

Abstract
ClC-K channels belong to the CLC family of chloride channels and chloride/proton antiporters. They contribute to sodium chloride reabsorption in Henle's loop of the kidney and to potassium secretion into the endolymph by the stria vascularis of the inner ear. Their accessory subunit barttin stabilizes the ClC-K/barttin complex, promotes its insertion into the surface membrane, and turns the pore-forming subunits into a conductive state. Barttin mutations cause Bartter syndrome type IV, a salt-wasting nephropathy with sensorineural deafness. Here, studying ClC-K/barttin channels heterologously expressed in MDCK-II and HEK293T cells with confocal imaging and patch-clamp recordings, we demonstrate that the eight-amino-acids-long barttin N terminus is required for channel trafficking and activation. Deletion of the complete N terminus (Δ2–8 barttin) retained barttin and human hClC-Ka channels in intracellular compartments. Partial N-terminal deletions did not compromise subcellular hClC-Ka trafficking but drastically reduced current amplitudes. Sequence deletions encompassing Thr-6, Phe-7, or Arg-8 in barttin completely failed to activate hClC-Ka. Analyses of protein expression and whole-cell current noise revealed that inactive channels reside in the plasma membrane. Substituting the deleted N terminus with a polyalanine sequence was insufficient for recovering chloride currents, and single amino acid substitutions highlighted that the correct sequence is required for proper function. Fast and slow gate activation curves obtained from rat V166E rClC-K1/barttin channels indicated that mutant barttin fails to constitutively open the slow gate. Increasing expression of barttin over that of ClC-K partially recovered this insufficiency, indicating that N-terminal modifications of barttin alter both binding affinities and gating properties.
Keywords: chloride channel, gating, intracellular trafficking, renal physiology, electrophysiology, patch clamp, confocal microscopy, protein expression, barttin, ClC-K
Introduction
Human hClC-Ka and hClC-Kb channels and their rat homologues rClC-K1 and rClC-K2 belong to the CLC family of chloride channels and chloride/proton antiporters. They exclusively reside in the kidney and the inner ear (1, 2). ClC-Ka/K1 channels contribute to the osmotic gradient in the renal interstitium by introducing a transepithelial chloride conductance to the thin ascending limb of the loop of Henle (3, 4). ClC-Kb/K2 channels are essential for sodium chloride reabsorption in the thick ascending limb of Henle's loop, but they also permit anion conductance in the distal convoluted tubule and intercalated cells of the collecting duct (5–8). In the inner ear, both isoforms of ClC-K channel contribute to potassium secretion into the endolymph by marginal cells of the stria vascularis. Renal and inner ear chloride channels are dimers that comprise two pore-forming subunits. Barttin is an accessory β-subunit that stabilizes the channel complex and promotes its insertion into the plasma membrane (9, 10). It additionally turns the channel into a conductive state (10, 11). With the exception of rClC-K1, ClC-K channels fail to conduct anions in absence of barttin. Certain mutations of hClC-Kb result in a salt-wasting nephropathy called Bartter syndrome type III (12–14). However, no disease-causing mutation for hClC-Ka has been described so far. Some mutations of the BSND gene, encoding for barttin, cause Bartter syndrome type IV with additional sensorineural deafness (1, 15). According to the different functions of barttin, the phenotype of Bartter syndrome type IV differs in severity and onset.
Barttin consists of 320 amino acids. Hydropathy modeling predicts a short cytoplasmic N terminus of eight amino acids (aa3 1–8), two transmembrane helices (aa 9–26 and 35–54) that are connected by a short extracellular linker (aa 27–34), and a long intracellular C terminus (aa 55–320) (15, 16). Intact transmembrane helices are mandatory for incorporation of ClC-K channels into the surface membrane. A naturally occurring disease-causing mutation, Q32X, truncates barttin behind its first helix and leads to retention of both barttin and ClC-K channels in the endoplasmic reticulum (10, 17). Investigation of further artificial truncations indicated that the two membrane domains and a short stretch of the C terminus (aa 55–72) are sufficient to promote surface membrane insertion and activation of ClC-K channels (1, 10). A tryptophan scan of both transmembrane domains identified a number of residues that are important for the interaction between barttin and the pore-forming subunits of ClC-K channels (18). The majority of mutations left intracellular trafficking unaffected but influenced the gating of ClC-K/barttin channels. E88X, another disease-causing mutation, truncates the main part of the C terminus but leaves both transmembrane helices intact. In this case, ClC-K and barttin are well-integrated in the surface membrane, but the channel complex fails to predominantly enter the basolateral membrane of Madin–Darby canine kidney II (MDCK-II) cells, indicating the existence of a sorting motif in the cytoplasmic tail (17). Two cysteine residues at the end of the second transmembrane helix, Cys-54 and Cys-56, have been identified as sites for posttranslational palmitoylation (19). Although palmitoylation is not necessary for proper intracellular trafficking, it is essential for channel activation.
In contrast to extensive studies on barttin helices and the C terminus, little is known about the impact of its N terminus on channel function. So far, two naturally occurring N-terminal mutations, R8L and R8W, have been reported to cause Bartter syndrome type IV (15). Both barttin mutations nearly abolish anion currents through ClC-K channels heterologously expressed in Xenopus oocytes (20) or HEK293T cells, although a sufficient number of channels is integrated in the surface membrane (17). In this study, we investigate the role of each amino acid of the short N terminus by analyzing the effects of numerous N-terminal barttin mutations on ClC-K/barttin expression, intracellular distribution, and function. We show that truncation of the entire N terminus impairs subcellular trafficking of ClC-K/barttin. Partial deletion of the N terminus or substitution of single amino acids marginally influences its intracellular localization but considerably impairs the function of ClC-K/barttin channels.
Results
N-terminal truncation of barttin impairs intracellular trafficking of hClC-Ka/barttin
Barttin consists of a short N terminus, two transmembrane domains that are connected by a short extracellular linker, and a long intracellular C terminus. We C-terminally fused monomeric cyan fluorescent protein (mCFP) to barttin to investigate its subcellular localization with confocal microscopy when transiently expressed in MDCK-II cells (Fig. 1A). MDCK-II cells grew as confluent monolayers. Nontransfected cells, which are included in each image due to incomplete transfection efficiency, remained dark, demonstrating negligible intensities of background fluorescence. Deletion of the whole N terminus (Δ2–8 barttin) or the N terminus together with the first transmembrane domain (Δ1–25 barttin) considerably altered intracellular distribution (Fig. 1C). In both cases, barttin was retained in intracellular compartments, whereas WT barttin was well transported to the surface membrane. When WT barttin was co-expressed with hClC-Ka, which was tagged with yellow fluorescent protein (YFP) at its N terminus (Fig. 1B), both fluorescent proteins displayed clear staining of the surface membrane (Fig. 1D, WT). However, lacking its N terminus, barttin failed to promote the transport of the hClC-Ka/barttin complex to the cell surface (Fig. 1D, Δ2–8). We next removed shorter parts of the barttin N terminus (Δ2, Δ2–3 through Δ2–7) to determine the relevant amino acids for proper trafficking. Each of these barttin proteins revealed clear plasma membrane staining and guided co-expressed hClC-Ka channels to the surface membrane. Deletion of the second part of the N terminus (Δ5–8, Δ6–8, Δ7–8, Δ8) also displayed intact subcellular trafficking of barttin and hClC-Ka/barttin. Representative confocal images are shown in Fig. 1D and Fig. S1. We conclude that binding of barttin to hClC-Ka is not impaired by partial N-terminal deletion of barttin. Only if the whole N terminus is missing, the hClC-Ka/barttin complex lacks a trafficking signal and is retained in intracellular compartments.
Figure 1.
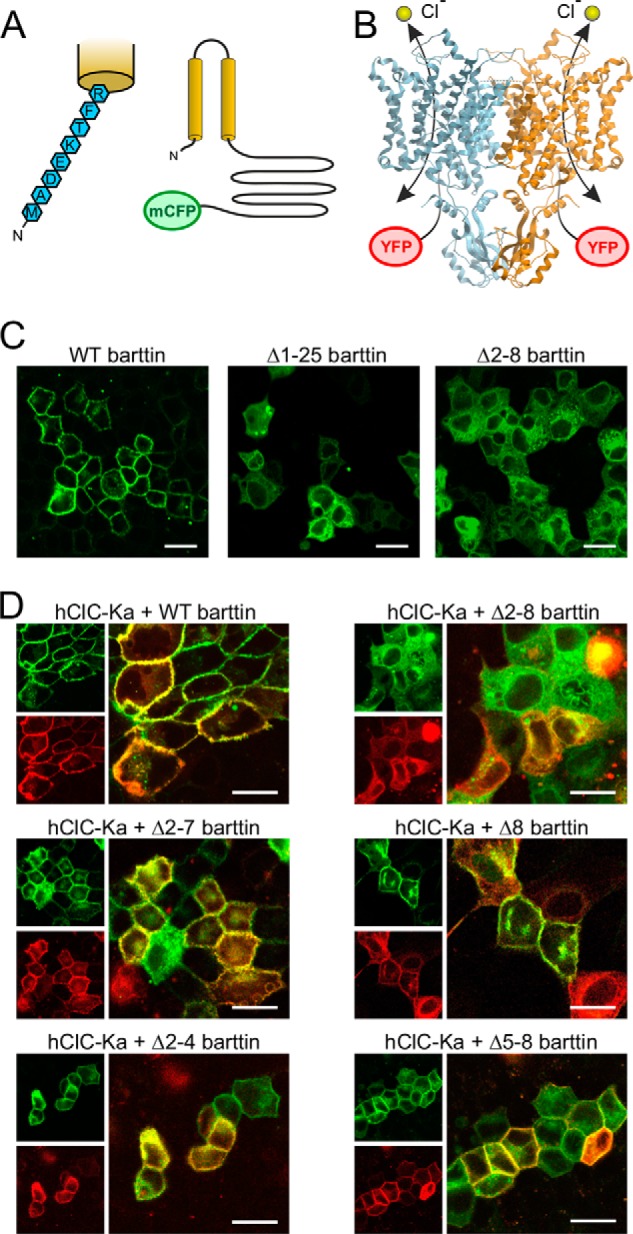
Total but not partial deletion of the barttin N terminus impairs subcellular trafficking of barttin and hClC-Ka channels. A, predicted structure of barttin with two transmembrane helices, a long intracellular C terminus, and a short N terminus comprising only eight amino acids that are magnified on the left. mCFP was fused to the C terminus. B, side view of the bovine ClC-K channel structure obtained by cryo-EM (Protein Data Bank code 5TQQ (29)), displaying the homodimer with two separate conductive pathways for chloride ions. In our studies, hClC-Ka subunits were tagged by YFP at their N terminus. C and D, confocal images of MDCK-II cells showing the subcellular distribution of transiently expressed WT barttin-mCFP or N-terminally mutated barttin-mCFP alone (C) or together with YFP-hClC-Ka channels (D). mCFP fluorescence is displayed in green, and YFP fluorescence is shown in red. Scale bar, 20 μm.
Partial deletion of the barttin N terminus reduces hClC-Ka/barttin current amplitudes
Fig. 2A shows representative whole-cell patch-clamp recordings of HEK293T cells heterologously expressing hClC-Ka channels in the presence or absence of WT barttin. Robust currents are generated only in the presence of barttin, when voltage steps between +185 and −255 mV are applied from a holding potential of 0 mV. This holding potential is close to the reversal potential (−4.8 mV) for nearly symmetrical chloride concentrations of the intra- and extracellular solutions. The current/voltage relationship is linear over a broad range. Only hyperpolarization below −100 mV reduces the channel open probability, providing the well-known hook-shaped appearance of the I-V curve (Fig. 2B) (11, 18, 19, 21). hClC-Ka currents are absent without barttin. Partial deletion of the barttin N terminus reduced or abolished hClC-Ka currents (Fig. 2C). Deletions of one or more amino acids in the first part of the N terminus (Δ2, Δ2–3, Δ2–4, Δ2–5) were tolerated, but current amplitudes significantly declined to about 50% as compared with currents in the presence of WT barttin. Each mutant that included the deletion of one of the residues, Thr-6, Phe-7, or Arg-8, failed to activate hClC-Ka (Fig. 2, C and D).
Figure 2.

Mutations of the barttin N terminus reduce or even abolish hClC-Ka/barttin currents. A, voltage protocol and representative whole-cell patch-clamp current recordings from HEK293T cells expressing human hClC-Ka channels in the presence or absence of WT barttin. B, voltage dependence of the mean current amplitudes of hClC-Ka channels in the presence or absence of WT barttin. C, representative current recordings of hClC-Ka channels in the presence of mutant barttin (the same voltage protocol as in A). D, hClC-Ka current amplitudes at +105 mV in the presence of WT (reference condition) or mutant barttin (statistical significance tested by one-way ANOVA and Holm–Sidak post hoc test versus WT barttin: F(13, 135) = 15.6 and p < 0.001; WT, n = 44; Δ2, n = 9; Δ2–3, n = 14; Δ2–4, n = 12; Δ2–5, n = 11; Δ2–6, n = 7; Δ2–7, n = 15; Δ2–8, n = 6; Δ5–7, n = 4; Δ5–8, n = 6; Δ7–8, n = 8; Δ8, n = 5; Δ1–25, n = 4; no barttin, n = 4). E–G, stationary noise analysis of hClC-Ka/barttin currents. E, representative plot of the current variance (σ2), normalized to the product of the mean current (I) and the electrical driving force (V − Vrev) versus the macroscopic conductance (I/(V − Vrev)) for hClC-Ka in the presence of WT or Δ2–4 barttin. A linear fit provides the gCl (unitary pore conductance) as the y axis intercept and the number of conductive pores as the inverse slope. F, unitary pore conductances (gCl). The broken line represents the reference condition in the presence of WT barttin (statistical significance tested by one-way ANOVA and Holm–Sidak post hoc test versus WT barttin: F(4, 58) = 2.52 and p = 0.051; WT, n = 28; Δ2, n = 4; Δ2–3, n = 11; Δ2–4, n = 8; Δ2–5, n = 12). G, voltage dependence of absolute open probabilities of hClC-Ka/barttin channels (WT, n = 20; Δ2, n = 3; Δ2–3, n = 10; Δ2–4, n = 6; Δ2–5, n = 12). Error bars, S.E. in B and G, S.D. in D and F.
Whole-cell current amplitudes (I) depend on the number of conductive pores (N), the single pore current amplitude (i), and the open probability (P) of the pores, as follows.
| (1) |
We investigated the single pore conductance and absolute open probability by stationary noise analysis of whole-cell currents. This analysis was restricted to mutants that allow recordings of robust hClC-Ka currents (Δ2, Δ2–3, Δ2–4, Δ2–5, and WT barttin). Current variance increased for all of these hClC-Ka/barttin channels, when the current amplitude declined at negative membrane potentials. Current variances (σ2) were normalized to the product of the mean current amplitude (I) and the electrical driving force (V − Vrev) and plotted versus the macroscopic conductance (I/(V − Vrev)) (Fig. 2E). Such plots usually display a linear relationship for hClC-Ka/barttin channels (18, 19, 21). The y axis intercept of a linear regression represents the unitary pore conductance (gCl = i/(V − Vrev)), and the slope corresponds to the number of active pores in the plasma membrane (N = −1/slope). Fig. 2E shows a representative analysis for WT and Δ2–4 barttin. Single-pore conductances are comparable (WT, 23.6 pS; Δ2–4, 22.6 pS), but the number of conductive hClC-Ka pores is considerably reduced with mutant barttin (WT, 3961; Δ2–4, 2053). Mean values confirm that single-pore conductance and absolute open probability of active hClC-Ka/barttin channels are not significantly altered by the barttin mutations (Fig. 2, F and G).
Because whole-cell current amplitudes depend on the number of active channels, we compared expression levels of YFP-hClC-Ka channels in the presence of WT and mutant barttin-mCFP. Cleared lysates of transiently transfected HEK293T cells were electrophoresed on SDS-polyacrylamide gels, and intensities of fluorescent YFP-hClC-Ka bands and barttin-mCFP bands were quantified (Fig. 3, A–F). As previously reported (17–19), hClC-K proteins separate into three bands that correspond to non-, core-, and complex-glycosylated channels (Fig. S2). Expression levels of co-expressed WT barttin-mCFP and YFP-hClC-Ka served as reference in each gel. Expression of mutant barttin was generally affected, when Thr-6 was part of the deleted section of the N terminus (Fig. 3G). Missing Thr-6 and adjacent residues significantly reduced barttin expression. In some cases, the reduction also affected the availability of YFP-hClC-Ka channels (Fig. 3H). This is as expected, because WT barttin stabilizes the ClC-K/barttin complex (9, 17). However, the moderate decline in mutant barttin and/or hClC-Ka expression cannot fully explain the dramatic reduction of current amplitudes.
Figure 3.

Mutations of the barttin N terminus slightly modify expression levels of barttin and hClC-Ka proteins in HEK293T cells. A–F, images of fluorescent SDS-polyacrylamide gels from cleared whole-cell lysates of HEK293T cells co-expressing YFP-hClC-Ka (top) together with WT or mutant barttin-mCFP (bottom). For all samples, total protein concentrations were adjusted to the same level. In SDS-polyacrylamide gels, three hClC-Ka bands are visible that correspond to nonglycosylated (○), core-glycosylated (●), and complex-glycosylated (#) channel protein. Expression levels of co-expressed WT barttin-mCFP and YFP-hClC-Ka served as reference in each gel. Some weak bands of cyan fluorescent protein fluorescence are visible below the barttin-mCFP protein in A–F that presumably result from low amounts of degraded protein fragments. G and H, relative expression levels of barttin (G) and hClC-Ka proteins (H) co-expressed in HEK293T cells (Δ2, n = 9; Δ2–3, n = 7; Δ2–4, n = 7; Δ2–5, n = 11; Δ2–6, n = 11; Δ2–7, n = 15; Δ2–8, n = 7; Δ5–7, n = 5; Δ5–8, n = 11; Δ7–8, n = 5; Δ8, n = 5; Δ1–25, n = 7; no barttin, n = 15). Broken lines, WT barttin expression in G (reference condition for statistical analysis with one-sample Student's t test) and hClC-Ka expression in the presence of WT barttin (top line, reference condition for statistical analysis with one-sample Student's t test) or in the absence of barttin (bottom line) in H. I and J, enhanced fluorescence images of SDS-polyacrylamide gels displaying YFP-hClC-Ka channels in total whole-cell lysates (t) and the biotinylated protein fractions of the surface membrane (s) of HEK293T cells that transiently co-expressed YFP-hClC-Ka and WT or mutant barttin-mCFP. Contamination of the surface membrane fraction by cytosolic proteins is excluded by Western blots using anti-GAPDH antibodies (WB) that exclusively reveal bands in the total cell lysates. Error bars, S.D.
We therefore asked whether the channel proteins are correctly integrated in the plasma membrane. Whereas confocal images clearly showed the trafficking of hClC-Ka/barttin to the plasma membrane (Fig. 1), they do not undoubtedly prove their insertion. Accordingly, we biotinylated membrane-integrated proteins from the extracellular side and purified these proteins after cell lysis using NeutrAvidin affinity chromatography. Fig. 3 (I and J) shows enhanced fluorescence images of SDS-polyacrylamide gels displaying total whole-cell lysates (t) and biotinylated channel proteins from the surface membrane fraction (s) of HEK293T cells that transiently co-expressed YFP-hClC-Ka and WT or mutant barttin-mCFP. Although the efficiency of biotinylation was weak, membrane integration of hClC-Ka channels was evident and comparable in the presence of WT and mutant barttin. Notably, some hClC-Ka channels were also inserted in the plasma membrane in the absence of barttin. We generally checked the membrane fraction for contamination with cytosolic proteins by Western blot analysis. Treatment with anti-GAPDH antibodies resulted in prominent bands in the whole-cell lysate, but no signal was detectable in the biotinylated protein fraction. Because confocal images displayed impaired subcellular trafficking of hClC-Ka channels for Δ2–8 barttin in MDCK-II cells (Fig. 1), we also repeated biotinylation experiments with this cell line and indeed found reduced membrane insertion for the Δ2–8 mutant, which was not evident with HEK293T cells (Fig. S3).
At a first glance, the results of our biochemical analyses of protein expression and membrane insertion might seem inconsistent with our results from noise analyses that showed reduced numbers of active pores (N) for certain mutants like Δ2–4 barttin. However, reduced numbers of active pores in noise analysis are not necessarily related to limited channel availability. Previous reports on palmitoylation-deficient barttin mutants in HEK293T cells and on mouse ClC-K/barttin channels in the distal nephron demonstrated a large portion of inactive ClC-K channels residing in the surface membrane (7, 19). In analogy, we suppose that many hClC-Ka channels remained quiescent in the presence of mutant barttin during the complete test pulse of noise analysis and therefore reduced the parameter N in noise analyses, although biochemical approaches and confocal imaging indicate well-expressed proteins in the plasma membrane. Consequently, macroscopic current amplitudes of hClC-Ka channels are most probably reduced, because only a limited number of channels are switched into an active state and display the high open probabilities. Certain mutations of the barttin N terminus even appear to fully prevent hClC-Ka activation.
Polyalanine insertions do not restore the function of N-terminally truncated barttin
Because the extent of N-terminal barttin deletion affected the magnitude of hClC-Ka current reduction, we asked whether stepwise reconstitution of the N terminus by insertion of polyalanine sequences could restore barttin function. This approach is based on Δ2–7 barttin, where Arg-8 remained untouched to guarantee proper subcellular trafficking of the channel complex. Two, four, or six alanine residues filled the gap between Met-1 and Arg-8. Indeed, these mutants allowed sufficient membrane insertion, as confirmed by confocal images, but current amplitudes of hClC-Ka/barttin channels remained extremely low (Fig. 4, A and B). Insertion of two alanines slightly ameliorated channel activity as compared with Δ2–7 barttin, but further alanine addition rendered channel function even worse (Fig. 4C). Protein expression of barttin and hClC-Ka was significantly reduced, as expected from our previous experiments, because all of these mutants are missing Thr-6 (Fig. 4, D and E). But again, current amplitudes are smaller than expected from biochemical analyses and confocal microscopy.
Figure 4.

Insertion of polyalanine sequences does not restore the function of Δ2–7 barttin. A, confocal images of MDCK-II cells showing the subcellular distribution of transiently expressed YFP-hClC-Ka channels (red) in the presence of mutant barttin-mCFP (green). Scale bar, 20 μm. B, sketch of N-terminal barttin mutations and representative current recordings of hClC-Ka/barttin channels (same voltage protocol as in Fig. 2A). C, hClC-Ka current amplitudes at +105 mV in the presence of WT (reference condition) or mutant barttin (statistical significance tested by one-way ANOVA and Holm–Sidak post hoc test versus WT barttin: F(4, 79) = 31.3 and p < 0.001; WT, n = 44; Δ2–7ins2A, n = 12; Δ2–7ins4A, n = 6; Δ2–7ins6A, n = 7). D and E, relative expression levels of mutant barttin (D) and hClC-Ka proteins (E) co-expressed in HEK293T cells (Δ2–7ins2A, n = 11; Δ2–7ins4A, n = 11; Δ2–7ins6A, n = 11). Broken lines, WT barttin expression in D (reference condition for statistical analysis with one-sample Student's t test) and hClC-Ka expression in the presence of WT barttin (top line, reference condition for statistical analysis with one-sample Student's t test) or in the absence of barttin (bottom line) in E. Error bars, S.D.
N-terminal exchange of single amino acids modifies hClC-Ka/barttin function
As polyalanine substitution of N-terminal amino acids failed to recover barttin functionality, the specific sequence of residues appears to be essential for proper activation of hClC-Ka channels. Accordingly, we next substituted each single amino acid of the barttin N terminus individually to identify determinants for accurate communication between hClC-Ka and its accessory subunit. Ala-2 was substituted by valine, another nonpolar residue (A2V). We removed the negatively charged Asp-3 and inserted neutral asparagine (D3N) or reduced the side-chain volume by alanine insertion (D3A). Similarly, glutamate was substituted by glutamine to neutralize the fourth amino acid (E4Q). With mutation E4R, we inverted the charge at this position, replacing glutamate by arginine. Lys-5 was shortened by alanine (K5A), or its charge was inverted by an exchange to glutamate (K5E). Threonine substitution by valine (T6V) removed the hydroxyl group, and mutation F7A removed the large aromatic ring of phenylalanine. We omitted further investigation of Arg-8, because R8L and R8W have been intensively studied as disease-causing mutations of barttin (15, 17, 19, 20). Confocal images of MDCK-II cells expressing YFP-hClC-Ka and mutant barttin-mCFP show accurate staining of the plasma membrane by mutant barttin and hClC-Ka channels (Fig. 5A and Fig. S1). However, varying degrees of intracellular YFP staining indicated that a fraction of YFP-hClC-Ka protein was retained in intracellular compartments in the presence of certain barttin mutants. Fig. 5B shows representative whole-cell current recordings from HEK293T cells co-expressing hClC-Ka and E4Q, T6V, or F7A barttin. Robust currents in the presence of T6V barttin indicate sufficient insertion of the hClC-Ka/barttin complex in the surface membrane despite intense intracellular YFP fluorescence. Conversely, F7A barttin promotes proper trafficking to the surface membrane, but anion currents are missing, indicating impaired hClC-Ka channel activation by this mutant. Fig. 5C compares the whole-cell current amplitudes of hClC-Ka at +105 mV in the presence of WT or mutant barttin. Substitution of Asp-3 by asparagine or alanine left current amplitudes unaffected, and E4Q insignificantly reduced currents, indicating that negative charges of the N terminus are less important for proper function. However, insertion of an additional positive charge by mutation E4R was not tolerated, as indicated by considerably decreased current amplitudes. Highly significant reduction of chloride currents was also observed for A2V, K5A, K5E, T6V, and F7A barttin. We again checked expression levels of barttin and hClC-Ka proteins (Fig. 5, D and E). Barttin expression significantly declined for E4R, K5A, and T6V. Decreased barttin expression was accompanied by lowered expression of hClC-Ka for K5A only. Nevertheless, expression levels were still too high to predict the drastic reduction in current amplitudes. The same holds true for A2V, K5E, and F7A barttin.
Figure 5.

Substitution of single amino acids of the barttin N terminus reduces hClC-Ka/barttin current amplitudes. A, confocal images of MDCK-II cells showing the subcellular distribution of transiently expressed YFP-hClC-Ka channels (red) in the presence of mutant barttin-mCFP (green). Scale bar, 20 μm. B, sketch of N-terminal barttin mutations and representative current recordings of hClC-Ka/barttin channels (same voltage protocol as in Fig. 2A). C, hClC-Ka current amplitudes at +105 mV in the presence of WT (reference condition) or mutant barttin (statistical significance tested by one-way ANOVA and Holm–Sidak post hoc test versus WT barttin: F(9, 140) = 13.3 and p < 0.001; WT, n = 44; A2V, n = 6; D3N, n = 9; D3A, n = 11; E4Q, n = 16; E4R, n = 14; K5A, n = 17; K5E, n = 12; T6V, n = 12; F7A, n = 9). D and E, relative expression levels of mutant barttin (D) and hClC-Ka proteins (E) co-expressed in HEK293T cells (A2V, n = 9; D3N, n = 3; D3A, n = 3; E4Q, n = 7; E4R, n = 7; K5A, n = 7; K5E, n = 9; T6V, n = 11; F7A, n = 9). Broken lines display WT barttin expression in D (reference condition for statistical analysis with one-sample Student's t test) and hClC-Ka expression in the presence of WT barttin (top line, reference condition for statistical analysis with one-sample Student's t test) or in the absence of barttin (bottom line) in E. F–H, stationary noise analysis of hClC-Ka/barttin currents. F, representative plot of the current variance (σ2), normalized to the product of the mean current (I) and the electrical driving force (V − Vrev), versus the macroscopic conductance (I/(V − Vrev)) for hClC-Ka in the presence of WT or A2V barttin. G, unitary pore conductances (gCl). The broken line represents the reference condition in the presence of WT barttin (statistical significance tested by one-way ANOVA and Holm–Sidak post hoc test versus WT barttin: F(7, 69) = 4.71 and p < 0.001; WT, n = 28; A2V, n = 4; D3N, n = 3; D3A, n = 6; E4Q, n = 9; E4R, n = 9; K5A, n = 11; T6V, n = 7). H, voltage dependence of absolute open probabilities of hClC-Ka/barttin channels (WT, n = 20; A2V, n = 7; D3N, n = 3; D3A, n = 7; E4Q, n = 9; E4R, n = 5; K5A, n = 10; T6V, n = 7). Error bars, S.D. (in C, D, G, and E); S.E. (in H).
Stationary noise analyses revealed unaffected single-pore conductance and absolute open probabilities for most hClC-Ka/barttin channel variants (Fig. 5, F–H). A minor reduction of conductance was observed in the presence of E4R barttin only. K5E and F7A barttin could not be tested, because current amplitudes were too small for reliable evaluation.
N-terminal barttin mutations reduce the slow gate open probability of V166E rClC-K1/barttin channels
The open probability of ClC-K channels is regulated by at least two processes (i.e. fast gating of the individual protopores of the dimer and common slow gating of both pores together) (22, 23). Whereas the so-called “gating glutamate” at the outer entrance of each conduction pathway appears to represent a correlate for the fast gate in most CLC proteins, this residue is missing in ClC-K channels, which nevertheless display fast protopore gating (1). Mutation V166E reconstitutes the gating glutamate in rClC-K1 channels, the rat homolog of hClC-Ka (10, 24). It inverts voltage dependence of fast and slow gating but does not introduce an additional gating process (11). Because it also increases the voltage dependence, we used V166E rClC-K1 to investigate the influence of N-terminally mutated barttin on fast and slow gating of ClC-K channels. Fig. 6A shows representative whole-cell current recordings from HEK293T cells transiently expressing V166E rClC-K1 alone or in the presence of WT barttin. V166E rClC-K1 channels conduct robust currents even in the absence of barttin with fast time-dependent activation at positive potentials and slow activation at negative potentials. Current amplitudes increase in the presence of WT barttin. Fast activation at positive potentials is preserved, but slow activation at negative potentials vanishes. Instead, fast deactivation is visible. A test voltage step to −125 mV after conditioning pulses between −155 and +125 mV (300 ms) allows calculation of relative open probabilities from instantaneous tail current amplitudes (Fig. 6, B and C), which is the product of fast gate open probability and slow gate open probability (Ptotal = Pfast·Pslow). Insertion of a short positive voltage step (+180 mV, 2–5 ms; not shown) between conditioning and test pulse fully opens the fast gate (Pfast = 1) without considerably affecting the slow gate open probability (Ptotal = Pslow), because the time constant for slow gate closure is longer than 40 ms at +180 mV. The procedure thus allows separation of fast and slow gate open probabilities, as described previously (11). In the absence of barttin, fast gate open probability increases with positive potentials but remains constant between −155 and 0 mV (Fig. 6B, open circles). Slow gate open probability increases with hyperpolarization and keeps constant at a low level with depolarization (Fig. 6B, gray circles). The total channel open probability is the product of fast and slow gate open probabilities (Fig. 6B, black circles) (23). WT barttin constitutively opens the slow gate, increases voltage dependence of the fast gate, and shifts the fast gate activation curve to the left (Fig. 6C). These results totally agree with previous publications (11, 18).
Figure 6.

N-terminal barttin mutations modify the slow gate open probability of V166E rClC-K1/barttin channels. A, voltage protocol and representative whole-cell current recordings from HEK293T cells expressing rat V166E rClC-K1 channels in the absence or presence of WT barttin. B and C, voltage dependence of relative open probabilities of the fast gates (white symbols), the slow gate (gray symbols), and the total channel (black symbols) in the absence of barttin (B, n = 20) and in the presence of WT barttin (C, n = 13). WT barttin constitutively opens the slow gate of V166E rClC-K1 channels. D, representative whole-cell current recordings from HEK293T cells expressing V166E rClC-K1 channels in the presence of mutant barttin (same voltage protocol as in A). E, voltage dependence of relative slow gate open probabilities of V166E rClC-K1 channels in the presence of mutant barttin. Activation curves of V166E rClC-K1 with WT barttin and without barttin are shown as gray lines. Experiments were conducted under standard conditions using the pRcCMV vector for V166E rClC-K1 protein expression. F and G, representative current recordings and slow gate activation curves for the same mutants as in D and E but using a low expression pSVL vector for V166E rClC-K1 channel proteins, resulting in extensive excess of barttin over ClC-K subunits. H, comparison of relative slow gate open probabilities at +105 mV for standard (black) and low expression (gray) of V166E rClC-K1 channels in the presence of WT or mutant barttin. Broken lines, relative open probabilities of the slow gate at +105 mV for WT barttin (top line, reference condition) or in the absence of barttin (bottom line). Data volume for standard expression/low expression: WT, n = 13/0; Δ2, n = 5/0; Δ2–3, n = 11/6; Δ2–4, n = 8/7; Δ2–5, n = 6/0; Δ2–6, n = 7/4; Δ2–7, n = 6/7; Δ2–8, n = 7/8; Δ5–7, n = 10/5; Δ5–8, n = 4/5; Δ7–8, n = 4/6; Δ8, n = 4/4; Δ1–25, n = 8/7; no barttin, n = 20/0; Δ2–7ins2A, n = 3/8; Δ2–7ins4A, n = 4/6; Δ2–7ins6A, n = 4/12; A2V, n = 8/6; D3N, n = 5/0; D3A, n = 6/7; E4Q, n = 7/6; E4R, n = 5/0; K5A, n = 6/10; K5E, n = 3/8; T6V, n = 10/6; F7A, n = 3/6. Statistical significance was tested by one-way ANOVA and Holm–Sidak post hoc test versus WT barttin: F(45, 265) = 28.7 and p < 0.001. Error bars, S.E. (in B, C, E, and G); S.D. (in H).
Fig. 6D shows representative current recordings of V166E rClC-K1 channels in the presence of mutant barttin. Voltage steps to positive potentials induce fast current activation, and steps to negative potentials elicit slow activation with varying degrees for the selected mutants. Fig. 6E displays voltage dependences of mean slow gate open probabilities. In contrast to WT barttin, mutant barttin fails to constitutively open the slow gate of V166E rClC-K1 channels. As a rule, the maximum relative open probability of the slow gate is found at negative potentials. But depolarization reduces slow gate open probabilities to a distinct level for each barttin mutant. Complete truncation of the N terminus (Δ2–8) most severely impairs slow gating. Single point mutations like K5A or F7A exert moderate effects.
Our observation may be interpreted as direct influence of barttin mutations on the gating process or as reduced binding affinity between V166E rClC-K1 channels and its mutated accessory subunit. In the latter case, whole-cell current signature would resemble a mixture of currents from V166E rClC-K1 channels without and with barttin bound. We tested this possibility by varying the channel expression level. A pRcCMV plasmid was used for standard expression of V166E rClC-K1 in HEK293T cells. We subcloned the cDNA in a weaker expressing pSVL vector and repeated the experiments without changing the amount of transfected barttin cDNA. Accordingly, the ratio of barttin proteins to channel proteins should be considerably increased. In fact, this procedure altered the appearance of V166E rClC-K1 current traces, which now seemed more comparable with those in the presence of WT barttin (Fig. 6F). However, the slow gate was not fully opened over the whole voltage range (Fig. 6G). We extended our investigation on the other barttin mutants that were previously studied with hClC-Ka and compared mean minimum open probabilities of the slow gate at +105 mV, as shown in Fig. 6H. Black bars display results from standard V166E rClC-K1 expression using the pRcCMV vector, and gray bars depict results from low channel expression using the pSVL vector. Among N-terminal deletions of barttin, those mutants that are missing Phe-7 and/or Arg-8 (Δ2–7, Δ2–8, Δ5–7, Δ5–8, Δ7–8, Δ8) most strongly reduce slow gate activation at positive potentials as compared with slow gate activation by WT barttin. However, increasing the barttin/channel ratio nearly completely restored the constitutive opening of the slow gate for some barttin mutants, including Δ2–7 and Δ5–7. Nevertheless, the slow gate open probability remained significantly reduced for Δ2–8, Δ5–8, Δ7–8, and Δ8 barttin, where Arg-8 was part of the deleted sequence. Moreover, insertions of polyalanine sequences in the Δ2–7 mutant reduced slow gate open probability. The longer the alanine inset was, the more compromised was the slow gate activation. Single point mutations weakly modulated slow gating. Significant reduction of minimum open probability was observed for F7A. We conclude that impaired ClC-K channel activation is caused to a certain extent by reduced binding affinities between the pore-forming subunit and N-terminally mutated barttin. Incomplete recovery despite large excess of barttin, however, suggests additional modification of gating properties for some mutants that alter the N-terminal structure in the vicinity of the first transmembrane domain of barttin.
Discussion
This study demonstrates a crucial role of the short barttin N terminus for trafficking and activation of renal and inner ear ClC-K chloride channels. Deletion of the complete sequence of eight amino acids retains barttin and hClC-Ka channels in intracellular compartments. Partial deletion allows insertion of the channel/barttin complex in the plasma membrane but leaves the channel in an inactive state for many N-terminal barttin mutations.
Previous studies underlined the importance of the transmembrane helices TM1 and TM2 of barttin for its function as chaperone for ClC-K channels, favoring their exit from the endoplasmic reticulum and incorporation into the plasma membrane (10). Truncated Q32X barttin solely consists of the N terminus and the first transmembrane domain, TM1. The shortened protein remained in intracellular compartments and insufficiently guided ClC-K channels to the surface membrane. In contrast, all C-terminal truncations longer than C54X were found to reside in the plasma membrane. The C terminus of barttin alone was soluble in the cytosol and presumably did not bind to the channel protein. Nonetheless, investigation of disease-causing mutations like E88X barttin demonstrated that appropriate sorting to the basolateral membrane requires a so far unknown sequence of the C terminus (17). In summary, the second transmembrane domain TM2 and parts of the C terminus are thought to obtain essential information for target-oriented subcellular trafficking. We here show, however, that the second transmembrane domain including the full C terminus also fails to traffic properly, because Δ1–25 barttin is retained intracellularly. The same holds true for exclusive deletion of the short N terminus, leaving both TM1 and TM2 intact. Thus, subcellular trafficking requires the cooperation of protein sequences that are located in disconnected regions of the barttin protein. In contrast to full removal of the N terminus (Δ2–8), partial deletions leave barttin trafficking and its chaperone function for ClC-K channels unaffected. Confocal microscopy and biotinylation of the channel protein from the extracellular side substantiate co-localization of these barttin mutants and hClC-Ka in the surface membrane of MDCK-II cells. We conclude that binding of the pore-forming subunit and its accessory subunit is not dependent on an intact N terminus of barttin. Moreover, the effect on slow gate activation even indicates efficient binding of Δ2–8 barttin to V166E rClC-K1 channels in the cell membrane, although its chaperone function is lost. Our study cannot fully clarify whether hClC-Ka interacts with Δ2–8 barttin too. According to previous investigations that identified binding sites in the transmembrane domains of barttin (18), we hypothesize that the channel/barttin complex may be formed but that barttin missing the N terminus is not able to activate the hClC-Ka pore.
Patch-clamp recordings of hClC-Ka currents in the presence of WT or mutant barttin revealed insights into the N-terminal determinants for channel activation. Because co-localization in the plasma membrane proved binding of most barttin mutants to the pore-forming subunit, low current amplitudes ought to be caused either by impaired expression and/or stability of the channel complex or by modified pore and/or gating properties. We frequently observed slightly decreased expression of mutant barttin, but the magnitude of expression reduction could not explain the strong decline in hClC-Ka current amplitudes. Deletion or substitution of Thr-6 generally coincided with reduced expression levels, rendering this position important for biosynthesis or stability of the barttin protein. However, we are not aware of any target sequence including this position or posttranslational modification of the amino acid. Instead, Tyr-98 is known to be part of a PY motif that forms a target site for the ubiquitin ligase Nedd4-2 and supports degradation of ClC-K/barttin channels (25). Accordingly, mutant Y98A barttin increased ClC-K/barttin abundance (16). Mutation T6V slightly reduced barttin expression and decreased hClC-Ka current amplitudes to about 50%. Even more remarkable is the inactivity of hClC-Ka, when Thr-6 is removed together with adjacent amino acids. Such inactivity is certainly not explained by diminished availability of mutant barttin. Δ7–8 and Δ8 barttin, for instance, are well expressed and co-localize perfectly with hClC-Ka in the surface membrane but fail to switch the channel into a conductive state. Partial deletions of the N terminus indicate that Thr-6, Phe-7, and Arg-8 are most important for channel activation, whereas mutations of the first amino acids cause milder effects. This observation fits to previous investigations of disease-causing mutations R8L and R8W, which result in a severe phenotype of Bartter syndrome type IV with early onset of renal salt wasting and deafness (1, 15, 17). When heterologously expressed in MDCK-II cells, R8L and R8W barttin insert into the cell membrane, but R8L exhibits additional intracellular staining. Both mutants interact with ClC-K subunits but fail to switch the pores into an active state, resulting in dramatically reduced current amplitudes for hClC-Ka and hClC-Kb channels expressed in mammalian HEK293T cells (17). Very similar results were obtained for rClC-K1 channels and R8L or R8W barttin expressed in Xenopus oocytes (20). Our findings in this study display exactly the same effects for Δ8 barttin. Δ8 barttin is well expressed and co-localizes with hClC-Ka in the plasma membrane, but anion currents are missing. Furthermore, slow gate activation of V166E rClC-K1 channels is impaired at positive potentials. This gating insufficiency cannot be recovered by increasing the barttin/rClC-K1 ratio using a pSVL vector for channel expression. Mutation of the adjacent amino acid (F7A) reveals very similar effects on channel trafficking and activation. This indicates a direct effect of these amino acids on gating properties and the residence of inactive channel complexes in the plasma membrane.
To date, the stoichiometry between pore-forming ClC-K subunits and their accessory subunit barttin is a matter of speculation. Co-immunoprecipitation studies and confocal microscopy using fragments of rat rClC-K2 channels indicate binding of barttin to the B- and/or J-helix at the outer surface of the pore-forming subunit (26). Accordingly, 2:4 stoichiometry with two ClC-K subunits and four barttin molecules could be a possible arrangement for the channel dimer. Nothing is known about affinities of the presumed binding sites. We here show that most deletion mutants of barttin bind to ClC-K channels but insufficiently activate the slow gate of V166E rClC-K1. Slow gate functionality was improved for some mutants by raising the abundance of barttin over the rClC-K1 channel protein, indicating that the barttin mutation diminished the affinity to at least one of the binding sites. We do not know whether one or both binding sites of each channel monomer have to be occupied by barttin to build functional complexes. However, physiological interaction seems to rely on the short N terminus of barttin in conjunction with an intact first transmembrane helix TM1. Wojciechowski et al. (18) demonstrated by tryptophan scanning analysis of the first and second transmembrane domain that binding and activation of hClC-Ka largely depends on a tight interaction with the TM1 helix. The minority of amino acids tolerated substitution by tryptophan. Most mutants failed to activate hClC-Ka but left expression and trafficking unchanged. In contrast, substitution of single amino acids of TM2 weakly affected channel function. Accordingly, an intact N terminus of barttin probably supports accurate orientation of the TM1 helix to efficiently interact with ClC-K subunits. Loose binding may allow correct subcellular trafficking but does not fulfill the requirements for functional activation of ClC-K channels.
Experimental procedures
Mutagenesis and heterologous expression
Mutant barttin cDNA was constructed using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). For all constructs, PCR-amplified sequences were verified by direct sequencing. MDCK-II or HEK293T cells were transiently transfected with plasmid DNA containing the coding region for fusion proteins of barttin and monomeric cyan fluorescent protein (pcDNA3.1(+) barttin-mCFP) alone or together with plasmid DNA encoding for ClC-K channel proteins tagged with yellow fluorescent protein (pcDNA3.1(−) YFP-hClC-Ka or pRcCMV YFP-V166E-rClC-K1). In some experiments, a pSVL vector for low expression of V166E rClC-K1 channels was used to extensively increase the excess of barttin over channel proteins. Lipofectamine 2000 (Invitrogen) was used for transfection of MDCK-II cells to study intracellular distribution of barttin and ClC-K channels, an established model system for the analyses of trafficking in epithelial cells (10, 17–19). MDCK-II cells originate from a canine kidney tubule and generate dense monolayers with tight cell contacts and an apical and basolateral polarity. MDCK cells, however, are less suitable for patch-clamp experiments, because their rough surface tends to form microvilli impeding the gigaseal formation of the cell membrane with the glass micropipette. We therefore chose HEK293T cells, which represent the preferred expression system for patch-clamp analyses of ClC-K/barttin currents (10, 11, 17–19, 21, 27). Due to the presence of the SV40 large T-antigen, they robustly express channel proteins, even if the proteins harbor severe disease-causing mutations. Calcium phosphate precipitation was used to transfect HEK293T cells (11, 19).
Biochemical analysis of protein expression
Expression of barttin and ClC-K proteins was analyzed 24 h after transient transfection of HEK293T cells. Cells were lysed by incubation in a buffer containing NaCl (150 mm), HEPES (10 mm), Triton X-100 (1%), and a protease inhibitor mix (1%; Sigma-Aldrich, Hamburg, Germany), pH 7.4. Cleared lysates were denatured for 15 min at room temperature in SDS sample buffer containing 100 mm DTT, 45.4 mm Tris/HCl, 1% SDS, 5% glycerol, and 0.01% bromophenol blue. Total protein concentration of each sample was determined by a BCA assay (Thermo Fisher Scientific, Bremen, Germany). Aliquots of equal protein amount were loaded on SDS-polyacrylamide gels (10% acrylamide) and electrophoresed in parallel with a fluorescent mass marker (Dual Color (Bio-Rad, München, Germany) and Spectra Multicolor Broad Range Protein Ladder (Thermo Fisher Scientific, Bremen, Germany)). The wet polyacrylamide gel was scanned with a fluorescence scanner (Fusion, Vilber Lourmat, Eberhardzell, Germany) to visualize barttin and hClC-Ka as identified by their mCFP and YFP tag, respectively. Fluorescence intensities of individual bands were quantified using Fiji software (ImageJ, Wayne Rasband, National Institutes of Health). Fluorescence intensities of lysates from HEK293T cells co-expressing WT barttin-mCFP and YFP-hClC-Ka served as reference in each gel.
Biochemical analysis of hClC-Ka surface membrane insertion
Plasma membrane expression of hClC-Ka was investigated in the presence of WT and mutant barttin using two cell lines. HEK293T cells were cultivated for 24 h and MDCK-II cells for 48 h after transfection and subsequently incubated with 0.375 mg of biotin (EZ-link sulfo-NHS-SS-biotin, Thermo Fisher Scientific) for 30 min. Adding 10 mm glycine for 20 min stopped the biotinylation process before cell lysis with radioimmune precipitation assay buffer (100 mm NaCl, 20 mm HEPES, 1 mm sodium orthovanadate, 1 mm NaF, 1 mm EDTA, 1% SDS, 1% deoxycholate, 1% protease inhibitor mix, pH 7.4). Biotinylated proteins were purified by NeutrAvidin affinity chromatography (High Capacity NeutrAvidin Agarose, Thermo Fisher Scientific) and eluted by 2× SDS sample buffer. Samples of total cell lysate and biotinylated protein fraction were electrophoresed on 10% SDS-polyacrylamide gels. Fluorescent bands of YFP-hClC-Ka in the biotinylated fraction disclosed surface membrane insertion of channel proteins in the presence of WT and mutant barttin. Western blot analysis of the gels using anti-GAPDH antibodies was performed to validate exclusive biotinylation of surface membrane proteins (primary antibody, GAPDH (A-3), sc-137179 (Santa Cruz Biotechnology, Heidelberg, Germany); secondary antibody, rabbit anti-mouse IgG Fc, horseradish peroxidase conjugate (Thermo Fisher Scientific)).
Confocal microscopy
Live cell confocal imaging was carried out to study the subcellular distribution of barttin and hClC-Ka channels in transiently transfected MDCK-II cells grown in special dishes for microscopy (Ibidi, Martinsried, Germany). Confocal images were obtained after 2 days of expression of fluorescent barttin-mCFP and YFP-hClC-Ka using a laser-scanning Zeiss LSM 780 microscope (Zeiss, Oberkochen, Germany). mCFP was excited at 440 nm and YFP at 514 nm. Emission was recorded between 454 and 520 nm for mCFP detection and between 520 and 620 nm for YFP detection. Fiji software was used to linearly adjust brightness and contrast in representative images.
Electrophysiology and data analysis
Standard patch-clamp recordings were performed typically 1 day after co-transfection of HEK293T cells with cDNA encoding for human barttin-mCFP and cDNA encoding for human YFP-hClC-Ka or rat YFP-V166E-rClC-K1 channels. Successfully transfected cells were selected for patch-clamp analysis by their mCFP and YFP fluorescence. mCFP fluorescence intensity was generally in excess over YFP fluorescence, according to the stronger expression of the barttin subunit compared with the channel subunit. When the pSVL vector was used for very low expression of the channel protein, only the mCFP fluorescence was usable for cell selection. The extracellular bath solution contained NaCl (140 mm), KCl (4 mm), CaCl2 (2 mm), MgCl2 (1 mm), and HEPES (5 mm). The intracellular pipette solution contained NaCl (120 mm), MgCl2 (2 mm), EGTA (5 mm), and HEPES (10 mm). pH was adjusted to 7.4 for both solutions. Pipettes were pulled from borosilicate glass (Harvard Apparatus, Holliston, MA) with resistances of 1–2 megaohms. For noise analyses, pipettes were coated with dental wax to reduce their capacitance. Whole-cell chloride currents were amplified and filtered (5–10 kHz) by an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). More than 85% of the series resistance was compensated by an analog procedure. Cells were clamped to 0 mV between test sweeps, which is close to the reversal potential of −4.8 mV for the nearly symmetrical chloride concentrations in the bath and pipette solution.
Stationary noise analysis from whole-cell currents was performed to determine single-pore conductance and absolute open probabilities of hClC-Ka/barttin channels, as previously described in detail (18, 19, 28). For this purpose, currents were filtered at 10 kHz and digitized with a high sampling rate of 50 kHz. Steady-state current variances (σ2) and steady-state current amplitudes (I) were measured at various voltages (V). Current variances were corrected for the background variance that was measured at the reversal potential (Vrev). The current variance was next normalized to the product of the mean current amplitude (I) and the electrical driving force (V − Vrev) and plotted versus the macroscopic conductance (I/(V − Vrev)).
| (2) |
In such a plot, the negative inverse slope of the linear regression reveals the number of conductive pores in the cell membrane (N), and the y axis intercept represents the unitary pore conductance (gCl = i/(V − Vrev)). Because channels of the CLC family are dimers, N/2 accords with the number of ClC-K channels, and 2i is the current amplitude of a single double-barreled channel. Knowing the number of pores and the single pore conductance allows calculation of the absolute open probability of ClC-K channels (P) from Equation 1.
Absolute open probabilities were calculated for voltages between −255 mV and +5 mV to compare the activation curve of hClC-Ka channels in the presence of WT and mutant barttin. A modified Boltzmann equation was fit to the activation curves to determine the minimum (Pmin) and maximum (Pmax) open probabilities, the midpoint of activation (V0.5), and the apparent gating charge (zδ).
| (3) |
To determine relative open probabilities of V166E rClC-K1/barttin channels, voltage steps to potentials between −155 and +145 mV were applied for 300 ms, followed by a constant test step to −125 mV. Instantaneous tail current amplitudes from the test potential were normalized to their maximum amplitude and plotted versus the potentials of the conditioning steps to display the channel activation curve. A short step to +180 mV (2–5 ms) was inserted before the test pulse to fully activate the fast protopore gates of V166E rClC-K1 channels without affecting the slow gate open probability. In this case, the relative tail current amplitudes represent the slow gate open probabilities. Dividing the channel open probabilities by the slow gate open probabilities reveals the fast gate open probabilities (11, 23).
Data analysis was performed with a software combination of Clampfit (Axon Instruments, Union City, CA) and SigmaPlot (Systat Software Inc., San Jose, CA). Scatter plots present individual data points combined with means ± S.D. One-way analysis of variance (ANOVA) was performed to test the statistical significance in multiple-group comparisons. The Holm–Sidak post hoc test was used to compare the effects of mutant barttin versus control conditions (WT barttin). One-sample Student's t test was performed to test significant changes of expression levels as compared with normalized reference conditions (co-expression of hClC-Ka and WT barttin). Levels of significance are indicated as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Author contributions
M. F. conceived and designed the experiments. Mutagenesis and expression studies were performed by B. B., T. B., C. S., and D. W. D. W., S. T., and A. R. performed live cell confocal imaging. B. B. and S. T. analyzed surface membrane insertion of hClC-Ka proteins by biotinylation. Electrophysiological experiments were conducted and analyzed by S. T., D. W., C. S., A. R., M. F., and J. d. l. R. M. F. and D. W. drafted the manuscript with input from the co-authors. All authors approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Christoph Fahlke for providing the expression constructs for hClC-Ka, rClC-K1, and barttin; Dr. Matthias Grieschat and Dr. Michael Schänzler for helpful discussions; and Petra Kilian for excellent technical assistance with cell culture.
Parts of this study have been reported in abstract form (30). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3.
- aa
- amino acids
- MDCK
- Madin–Darby canine kidney
- mCFP
- monomeric cyan fluorescent protein
- YFP
- yellow fluorescent protein
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- ANOVA
- analysis of variance
- pS
- picosiemens.
References
- 1. Fahlke C., and Fischer M. (2010) Physiology and pathophysiology of ClC-K/barttin channels. Front. Physiol. 1, 155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stölting G., Fischer M., and Fahlke C. (2014) CLC channel function and dysfunction in health and disease. Front. Physiol. 5, 378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Uchida S. (2000) In vivo role of CLC chloride channels in the kidney. Am. J. Physiol. Renal Physiol. 279, F802–F808 10.1152/ajprenal.2000.279.5.F802 [DOI] [PubMed] [Google Scholar]
- 4. Uchida S., Sasaki S., Nitta K., Uchida K., Horita S., Nihei H., and Marumo F. (1995) Localization and functional characterization of rat kidney-specific chloride channel, ClC-K1. J. Clin. Invest. 95, 104–113 10.1172/JCI117626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kobayashi K., Uchida S., Okamura H. O., Marumo F., and Sasaki S. (2002) Human CLC-KB gene promoter drives the EGFP expression in the specific distal nephron segments and inner ear. J. Am. Soc. Nephrol. 13, 1992–1998 10.1097/01.ASN.0000023434.47132.3D [DOI] [PubMed] [Google Scholar]
- 6. Krämer B. K., Bergler T., Stoelcker B., and Waldegger S. (2008) Mechanisms of disease: the kidney-specific chloride channels ClCKA and ClCKB, the Barttin subunit, and their clinical relevance. Nat. Clin. Pract. Nephrol. 4, 38–46 10.1038/ncpneph0689 [DOI] [PubMed] [Google Scholar]
- 7. Pinelli L., Nissant A., Edwards A., Lourdel S., Teulon J., and Paulais M. (2016) Dual regulation of the native ClC-K2 chloride channel in the distal nephron by voltage and pH. J. Gen. Physiol. 148, 213–226 10.1085/jgp.201611623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stölting G., and Fahlke Ch. (2017) Chloride channels in renal salt and water transport. Acta Physiol. 219, 11–13 10.1111/apha.12802 [DOI] [PubMed] [Google Scholar]
- 9. Hayama A., Rai T., Sasaki S., and Uchida S. (2003) Molecular mechanisms of Bartter syndrome caused by mutations in the BSND gene. Histochem. Cell Biol. 119, 485–493 10.1007/s00418-003-0535-2 [DOI] [PubMed] [Google Scholar]
- 10. Scholl U., Hebeisen S., Janssen A. G., Müller-Newen G., Alekov A., and Fahlke C. (2006) Barttin modulates trafficking and function of ClC-K channels. Proc. Natl. Acad. Sci. U.S.A. 103, 11411–11416 10.1073/pnas.0601631103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fischer M., Janssen A. G., and Fahlke C. (2010) Barttin activates ClC-K channel function by modulating gating. J. Am. Soc. Nephrol. 21, 1281–1289 10.1681/ASN.2009121274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hebert S. C. (2003) Bartter syndrome. Curr. Opin. Nephrol. Hypertens. 12, 527–532 10.1097/00041552-200309000-00008 [DOI] [PubMed] [Google Scholar]
- 13. Seyberth H. W. (2008) An improved terminology and classification of Bartter-like syndromes. Nat. Clin. Pract. Nephrol. 4, 560–567 10.1038/ncpneph0912 [DOI] [PubMed] [Google Scholar]
- 14. Simon D. B., Bindra R. S., Mansfield T. A., Nelson-Williams C., Mendonca E., Stone R., Schurman S., Nayir A., Alpay H., Bakkaloglu A., Rodriguez-Soriano J., Morales J. M., Sanjad S. A., Taylor C. M., Pilz D., et al. (1997) Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat. Genet. 17, 171–178 10.1038/ng1097-171 [DOI] [PubMed] [Google Scholar]
- 15. Birkenhäger R., Otto E., Schürmann M. J., Vollmer M., Ruf E. M., Maier-Lutz I., Beekmann F., Fekete A., Omran H., Feldmann D., Milford D. V., Jeck N., Konrad M., Landau D., Knoers N. V., et al. (2001) Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 29, 310–314 10.1038/ng752 [DOI] [PubMed] [Google Scholar]
- 16. Estévez R., Boettger T., Stein V., Birkenhäger R., Otto E., Hildebrandt F., and Jentsch T. J. (2001) Barttin is a Cl− channel β-subunit crucial for renal Cl− reabsorption and inner ear K+ secretion. Nature 414, 558–561 10.1038/35107099 [DOI] [PubMed] [Google Scholar]
- 17. Janssen A. G., Scholl U., Domeyer C., Nothmann D., Leinenweber A., and Fahlke C. (2009) Disease-causing dysfunctions of barttin in Bartter syndrome type IV. J. Am. Soc. Nephrol. 20, 145–153 10.1681/ASN.2008010102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wojciechowski D., Fischer M., and and Fahlke C. (2015) Tryptophan scanning mutagenesis identifies the molecular determinants of distinct barttin functions. J. Biol. Chem. 290, 18732–18743 10.1074/jbc.M114.625376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Steinke K. V., Gorinski N., Wojciechowski D., Todorov V., Guseva D., Ponimaskin E., Fahlke C., and Fischer M. (2015) Human CLC-K channels require palmitoylation of their accessory subunit barttin to be functional. J. Biol. Chem. 290, 17390–17400 10.1074/jbc.M114.631705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waldegger S., Jeck N., Barth P., Peters M., Vitzthum H., Wolf K., Kurtz A., Konrad M., and Seyberth H. W. (2002) Barttin increases surface expression and changes current properties of ClC-K channels. Pflugers Arch. 444, 411–418 10.1007/s00424-002-0819-8 [DOI] [PubMed] [Google Scholar]
- 21. Riazuddin S., Anwar S., Fischer M., Ahmed Z. M., Khan S. Y., Janssen A. G., Zafar A. U., Scholl U., Husnain T., Belyantseva I. A., Friedman P. L., Riazuddin S., Friedman T. B., and Fahlke C. (2009) Molecular basis of DFNB73: mutations of BSND can cause nonsyndromic deafness or Bartter syndrome. Am. J. Hum. Genet. 85, 273–280 10.1016/j.ajhg.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Accardi A. (2015) Structure and gating of CLC channels and exchangers: structure and gating of CLC channels and exchangers. J. Physiol. 593, 4129–4138 10.1113/JP270575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Accardi A., and Pusch M. (2000) Fast and slow gating relaxations in the muscle chloride channel CLC-1. J. Gen. Physiol. 116, 433–444 10.1085/jgp.116.3.433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Waldegger S., and Jentsch T. J. (2000) Functional and structural analysis of ClC-K chloride channels involved in renal disease. J. Biol. Chem. 275, 24527–24533 10.1074/jbc.M001987200 [DOI] [PubMed] [Google Scholar]
- 25. Embark H. M., Böhmer C., Palmada M., Rajamanickam J., Wyatt A. W., Wallisch S., Capasso G., Waldegger P., Seyberth H. W., Waldegger S., and Lang F. (2004) Regulation of CLC-Ka/barttin by the ubiquitin ligase Nedd4-2 and the serum- and glucocorticoid-dependent kinases. Kidney Int. 66, 1918–1925 10.1111/j.1523-1755.2004.00966.x [DOI] [PubMed] [Google Scholar]
- 26. Tajima M., Hayama A., Rai T., Sasaki S., and Uchida S. (2007) Barttin binds to the outer lateral surface of the ClC-K2 chloride channel. Biochem. Biophys. Res. Commun. 362, 858–864 10.1016/j.bbrc.2007.08.097 [DOI] [PubMed] [Google Scholar]
- 27. Imbrici P., Liantonio A., Gradogna A., Pusch M., and Camerino D. C. (2014) Targeting kidney CLC-K channels: pharmacological profile in a human cell line versus Xenopus oocytes. Biochim. Biophys. Acta 1838, 2484–2491 10.1016/j.bbamem.2014.05.017 [DOI] [PubMed] [Google Scholar]
- 28. Sesti F., and Goldstein S. A. (1998) Single-channel characteristics of wild-type IKs channels and channels formed with two minK mutants that cause long QT syndrome. J. Gen. Physiol. 112, 651–663 10.1085/jgp.112.6.651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Park E., Campbell E. B., and MacKinnon R. (2017) Structure of a CLC chloride ion channel by cryo-electron microscopy. Nature 541, 500–505 10.1038/nature20812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fischer M., Thiemann S., Schaal C., Rahtz A., de la Roche J., and Wojciechowski D. (2017) Activation of renal ClC-K chloride channels is dependent on an intact N-terminus of their accessory subunit barttin. in Proceedings of the 12th Göttingen Meeting of the German Neuroscience Society. Neuroforum 23, (suppl.) T6-2A [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.