

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a burgeoning health problem worldwide, ranging from nonalcoholic fatty liver (NAFL, steatosis without hepatocellular injury) to the more aggressive nonalcoholic steatohepatitis (NASH, steatosis with ballooning, inflammation, or fibrosis). Although many studies have greatly contributed to the elucidation of NAFLD pathogenesis, the disease progression from NAFL to NASH remains incompletely understood. Nuclear receptor small heterodimer partner (Nr0b2, SHP) is a transcriptional regulator critical for the regulation of bile acid, glucose, and lipid metabolism. Here, we show that SHP levels are decreased in the livers of patients with NASH and in diet-induced mouse NASH. Exposing primary mouse hepatocytes to palmitic acid and lipopolysaccharide in vitro, we demonstrated that the suppression of Shp expression in hepatocytes is due to c-Jun N-terminal kinase (JNK) activation, which stimulates c-Jun–mediated transcriptional repression of Shp. Interestingly, in vivo induction of hepatocyte-specific SHP in steatotic mouse liver ameliorated NASH progression by attenuating liver inflammation and fibrosis, but not steatosis. Moreover, a key mechanism linking the anti-inflammatory role of hepatocyte-specific SHP expression to inflammation involved SHP-induced suppression of NF-κB p65-mediated induction of chemokine (C–C motif) ligand 2 (CCL2), which activates macrophage proinflammatory polarization and migration. In summary, our results indicate that a JNK/SHP/NF-κB/CCL2 regulatory network controls communications between hepatocytes and macrophages and contributes to the disease progression from NAFL to NASH. Our findings may benefit the development of new management or prevention strategies for NASH.
Keywords: nuclear receptor, inflammation, liver injury, fibrosis, chemokine, nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, small heterodimer partner
Introduction
Nonalcoholic fatty liver disease (NAFLD)3 affects 25.24% (95% confidence interval: 22.10–28.65) of the general population (1) and is rapidly becoming a major health concern because of significant increases in the prevalence of obesity, insulin resistance diabetes, and hyperlipidemia (2, 3). Encompassing the entire spectrum of fatty liver disease in individuals without significant alcohol consumption, NAFLD is histologically categorized into nonalcoholic fatty liver (NAFL; steatosis without hepatocellular injury) and nonalcoholic steatohepatitis (NASH; steatosis with ballooning, inflammation, with or without fibrosis) (4). The chances of developing more serious diseases such as cirrhosis, hepatocellular carcinoma, and cardiovascular diseases are increased in patients with NASH (5). NASH is characterized by hepatocyte damage due to lipotoxicity as well as macrophage-associated liver inflammation, a process in which the cross-talk between hepatocytes and macrophages is crucial. Emerging evidence highlights that lipid accumulation in hepatocytes stimulates the production of proinflammatory cytokines and chemokines, thereby potentially contributing to the initiation of hepatic inflammation and subsequent liver injury (6, 7). Despite this knowledge, what controls the release of proinflammatory cytokines and chemokines leading to the NASH transition is still obscure and requires elucidation.
Nuclear receptor small heterodimer partner (Nr0b2, Homo sapiens SHP; Mus musculus Shp) is highly expressed in normal hepatocytes and acts as an important transcriptional regulator for bile acid, glucose, and lipid metabolism (8). In support of a critical role for SHP in metabolic diseases, SHP mutation is associated with an increase in body weight and morbidity risk of type 2 diabetes in Japanese populations (9). More recently, SHP has been shown to suppress toll-like receptor 4 (TLR4)-induced (10) and NLRP3 inflammasome-mediated (11) inflammatory responses in monocytes, which suggests a connection between SHP and inflammation. Now, hepatocytes are gradually being recognized as important players involved in the initiation of inflammation in NASH (6, 7). However, whether hepatocyte SHP plays a role in this process remains unexplored.
Inflammatory chemokine (C–C motif) ligand 2 (CCL2; or monocyte chemoattractant protein 1 (MCP1)) is responsible for attracting monocytes and T cells during liver injury (12). There are many types of liver cells that produce CCL2, including hepatocytes, stellate cells, and Kupffer cells (13, 14). Studies have shown that high levels of CCL2 in NAFLD contribute to the conversion of NAFL to NASH (15), and pharmacological inhibition of CCL2 reduces liver macrophage infiltration in NASH (16), which makes CCL2 a good therapeutic target for NASH prevention and treatment. However, how CCL2 is produced during the disease progression from NAFL to NASH is incompletely understood. A recent study has demonstrated that the activation of SHP by a small-molecule activator inhibits liver cancer cell migration by blocking CCL2 signaling (17), which potentially links SHP to CCL2 production. Here, we show that SHP was markedly decreased in the livers of patients with NASH and in diet-induced mouse NASH. The loss of SHP in hepatocytes resulted in NF-κB p65-mediated induction of CCL2, leading to macrophage activation. Meanwhile, overexpressing SHP in hepatocytes prevented NAFL progression to NASH by attenuating liver Inflammation and fibrosis. Taken together, our study has uncovered a novel regulatory network in hepatocytes consisting of JNK/SHP/NF-κB/CCL2, which controls macrophage recruitment during the disease progression from NAFL to NASH. The findings from our study may benefit the development of new management or prevention strategies for NASH.
Results
Decrease of SHP in livers of human NASH
To determine whether the expression of SHP is associated with NAFLD pathogenesis, we examined SHP mRNA levels in two sets of human liver specimens. The first set was obtained through the University of Kansas Liver Center. The liver histology of human NAFL and NASH is shown in Fig. 1A. NAFL is characterized by the deposition of triglycerides as lipid droplets in hepatocytes. NASH is distinguished from NAFL by the presence of hepatocyte injury (hepatocyte ballooning and cell death), inflammation, and/or collagen deposition (fibrosis). Perisinusoidal/pericellular (chicken wire) fibrosis is the characteristic pattern of liver fibrosis in NASH, which typically begins in zone 3 due to the deposition of collagen along the sinusoids and around the hepatocytes. As shown in Fig. 1A, picrosirius red staining showed the chicken-wire pattern of perisinusoidal/pericellular fibrosis and periportal fibrosis in human NASH. Although there were similar SHP mRNA levels in the liver of normal and NAFL samples, a significant decrease in SHP mRNA was observed in NASH samples compared with NAFL samples (Fig. 1A). Consistently, the analysis of microarray data set GSE48452 also revealed a significant decrease in SHP mRNA levels in patients with NASH compared with NAFL and normal controls (Fig. 1B). Western blot analysis confirmed the decrease in SHP protein in NASH samples compared with NAFL and normal livers (Fig. 1C). Collectively, our results strongly suggest the biological relevance of SHP down-regulation during the disease progression from NAFL to NASH in humans.
Figure 1.

Decrease of SHP in the livers of human NASH compared with NAFL. A, human liver specimens were obtained from the University of Kansas Liver Center, including 12 normal, 12 NAFL, and 8 NASH specimens. Left, representative images of liver sections stained with H&E or Picrosirius red from patients with NAFL or NASH. Original magnification, ×10. Right, relative level of SHP mRNA was determined by qPCR. *, p < 0.05. B, analysis of SHP mRNA expression in a microarray data set GSE48452. The number of specimens in each group was as follows: normal (n = 14), steatosis (n = 14), and NASH (n = 18). Data are represented as mean ± S.D. *, p < 0.05. C, left, Western blot analysis of SHP in human livers. Right, band intensities were calculated using ImageJ software. The relative expression of SHP was normalized to the expression of the loading control, β-actin. Data are presented as -fold change relative to that of the control. NS, no significance. *, p < 0.05.
Developing a mouse model of NAFL progression to NASH
To more precisely examine SHP expression during the development of NAFLD, we developed a mouse model that carries the disease progression from NAFL to NASH with obesity and insulin resistance, the two common features of NAFLD in humans. Diet enriched in high fat and fructose has been implicated in the development of obesity and NASH in humans (18, 19). Recently, a diet enriched in high fat, cholesterol, and fructose (research diet D09100301: 40 kcal% fat, 2% cholesterol, 20 kcal% fructose; hereafter referred to as HFCF diet) was utilized to induce mouse NASH (20, 21). In this diet, excess fat alone contributes to the development of mild steatosis, whereas the addition of elevated fructose and cholesterol levels increases hepatic oxidative stress; combined, these dietary components predispose animals to necroinflammation and fibrogenesis (22). We fed 2-month-old C57Bl/6J male mice with either a chow or HFCF diet for 1 and 5 months. We chose to study male mice based on our previous observation that male but not female mice developed NASH after 5 months of HFCF diet,4 which also has been reported by another group (23). The observation that males are more susceptible to NASH is supported by human epidemiology studies showing that NAFLD cases more commonly arise and frequently progress in males, as females possess a resistance to NAFLD attributed to higher levels of estrogen (24).
Mice on the HFCF diet developed rapid weight gain and obesity compared with chow-fed controls (Fig. 2A). Although 1 month of HFCF feeding did not significantly change the liver weight compared with controls, the liver weight was significantly increased in mice on the HFCF diet for 5 months (Fig. 2B). This was accompanied by an increase in the liver to body weight ratio (Fig. 2B). Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST), two markers of liver injury, both increased after mice were fed the HFCF diet for 5 months (Fig. 2C). Additionally, 5 months of HFCF feeding led to a significant increase in fasting glucose levels and total cholesterol levels (Fig. 2D). Meanwhile, mice fed for 1 month on the HFCF diet developed hypertriglyceridemia, but the levels of serum triglycerides (TG) at the 5-month time point declined to levels that were similar to those noted in chow-fed controls (Fig. 2D). Moreover, the mice displayed glucose intolerance after 5 months on the HFCF diet (Fig. 2E). Collectively, these results indicated that mice fed a HFCF diet developed liver injury with obesity, dyslipidemia, and hyperglycemia with impaired glucose tolerance.
Figure 2.

A mouse model carries NAFL progression to NASH. Two-month-old C57BL/6J male mice were fed a chow or HFCF diet for 1 and 5 months. n = 5/group. *, p < 0.05 HFCF versus chow-fed. A, body weight change over time. B, liver weight and liver to body weight ratio. C, serum levels of ALT and AST. D, serum levels of glucose, cholesterol, and triglycerides. E, glucose tolerance test. F, representative images of liver sections stained with H&E, oil red O, TUNEL, F4/80, or Picrosirius Red. Original magnification, ×40. G, histology scores of steatosis, inflammation, cell death, and fibrosis. Data are presented as mean ± S.D. *, p < 0.05 versus respective controls.
We next examined the extent of steatosis, cell death, inflammation, and fibrosis in the livers of mice fed chow or HFCF diets. Liver sections stained with hematoxylin and eosin (H&E) and oil red O revealed liver steatosis in mice fed a HFCF diet for 1 and 5 months, which was not observed in chow-fed controls (Fig. 2F). However, the cell death detected by TUNEL staining was only observed in the livers of mice fed a HFCF diet for 5 months (Fig. 2F). Consistently, immunohistochemistry staining with the macrophage-specific antibody F4/80 showed a dramatic increase in macrophage infiltration in the livers of mice on the HFCF diet for 5 months (Fig. 2F). Moreover, liver sections stained with Picosirius red detected an apparent collagen deposition in 5-month HFCF diet-fed mice, indicative of fibrosis development (Fig. 2F). The summary in Fig. 2G shows that the grades of steatosis, inflammation, necrosis, and fibrosis became apparent in the livers of mice fed the HFCF diet for 5 months.
At the mRNA levels, peroxisome proliferator-activated receptor γ (Pparγ), a master control of lipid synthesis, sustained an increase in the livers of mice fed a HFCF diet (Fig. 3A). However, the expression of both apolipoprotein B (ApoB) and microsomal triglyceride transfer protein (Mttp), two genes involved in the very-low-density lipoprotein (VLDL) synthesis that carries TG in the plasma, was decreased in mice fed the HFCF diet for 5 months (Fig. 3A), consistent with the changes in serum TG levels shown in Fig. 2D. Tumor necrosis factor α (Tnfα) and chemokine Ccl2, two important inflammatory mediators contributing to the inflammatory cell infiltration in the liver, were both significantly increased over time in mice on the HFCF diet (Fig. 3A). In addition, a progressive increase in the expression of macrophage M1 marker nitric-oxide synthase 2 (Nos2) and a decrease in the expression of M2 markers arginase-1 (Agr1) and CD163 were observed in the livers of mice on the HFCF diet for 5 months (Fig. 3A). Similarly, the liver expression of collagen 1α1 (Col1A1) increased in 5-month HFCF diet-fed animals (Fig. 3A), supporting the increase in collagen deposition shown in Fig. 2F. Additionally, the serum levels of CCL2 were markedly elevated in the 5-month HFCF diet-fed mice (Fig. 3B). Overall, these results indicate the progression of NAFL to NASH in mice on the HFCF diet for 1 and 5 months as evidenced by increases in hepatic cell death, macrophage infiltration, and liver fibrosis.
Figure 3.

SHP level is decreased in the livers of mice NASH. A, qPCR analysis of relative mRNA levels of genes related to lipid metabolism, inflammation, and fibrosis in the livers of mice fed chow or HFCF for 1 and 5 moths. n = 5 mice/group. *, p < 0.05 HFCF versus chow-fed. B, serum level of CCL2 was measured by ELISA. n = 5 mice/group. *, p < 0.05 HFCF versus chow-fed. C, qPCR analysis of Shp mRNA level in the livers of mice fed chow or HFCF. n = 5 mice/group. *, p < 0.05 HFCF versus chow-fed. D, left, Western blot analysis of SHP protein in the livers of mice fed chow or HFCF for 5 months. SHP H-160 is a rabbit polyclonal antibody and SHP H-5 is a mouse mAb. Both antibodies recognize the epitope corresponding to amino acid 1–160 mapping at the N terminus of SHP protein. Right, band intensities were calculated using ImageJ software. The level of SHP was normalized to the expression of loading control β-actin, and -fold changes relative to that of the controls are plotted. n = 5 mice/group. *, p < 0.05 HFCF versus chow-fed. E, representative images of liver sections stained with H&E in mice fed chow or MCD diet for 1 month. Original magnification, ×40. F, qPCR analysis of gene expression in the livers of mice fed chow or MCD diet for 1 month. n = 5 mice/group. Data are presented as mean ± S.D. *, p < 0.05 versus respective controls.
Decrease of SHP in diet-induced mouse NASH
We next examined SHP expression in the liver of the HFCF-dietary mouse model. As shown in Fig. 3C, 1 month of HFCF feeding did not change liver Shp mRNA levels compared with those of the chow-fed controls. However, a significant decrease in Shp mRNA was observed after mice were on the HFCF diet for 5 months. SHP protein is a rapidly degraded protein with a very short half-life (25). We employed two anti-SHP antibodies in Western blotting to determine SHP protein levels in the liver. SHP (H-160) is a rabbit polyclonal antibody, whereas SHP (H-5) is a mouse mAb. Both antibodies recognized the epitope corresponding to amino acids 1–160 mapping at the N terminus of SHP protein. As shown in Fig. 3D, both antibodies detected a significant decrease in SHP protein expression in the livers of mice fed a HFCF diet for 5 months.
A methionine/choline–deficient (MCD) diet induces NASH-like liver pathology including liver steatosis, inflammation, and fibrosis, despite weight loss and insulin sensitivity (26). Next, we explored SHP expression in the livers of mice fed an MCD diet. As shown in Fig. 3E, the liver morphology indicated the development of liver steatosis, inflammation, and fibrosis in mice fed an MCD diet for 1 month. Consistently, the expression of genes involved in liver inflammation and fibrosis such as F4/80, Tnfα, Ccl2, IL-1β, and Col1A1 was increased in mice fed an MCD diet (Fig. 3F). Importantly, we observed a significant decrease in Shp mRNA level in the livers of mice fed an MCD diet compared with chow-fed controls (Fig. 3F). Collectively, our results indicate that the expression of Shp is decreased dramatically in the liver of mouse NASH. Moreover, our HFCF dietary mouse studies provide convincing evidence that SHP is suppressed during NAFL progression to NASH.
c-Jun N-terminal kinase (JNK) is activated during NAFLD progression and inhibits SHP expression in hepatocytes
Because SHP is highly expressed in hepatocytes (27), we speculated that SHP suppression during NAFL transition to NASH results mainly from the decrease of SHP in hepatocytes. To begin to test our hypothesis, we isolated hepatocytes, hepatic stellate cells (HSC), and resident macrophage cells (KC) from mouse liver and compared Shp expression among these cells. Cell purification was confirmed by the detection of various cell-specific markers by quantitative PCR (qPCR), including hepatocyte marker albumin (Alb), quiescent HSC marker Hh-interacting protein (Hhip), and KC marker F4/80. As expected, Shp mRNA is abundantly expressed in hepatocytes and low is in KCs and HSCs (Fig. 4A).
Figure 4.

Activation of JNK induces c-Jun targeting of Shp promoter leading to Shp suppression. A, qPCR analysis of gene expression in primary hepatocytes (Hepa), HSC, and resident macrophage KC isolated from mouse liver. Data are represented as mean ± S.D. *, p < 0.05. B, qPCR analysis of Shp mRNA expression in mouse hepatocytes. Hepatocytes were incubated with 0.5 mm PA or 100 ng/ml LPS for 6 h in the presence or absence of various inhibitors such as JNK inhibitor SP600125 (50 μm), NF-κB inhibitor BAY 11-7082 (5 μm), and PI3K inhibitor LY294002 (50 μm). The relative expression of Shp is normalized to the expression of internal control HPRT1. The -fold changes relative to that of the controls are plotted and presented as mean ± S.D. *, p < 0.05. C, Western blot analysis in the livers of mice fed chow or HFCF for 1 and 5 months. D, diagram shows the location of the TRE (core sequence TGAGTCA) site on the Shp promoter/reporter (Shp-Luc) and Shp-Luc mutant with a mutated TRE site. Three LRH1 binding sites are close to the TRE site. E, left, AML12 cells were transfected with Shp-Luc or its mutant with or without various expression plasmids. Luciferase activities were determined at 24 h post-transfection. Right, AML12 cells were transfected with Shp-Luc or its mutant for 24 h followed by incubation with SP600125 (50 μm) for 6 h. Data are displayed as the ratio of firefly luminescence divided by Renilla luminescence and represented as mean ± S.D. for triplicate experiments/group. *, p < 0.05. F, ChIP assay to determine the enrichment of c-Jun to Shp promoter. Left, AML12 cells overexpressed with c-Jun were harvested at 24 h post-transfection. pcDNA served as a transfection control. Right, AML12 cells were incubated with BSA control or 0.5 mm PA with or without JNK inhibitor SP600125 (50 μm) for 6 h. The cross-linked chromatin was immunoprecipitated by an antibody against c-Jun. The enriched DNA was amplified by qPCR and normalized to the input. -Fold changes relative to that of the controls are plotted and represented as mean ± S.D. *, p < 0.05. G, the enrichment of LRH1 to Shp promoter was revealed by ChIP assay. Left, AML12 cells overexpressed with c-Jun were harvested at 24 h post-transfection. Right, AML12 cells were incubated with 0.5 mm PA or BSA control for 6 h. The cross-linked chromatin was immunoprecipitated by an antibody against LRH1. The enriched DNA was amplified by qPCR and normalized to the input. The -fold changes relative to that of the controls are plotted and represented as mean ± S.D. *, p < 0.05.
We next sought to investigate the potential mechanisms of SHP suppression in NASH. Lipotoxicity, the major mechanism underlying hepatocyte dysfunction in NAFLD, occurs in the setting of excessive free fatty acid traffic in hepatocytes, especially saturated fatty acids (28). Palmitic acid (PA) is one of the most abundant of the saturated fatty acids presented in diets and in serum (29). PA binds to TLR4 leading to JNK activation (30, 31). We next employed a primary mouse hepatocyte culture as an in vitro model and explored whether PA or TLR4 ligand lipopolysaccharide (LPS) could alter SHP expression. Treatment with PA (0.5 mm) or LPS (100 ng/ml) for 6 h significantly decreased Shp mRNA expression (Fig. 4B). To further investigate the potential mechanisms of SHP suppression by PA and LPS, we superimposed various signaling pathway inhibitors, including JNK inhibitor SP600125 (50 μm), NF-κB inhibitor BAY 11-7082 (5 μm), and phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002 (50 μm) on a PA or LPS treatment regimen. Interestingly, co-treatment with the JNK inhibitor completely obviated the decrease of Shp mRNA by PA or LPS (Fig. 4B). Thus, our results indicate that JNK activation mediates the suppression of SHP by PA and LPS in hepatocytes. Moreover, JNK activation was observed in the livers of mice fed a HFCF diet for 5 months as evidenced by the induction of phosphorylated JNK (Fig. 4C, p-JNK, activated form of JNK). Importantly, the activation of JNK correlated positively with SHP suppression in the livers of 5-month HFCF diet-fed mice (Fig. 3D). Collectively, our in vitro and in vivo results indicate, for the first time, that JNK activation suppresses Shp expression in NASH.
c-Jun is activated by JNK and targets Shp promoter for Shp suppression
JNK was originally identified because of its capability of specifically phosphorylating c-Jun on its N-terminal transactivation domain at two serine residues, Ser-63 and Ser-73 (32). In our study, HFCF feeding for 5 months led to JNK activation, which was accompanied by c-Jun phosphorylation at Ser-73 (Fig. 4C), potentially connecting JNK activation and c-Jun phosphorylation to Shp suppression during NAFLD progression. The SHP gene contains a consensus 12-O-tetradecanoylphorbol-13-acetate response element (TRE; core sequence TGAGTCA) located on its promoter region (−295 to −289 on human SHP gene and −333 to −326 on mouse Shp gene), which is predicted to be a c-Jun response element (Fig. 4D). We cloned the mouse Shp proximal promoter (2 kb) into a luciferase reporter (Shp-Luc) and examined the effect of c-Jun on Shp promoter activity in the mouse hepatocyte AML12 cell line. Nuclear receptor liver receptor homolog-1 (LRH1) binds to the Shp promoter and induces Shp-Luc activity (33); thus, LRH1 was included as a positive activator. Overexpressing c-Jun decreased the basal activity as well as the induction of Shp-Luc by LRH1, which was completely blocked in the reporter construct containing a mutated TRE site (Shp-Luc Mut) (Fig. 4E), suggesting that c-Jun inhibits Shp promoter activity through the TRE site. Moreover, blocking JNK activation by a specific inhibitor, SP600125, increased Shp-Luc activity, which was completely disrupted in the Shp-Luc Mut (Fig. 4E), indicating for the first time that the suppression of Shp by JNK is facilitated primarily by c-Jun.
A chromatin immunoprecipitation (ChIP) assay was performed in AML12 cells overexpressed with c-Jun, confirming the recruitment of c-Jun to the Shp promoter (Fig. 4F). Additionally, PA (0.5 mm) treatment in AML12 cells dramatically stimulated the recruitment of endogenous c-Jun to the Shp promoter, which was completely abrogated by the co-treatment of a specific JNK inhibitor, SP600125 (Fig. 4F). The mouse Shp proximal promoter contains three LRH1 binding sites (Fig. 4D, GAGACCTTGG at −383 to −374, TCAAGGTTG at −116 to −108, and TCAAGGATA at −83 to −75), which are close to the TRE site. To understand how c-Jun inhibits LRH1-induced Shp-Luc activity, we performed a ChIP assay that examined whether c-Jun would interfere with the recruitment of LRH1 to Shp promoter. As shown in Fig. 4G, overexpressing c-Jun in AML12 cells dramatically decreased the recruitment of LRH1 to all three LRH1 sites on the Shp promoter. Similarly, PA treatment in AML12 cells also significantly decreased the recruitment of LRH1 to the Shp promoter (Fig. 4G). Thus, our data suggest that the binding of c-Jun to the Shp promoter inhibits the recruitment of LRH1 to the Shp promoter. Taken together, these results indicate that JNK activation induces the binding of c-Jun to the Shp promoter, leading to suppression of Shp transcription.
Increasing hepatocyte SHP levels in steatotic livers does not alter liver steatosis
We next asked whether treating mice with SHP overexpression in hepatocytes could prevent the progression of NAFL to NASH. For this purpose, we used adeno-associated virus type 8 (AAV8) in which FLAG-tagged SHP was driven by a thyroxine-binding globulin (Tbg) promoter (AAV8-Tbg-FlagSHP). Use of the Tbg promoter ensures that AAV8 specifically targets hepatocytes (34). C57Bl/6J mice were placed on the HFCF diet for 1 month to induce liver steatosis followed by tail vein administration of AAV8-Tbg-FlagSHP or control vector AAV8-Tbg-GFP. The mice remained on the HFCF diet for an additional 3 months (Fig. 5A). The overexpression of SHP in hepatocytes was confirmed by qPCR (Fig. 5A) and immunohistochemistry staining of FLAG-tagged SHP protein (Fig. 5E). The body weight, liver weight, liver to body weight ratio, and serum levels of fasting cholesterol and TG were similar in SHP and GFP mice (Fig. 5, B and C). However, serum ALT and AST, two markers of liver injury, were significantly decreased in SHP-overexpressing mice compared with GFP control mice (Fig. 5C). To investigate whether over-expression of SHP could affect glucose metabolism, we performed a glucose tolerance test. The results showed that SHP-overexpressing mice displayed no differences in glucose tolerance when compared with GFP control mice (Fig. 5D). Further quantification of areas under the curve of the glucose tolerance test (GTT) showed no statistical differences in GFP control and SHP-overexpressing groups (Fig. 5D). We next examined liver histology. Liver steatosis and inflammation were apparent in the livers of GFP mice fed a HFCF diet (Fig. 5E). A similar level of lipid accumulation was observed in the livers of SHP-overexpressing mice on the HFCF diet compared with GFP controls; however, there was less inflammation in SHP-overexpressing mice (Fig. 5E). Consistently, liver TG and cholesterol levels were similar in GFP and SHP mice fed the HFCF diet (Fig. 5F). Taken together, our results suggest that overexpressing SHP in steatotic livers improves liver injury without altering liver steatosis and glucose tolerance.
Figure 5.

Increasing hepatocyte SHP levels in steatotic liver does not change liver steatosis. Two-month-old male C57BL/6J mice were fed a HFCF diet for 1 month to develop liver steatosis followed by tail vein administration of AAV8-Tbg-FlagSHP or control vector AAV8-Tbg-GFP. The mice remained on the HFCF diet for an additional 3 months. A, left, schematic diagram showing experimental design. Right, qPCR analysis of Shp mRNA levels in the liver. n = 5 mice/group. *, p < 0.05 SHP versus GFP. B, left, body weight change over time; middle, liver weight; right, liver weight to body weight ratio. n = 5 mice/group. C, serum levels of cholesterol, triglycerides, ALT, and AST. n = 5 mice/group. *, p < 0.05 SHP versus GFP. D, left, GTT. Right, the area under the curve (AUC) of GTT was calculated. n = 5 mice/group. E, representative images of liver sections stained with H&E, immunohistochemistry staining (IHC) of FLAG-SHP, and oil red O. Original magnification, ×40. n = 5 mice/group. F, liver triglycerides and cholesterol content. n = 5 mice/group.
Hepatocyte SHP overexpression prevents NAFL progression to NASH by attenuating liver inflammation and fibrosis
Liver inflammation and fibrosis exacerbate NASH progression (35). Next, we evaluated the extent of liver inflammation and fibrosis in GFP and SHP-overexpressing mice. The results showed that increasing the hepatocyte SHP levels dramatically attenuated liver inflammation and fibrosis in mice fed a HFCF diet, as evidenced by the marked reduction in the staining of F4/80-positive KCs and collagen deposition in the livers of SHP-overexpressing mice (Fig. 6A). Consistently, the liver hydroxyproline level was reduced in SHP mice (Fig. 6B). Overexpressing SHP in HFCF diet-fed mice led to a robust reduction in liver expression of genes related to inflammation, such as IL-6, Tnfα, and Ccl2 (Fig. 6C). Meanwhile, overexpressing SHP altered macrophage polarization in the liver with a decrease in the expression of the proinflammatory M1 marker Nos2 and increases in the expression of anti-inflammatory M2 markers such as Arg1 and CD163 (Fig. 6C). Furthermore, overexpressing SHP significantly decreased the expression of genes related to fibrosis, including Tgfβ1, Ctgf, Col1A1, and Col1A2 (Fig. 6C), without altering the mRNAs related to lipid metabolism such as fatty acid biosynthesis (Pparγ, Srebp-1c, Me1, ACC1, and Acly), fatty acid oxidation (Cpt1a and ACC2), VLDL secretion (Mttp and ApoB), and cholesterol metabolism (Srebp2, Abca1, and Abcg5) (Fig. 6C). Thus, hepatic SHP overexpression improved the key parameters of NASH progression that are related to liver inflammation and fibrosis without affecting liver steatosis.
Figure 6.

Hepatocyte SHP overexpression attenuates liver inflammation and fibrosis. Two-month-old male C57BL/6J mice were fed a HFCF diet for 1 month to develop liver steatosis followed by tail vein administration of AAV8-Tbg-FlagSHP or control vector AAV8-Tbg-GFP. Mice were continued on the HFCF diet for an additional 3 months. A, representative images of liver sections stained with F4/80 and Picrosirius red. Original magnification, ×40. n = 5 mice/group. B, liver collagen content was determined by hydroxyproline assay. n = 5 mice/group. Data are represented as mean ± S.D.; *, p < 0.05 SHP versus GFP. C, relative mRNA levels of genes related to inflammation, fibrosis, and lipid metabolism in the liver were determined by qPCR. Data are represented as mean ± S.D. for 5 mice/group; *, p < 0.05 SHP versus GFP. D, left, Western blot analysis of cytosolic and nuclear proteins (Pt) in the liver. Middle and right, band intensities were calculated using ImageJ software, and the intensities relative to that of the control were plotted. Data are represented as mean ± S.D.; *, p < 0.05 SHP versus GFP.
We next sought to explore potential mechanisms by which hepatocyte SHP inhibits liver inflammation. NF-κB signaling controls inflammation (36), and growing evidence indicates that NF-κB p65 activation contributes to the pathogenesis of NASH (37). SHP has been shown to inhibit TLR4-triggered activation of NF-κB p65 signaling in monocytes (10). We therefore hypothesized that increasing SHP in hepatocytes may lead to a repression of NF-κB p65 signaling and inhibit subsequent liver inflammation. As expected, overexpressing SHP in hepatocytes dramatically decreased HFCF diet feeding-induced nuclear translocation of total p65 and phospho-p65 (Ser-536) (Fig. 6D), an active form of p65, supporting the overall decrease in liver inflammation in SHP-overexpressing mice compared with GFP controls.
In vitro deletion of Shp increases CCL2 production leading to macrophage proinflammatory M1 polarization
To understand how hepatocyte SHP regulates liver inflammation, we isolated hepatocytes from Shpfl/fl mice and deleted Shp using adenovirus expressing Cre recombinase (Ad-Cre). We sought to assess the effect of hepatocyte conditioned medium (CM) on macrophage M1or M2 polarization and migration (Fig. 7A). We chose Ad-Cre for in vitro Shpfl/fl hepatocyte infection based on our experience that Ad-Cre is more potent than AAV8-Cre in knocking down Shp in vitro. The knockdown of Shp mRNA in hepatocytes was confirmed by real-time PCR (Fig. 7B). Interestingly, the loss of SHP in hepatocytes dramatically induced p65 nuclear translocation (Fig. 7B), suggesting an activation of p65 in Shp-deficient hepatocytes. A significant increase in CCL2 was subsequently detected in Shp-deficient hepatocytes and in its CM (Fig. 7C), indicating that loss of Shp in hepatocytes induces the secretion of CCL2. Next, we treated RAW 264.7 cells, a mouse macrophage cell line, with CM for 6 h and assessed the polarization of macrophages. The CM from Shp-deficient hepatocytes increased the expression of M1 markers such as Nos2, IL1, IL6, and Tnfα and repressed the expression of the M2 marker Arg1 in RAW 264.7 cells (Fig. 7D), suggesting that Shp-deficient CM stimulated macrophage proinflammatory M1 polarization. In addition, CM from Shp-deficient hepatocytes stimulated a significant increase in migration of RAW 264.7 cell (Fig. 7E), which was mimicked by exposure to control CM supplemented with 40 ng/ml recombinant mouse CCL2 protein or inhibited by blocking of CCL2 action by a CCL2 antibody (500 ng/ml) (Fig. 7E). Thus, our findings suggest a novel mechanism in which the loss of Shp in hepatocytes stimulates macrophage proinflammatory M1 polarization mediated through CCL2 secretion from hepatocytes.
Figure 7.

Deletion of Shp in hepatocytes increases CCL2 production leading to macrophage proinflammatory polarization. A, schematic diagram shows experimental design. Primary hepatocytes from Shpflox/flox mice were infected with adenovirus expressing Cre recombinase (Ad-Cre) or vector control (Ad-Null). CM was collected at 24 h post-adenoviral vector infection and used for RAW cell treatment. B, left, relative Shp mRNA levels were determined by qPCR. Data are represented as mean ± S.D. for triplicate experiments/group; *, p < 0.05. Right, representative images of immunofluorescence (IF) staining of p65 in hepatocytes. C, left, qPCR analysis of relative Ccl2 mRNA levels in hepatocytes. Right, CCL2 protein level in CM measured by ELISA. Data are represented as mean ± S.D. for experiments/group; *p < 0.05. D, RAW cells were incubated with CM from hepatocyte culture for 6 h, and the relative expression of genes involved in inflammation was determined by qPCR. Data are represented as mean ± S.D. for triplicate experiments/group; *, p < 0.05. E, left, representative images of RAW cell migration. RAW cells were incubated with CM in the presence or absence of recombinant mouse CCL2 (40 ng/ml) or anti-mouse CCL2 antibody (500 ng/ml). Cell migration was assessed after incubation for 12 and 24 h, respectively. Right, quantitation of cell migration was determined by measuring the pixel density of crystal violet–stained cells using ImageJ software. Data are represented as mean ± S.D. for five fields/sample. *, p < 0.05. F, left, diagram shows the location of two IκB sites on the mouse Ccl2 promoter reporter (Ccl2-Luc). Right, AML12 cells were transfected with Ccl2-Luc with various expression plasmids. Luciferase activities were determined at 24 h post-plasmid transfection. Data are calculated as the ratio of firefly luminescence divided by Renilla luminescence and presented as mean ± S.D. for triplicate experiments/group. *, p < 0.05. G, AML12 cells were overexpressed with FLAG-SHP or HA-p65 and harvested at 24 h post-plasmid transfection. Immunoprecipitation (IP) followed by Western blotting (WB) was employed to detect protein-protein interactions between FLAG-SHP and HA-p65.
SHP inhibits Ccl2 transcription in hepatocytes
To understand how Shp represses Ccl2 expression, we analyzed the proximal promoter of mouse Ccl2 and identified two IκB sites (Fig. 7F). We cloned a 3-kb mouse Ccl2 proximal promoter fragment into a luciferase reporter and examined the effect of SHP on Ccl2 promoter activity in AML12 cells. Overexpression of p65 increased Ccl2 promoter activity, which was inhibited by co-expression of SHP (Fig. 7F). EIA-like inhibitor of differentiation 1 (EID1) is a common co-repressor that interacts with SHP (38). Co-expression of SHP and EID1 synergistically diminished p65-induced activation of the Ccl2 promoter (Fig. 7F). Additionally, we overexpressed FLAG-SHP and HA-p65 in AML12 cells, and the immunoprecipitation results showed the protein-protein interactions between SHP and p65 (Fig. 7G). Taken together, the results suggest that SHP interacts with p65 and inhibits p65-induced Ccl2 transcription.
Discussion
The pathogenesis of NASH is not yet entirely understood, and the mechanisms leading to NASH appear multifactorial. Recent retrospective studies using liver biopsies from patients with NAFL or NASH highlight the possibility that rather than being distinct entities, NAFL and NASH represent different stages in the progression of NAFLD (39–41). However, the mechanisms underlying the disease progression from NAFL to NASH remain poorly understood. No pharmacological treatment has been proven effective for NASH. Thus, a better understanding of what drives the transition of NAFL to NASH to develop effective prophylactic and therapeutic strategies is an urgent medical need. Here we have identified a novel regulatory network in hepatocytes consisting of JNK/SHP/NF-κB/CCL2, which regulates macrophage recruitment and initiation of inflammation for NAFLD progression (Fig. 8).
Figure 8.
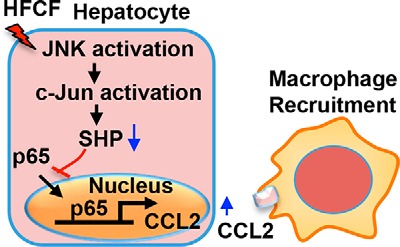
Hepatic cascade JNK/SHP/NF-κB/CCL2 regulates liver inflammation in NAFLD. Schematic diagram illustrates a novel regulatory network in hepatocytes, consisting of JNK/SHP/NF-κB/CCL2, that regulates macrophage recruitment and inflammation initiation critical for NAFLD progression.
An important finding of our study is the demonstration that SHP is markedly decreased during the progression of NAFL to NASH, which was observed in the livers of patients with NAFLD and in diet-induced mouse NAFLD. The study of SHP expression in NAFLD has been inconsistently reported in literature. An earlier study showed that SHP protein levels are reduced in NASH, whereas mRNA levels are not affected (42). Another study demonstrates that SHP mRNA is similarly reduced in obese patients with NAFL or NASH compared with the lean NAFLD (43). These discrepancies may be because SHP protein is not stable and degrades rapidly (25); thus, protein expression and mRNA levels may not change in parallel. Additionally, not all commercial SHP antibodies are good for detecting SHP protein, which may explain the inconsistent results reported. In a recent study, Benet et al. (44) compared SHP expression levels in different mouse models of NAFLD, demonstrating that SHP is repressed in advanced NAFLD livers (NASH with fibrosis) such as tetracycline-treated rat livers, MCD diet livers, and glycine N-methyltransferase–deficient (Gnmt−/−) mice livers but is not suppressed in the steatotic livers of methionine adenosyltransferase 1A–deficient (Mat1a−/−) mice. Similarly, our study showed that SHP is not suppressed in NAFL but is suppressed in NASH, suggesting that a gradual decrease in SHP occurs during the disease progression.
Our findings also indicate that the activation of particular signals during NAFLD progression is required for SHP suppression. Several signaling pathways have been implicated in the regulation of SHP. For instance, the mitogen-activated protein kinase kinase 1/2 (MAPKK 1/2) pathway activates SHP expression, whereas PI3K pathway suppresses it (44). In the present study, we uncovered a new pathway governed by JNK activation of c-Jun that binds to the Shp promoter and suppresses Shp transcription in NASH. This is supported by several pieces of evidence: 1) in vitro hepatocyte culture with PA or LPS suppresses SHP expression mediated by JNK activation; 2) JNK activation promotes c-Jun binding to the Shp promoter thereby suppressing Shp transcription; and 3) a positive correlation between JNK activation and SHP suppression is observed during NAFL transition to NASH. Consistent with our data is the observation that sustained JNK activation by saturated fatty acids plays a key role in lipotoxicity and the pathogenesis of NASH (31). Further investigation is warranted to explore whether the inhibition of JNK could rescue SHP suppression and ameliorate the disease progression of NAFLD.
In our HFCF diet-induced NAFLD model, overexpressing SHP in steatotic liver did not affect liver lipid content. This is a somewhat unexpected finding, because SHP is known to regulate hepatic lipid metabolism (8). An earlier study showed that SHP is essential for bile acid–mediated suppression of hepatic lipogenesis, as it inhibits the expression of Srebp-1c, a key lipogenic activator (45). Consistently, acute liver-specific SHP overexpression in C57BL/6J mice via adenovirus-mediated gene delivery decreases hepatic TG levels (46). These studies suggest that SHP plays a role in alleviating lipid accumulation in the liver. In contrast, increasing hepatic lipid accumulation by SHP has been reported in transgenic mice with chronically elevated hepatic SHP (47). In addition, studies show that whole-body Shp-deficient (Shp-KO) mice display resistance to high-fat diet–induced fatty liver (48–50). Thus, the role of SHP in NAFLD is very controversial. These discrepancies may be because of the acute versus chronic manipulation of SHP levels in mice or the effects of nonhepatic tissues on the overall energy balance and metabolic outcomes. It is also crucial to note that hepatic SHP is a critical suppressor of bile acid synthesis through the direct regulation of Cyp7a1 (51, 52), a critical rate-limiting enzyme in bile acid synthesis (53). Accumulated evidence highlights that changes in bile acid synthesis and composition can alter gut microbiota, which may affect liver physiology and be involved in NAFLD development (54). Thus, it is possible that SHP deletion or SHP overexpression may affect bile acid synthesis, composition, and gut microbiota differently, which may result in distinct roles for SHP in NAFLD.
Here, we have shown that SHP was decreased during NAFL transition to NASH (Figs. 1 and 3), indicating that SHP may play an important role during NAFLD progression. To test our hypothesis, we employed a mouse model of NAFLD that carries the progression of NAFL to NASH and overexpressed SHP specifically in hepatocytes. Increasing the hepatocyte SHP levels in steatotic liver did not change the hepatic lipid content but dramatically attenuated liver inflammation and fibrosis, two critical events responsible for NAFLD progression, indicating that SHP plays a protective role during NAFLD progression. Because these mice had been on a HFCF diet for 1 month and developed liver steatosis at the time we overexpressed SHP in hepatocytes, the unchanged hepatic lipid content may be because manipulating the SHP levels in the steatotic livers did not change the expression of genes involved in fatty acid biosynthesis, oxidation, and TG secretion. Combined, these studies indicate that SHP plays distinct roles in different stages of NAFLD, and the sustained decrease in SHP expression could worsen the disease progression. Supporting this summary is an earlier study showing that the prevention of liver fibrosis in NASH by AMP-activated protein kinase (AMPK) is SHP-dependent, as the protective effect of AMPK is absent in Shp−/− mice (55).
Another important finding from our study is that the expression of hepatic SHP attenuates chemokine CCL2 production. Overnutrient-induced aberrant synthesis and accumulation of proinflammatory mediators within the liver are major culprits in the pathogenesis of NASH (35). More specifically, an increased level of CCL2 is associated with a high risk of NASH in animal models and humans (15). Here, we have identified that the expression of SHP in hepatocytes is essential to suppress overnutrition-induced CCL2 production. More importantly, our study demonstrates that the cross-talk between p65 and SHP in hepatocytes is one of the key events in regulating CCL2 production. The identification of this regulatory axis is important, as it will shed light on our understanding of farnesoid X–activated receptor (FXR; nuclear receptor subfamily 1, group H, member 4), a very well-known up-stream regulator of SHP (56–60), in the alleviation of hepatic inflammation. FXR has been recognized as an interesting target for NAFLD treatment, and the FXR agonist obeticholic acid has been used in NAFLD treatment (61). However, the mechanism underlying FXR-mediated reduction of hepatic inflammation remains undetermined. Therefore, future research is warranted to determine whether the mechanism governed by SHP/p65/CCL2 could contribute to FXR-mediated alleviation of hepatic inflammation in NAFLD.
In summary, we have provided compelling evidence that a novel regulatory network in hepatocytes consisting of JNK/SHP/NF-κB/CCL2, is critical to the regulation of macrophage recruitment and initiation of inflammation for NAFLD progression. Additionally, we have demonstrated that overexpression of SHP in steatotic liver prevents NAFL progression to NASH, suggesting that specifically targeting a component of this network is beneficial for NASH prevention and treatment.
Experimental procedures
Cell lines, chemicals, plasmids, adenoviruses, and antibodies
Mouse macrophage RAW 264.7 cells (ATCC TIB-71) were maintained in Dulbecco's modified Eagle's medium with 100 units of penicillin G–streptomycin sulfate/ml and 10% heat-inactivated fetal bovine serum (FBS). AML12 (ATCC CRL-2254), a cell line established from hepatocytes from a mouse transgenic for human TGFα, was maintained in Dulbecco's modified Eagle's medium containing 10% FBS supplemented with 0.005 mg/ml insulin, 0.005 mg/ml transferrin, 5 ng/ml selenium, and 40 ng/ml dexamethasone. Reagents including palmitic acid (P5585), lipopolysaccharide (L2654), JNK inhibitor SP600125 (S5567), NF-κB inhibitor BAY 11-7082 (B5556), PI3K inhibitor LY294002 (catalog no. 440202), and d-glucose anhydrous (catalog no. 346351) were purchased from Sigma. Luciferase reporters containing a 2-kb mouse Shp proximal promoter fragment (Shp-Luc) and a 3-kb mouse Ccl2 proximal promoter fragment (Ccl2-Luc) were engineered in our laboratory and confirmed by sequencing. Expression plasmids for c-Jun, FLAG-LRH1, FLAG-SHP, HA-p65, and EID1 and adenoviruses for GFP and SHP were described previously (62). The following antibodies were used for Western blotting, immunofluorescent staining, and immunoprecipitation: SHP (Santa Cruz Biotechnology, sc-30169 and sc-271511), β-actin (Sigma, A-1978), phospho-JNK (Thr-183/Tyr-185) (Cell Signaling Technology, 4668), JNK (Cell Signaling Technology, 9252), phospho-c-Jun (Ser-73) (Cell Signaling Technology, 3270), FLAG-HRP (Sigma, A8592), α-tubulin (Sigma, T6074), histone H3 (Cell Signaling Technology, 14269), NF-κB p65 (Cell Signaling Technology, 8242), phospho-p65 (Ser-536) (Cell Signaling Technology, 4764), LRH1 (R&D Systems, PP-H2325-00). Recombinant mouse CCL2 protein (479-JE-010), and anti-mouse CCL2 antibody (AF-479-SP) for neutralization were obtained from R&D Systems.
Human liver samples
Two sets of human liver specimens were included. The first set was obtained through the Liver Center at University of Kansas, including 12 normal, 12 NAFL, and 8 NASH specimens. The exclusion criteria were excessive alcohol use (>20 g of alcohol daily for women and >30 g for men) and chronic liver diseases, including chronic viral hepatitis, autoimmune, Wilson's disease, and drug-induced hepatitis. A NAFLD activity score (NAS) representing the sum of scores for steatosis, lobular inflammation, and ballooning (20) was applied in our study. Cases with NAS 0 were classified as normal. Cases with a steatosis score ≥ 1 without inflammation, ballooning, or fibrosis were defined as NAFL. Cases with NAS ≥ 5 were classified as NASH. The studies were approved by the University of Kansas Medical Center Institutional Review Board for Human Research Committee and abide by the Declaration of Helsinki principles. All liver samples were deidentified, and the researchers were not able to ascertain individual identities associated with the samples. The second set was obtained through GEO data set GSE48452, which contained 14 normal, 14 NAFL, and 18 NASH samples.
Animal studies
C57BL/6J mice (stock no. 000664) were obtained from The Jackson Laboratory. Shpfl/fl mice were generously provided by Drs. Johan Auwerx and Kristina Schoonjans at the Ecole Polytechnique de Lausanne and back-crossed into C57BL/6J background for 10 generations. Mice were maintained in a 12-h light/dark cycle (light on 6 a.m. to 6 p.m.), temperature-controlled (23 °C), and virus-free facility with free access to food and water. Experiments on the mice were performed on males at the age of 8–10 weeks unless stated otherwise (n = 5/group). For dietary NAFLD models, C57BL/6J mice were either placed on a MCD diet (Harlan Laboratories, TD.90262) for 1 month or on a diet enriched in high fat, cholesterol, and fructose (Research Diet, D09100301; 40 kcal% fat, 2% cholesterol, 20 kcal% fructose) for 1 or 5 months. Mice fed normal chow served as controls. In the SHP overexpression experiment, AAV8, including AAV8-Tbg-FlagSHP and AAV8-Tbg-GFP, was obtained from the University of Pennsylvania Vector Core. C57BL/6J mice were on the HFCF diet for 1 month followed by administration of either AAV8-Tbg-FlagSHP or control AAV8-Tbg-GFP at a dose of 2 × 1011 genome copies/mouse through tail vein injection. The mice remained on the HFCF diet for 3 months. Samples were collected after the mice had been fasted for 16 h. All experiments were performed in accordance with relevant guidelines and regulations approved by the Institutional Animal Care and Use Committee (ICAUC) at the University of Kansas Medical Center.
Glucose tolerance test
Mice were fasted for 16 h followed by intraperitoneal injection of glucose at 2g/kg body weight. Blood was collected by tail vein puncture. Glucose levels were determined before and at different times after glucose administration.
Serum analyses
Blood was collected from anesthetized animals via cardiac puncture and deposited into blood collection tubes (Fisher Scientific, 22-225516). Serum was isolated to measure triglycerides (Pointe Scientific, T7532), total cholesterol (Pointe Scientific, C7510), AST (Pointe Scientific, A7561), ALT (Pointe Scientific, A7526), and glucose (Pointe Scientific, G7521) according to the manufacturer's suggestions.
Liver histology
Fresh liver tissues were fixed with 10% formalin (Fisher Scientific, SF100). Paraffin sections at 4 μm were stained with H&E. The severity of steatosis, inflammation, cell death, and fibrosis were assessed and scored by a pathologist according to the NASH Clinical Research Network criteria (63, 64). Specifically, the amount of steatosis was scored as 0 (<5%), 1 (5–33%), 2 (>33–66%), and 3 (>66%). Hepatic cell death was scored as 0 (none), 1 (few cells), and 2 (many cells). Foci of lobular inflammation were scored as 0 (no foci), 1 (<2 foci), 2 (2–4 foci), and 3 (>4 foci). Fibrosis was scored as 0 (no fibrosis), 1 (perisinusoidal or periportal fibrosis), 2 (perisinusoidal and portal/periportal fibrosis), 3 (bridging fibrosis), and 4 (cirrhosis).
Oil red O staining of liver lipids
Fresh liver tissues were immediately embedded in Tissue-Tek O.C.T. compound (VWR 25608-930). To visualize lipid in the liver, frozen sections were cut at 8 μm, fixed by 10% formalin, and stained with oil red O (Sigma O0625) followed by counterstaining with hematoxylin. Images were acquired with a BX60 microscope (Olympus, Lake Success, NY).
Immunohistochemistry
For immunohistochemistry staining of F4/80, 4-μm paraffin sections were rehydrated and treated with 0.3% H2O2 in methanol for 15 min to block endogenous peroxidase activity. Antigen retrieval was acquired by incubation of slides in 20 μg/ml proteinase K solution (Fisher Scientific, PRMC5005) for 3 min at room temperature. Slides were then treated with 10% normal serum for 30 min followed by incubation with rat anti-mouse F4/80 antibody (Abd-Serotec, MCA497R) overnight at 4 °C. An ImmPRESS peroxidase polymer detection kit (Vector Laboratories, MP-7444) and ImmPACT 3,3′-diaminobenzidine peroxidase substrate (Vector Laboratories, SK-4105) were used for the final detection. Sections were then counterstained with hematoxylin, dehydrated, cleared, and mounted. Images were acquired with a BX60 microscope.
Picrosirius red staining of liver fibrosis
Paraffin sections were cut at 4 μm, rehydrated, and incubated in 0.1% Sirius red F3B (Sigma, direct red 80, 365548) containing saturated picric acid (Sigma, p6744) for 1 h. After washing three times in 0.5% glacial acetic acid, sections were briefly dehydrated, cleared, and mounted. Images were acquired with a BX60 microscope and collagen density was quantified using ImageJ software.
Detection of cell death by TUNEL staining
TUNEL staining for detection of cell death in the liver was performed using an in situ alkaline phosphatase cell death detection kit (Sigma 11684809910) according to the manufacturer's suggestions.
Hepatic TG and cholesterol content
Liver tissues (100 mg) were homogenized in 300 μl of chloroform:methanol (1:2 volume) for 2 min followed by a second homogenization for 30 s with the addition of 300 μl of chloroform. The homogenates were mixed with 100 μl of H2O and homogenized again for 30 s. The lipid layer (∼600 μl) was separated via centrifugation at 800 × g for 10 min at room temperature. The lower phase, enriched in lipid, was transferred and dried using nitrogen gas. The lipid extract was suspended in 300 μl of 5% Triton X-100 in PBS, pH 7.4. The measurement was performed using respective kits for triglycerides (Pointe Scientific, T7532) and cholesterol (Pointe Scientific, C7510). The hepatic TG or cholesterol content was defined as μg of TG or cholesterol/mg of liver tissue.
Hepatic collagen determination by hydroxyproline assay
Liver tissues (10 mg) were homogenized in 100 μl of H2O. The homogenates were mixed with 100 μl of 12 m HCl and incubated at 120 °C for 3 h for acid hydrolysis. The homogenates were then centrifuged at 10,000 × g for 10 min. Aliquots of the hydrolyzed samples (10 μl) were incubated with 100 μl of chloramine T solution (1.27% chloramine T and 10% isopropanol in acetate-citrate buffer, pH 6.0) at room temperature for 25 min followed by a second incubation with 100 μl of Ehrlich's solution (Sigma, 03891) at 60 °C for 35 min. A plate reader measured the sample absorbance at 550 nm. The hepatic collagen content was defined as μg of collagen/mg of liver tissue.
Western blotting and immunoprecipitation
Mouse liver tissues were homogenized using a PowerGen 700 homogenizer (Fisher Scientific) in lysis buffer (50 mm Tris, pH 7.5, 1% Nonidet P-40, 150 mm NaCl, 0.5% sodium deoxycholate, and 0.1% SDS) containing protease inhibitors (Fisher Scientific, protease inhibitor mixture PI78410) to obtain whole protein lysates. Nuclear and cytoplasmic protein extraction was carried out using a commercial kit (Fisher Scientific, PI78833). Protein lysates (60 μg) were resolved by SDS-PAGE and transferred to nitrocellulose membranes according to standard procedures. Membranes were blocked and incubated with primary antibodies followed by incubation with the corresponding horseradish peroxidase–conjugated secondary antibody. Antibody binding was visualized using either SuperSignal West Pico Plus chemiluminescent substrate (Fisher Scientific, PI34580) or SuperSignal West Femto chemiluminescent substrate (Fisher Scientific, PI34094). Equal loading of protein was verified by loading controls such as β-actin, α-tubulin, and histone H3. Quantitative analysis of band intensity was performed by ImageJ software, and relative expression levels were normalized to loading controls. For immunoprecipitation experiment, 800 μg of whole protein lysates from AML12 cells overexpressed with FLAG-SHP or HA-p65 were incubated with 2 μg of anti-p65 antibody, and immune complexes were captured by sheep anti-rabbit IgG M-280 Dynabeads (Fisher Scientific, 11-203-D). The immune complexes were then eluted by 2× SDS loading buffer. The pulldown of p65 and FLAG-SHP were detected by Western blotting. A TrueBlot® anti-rabbit IgG HRP (Rockland, RL18-8816-33) was used as a secondary antibody to detect p65; this antibody does not interfere with immunoprecipitating immunoglobulin heavy and light chains.
RNA isolation and real-time qPCR
Total RNA was isolated using TRIzol reagent (Invitrogen), and cDNA was synthesized as described (65). Real-time PCR was carried out using SYBR Green PCR Master Mix (Applied Biosystems). The specific primers used are shown in Table S1. The amount of PCR product was measured by threshold cycle (Ct) values, and the relative ratio of specific genes to HPRT1 was calculated and presented as the -fold change in the tested group relative to the control group.
Transient transfection and promoter activity assays
AML12 cells in 24-well plates were transfected with luciferase reporters along with various expression plasmids using Lipofectamine 2000 (Invitrogen). Firefly luciferase and Renilla luciferase were examined in cells after a 24-h transfection using luciferase and Renilla assay systems (Promega, Madison, WI). Data are displayed as the ratio of firefly luminescence divided by Renilla luminescence. Each point is the average of triplicate experiments, of which one representative is shown.
ChIP assays
AML12 cells were fixed by 1% formaldehyde followed by nuclei isolation and sonication. Chromatin samples were precleared and subjected to immunoprecipitation with specific antibodies or rabbit normal IgG, respectively. The chromatin immune complexes were captured by sheep anti-rabbit IgG Dynabeads (Fisher Scientific, 11-203-D) and eluted. After reversing the cross-links, DNA fragments were purified and used as templates in qPCR. Four sets of primers (Table S1) were designed for ChIP assays. P1 was specific for the TRE site (c-Jun binding site) on mouse Shp promoter, whereas P2 was located 4 kb upstream from TSS, thus serving as a negative control. L1 and L2 + 3 were specifically to amplify LRH1 sites on mouse Shp promoter.
Perfusion and separation of hepatocytes, HSCs, and KCs
Cell isolation and purification was performed at the Kansas University Medical Center Cell Isolation Core using the method described previously (66) with a slight modification. Mouse liver was perfused with 25 ml of solution I (9.5 g/liter Hanks' balanced salt solution, 0.5 mmol/liter EGTA, pH 7.2) followed by 50 ml of solution II (9.5 g/liter Hanks' balanced salt solution, 0.14 g/liter collagenase IV, and 40 mg/liter trypsin inhibitor, pH 7.5). After digestion, a single-cell suspension was filtered through a 100-μm Falcon cell strainer (Fisher Scientific, 08-771-19), and the cells were centrifuged at 50 × g for 5 min at 4 °C to pellet hepatocytes. The supernatant was then centrifuged at 300 × g for 10 min at 4 °C to enrich nonparenchymal cells. To isolate HSCs and KCs, the nonparenchymal cells were gently resuspended in PBS-BSA solution and mixed with Percoll (Sigma, P1644) followed by a two-step Percoll gradient (50 and 35% Percoll) and centrifugation at 900 × g for 30 min at 4 °C. Three different cell bands were obtained following centrifugation. HSCs were enriched at the top of the Percoll gradient, and KCs were located near the bottom of the centrifugation tube. Cell fractions were then harvested and washed twice. KCs were stained with Alexa Fluor 488 anti-mouse F4/80 antibody (Thermo Fisher Scientific, MF48020) and sorted by FACSAria II (BD Biosciences) at the Kansas University Medical Center Flow Cytometry Core Facility.
Adenovirus infection of mouse primary hepatocyte
Hepatocytes were seeded in collagen type 1–coated dishes. After a 2-h incubation, the medium was replaced by fresh William's E medium (Sigma, W4128) with 10% FBS. On the second day, hepatocytes were infected for 2 h with viral supernatant at a multiplicity of infection of 20. After 24 h the hepatocytes were collected for RNA isolation, Western blotting, and immunofluorescence staining.
RAW cell migration assay
RAW cells were serum-starved for 24 h, and 200,000 cells were seeded on Transwell inserts (8-μm-pore size, Corning 24-well format, Thermo Fisher Scientific, 07-20-150) in FBS-free medium. The lower chamber of the Transwell contained conditioned medium from hepatocyte culture supplemented with or without recombinant mouse CCL2 (40 ng/ml) or anti-mouse CCL2 antibody (500 ng/ml). The cells were cultured at 37 °C for 12 or 24 h. The migrated cells were fixed in 10% neutral formalin (Thermo Fisher Scientific, SF100) for 10 min before staining with 0.1% crystal violet (Thermo Fisher Scientific, AC21212-0250) for 10 min followed by washing with PBS. A cotton swab removed the cells on the top side of the filter. The pictures of migrated cells were taken using a Microfire/Qcam CCD Olympus BX60 microscope. Five fields/sample were captured at ×10 magnifications. The quantitation of cell migration was determined by measuring the pixel density of crystal violet–stained cells using ImageJ software.
Statistical analysis
Data are expressed as mean ± S.D. Statistical analyses were carried out using one-way analysis of variance followed by Student's t test, and p < 0.05 was considered statistically significant.
Author contributions
A. Z., N. M., F. D., S. L., C. Z., and Y. Z. data curation; A. Z., N. M., and Y. Z. investigation; A. Z., N. M., and Y. Z. methodology; N. M. formal analysis; N. M. and Y. Z. writing-review and editing; C. Z. software; Y. Z. supervision; Y. Z. funding acquisition; Y. Z. validation; Y. Z. writing-original draft.
Supplementary Material
Acknowledgment
We thank Dr. Maura F O'Neil for the evaluation of liver histology.
This work was supported by National Institutes of Health Grants NCI K22CA184146, P20GM103549, P30GM11824, P20GM103418, and T32ES007079. The authors declare that they have no conflicts of interest with contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1.
A. Zou, N. Magee, F. Deng, S. Lehn, C. Zhong, and Y. Zhang, unpublished data.
- NAFLD
- nonalcoholic fatty liver disease
- NAFL
- nonalcoholic fatty liver
- NASH
- nonalcoholic steatohepatitis
- SHP
- small heterodimer partner
- HFCF
- high fat, cholesterol, and fructose
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- TG
- triglyceride
- H&E
- hematoxylin and eosin
- TUNEL
- terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling
- VLDL
- very-low-density lipoprotein
- MCD
- methionine/choline–deficient
- JNK
- c-Jun N-terminal kinase
- HSC
- hepatic stellate cell
- KC
- Kupffer cell
- qPCR
- quantitative PCR
- PA
- palmitic acid
- LPS
- lipopolysaccharide
- PI3K
- phosphatidylinositol 3-kinase
- TRE
- 12-O-tetradecanoylphorbol-13-acetate response element
- CM
- conditioned medium
- FXR
- farnesoid X–activated receptor
- FBS
- fetal bovine serum
- NAS
- NAFLD activity score
- GTT
- glucose tolerance test.
References
- 1. Younossi Z. M., Koenig A. B., Abdelatif D., Fazel Y., Henry L., and Wymer M. (2016) Global epidemiology of nonalcoholic fatty liver disease: Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 10.1002/hep.28431 [DOI] [PubMed] [Google Scholar]
- 2. Bettermann K., Hohensee T., and Haybaeck J. (2014) Steatosis and steatohepatitis: Complex disorders. Int. J. Mol. Sci. 15, 9924–9944 10.3390/ijms15069924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Temple J. L., Cordero P., Li J., Nguyen V., and Oben J. A. (2016) A guide to non-alcoholic fatty liver disease in childhood and adolescence. Int. J. Mol. Sci. 17, E974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chalasani N., Younossi Z., Lavine J. E., Diehl A. M., Brunt E. M., Cusi K., Charlton M., and Sanyal A. J. (2012) The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 55, 2005–2023 10.1002/hep.25762 [DOI] [PubMed] [Google Scholar]
- 5. Farrell G. C., van Rooyen D., Gan L., and Chitturi S. (2012) NASH is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liver 6, 149–171 10.5009/gnl.2012.6.2.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ibrahim S. H., Hirsova P., Tomita K., Bronk S. F., Werneburg N. W., Harrison S. A., Goodfellow V. S., Malhi H., and Gores G. J. (2016) Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 63, 731–744 10.1002/hep.28252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Joshi-Barve S., Barve S. S., Amancherla K., Gobejishvili L., Hill D., Cave M., Hote P., and McClain C. J. (2007) Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 46, 823–830 10.1002/hep.21752 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Y., Hagedorn C. H., and Wang L. (2011) Role of nuclear receptor SHP in metabolism and cancer. Biochim. Biophys. Acta 1812, 893–908 10.1016/j.bbadis.2010.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nishigori H., Tomura H., Tonooka N., Kanamori M., Yamada S., Sho K., Inoue I., Kikuchi N., Onigata K., Kojima I., Kohama T., Yamagata K., Yang Q., Matsuzawa Y., Miki T., et al. (2001) Mutations in the small heterodimer partner gene are associated with mild obesity in Japanese subjects. Proc. Natl. Acad. Sci. U.S.A. 98, 575–580 10.1073/pnas.98.2.575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuk J. M., Shin D. M., Lee H. M., Kim J. J., Kim S. W., Jin H. S., Yang C. S., Park K. A., Chanda D., Kim D. K., Huang S. M., Lee S. K., Lee C. H., Kim J. M., Song C. H., et al. (2011) The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat. Immunol. 12, 742–751 10.1038/ni.2064 [DOI] [PubMed] [Google Scholar]
- 11. Yang C. S., Kim J. J., Kim T. S., Lee P. Y., Kim S. Y., Lee H. M., Shin D. M., Nguyen L. T., Lee M. S., Jin H. S., Kim K. K., Lee C. H., Kim M. H., Park S. G., Kim J. M., et al. (2015) Small heterodimer partner interacts with NLRP3 and negatively regulates activation of the NLRP3 inflammasome. Nat. Commun. 6, 6115 10.1038/ncomms7115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ansari A. W., Heiken H., Meyer-Olson D., and Schmidt R. E. (2011) CCL2: A potential prognostic marker and target of anti-inflammatory strategy in HIV/AIDS pathogenesis. Eur. J. Immunol. 41, 3412–3418 10.1002/eji.201141676 [DOI] [PubMed] [Google Scholar]
- 13. Saiman Y., and Friedman S. L. (2012) The role of chemokines in acute liver injury. Front. Physiol. 3, 213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weiskirchen R., and Tacke F. (2014) Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr. 3, 344–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haukeland J. W., Damås J. K., Konopski Z., Løberg E. M., Haaland T., Goverud I., Torjesen P. A., Birkeland K., Bjøro K., and Aukrust P. (2006) Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol. 44, 1167–1174 10.1016/j.jhep.2006.02.011 [DOI] [PubMed] [Google Scholar]
- 16. Baeck C., Wehr A., Karlmark K. R., Heymann F., Vucur M., Gassler N., Huss S., Klussmann S., Eulberg D., Luedde T., Trautwein C., and Tacke F. (2012) Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 61, 416–426 10.1136/gutjnl-2011-300304 [DOI] [PubMed] [Google Scholar]
- 17. Yang Z., Koehler A. N., and Wang L. (2016) A novel small molecule activator of nuclear receptor SHP inhibits HCC cell migration via suppressing Ccl2. Mol. Cancer Ther. 15, 2294–2301 10.1158/1535-7163.MCT-16-0153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ouyang X., Cirillo P., Sautin Y., McCall S., Bruchette J. L., Diehl A. M., Johnson R. J., and Abdelmalek M. F. (2008) Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 48, 993–999 10.1016/j.jhep.2008.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abdelmalek M. F., Suzuki A., Guy C., Unalp-Arida A., Colvin R., Johnson R. J., Diehl A. M., and Nonalcoholic Steatohepatitis Clinical Research Network (2010) Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 51, 1961–1971 10.1002/hep.23535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Trevaskis J. L., Griffin P. S., Wittmer C., Neuschwander-Tetri B. A., Brunt E. M., Dolman C. S., Erickson M. R., Napora J., Parkes D. G., and Roth J. D. (2012) Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G762–G772 10.1152/ajpgi.00476.2011 [DOI] [PubMed] [Google Scholar]
- 21. Clapper J. R., Hendricks M. D., Gu G., Wittmer C., Dolman C. S., Herich J., Athanacio J., Villescaz C., Ghosh S. S., Heilig J. S., Lowe C., and Roth J. D. (2013) Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G483–G495 10.1152/ajpgi.00079.2013 [DOI] [PubMed] [Google Scholar]
- 22. Kohli R., Kirby M., Xanthakos S. A., Softic S., Feldstein A. E., Saxena V., Tang P. H., Miles L., Miles M. V., Balistreri W. F., Woods S. C., and Seeley R. J. (2010) High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 52, 934–944 10.1002/hep.23797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ganz M., Bukong T. N., Csak T., Saha B., Park J. K., Ambade A., Kodys K., and Szabo G. (2015) Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice. J. Transl. Med. 13, 193 10.1186/s12967-015-0552-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suzuki A., and Abdelmalek M. F. (2009) Nonalcoholic fatty liver disease in women. Women's Health (Lond.) 5, 191–203 [DOI] [PubMed] [Google Scholar]
- 25. Miao J., Xiao Z., Kanamaluru D., Min G., Yau P. M., Veenstra T. D., Ellis E., Strom S., Suino-Powell K., Xu H. E., and Kemper J. K. (2009) Bile acid signaling pathways increase stability of small heterodimer partner (SHP) by inhibiting ubiquitin-proteasomal degradation. Genes Dev. 23, 986–996 10.1101/gad.1773909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hebbard L., and George J. (2011) Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 8, 35–44 10.1038/nrgastro.2010.191 [DOI] [PubMed] [Google Scholar]
- 27. Suh J. H., Huang J., Park Y. Y., Seong H. A., Kim D., Shong M., Ha H., Lee I. K., Lee K., Wang L., and Choi H. S. (2006) Orphan nuclear receptor small heterodimer partner inhibits transforming growth factor-β signaling by repressing SMAD3 transactivation. J. Biol. Chem. 281, 39169–39178 10.1074/jbc.M605947200 [DOI] [PubMed] [Google Scholar]
- 28. Leamy A. K., Egnatchik R. A., and Young J. D. (2013) Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 52, 165–174 10.1016/j.plipres.2012.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baylin A., Kabagambe E. K., Siles X., and Campos H. (2002) Adipose tissue biomarkers of fatty acid intake. Am. J. Clin. Nutr. 76, 750–757 10.1093/ajcn/76.4.750 [DOI] [PubMed] [Google Scholar]
- 30. Huang S., Rutkowsky J. M., Snodgrass R. G., Ono-Moore K. D., Schneider D. A., Newman J. W., Adams S. H., and Hwang D. H. (2012) Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res. 53, 2002–2013 10.1194/jlr.D029546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Win S., Than T. A., Le B. H., García-Ruiz C., Fernandez-Checa J. C., and Kaplowitz N. (2015) Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J. Hepatol. 62, 1367–1374 10.1016/j.jhep.2015.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hibi M., Lin A., Smeal T., Minden A., and Karin M. (1993) Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 7, 2135–2148 10.1101/gad.7.11.2135 [DOI] [PubMed] [Google Scholar]
- 33. Zhang Y., Soto J., Park K., Viswanath G., Kuwada S., Abel E. D., and Wang L. (2010) Nuclear receptor SHP, a death receptor that targets mitochondria, induces apoptosis and inhibits tumor growth. Mol. Cell. Biol. 30, 1341–1356 10.1128/MCB.01076-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun Z., Miller R. A., Patel R. T., Chen J., Dhir R., Wang H., Zhang D., Graham M. J., Unterman T. G., Shulman G. I., Sztalryd C., Bennett M. J., Ahima R. S., Birnbaum M. J., and Lazar M. A. (2012) Hepatic Hdac3 promotes gluconeogenesis by repressing lipid synthesis and sequestration. Nat. Med. 18, 934–942 10.1038/nm.2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Magee N., Zou A., and Zhang Y. (2016) Pathogenesis of nonalcoholic steatohepatitis: Interactions between liver parenchymal and nonparenchymal cells. BioMed Res. Int. 2016, 5170402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lawrence T. (2009) The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1, a001651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dela Peña A., Leclercq I., Field J., George J., Jones B., and Farrell G. (2005) NF-κB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology 129, 1663–1674 10.1053/j.gastro.2005.09.004 [DOI] [PubMed] [Google Scholar]
- 38. Zhang Y., Xu N., Xu J., Kong B., Copple B., Guo G. L., and Wang L. (2014) E2F1 is a novel fibrogenic gene that regulates cholestatic liver fibrosis through the Egr-1/SHP/EID1 network. Hepatology 60, 919–930 10.1002/hep.27121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wong V. W., Wong G. L., Choi P. C., Chan A. W., Li M. K., Chan H. Y., Chim A. M., Yu J., Sung J. J., and Chan H. L. (2010) Disease progression of non-alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut 59, 969–974 10.1136/gut.2009.205088 [DOI] [PubMed] [Google Scholar]
- 40. Pais R., Charlotte F., Fedchuk L., Bedossa P., Lebray P., Poynard T., Ratziu V., and LIDO Study Group (2013) A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 59, 550–556 10.1016/j.jhep.2013.04.027 [DOI] [PubMed] [Google Scholar]
- 41. McPherson S., Hardy T., Henderson E., Burt A. D., Day C. P., and Anstee Q. M. (2015) Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 62, 1148–1155 10.1016/j.jhep.2014.11.034 [DOI] [PubMed] [Google Scholar]
- 42. Aguilar-Olivos N. E., Carrillo-Córdova D., Oria-Hernández J., Sánchez-Valle V., Ponciano-Rodríguez G., Ramírez-Jaramillo M., Chablé-Montero F., Chávez-Tapia N. C., Uribe M., and Méndez-Sánchez N. (2015) The nuclear receptor FXR, but not LXR, up-regulates bile acid transporter expression in non-alcoholic fatty liver disease. Ann. Hepatol. 14, 487–493 [PubMed] [Google Scholar]
- 43. Bechmann L. P., Kocabayoglu P., Sowa J. P., Sydor S., Best J., Schlattjan M., Beilfuss A., Schmitt J., Hannivoort R. A., Kilicarslan A., Rust C., Berr F., Tschopp O., Gerken G., Friedman S. L., Geier A., and Canbay A. (2013) Free fatty acids repress small heterodimer partner (SHP) activation and adiponectin counteracts bile acid-induced liver injury in superobese patients with nonalcoholic steatohepatitis. Hepatology 57, 1394–1406 10.1002/hep.26225 [DOI] [PubMed] [Google Scholar]
- 44. Benet M., Guzmán C., Pisonero-Vaquero S., García-Mediavilla M. V., Sánchez-Campos S., Martínez-Chantar M. L., Donato M. T., Castell J. V., and Jover R. (2015) Repression of the nuclear receptor small heterodimer partner by steatotic drugs and in advanced nonalcoholic fatty liver disease. Mol. Pharmacol. 87, 582–594 10.1124/mol.114.096313 [DOI] [PubMed] [Google Scholar]
- 45. Watanabe M., Houten S. M., Wang L., Moschetta A., Mangelsdorf D. J., Heyman R. A., Moore D. D., and Auwerx J. (2004) Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 113, 1408–1418 10.1172/JCI21025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Seok S., Kanamaluru D., Xiao Z., Ryerson D., Choi S. E., Suino-Powell K., Xu H. E., Veenstra T. D., and Kemper J. K. (2013) Bile acid signal-induced phosphorylation of small heterodimer partner by protein kinase Czeta is critical for epigenomic regulation of liver metabolic genes. J. Biol. Chem. 288, 23252–23263 10.1074/jbc.M113.452037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boulias K., Katrakili N., Bamberg K., Underhill P., Greenfield A., and Talianidis I. (2005) Regulation of hepatic metabolic pathways by the orphan nuclear receptor SHP. EMBO J. 24, 2624–2633 10.1038/sj.emboj.7600728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang L., Liu J., Saha P., Huang J., Chan L., Spiegelman B., and Moore D. D. (2005) The orphan nuclear receptor SHP regulates PGC-1α expression and energy production in brown adipocytes. Cell Metab. 2, 227–238 10.1016/j.cmet.2005.08.010 [DOI] [PubMed] [Google Scholar]
- 49. Huang J., Iqbal J., Saha P. K., Liu J., Chan L., Hussain M. M., Moore D. D., and Wang L. (2007) Molecular characterization of the role of orphan receptor small heterodimer partner in development of fatty liver. Hepatology 46, 147–157 10.1002/hep.21632 [DOI] [PubMed] [Google Scholar]
- 50. Park Y. J., Kim S. C., Kim J., Anakk S., Lee J. M., Tseng H. T., Yechoor V., Park J., Choi J. S., Jang H. C., Lee K. U., Novak C. M., Moore D. D., and Lee Y. K. (2011) Dissociation of diabetes and obesity in mice lacking orphan nuclear receptor small heterodimer partner. J. Lipid Res. 52, 2234–2244 10.1194/jlr.M016048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu T. T., Makishima M., Repa J. J., Schoonjans K., Kerr T. A., Auwerx J., and Mangelsdorf D. J. (2000) Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 6, 507–515 10.1016/S1097-2765(00)00050-2 [DOI] [PubMed] [Google Scholar]
- 52. Goodwin B., Jones S. A., Price R. R., Watson M. A., McKee D. D., Moore L. B., Galardi C., Wilson J. G., Lewis M. C., Roth M. E., Maloney P. R., Willson T. M., and Kliewer S. A. (2000) A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 6, 517–526 10.1016/S1097-2765(00)00051-4 [DOI] [PubMed] [Google Scholar]
- 53. Chiang J. Y. (2009) Bile acids: Regulation of synthesis. J. Lipid Res. 50, 1955–1966 10.1194/jlr.R900010-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Janssen A. W. F., Houben T., Katiraei S., Dijk W., Boutens L., van der Bolt N., Wang Z., Brown J. M., Hazen S. L., Mandard S., Shiri-Sverdlov R., Kuipers F., Willems van Dijk K., Vervoort J., Stienstra R., et al. (2017) Modulation of the gut microbiota impacts nonalcoholic fatty liver disease: A potential role for bile acids. J. Lipid Res. 58, 1399–1416 10.1194/jlr.M075713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chanda D., Lee C. H., Kim Y. H., Noh J. R., Kim D. K., Park J. H., Hwang J. H., Lee M. R., Jeong K. H., Lee I. K., Kweon G. R., Shong M., Oh G. T., Chiang J. Y., and Choi H. S. (2009) Fenofibrate differentially regulates plasminogen activator inhibitor-1 gene expression via adenosine monophosphate-activated protein kinase-dependent induction of orphan nuclear receptor small heterodimer partner. Hepatology 50, 880–892 10.1002/hep.23049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zou A., Lehn S., Magee N., and Zhang Y. (2015) New insights into orphan nuclear receptor SHP in liver cancer. Nucl. Receptor Res. 2, 101162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hoeke M. O., Heegsma J., Hoekstra M., Moshage H., and Faber K. N. (2014) Human FXR regulates SHP expression through direct binding to an LRH-1 binding site, independent of an IR-1 and LRH-1. PLoS ONE 9, e88011 10.1371/journal.pone.0088011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Li G., Kong B., Zhu Y., Zhan L., Williams J. A., Tawfik O., Kassel K. M., Luyendyk J. P., Wang L., and Guo G. L. (2013) Small heterodimer partner overexpression partially protects against liver tumor development in farnesoid X receptor knockout mice. Toxicol. Appl. Pharmacol. 272, 299–305 10.1016/j.taap.2013.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Anakk S., Watanabe M., Ochsner S. A., McKenna N. J., Finegold M. J., and Moore D. D. (2011) Combined deletion of Fxr and Shp in mice induces Cyp17a1 and results in juvenile onset cholestasis. J. Clin. Invest. 121, 86–95 10.1172/JCI42846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ding L., Yang L., Wang Z., and Huang W. (2015) Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 5, 135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ali A. H., Carey E. J., and Lindor K. D. (2015) Recent advances in the development of farnesoid X receptor agonists. Ann. Transl. Med. 3, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang Y., Liu C., Barbier O., Smalling R., Tsuchiya H., Lee S., Delker D., Zou A., Hagedorn C. H., and Wang L. (2016) Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci. Rep. 6, 20559 10.1038/srep20559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Asgharpour A., Cazanave S. C., Pacana T., Seneshaw M., Vincent R., Banini B. A., Kumar D. P., Daita K., Min H. K., Mirshahi F., Bedossa P., Sun X., Hoshida Y., Koduru S. V., Contaifer D. Jr., et al. (2016) A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 65, 579–588 10.1016/j.jhep.2016.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kleiner D. E., Brunt E. M., Van Natta M., Behling C., Contos M. J., Cummings O. W., Ferrell L. D., Liu Y. C., Torbenson M. S., Unalp-Arida A., Yeh M., McCullough A. J., Sanyal A. J., and Nonalcoholic Steatohepatitis Clinical Research Network (2005) Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–1321 10.1002/hep.20701 [DOI] [PubMed] [Google Scholar]
- 65. Smalling R. L., Delker D. A., Zhang Y., Nieto N., McGuiness M. S., Liu S., Friedman S. L., Hagedorn C. H., and Wang L. (2013) Genome-wide transcriptome analysis identifies novel gene signatures implicated in human chronic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 305, G364–G374 10.1152/ajpgi.00077.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Vrochides D., Papanikolaou V., Pertoft H., Antoniades A. A., and Heldin P. (1996) Biosynthesis and degradation of hyaluronan by nonparenchymal liver cells during liver regeneration. Hepatology 23, 1650–1655 10.1002/hep.510230648 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.