forced expiratory volume in 1 min
- FVC
forced viral capacity
- GAG
glycosaminoglycan
- GALNS
N-acetylgalactosamine-6-sulfate sulfatase
- GLP
good laboratory practice
- GUSB
β-glucuronidase
- GVHD
graft-versus-host disease
- HA
hydroxyapatite
- hARSB
human arylsulfatase B
- hIDS
human iduronate 2-sulfatase
- HIV-1
human immunodeficiency virus – 1
- hNAGLU
human N-acetyl-alpha-glucosaminidase
- HS
heparan sulfate
- HSCT
hematopoietic stem cell therapy
- hSGSH
human N-sulphoglucosamine sulphohydrolase
- HSPG
heparan sulfate proteoglycans
- IC
intracerebral
- ICS
intracisternal
- ICV
intracerebroventricular
- IDUA
α-L-iduronidase
- IDS
iduronate-2-sulfatase
- IND
investigational new drug
- IP
intraparenchymal
- IT
intrathecal
- IV
intravenous
- LSDs
lysosomal storage disorders
- MPS
mucopolysaccharidoses
- MRI
magnetic resonance imaging
- NAGLU
α-N-acetylglucosaminidase
- rAAV
recombinant adeno-associated virus
- SA
sialic acid
- sc
self-complementary
- SGSH
N-sulfoglucosamine sulfohydrolase
- SRT
substrate reduction therapy
- ss
single strand
- SUMF1
Sulfatase Modifying Factor 1
- TBG
thyroxine-binding globulin
- VABS
Vineland Adaptive Behavior Scales
- ZFN
zinc finger nuclease
Footnotes
Conflict of interest
All the authors contributed to the Review Article and had no conflict of interest with any other party.
Contributions to the project
Kazuki Sawamoto is the primary author of this review article and an expert in pharmacokinetics. He has 10 years of research experience in model mice for translational research and drug development. He has contributed to the concept, the planning, data analysis, and reporting of the work described.
Hui-Hsuan Chen is the primary author of this article and expert in biology. She has contributed to the planning, data analysis, and reporting of the work described.
Carlos J. Alméciga-Díaz is expert in pharmacology, ERT, and gene therapy. He has contributed to the planning, data analysis, and reporting of the work described.
Robert W. Mason has contributed to data analysis, and reporting of the work described.
Shunji Tomatsu is a Principal Investigator and has 30 years of clinical and research experience in Morquio A, publishing over 70 articles in this field. He has contributed to the concept of the project, planning, analysis of data, and reporting of the work described in the review.
References
- 1.Neufeld E, Muenzer J, Scriver C, Beaudet A, Sly W, Valle D. The mucopolysaccharidoses. The Metabolic and Molecular Bases of Inherited Disease. 2001;136:3421–3452. [Google Scholar]
- 2.Turbeville S, Nicely H, Rizzo JD, Pedersen TL, Orchard PJ, Horwitz ME, Horwitz EM, Veys P, Bonfim C, Al-Seraihy A. Clinical outcomes following hematopoietic stem cell transplantation for the treatment of mucopolysaccharidosis VI. Mol Genet Metab. 2011;102:111–115. doi: 10.1016/j.ymgme.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aldenhoven M, Jones SA, Bonney D, Borrill RE, Coussons M, Mercer J, Bierings MB, Versluys B, van Hasselt PM, Wijburg FA, van der Ploeg AT, Wynn RF, Boelens JJ. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: results after implementation of international guidelines. Biol Blood Marrow Transplant. 2015;21:1106–1109. doi: 10.1016/j.bbmt.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Luan Z, Jiang H, Fang J, Qin M, Lee V, Chen J. Allogeneic hematopoietic stem cell transplantation in thirty-four pediatric cases of mucopolysaccharidosis-a ten-year report from the China children transplant group. Biol Blood Marrow Transplant. 2016;22:2104–2108. doi: 10.1016/j.bbmt.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 5.Yabe H, Tanaka A, Chinen Y, Kato S, Sawamoto K, Yasuda E, Shintaku H, Suzuki Y, Orii T, Tomatsu S. Hematopoietic stem cell transplantation for Morquio A syndrome. Mol Genet Metab. 2016;117:84–94. doi: 10.1016/j.ymgme.2015.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barth AL, de Magalhães TSPC, Reis ABR, de Oliveira ML, Scalco FB, Cavalcanti NC, Silva DSE, Torres DA, Costa AAP, Bonfim C, Giugliani R, Llerena JC, Jr, Horovitz DGG. Early hematopoietic stem cell transplantation in a patient with severe mucopolysaccharidosis II: a 7 years follow-up. Mol Genet Metab Rep. 2017;12:62–68. doi: 10.1016/j.ymgmr.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kubaski F, Yabe H, Suzuki Y, Seto T, Hamazaki T, Mason RW, Xie L, Onsten TGH, Leistner-Segal S, Giugliani R, Dũng VC, Ngoc CTB, Yamaguchi S, Montaño AM, Orii KE, Fukao T, Shintaku H, Orii T, Tomatsu S. Hematopoietic stem cell transplantation for patients with mucopolysaccharidosis II. Biol Blood Marrow Transplant. 2017;23:1795–1803. doi: 10.1016/j.bbmt.2017.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomatsu S, Sawamoto K, Almeciga-Diaz CJ, Shimada T, Bober MB, Chinen Y, Yabe H, Montano AM, Giugliani R, Kubaski F, Yasuda E, Rodriguez-Lopez A, Espejo-Mojica AJ, Sanchez OF, Mason RW, Barrera LA, Mackenzie WG, Orii T. Impact of enzyme replacement therapy and hematopoietic stem cell transplantation in patients with Morquio A syndrome. Drug Des Devel Ther. 2015;9:1937–1953. doi: 10.2147/DDDT.S68562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomatsu S, Sawamoto K, Shimada T, Bober MB, Kubaski F, Yasuda Y, Mason RW, Khan S, Almeciga-Diaz CJ, Barrera LA, Mackenzie WG, Orii T. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): effect and limitations. Expert Opin Orphan Drugs. 2015;3:1279–1290. doi: 10.1517/21678707.2015.1086640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sawamoto K, Suzuki Y, Mackenzie WG, Theroux MC, Pizarro C, Yabe H, Orii K, Mason R, Orii T, Tomatsu S. Current therapies for Morquio A syndrome and their clinical outcomes. Expert Opin Orphan Drugs. 2016;4:941–951. doi: 10.1080/21678707.2016.1214572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khan S, Alméciga-Díaz CJ, Sawamoto K, Mackenzie WG, Theroux MC, Pizarro C, Mason RW, Orii T, Tomatsu S. Mucopolysaccharidosis IVA and glycosaminoglycans. Mol Genet Metab. 2017;120:78–95. doi: 10.1016/j.ymgme.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Majhail NS, Mau EM, Denzen LW, Arneson TJ. Costs of autologous and allogeneic hematopoietic cell transplantation in the United States: a study using a large national private claims database. Bone Marrow Transplant. 2013;48:294–300. doi: 10.1038/bmt.2012.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitazawa T, Matsumoto K, Fujita S, Seto K, Hasegawa T. Cost analysis of transplantation in Japan, performed with the use of the national database. Transplant Proc. 2017;49:4–9. doi: 10.1016/j.transproceed.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 14.ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02702115?term=sangamo&draw=1&rank=2.
- 15.ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/study/NCT03041324?term=sangamo&draw=1&rank=1.
- 16.ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01474343?term=Lysogene&rank=3.
- 17.ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02053064?term=Lysogene&rank=2.
- 18.ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02716246?term=Abeona&rank=1.
- 19.ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT03173521?term=tigem&rank=1.
- 20.Shi J, Zheng D. An update on gene therapy in China. Curr Opin Mol Ther. 2009;11:547–553. [PubMed] [Google Scholar]
- 21.Willyard C. Limb-saving medicines sought to prevent amputations. Nat Med. 2012;18:328. doi: 10.1038/nm0312-328. [DOI] [PubMed] [Google Scholar]
- 22.Gruber K. Europe gives gene therapy the green light. Lancet. 2012;380:e10. doi: 10.1016/s0140-6736(12)61992-8. [DOI] [PubMed] [Google Scholar]
- 23.U.S. Food and Drug Administration. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm574058.htm.
- 24.McCarty DM, Young SM, Jr, Samulski RJ. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu Rev Genet. 2004;38:819–845. doi: 10.1146/annurev.genet.37.110801.143717. [DOI] [PubMed] [Google Scholar]
- 25.Fu H, McCarty DM. Crossing the blood-brain-barrier with viral vectors. Curr Opin Virol. 2016;21:87–92. doi: 10.1016/j.coviro.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Kurian KM, Watson CJ, Wyllie AH. Retroviral vectors. Mol Pathol. 2000;53:173–176. doi: 10.1136/mp.53.4.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doi K, Takeuchi Y. Gene therapy using retrovirus vectors: vector development and biosafety at clinical trials. Uirusu. 2015;65:27–36. doi: 10.2222/jsv.65.27. [DOI] [PubMed] [Google Scholar]
- 28.Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 29.Sakuma T, Barry MA, Ikeda Y. Lentiviral vectors: basic to translational. Biochem J. 2012;443:603–618. doi: 10.1042/BJ20120146. [DOI] [PubMed] [Google Scholar]
- 30.Parks RJ. Improvements in adenoviral vector technology: overcoming barriers for gene therapy. Clin Genet. 2000;58:1–11. doi: 10.1034/j.1399-0004.2000.580101.x. [DOI] [PubMed] [Google Scholar]
- 31.Peters AH, Drumm J, Ferrell C, Roth DA, Roth DM, McCaman M, Novak PL, Friedman J, Engler R, Braun RE. Absence of germline infection in male mice following intraventricular injection of adenovirus. Mol Ther. 2001;4:603–613. doi: 10.1006/mthe.2001.0500. [DOI] [PubMed] [Google Scholar]
- 32.Ghodsi A, Stein C, Derksen T, Yang G, Anderson RD, Davidson BL. Extensive beta-glucuronidase activity in murine central nervous system after adenovirus-mediated gene transfer to brain. Hum Gene Ther. 1998;9:2331–2340. doi: 10.1089/hum.1998.9.16-2331. [DOI] [PubMed] [Google Scholar]
- 33.Kamata Y, Tanabe A, Kanaji A, Kosuga M, Fukuhara Y, Li XK, Suzuki S, Yamada M, Azuma N, Okuyama T. Long-term normalization in the central nervous system, ocular manifestations, and skeletal deformities by a single systemic adenovirus injection into neonatal mice with mucopolysaccharidosis VII. Gene Ther. 2003;10:406–414. doi: 10.1038/sj.gt.3301869. [DOI] [PubMed] [Google Scholar]
- 34.Kanaji A, Kosuga M, Li XK, Fukuhara Y, Tanabe A, Kamata Y, Azuma N, Yamada M, Sakamaki T, Toyama Y, Okuyama T. Improvement of skeletal lesions in mice with mucopolysaccharidosis type VII by neonatal adenoviral gene transfer. Mol Ther. 2003;8:718–725. doi: 10.1016/j.ymthe.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 35.Donsante A, Vogler C, Muzyczka N, Crawford JM, Barker J, Flotte T, Campbell-Thompson M, Daly T, Sands MS. Observed incidence of tumorigenesis in long-term rodent studies of rAAV vectors. Gene Ther. 2001;8:1343–1346. doi: 10.1038/sj.gt.3301541. [DOI] [PubMed] [Google Scholar]
- 36.Donsante A, Miller DG, Li Y, Vogler C, Brunt EM, Russell DW, Sands MS. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:447. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- 37.Rosas LE, Grieves JL, Zaraspe K, La Perle KM, Fu H, McCarty DM. Patterns of scAAV vector insertion associated with oncogenic events in a mouse model for genotoxicity. Mol Ther. 2012;20:2098–2110. doi: 10.1038/mt.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. 1996;70:3227–3234. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ. Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step transduction in vivo. Gene Ther. 2003;10:2112–2118. doi: 10.1038/sj.gt.3302134. [DOI] [PubMed] [Google Scholar]
- 40.McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther. 2008;16:1648–1656. doi: 10.1038/mt.2008.171. [DOI] [PubMed] [Google Scholar]
- 41.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walters RW, Yi SM, Keshavjee S, Brown KE, Welsh MJ, Chiorini JA, Zabner J. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J Biol Chem. 2001;276:20610–20616. doi: 10.1074/jbc.M101559200. [DOI] [PubMed] [Google Scholar]
- 44.Wu Z, Miller E, Agbandje-McKenna M, Samulski RJ. Alpha2,3 and alpha2,6 N-linked sialic acids facilitate efficient binding and transduction by adeno-associated virus types 1 and 6. J Virol. 2006;80:9093–9103. doi: 10.1128/JVI.00895-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Handa A, Muramatsu S, Qiu J, Mizukami H, Brown KE. Adeno-associated virus (AAV)-3-based vectors transduce haematopoietic cells not susceptible to transduction with AAV-2-based vectors. J Gen Virol. 2000;81:2077–2084. doi: 10.1099/0022-1317-81-8-2077. [DOI] [PubMed] [Google Scholar]
- 47.Kaludov N, Brown KE, Walters RW, Zabner J, Chiorini JA. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J Virol. 2001;75:6884–6893. doi: 10.1128/JVI.75.15.6884-6893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bell CL, Vandenberghe LH, Bell P, Limberis MP, Gao GP, Van Vliet K, Agbandje-McKenna M, Wilson JM. The AAV9 receptor and its modification to improve in vivo lung gene transfer in mice. J Clin Invest. 2011;121:2427–2435. doi: 10.1172/JCI57367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen S, Bryant KD, Brown SM, Randell SH, Asokan A. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J Biol Chem. 2011;286:13532–13540. doi: 10.1074/jbc.M110.210922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akache B, Grimm D, Pandey K, Yant SR, Xu H, Kay MA. The 37/67-kilodalton laminin receptor is a receptor for adeno-associated virus serotypes 8, 2, 3, and 9. J Virol. 2006;80:9831–9836. doi: 10.1128/JVI.00878-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 52.Desmaris N, Verot L, Puech JP, Caillaud C, Vanier MT, Heard JM. Prevention of neuropathology in the mouse model of Hurler syndrome. Ann Neurol. 2004;56:68–76. doi: 10.1002/ana.20150. [DOI] [PubMed] [Google Scholar]
- 53.Winner LK, Beard H, Hassiotis S, Lau AA, Luck AJ, Hopwood JJ, Hemsley KM. A preclinical study evaluating AAVrh10-based gene therapy for Sanfilippo syndrome. Hum Gene Ther. 2016;27:363–375. doi: 10.1089/hum.2015.170. [DOI] [PubMed] [Google Scholar]
- 54.Fraldi A, Hemsley K, Crawley A, Lombardi A, Lau A, Sutherland L, Auricchio A, Ballabio A, Hopwood JJ. Functional correction of CNS lesions in an MPS-IIIA mouse model by intracerebral AAV-mediated delivery of sulfamidase and SUMF1 genes. Hum Mol Genet. 2007;16:2693–2702. doi: 10.1093/hmg/ddm223. [DOI] [PubMed] [Google Scholar]
- 55.Fu H, Samulski RJ, McCown TJ, Picornell YJ, Fletcher D, Muenzer J. Neurological correction of lysosomal storage in a mucopolysaccharidosis IIIB mouse model by adeno-associated virus-mediated gene delivery. Mol Ther. 2002;5:42–49. doi: 10.1006/mthe.2001.0514. [DOI] [PubMed] [Google Scholar]
- 56.Cressant A, Desmaris N, Verot L, Bréjot T, Froissart R, Vanier MT, Maire I, Heard JM. Improved behavior and neuropathology in the mouse model of Sanfilippo type IIIB disease after adeno-associated virus-mediated gene transfer in the striatum. J Neurosci. 2004;24:10229–10239. doi: 10.1523/JNEUROSCI.3558-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Skorupa AF, Fisher KJ, Wilson JM, Parente MK, Wolfe JH. Sustained production of beta-glucuronidase from localized sites after AAV vector gene transfer results in widespread distribution of enzyme and reversal of lysosomal storage lesions in a large volume of brain in mucopolysaccharidosis VII mice. Exp Neurol. 1999;160:17–27. doi: 10.1006/exnr.1999.7176. [DOI] [PubMed] [Google Scholar]
- 58.Bosch A, Perret E, Desmaris N, Heard JM. Long-term and significant correction of brain lesions in adult mucopolysaccharidosis type VII mice using recombinant AAV vectors. Mol Ther. 2000;1:63–70. doi: 10.1006/mthe.1999.0005. [DOI] [PubMed] [Google Scholar]
- 59.Frisella WA, O’Connor LH, Vogler CA, Roberts M, Walkley S, Levy B, Daly TM, Sands MS. Intracranial injection of recombinant adeno-associated virus improves cognitive function in a murine model of mucopolysaccharidosis type VII. Mol Ther. 2001;3:351–358. doi: 10.1006/mthe.2001.0274. [DOI] [PubMed] [Google Scholar]
- 60.Sferra TJ, Qu G, McNeely D, Rennard R, Clark KR, Lo WD, Johnson PR. Recombinant adeno-associated virus-mediated correction of lysosomal storage within the central nervous system of the adult mucopolysaccharidosis type VII mouse. Hum Gene Ther. 2000;11:507–519. doi: 10.1089/10430340050015707. [DOI] [PubMed] [Google Scholar]
- 61.Cearley CN, Wolfe JH. A single injection of an adeno-associated virus vector into nuclei with divergent connections results in widespread vector distribution in the brain and global correction of a neurogenetic disease. Neuroscience. 2007;27:9928–9940. doi: 10.1523/JNEUROSCI.2185-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Manfredsson FP, Rising AC, Mandel RJ. AAV9: a potential blood-brain barrier buster. Mol Ther. 2009;17:403–405. doi: 10.1038/mt.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gray SJ, Nagabhushan Kalburgi S, McCown TJ, Jude Samulski R. Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther. 2013;20:450–459. doi: 10.1038/gt.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samaranch L, Salegio EA, San Sebastian W, Kells AP, Bringas JR, Forsayeth J, Bankiewicz KS. Strong cortical and spinal cord transduction after AAV7 and AAV9 delivery into the cerebrospinal fluid of nonhuman primates. Hum Gene Ther. 2013;24:526–532. doi: 10.1089/hum.2013.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sorrentino NC, Maffia V, Strollo S, Cacace V, Romagnoli N, Manfredi A, Ventrella D, Dondi F, Barone F, Giunti M, Graham AR, Huang Y, Kalled SL, Auricchio A, Bacci ML, Surace EM, Fraldi A. A comprehensive map of CNS transduction by eight recombinant adeno-associated virus serotypes upon cerebrospinal fluid administration in pigs. Mol Ther. 2016;24:276–286. doi: 10.1038/mt.2015.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang H, Yang B, Mu X, Ahmed SS, Su Q, He R, Wang H, Mueller C, Sena-Esteves M, Brown R, Xu Z, Gao G. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol Ther. 2011;19:1440–1448. doi: 10.1038/mt.2011.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nishioka T, Tomatsu S, Gutierrez MA, Miyamoto K, Trandafirescu GG, Lopez PL, Grubb JH, Kanai R, Kobayashi H, Yamaguchi S, Gottesman GS, Cahill R, Noguchi A, Sly WS. Enhancement of drug delivery to bone: characterization of human tissue-nonspecific alkaline phosphatase tagged with an acidic oligopeptide. Mol Genet Metab. 2006;88:244–255. doi: 10.1016/j.ymgme.2006.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Montaño AM, Oikawa H, Tomatsu S, Nishioka T, Vogler C, Gutierrez MA, Oguma T, Tan Y, Grubb JH, Dung VC, Ohashi A, Miyamoto K, Orii T, Yoneda Y, Sly WS. Acidic amino acid tag enhances response to enzyme replacement in mucopolysaccharidosis type VII mice. Mol Genet Metab. 2008;94:178–189. doi: 10.1016/j.ymgme.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 69.Tomatsu S, Montaño AM, Dung VC, Ohashi A, Oikawa H, Oguma T, Orii T, Barrera L, Sly WS. Enhancement of drug delivery: enzyme-replacement therapy for murine Morquio A syndrome. Mol Ther. 2010;18:1094–1102. doi: 10.1038/mt.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomatsu S, Mackenzie WG, Theroux MC, Mason RW, Thacker MM, Shaffer TH, Montaño AM, Rowan D, Sly W, Alméciga-Díaz CJ, Barrera LA, Chinen Y, Yasuda E, Ruhnke K, Suzuki Y, Orii T. Current and emerging treatments and surgical interventions for Morquio A syndrome: a review. Res Rep Endocr Disord. 2012:65–77. doi: 10.2147/RRED.S37278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duque S, Joussemet B, Riviere C, Marais T, Dubreil L, Douar AM, Fyfe J, Moullier P, Colle MA, Barkats M. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17:1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schuster DJ, Dykstra JA, Riedl MS, Kitto KF, Belur LR, McIvor RS, Elde RP, Fairbanks CA, Vulchanova L. Biodistribution of adeno-associated virus serotype 9 (AAV9) vector after intrathecal and intravenous delivery in mouse. Front Neuroanat. 2014;8:42. doi: 10.3389/fnana.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bey K, Ciron C, Dubreil L, Deniaud J, Ledevin M, Cristini J, Blouin V, Aubourg P, Colle MA. Efficient CNS targeting in adult mice by intrathecal infusion of single-stranded AAV9-GFP for gene therapy of neurological disorders. Gene Ther. 2017;24:325–332. doi: 10.1038/gt.2017.18. [DOI] [PubMed] [Google Scholar]
- 75.Bucher T, Dubreil L, Colle MA, Maquigneau M, Deniaud J, Ledevin M, Moullier P, Joussemet B. Intracisternal delivery of AAV9 results in oligodendrocyte and motor neuron transduction in the whole central nervous system of cats. Gene Ther. 2014;21:522–528. doi: 10.1038/gt.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chirmule N, Propert K, Magosin S, Qian Y, Qian R, Wilson J. Immune responses to adenovirus and adeno-associated virus in humans. Gene Ther. 1999;6:1574–1583. doi: 10.1038/sj.gt.3300994. [DOI] [PubMed] [Google Scholar]
- 77.Halbert CL, Miller AD, McNamara S, Emerson J, Gibson RL, Ramsey B, Aitken ML. Prevalence of neutralizing antibodies against adeno-associated virus (AAV) types 2, 5, and 6 in cystic fibrosis and normal populations: implications for gene therapy using AAV vectors. Hum Gene Ther. 2006;17:440–447. doi: 10.1089/hum.2006.17.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J Infect Dis. 2009;199:381–390. doi: 10.1086/595830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, Masurier C. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- 80.Ferla R, O’Malley T, Calcedo R, O’Donnell P, Wang P, Cotugno G, Claudiani P, Wilson JM, Haskins M, Auricchio A. Gene therapy for mucopolysaccharidosis type VI is effective in cats without pre-existing immunity to AAV8. Hum Gene Ther. 2013;24:163–169. doi: 10.1089/hum.2012.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ferla R, Claudiani P, Savarese M, Kozarsky K, Parini R, Scarpa M, Donati MA, Sorge G, Hopwood JJ, Parenti G, Fecarotte S, Nigro V, Sivri HS, Van Der Ploeg A, Andria G, Brunetti-Pierri N, Auricchio A. Prevalence of anti-adeno-associated virus serotype 8 neutralizing antibodies and arylsulfatase B cross-reactive immunologic material in mucopolysaccharidosis VI patient candidates for a gene therapy trial. Hum Gene Ther. 2015;26:145–152. doi: 10.1089/hum.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murrey DA, Naughton BJ, Duncan FJ, Meadows AS, Ware TA, Campbell KJ, Bremer WG, Walker CM, Goodchild L, Bolon B, La Perle K, Flanigan KM, McBride KL, McCarty DM, Fu H. Feasibility and safety of systemic rAAV9-hNAGLU delivery for treating mucopolysaccharidosis IIIB: toxicology, biodistribution, and immunological assessments in primates. Hum Gene Ther Clin Dev. 2014;25:72–84. doi: 10.1089/humc.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hinderer C, Bell P, Gurda BL, Wang Q, Louboutin JP, Zhu Y, Bagel J, O’Donnell P, Sikora T, Ruane T, Wang P, Haskins ME, Wilson JM. Intrathecal gene therapy corrects CNS pathology in a feline model of mucopolysaccharidosis I. Mol Ther. 2014;22:2018–2027. doi: 10.1038/mt.2014.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hinderer C, Bell P, Louboutin JP, Katz N, Zhu Y, Lin G, Choa R, Bagel J, O’Donnell P, Fitzgerald CA, Langan T, Wang P, Casal ML, Haskins ME, Wilson JM. Neonatal tolerance induction enables accurate evaluation of gene therapy for MPS I in a canine model. Mol Genet Metab. 2016;119:124–130. doi: 10.1016/j.ymgme.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ellinwood NM, Ausseil J, Desmaris N, Bigou S, Liu S, Jens JK, Snella EM, Mohammed EE, Thompson CB, Raoul S, Joussemet B, Roux F, Chérel Y, Lajat Y, Piraud M, Benchaouir R, Hermening S, Petry H, Froissart R, Tardisu M, Ciron C, Moullier P, Parkes J, Kline KL, Maire I, Vanier MT, Heard JM, Colle MA. Safe, efficient, and reproducible gene therapy of the brain in the dog models of Sanfilippo and Hurler syndromes. Mol Ther. 2011;19:251–259. doi: 10.1038/mt.2010.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vance M, Llanga T, Bennett W, Woodard K, Murlidharan G, Chungfat N, Asokan A, Gilger B, Kurtzberg J, Samulski RJ, Hirsch ML. AAV gene therapy for MPS1-associated corneal blindness. Sci Rep. 2016;22:22131. doi: 10.1038/srep22131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Motas S, Haurigot V, Garcia M, Marcó S, Ribera A, Roca C, Sánchez X, Sánchez V, Molas M, Bertolin J, Maggioni L, León X, Ruberte J, Bosch F. CNS-directed gene therapy for the treatment of neurologic and somatic mucopolysaccharidosis type II (Hunter syndrome) JCI Insight. 2016;1:e86696. doi: 10.1172/jci.insight.86696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hinderer C, Katz N, Louboutin JP, Bell P, Yu H, Nayal M, Kozarsky K, O’Brien WT, Goode T, Wilson JM. Delivery of an adeno-associated virus vector into cerebrospinal fluid attenuates central nervous system disease in mucopolysaccharidosis type II mice. Hum Gene Ther. 2016;27:906–915. doi: 10.1089/hum.2016.101. [DOI] [PubMed] [Google Scholar]
- 89.Laoharawee K, Podetz-Pedersen KM, Nguyen TT, Evenstar LB, Kitto KF, Nan Z, Fairbanks CA, Low WC, Kozarsky KF, McIvor RS. Prevention of neurocognitive deficiency in mucopolysaccharidosis type II mice by central nervous system-directed, AAV9-mediated Iduronate sulfatase gene transfer. Hum Gene Ther. 2017;28:626–638. doi: 10.1089/hum.2016.184. [DOI] [PubMed] [Google Scholar]
- 90.Ruzo A, Garcia M, Ribera A, Villacampa P, Haurigot V, Marcó S, Ayuso E, Anguela XM, Roca C, Agudo J, Ramos D, Ruberte J, Bosch F. Liver production of sulfamidase reverses peripheral and ameliorates CNS pathology in mucopolysaccharidosis IIIA mice. Mol Ther. 2012;20:254–266. doi: 10.1038/mt.2011.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ruzo A, Marcó S, García M, Villacampa P, Ribera A, Ayuso E, Maggioni L, Mingozzi F, Haurigot V, Bosch F. Correction of pathological accumulation of glycosaminoglycans in central nervous system and peripheral tissues of MPSIIIA mice through systemic AAV9 gene transfer. Hum Gene Ther. 2012;23:1237–1246. doi: 10.1089/hum.2012.029. [DOI] [PubMed] [Google Scholar]
- 92.Sorrentino NC, D’Orsi L, Sambri I, Nusco E, Monaco C, Spampanato C, Polishchuk E, Saccone P, De Leonibus E, Ballabio A, Fraldi A. A highly secreted sulphamidase engineered to cross the blood-brain barrier corrects brain lesions of mice with mucopolysaccharidoses type IIIA. EMBO Mol Med. 2013;5:675–690. doi: 10.1002/emmm.201202083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fu H, Cataldi MP, Ware TA, Zaraspe K, Meadows AS, Murrey DA, McCarty DM. Functional correction of neurological and somatic disorders at later stages of disease in MPS IIIA mice by systemic scAAV9-hSGSH gene delivery. Mol Ther Methods Clin Dev. 2016;3:16036. doi: 10.1038/mtm.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fu H, Dirosario J, Killedar S, Zaraspe K, McCarty DM. Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol Ther. 2011;19:1025–1033. doi: 10.1038/mt.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meadows AS, Duncan FJ, Camboni M, Waligura K, Montgomery C, Zaraspe K, Naughton BJ, Bremer WG, Shilling C, Walker CM, Bolon B, Flanigan KM, McBride KL, McCarty DM, Fu H. A GLP-compliant toxicology and biodistribution study: systemic delivery of an rAAV9 vector for the treatment of mucopolysaccharidosis IIIB. Hum Gene Ther Clin Dev. 2015;26:228–242. doi: 10.1089/humc.2015.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fu H, Meadows AS, Ware T, Mohney RP, McCarty DM. Near-complete correction of profound metabolomic impairments corresponding to functional benefit in MPS IIIB mice after IV rAAV9-hNAGLU gene delivery. Mol Ther. 2017;25:792–802. doi: 10.1016/j.ymthe.2016.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tardieu M, Zérah M, Husson B, de Bournonville S, Deiva K, Adamsbaum C, Vincent F, Hocquemiller M, Broissand C, Furlan V, Ballabio A, Fraldi A, Crystal RG, Baugnon T, Roujeau T, Heard JM, Danos O. Intracerebral administration of adeno-associated viral vector serotype rh.10 carrying human SGSH and SUMF1 cDNAs in children with mucopolysaccharidosis type IIIA disease: results of a phase I/II trial. Hum Gene Ther. 2014;25:506–516. doi: 10.1089/hum.2013.238. [DOI] [PubMed] [Google Scholar]
- 98.Tardieu M, Zérah M, Gougeon ML, Ausseil J, de Bournonville S, Husson B, Zafeiriou D, Parenti G, Bourget P, Poirier B, Furlan V, Artaud C, Baugnon T, Roujeau T, Crystal RG, Meyer C, Deiva K, Heard JM. Intracerebral gene therapy in children with mucopolysaccharidosis type IIIB syndrome: an uncontrolled phase 1/2 clinical trial. Lancet Neurol. 2017;16:712–720. doi: 10.1016/S1474-4422(17)30169-2. [DOI] [PubMed] [Google Scholar]
- 99.Tessitore A, Faella A, O’Malley T, Cotugno G, Doria M, Kunieda T, Matarese G, Haskins M, Auricchio A. Biochemical, pathological, and skeletal improvement of mucopolysaccharidosis VI after gene transfer to liver but not to muscle. Mol Ther. 2008;16:30–37. doi: 10.1038/sj.mt.6300325. [DOI] [PubMed] [Google Scholar]
- 100.Cotugno G, Annunziata P, Tessitore A, O’Malley T, Capalbo A, Faella A, Bartolomeo R, O’Donnell P, Wang P, Russo F, Sleeper MM, Knox VW, Fernandez S, Levanduski L, Hopwood J, De Leonibus E, Haskins M, Auricchio A. Long-term amelioration of feline Mucopolysaccharidosis VI after AAV-mediated liver gene transfer. Mol Ther. 2011;19:461–469. doi: 10.1038/mt.2010.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen YH, Claflin K, Geoghegan JC, Davidson BL. Sialic acid deposition impairs the utility of AAV9, but not peptide-modified AAVs for brain gene therapy in a mouse model of lysosomal storage disease. Mol Ther. 2012;20:1393–1399. doi: 10.1038/mt.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gurda BL, de Guilhem De Lataillade A, Bell P, Zhu Y, Yu H, Wang P, Bagel J, Vite CH, Sikora T, Hinderer C, Calcedo R, Yox AD, Steet RA, Ruane T, O’Donnell P, Gao G, Wilson JM, Casal M, Ponder KP, Haskins ME. Evaluation of AAV-mediated gene therapy for central nervous system disease in canine mucopolysaccharidosis VII. Mol Ther. 2016;24:206–216. doi: 10.1038/mt.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sharma R, Anguela XM, Doyon Y, Wechsler T, DeKelver RC, Sproul S, Paschon DE, Miller JC, Davidson RJ, Shivak D, Zhou S, Rieders J, Gregory PD, Holmes MC, Rebar EJ, High KA. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–1784. doi: 10.1182/blood-2014-12-615492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dekeler R, Ou L, Laoharawee K, Tom S, Radeke R, Rohde M, Sproul S, Przybilla M, Koniar B, Podetz-Pedersen K, Cooksley R, Holmes M, Mcivor RS, Whitley CB, Wechsler T. 13th International Congress of Inborn Errors of Metabolism. ZFN-mediated in vivo genome editing results in phenotypic correction in MPS I and MPS II mouse models. 2017 [Google Scholar]
- 105.RegenxBio. http://www.regenxbio.com/pages/programs/index.htm?panel=5.
- 106.Abeona Therapeutics. https://abeonatherapeutics.com/main/wp-content/uploads/2017/08/ABEO-Company-Presentation.pdf.
- 107.Esteve. https://www.esteve.es/en/research-development/pipeline-portfolio/gene-therapy-platform.
- 108.Abeona. http://investors.abeonatherapeutics.com/phoenix.zhtml?c=63510&p=irol-newsArticle&ID=2238337.
- 109.Chandler RJ, LaFave MC, Varshney GK, Trivedi NS, Carrillo-Carrasco N, Senac JS, Wu W, Hoffmann V, Elkahloun AG, Burgess SM, Venditti CP. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J Clin Invest. 2015;125:870–880. doi: 10.1172/JCI79213. [DOI] [PMC free article] [PubMed] [Google Scholar]