

Abstract
Differentiation of smooth muscle cells (SMCs) is critical for proper vasculogenesis and angiogenesis. However, the molecular mechanisms controlling SMC differentiation are not completely understood. During embryogenesis, the transcription factor mesenchyme homeobox 1 (Meox1) is expressed in the early developing somite, which is one of the origins of SMCs. In the present study, we identified Meox1 as a positive regulator of SMC differentiation. We found that transforming growth factor-β (TGF-β) induces Meox1 expression in the initial phase of SMC differentiation of pluripotent murine C3H10T1/2 cells. shRNA-mediated Meox1 knockdown suppressed TGF-β–induced expression of SMC early markers, whereas Meox1 overexpression increased expression of these markers. Mechanistically, Meox1 promoted SMAD family member 3 (Smad3) nuclear retention during the early stage of TGF-β stimulation because Meox1 inhibited protein phosphatase Mg2+/Mn2+-dependent 1A (PPM1A) and thereby prevented PPM1A-mediated Smad3 dephosphorylation. Meox1 appears to promote PPM1A degradation, leading to sustained Smad3 phosphorylation, thus allowing Smad3 to stimulate SMC gene transcription. In vivo, Meox1 knockdown in mouse embryos impaired SMC marker expression in the descending aorta of neonatal mice, indicating that Meox1 is essential for SMC differentiation during embryonic development. In summary, the transcriptional regulator Meox1 controls TGF-β–induced SMC differentiation from mesenchymal progenitor cells by preventing PPM1A-mediated Smad3 dephosphorylation, thereby supporting SMC gene expression.
Keywords: signal transduction, smooth muscle, cell differentiation, transforming growth factor beta (TGF-β), SMAD transcription factor, mesenchyme homeobox 1 (Meox1), SMAD family member 3 (Smad3)
Introduction
Smooth muscle cell (SMC)2 is mainly involved in blood vessel contraction, pressure regulation, and blood flow distribution (1). SMCs in different segments of arteries are derived from a number of precursors such as neural crest, proepicardium, mesothelium, somites, mesoangioblast, etc. For example, smooth muscles located in dorsal aorta originate from somite (2). A number of studies have suggested that several cardiovascular diseases including atherosclerosis are associated with the diversity of SMCs originating from different sources (3). However, the underlying mechanisms remain largely unknown. Several in vitro models are available for exploring mechanisms controlling SMC differentiation from different progenitors. For instance, Monc-1 cells originating from mouse neural crest can be induced into contractile SMC by TGF-β treatment (4), mouse pluripotent embryonal carcinoma–derived A404 cells can be induced to express several SMC markers with the treatment of all-trans–retinoic acid followed by puromycin (5), and the mesoderm-derived C3H10T1/2 cells can be induced by TGF-β to express SMC markers such as α-SMA, calponin, and SM22α (6).
Mesenchyme homeobox 1 (Meox1) belongs to a diverged subfamily of homeobox transcription factors (7). Meox1 is expressed in early developing somite and plays an important role in somitogenesis during embryogenesis (8). In adults, Meox1 is expressed in a variety of cells including endothelial cells (9) and vascular SMCs (10). Recent studies show that Meox1 is critical for the specification of endothelial cells in the endotome of the somites, which further give rise to hematopoietic stem cells (11). Because Meox1 is expressed in somite, and somite is one of the SMC precursors, we hypothesized that Meox1 may be involved in SMC differentiation. Indeed, Meox1 was significantly up-regulated in TGF-β–treated C3H10T1/2 cells. Thus we sought to determine whether Meox1 plays a role in TGF-β–induced SMC differentiation.
In the present study, we found that Meox1 was up-regulated along with SMC early markers in 10T1/2 cells by TGF-β induction. Knockdown or overexpression of Meox1 dramatically altered SMC marker gene expression. Mechanistically, Meox1 prevented nuclear Smad3 from shuttling back to cytoplasm during the early stage of TGF-β stimulation. Meox1 appeared to promote Smad3 nuclear retention through inhibiting the expression of the protein phosphatase, Mg2+/Mn2+-dependent 1A (PPM1A) as well as promoting its degradation. Most importantly, knockdown of Meox1 impaired SMC differentiation during embryonic development in mice.
Results
Meox1 expression was up-regulated in TGF-β–induced SMC differentiation
TGF-β–treated 10T1/2 cells are a widely used model for studying SMC differentiation. We confirmed that TGF-β induced the expression of SMC early differentiation markers α-SMA, calponin, and SM22α in 10T1/2 cells at both the mRNA (Fig. 1, A–D) and protein levels (Fig. 1, E–I). TGF-β induced Meox1 expression along with the SMC marker expression. Importantly, Meox1 expression was induced as early as 2 h following TGF-β stimulation, suggesting that Meox1 may be involved in TGF-β–induced SMC differentiation.
Figure 1.

TGF-β induced Meox1 expression along with SMC early differentiation markers. A–D, TGF-β induced Meox1 and SMC marker mRNA expression in 10T1/2 cells. Serum-starved 10T1/2 cells were treated with vehicle (0 h) or TGF-β (5 ng/ml) for the times indicated. mRNA levels were detected by qPCR. *, p < 0.05 compared with the vehicle-treated group (0 h), n = 3. E, Meox1 protein expression was induced in the initial stage of TGF-β stimulation in 10T1/2 cells. 10T1/2 cells were starved for 48 h followed by vehicle (−) or TGF-β (5 ng/ml) induction for the times indicated. Meox1 and SMC marker protein expression was detected by Western blotting. F–I, quantification of Meox1 and SMC marker levels shown in E normalized to the α-tubulin level. Shown are the relative protein levels with Ctrl (0 h) set as 1. *, p < 0.05 compared with the vehicle-treated group (0 h), n = 3. Error bars indicate S.D.
Meox1 was essential for TGF-β–induced SMC differentiation
To test if Meox1 plays a role in TGF-β–induced SMC differentiation, we knocked down Meox1 expression using its specific shRNA delivered via adenoviral vector prior to the TGF-β induction. As shown in Fig. 2A, Meox1 and SMC markers were induced simultaneously by TGF-β, and Meox1 expression was effectively blocked by its shRNA (Fig. 2, A and B). Importantly, knockdown of Meox1 blocked TGF-β–induced expression of SMC early markers α-SMA, calponin, and SM22α by 47, 84, and 78%, respectively (Fig. 2, C–E). Meox1 reduction appeared not to alter the basal level expression of SMC markers. These results indicated that Meox1 was essential for TGF-β–induced SMC differentiation.
Figure 2.

Meox1 was essential for TGF-β–induced SMC differentiation. A, Meox1 knockdown decreased TGF-β–induced SMC marker protein expression in 10T1/2 cells. 10T1/2 cells were transduced with control (shCtrl) or Meox1 shRNA (shMeox1) adenoviral vector. Following starvation for 48 h, 10T1/2 cells were treated with TGF-β for 24 h. Western blotting was performed to detect Meox1 and SMC marker protein expression. B–E, quantification of the protein expression shown in A. Meox1 and SMC marker protein levels were normalized to α-tubulin. *, p < 0.05 compared with the vehicle-treated group (−); #, p < 0.05 compared with TGF-β–treated cells transduced with Ctrl adenoviral vector (−); &, p < 0.05 compared with shCtrl group without TGF-β treatment; $, p > 0.05 compared with shCtrl group without TGF-β treatment; n = 3. Error bars indicate S.D.
Meox1 is sufficient for SMC differentiation of 10T1/2 cells
To further investigate the role of Meox1 in SMC differentiation, we tested if Meox1 alone can induce SMC marker gene expression. Thus, we forcefully expressed Meox1 in 10T1/2 cells via adenoviral transduction of Meox1 cDNA and detected the expression of SMC early differentiation markers. As shown in Fig. 3, Meox1 was robustly expressed in 10T1/2 cells with a 5-fold increase compared with the control vector transduction, and notably, overexpression of Meox1 promoted the protein expression of α-SMA and calponin, indicating that Meox1 was able to stimulate 10T1/2 cells to differentiate to a SMC lineage.
Figure 3.

Meox1 alone was able to induce SMC early differentiation marker expression. A, Meox1 increased SMC marker expression. 10T1/2 cells were transduced with control (Ctrl, GFP adenoviral vector) or Meox1 adenoviral vectors (AdMeox1) for 48 h. Meox1 and SMC marker protein expression was analyzed by Western blotting. B–D, quantitative analysis of Meox1 and SMC marker protein levels shown in A by normalizing to α-tubulin. *, p < 0.05 compared with the Ctrl group (n = 3). Error bars indicate S.D. E, Meox1 promoted the contractile SMC phenotype. 10T1/2 cells were transduced with control (Ctrl) or Meox1 shRNA adenoviral vector (Ad-shMeox1) for 24 h and then starved for 24 h before treatment with vehicle or 5 ng/ml of TGF-β for 36 h. Cell morphology was captured with a Nikon microscope. Knockdown of Meox1 prevented the formation of TGF-β–induced spindle-shaped SMC morphology.
In addition to SMC marker gene expression, Meox1 also affected SMC morphology. TGF-β induced a spindle-shaped SMC morphology. However, knockdown of Meox1 reversed the cell morphology to its original appearance (Fig. 3E), suggesting that Meox1 may be essential for the progenitor cells to differentiate to mature SMCs.
Meox1 regulated SMC marker gene transcription and mediated Smad3 function
Transcriptional activation of SMC genes is one of the molecular mechanisms controlling SMC differentiation (12–14). Therefore, we sought to test if Meox1 regulates SMC differentiation by mediating SMC marker gene transcription. We first detected whether Meox1 regulates SMC marker expression at the mRNA level and found that knockdown of Meox1 by its shRNA dramatically attenuated TGF-β–induced mRNA expression of α-SMA and SM22α without changing their levels at the basal state (Fig. 4, A–D), suggesting that Meox1 may be involved in SMC marker gene transcription induced by TGF-β. Indeed, knockdown of Meox1 inhibited TGF-β induction of α-SMA and SM22α promoter activities, as shown by SMC promoter-reporter luciferase assays (Fig. 4, E and F). Remarkably, Meox1 shRNA reduced α-SMA and SM22α promoter activities by 58.6 and 43%, respectively, demonstrating that Meox1 was a critical regulator for SMC gene transcription.
Figure 4.
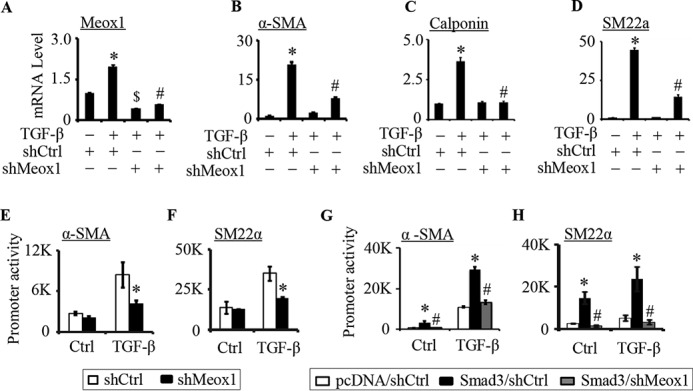
Meox1 was required for the activation of SMC marker gene promoters by TGF-β/Smad3. A–D, knockdown of Meox1 diminished SMC marker mRNA expression in 10T1/2 cells. 10T1/2 cells were transduced with control (shCtrl) or Meox1 shRNA adenoviral vector (shMeox1). 24 h later, the cells were serum-starved for 48 h followed by vehicle (−) or TGF-β (5 ng/ml) treatment for 8 h. Meox1 (A), α-SMA (B), calponin (C), and SM22α (D) mRNA expression was detected by qPCR. *, p < 0.05 compared with the shCtrl group without TGF-β treatment; #, p < 0.01 compared with the TGF-β–treated group transfected with shCtrl; $, p < 0.05 compared with the shCtrl group without TGF-β treatment; n = 3. E and F, knockdown of Meox1 blocked TGF-β–induced promoter activities of SMC marker genes. 10T1/2 cells were co-transfected with α-SMA (E) or SM22α (F) promoter construct with shCtrl or shMeox1 followed by TGF-β treatment for 8 h. Luciferase assays were performed. *, p < 0.01 compared with the shCtrl group (n = 3). G and H, knockdown of Meox1 blocked Smad3 function in enhancing TGF-β–induced SMC marker promoter activities. 10T1/2 cells were transduced with shCtrl or shMeox1 adenoviral vector followed by co-transfection of pcDNA or Smad3 plasmids with α-SMA (G) or SM22α (H) promoter construct. Following starvation for 24 h, cells were treated with vehicle (Ctrl) or TGF-β for 8 h followed by luciferase assays. *, p < 0.05 compared with the pcDNA/shCtrl groups; #, p < 0.05 compared with the Smad3/shCtrl groups; n = 3. Error bars indicate S.D.
We and others have reported previously that Smad3 is the key factor mediating TGF-β function in SMC differentiation of 10T1/2 cells (6, 15, 16). Smad3 mainly regulates the transcription of SMC differentiation marker genes in the initial stage of SMC differentiation (15). Therefore, we sought to determine whether Meox1 is involved in Smad3-mediated early SMC marker gene transcription. As shown in Fig. 4, G and H, forced expression of Smad3 markedly up-regulated SMC marker α-SMA and SM22α promoter activities. However, knockdown of Meox1 blocked Smad3 function in increasing the activities of both α-SMA and SM22α promoters. Interestingly, Smad3 increased the promoter activities even in the basal state, i.e. without TGF-β treatment, whereas knockdown of Meox1 completely blocked the Smad3-mediated promoter activities, suggesting that in addition to TGF-β–treated cells, Meox1 was also important for Smad3 activity in the quiescent SMC.
Meox1 was essential for Smad3 nuclear location
Because Smad3 is continuously shuttling back and forth between cytoplasm and nuclei with or without TGF-β treatment (17) and Smad3 nuclear translocation is required for its transcription activity in SMC differentiation, we tested if Meox1 affects Smad3 nuclear localization. Immunostaining showed that TGF-β treatment for 30 min caused the majority of Smad3 translocated into the nuclei of 10T1/2 cells (Fig. 5A). However, knockdown of Meox1 by its shRNA caused less Smad3 located in the nuclei (Fig. 5A). Because Meox1 is a nuclear protein, these data suggest that Meox1 may be important for Smad3 nuclear retention. To confirm the Meox1 function in Smad3 nuclear location, we detected cytoplasmic and nuclear Smad3 levels in TGF-β–treated cells when the Meox1 expression was manipulated. As shown in Fig. 5, B and C, TGF-β induction for 2 h caused a significantly higher level of nuclear Smad3 compared with the untreated cells along with a reduction of cytoplasmic Smad3. Knockdown of Meox1, however, significantly reduced the nuclear Smad3 level by 48.7% compared with TGF-β–treated cells with intact Meox1 (Fig. 5, B and C). These data further demonstrated that Meox1 was essential for Smad3 nuclear retention.
Figure 5.

Meox1 was critical for TGF-β–induced Smad3 nuclear localization. A, knockdown of Meox1 reduced Smad3 nuclear location. 10T1/2 cells were transduced with control (Ctrl) or Meox1 shRNA (shMeox1) adenoviral vectors for 1 day followed by 2-day starvation. The cells were then treated with vehicle (0 h) or TGF-β (5 ng/ml) for 30 min. Immunostaining was performed to detect Smad3 cellular location. DAPI stains nuclei. Scale bar = 20 μm. B and C, knockdown of Meox1 blocked TGF-β–induced Smad3 nuclear accumulation.10T1/2 cells were transduced with Ctrl or shMeox1 similarly as in A followed by treatment with vehicle (−) or TGF-β (5 ng/ml) for 1 h. B, nuclear and cytoplasmic proteins were extracted to perform Western blotting using antibodies as indicated. C, quantification of Smad3 levels by normalized to α-tubulin (cytoplasmic portion) or Lamin B (nuclear portion). *, p < 0.05 compared with shCtrl without TGF-β treatment; #, p > 0.05 compared with shMeox1 with vehicle treatment for cytoplasmic Smad3; $, p < 0.05 compared with all other groups for nuclear Smad3; n = 3. Error bars indicate S.D.
Meox1 maintained nuclear Smad3 phosphorylation by limiting PPM1A expression
To determine the mechanism underlying Meox1 function in Smad3 nuclear retention, we tested whether Meox1 regulates Smad3 phosphorylation status because Smad3 phosphorylation leads to the nuclear location whereas dephosphorylation of the protein causes Smad3 export from the nuclei (18). Thus, the phosphorylated Smad3 reflects the nuclear Smad3 protein level. Time-dependent study showed that Smad3 was phosphorylated 10 min after TGF-β induction, and the phospho-Smad3 reached the highest level at 10–30 min but gradually decreased after 2 h of TGF-β treatment (Fig. 6, A and B). However, knockdown of Meox1 by shRNA significantly or completely diminished the phospho-Smad3 level during TGF-β induction. Because Meox1 is not located in the cytoplasm, it is unlikely that Meox1 blocks TGF-β–induced Smad3 phosphorylation process. Instead, we hypothesized that Meox1 may be essential in preserving the phosphorylation of nuclear Smad3, which can be removed by PPM1A (19). PPM1A is a phosphatase belonging to the protein serine/threonine phosphatase family, which specifically removes phosphates on serine/threonine residues (20, 21). Among 49 family members, PPM1A is the only phosphatase that dephosphorylates Smad2/3 on C-terminal and terminates TGF-β signaling (19). PPM1A-mediated Smad3 dephosphorylation is known to reduce Smad3 phosphorylation and thus facilitate Smad3 nuclear export. Knockdown of Meox1 significantly enhanced the expression of PPM1A in both vehicle and TGF-β–treated cells (Fig. 6, C and D). To determine how Meox1 inhibited the PPM1A level, we tested if Meox1 affected PPM1A protein stability when the protein synthesis was blocked by cycloheximide. Time-dependent study indicated that knockdown of Meox1 increased the stability of PPM1A protein as shown by the reduced degradation rate or prolonged half-life of PPM1A as compared with the control vector–treated cells (Fig. 6, E and F). These results indicated that Meox1 may decrease PPM1A level by increasing its degradation. To further confirm the role of Meox1 in PPM1A expression, we overexpressed Meox1 in 10T1/2 cells and found that forced expression of Meox1 could inhibit PPM1A expression (Fig. 6, G and H). Taken together, these data support a novel concept that Meox1 confines PPM1A level to preserve Smad3 phosphorylation and thus facilitates Smad3 nuclear location.
Figure 6.

Meox1 preserved Smad3 phosphorylation by inhibiting PPM1A expression through increasing its degradation. A, knockdown of Meox1 decreased Smad3 phosphorylation (p-Smad3). 10T1/2 cells were transduced with control (shCtrl) or Meox1 shRNA (shMeox1) adenoviral vectors for 1 day followed by 2-day starvation. The cells were then treated with TGF-β (5 ng/ml) for the times indicated. Total and p-Smad3 levels were analyzed by Western blotting. B, p-Smad3 level shown in A was quantified by normalizing to GAPDH level for each time point, respectively. *, p < 0.05 compared with shMeox1-treated group in each time point (n = 3). C, knockdown of Meox1 increased PPM1A level. 10T1/2 cells were transduced with shCtrl or shMeox1 for 1 day followed by 2-day serum starvation. The cells were then treated with TGF-β (5 ng/ml) for the times indicated. PPM1A protein level was measured by Western blotting. D, quantification of PPM1A levels shown in C by normalized to α-tubulin for each time point, respectively. *, p < 0.05 compared with shCtrl group for each time point (n = 3). E, knockdown of Meox1 reduced the degradation of PPM1A. 10T1/2 cells were transduced with shCtrl or shMeox1 adenoviral vectors for 3 days followed by 30 μg/ml cycloheximide (CHX) treatment for the times indicated. PPM1A level was measured by Western blotting. F, the percentage of remaining PPM1A was quantified by normalizing to α-tubulin. *, p < 0.05 compared with shCtrl-treated group for each time point (n = 3). G, overexpression of Meox1 decreased PPM1A expression. 10T1/2 cells were transduced with control (Ctrl, GFP adenoviral vector) or Meox1 adenoviral vectors (AdMeox1) for 48 h. PPM1A expression was analyzed by Western blotting. H, PPM1A level was quantified by normalizing to α-tubulin. *, p < 0.05 compared with the Ctrl group (n = 3). Error bars indicate S.D.
Knockdown of Meox1 impeded SMC differentiation in vivo
To test if Meox1 is important for SMC differentiation in vivo, we administered adenoviral vector-expressing control and Meox1 shRNA intraplacentally into C57BL/6J mouse embryos (E12.5) and collected the descending aortas of mice that were born on postnatal day 1 (P1). As shown in Fig. 7A, Meox1 expression in the descending aorta was significantly knocked down by its shRNA, which was confirmed by immunostaining of Meox1 in the descending aorta frozen section (Fig. 7B). Importantly, knockdown of Meox1 significantly inhibited the expression of SMC markers α-SMA and calponin, as shown by the reduction of both their mRNA and protein expression in the descending aorta (Fig. 7, A–E). In addition, the SMC numbers were also reduced when the Meox1 was knocked down (Fig. 7F). These data indicated that Meox1 played an important role in SMC differentiation during embryonic development.
Figure 7.
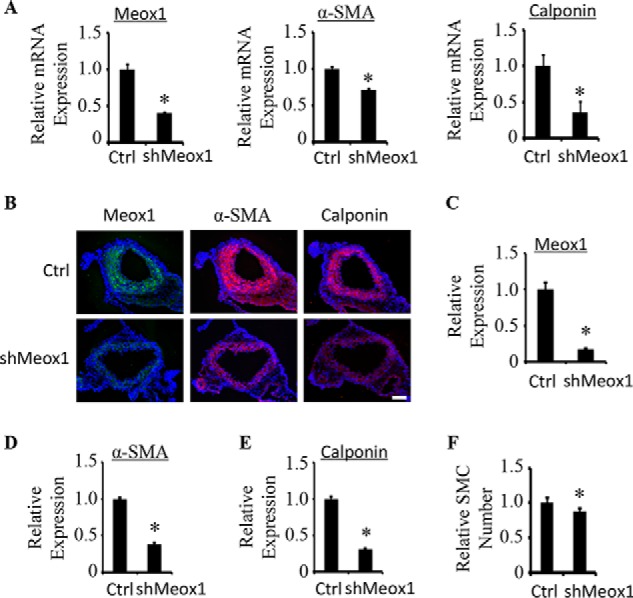
Knockdown of Meox1 attenuated SMC differentiation during mouse embryonic development. A, intraplacental administration of Meox1 shRNA (shMeox1) via adenoviral delivery into mouse E12.5 embryo attenuated Meox1 and inhibited SMC marker expression in descending aortas of postnatal mice (P1) as measured by qPCR. *, p < 0.05 compared with the Ctrl group, n = 5. B, knockdown of Meox1 attenuated SMC marker expression in aorta of newborn mice. Meox1 (green), α-SMA (red), and calponin (red) protein expression were detected by immunofluorescent staining. DAPI stains the nuclei (200×). Scale bar = 50 μm. C–E, quantification of the relative Meox1, α-SMA, and calponin levels shown in B by calibrating the positive staining intensity to the mean signal in aorta and by setting the value in control shRNA group (Ctrl) as 1. *, p < 0.05 compared with the Ctrl group, n = 10. F, quantification of the relative smooth muscle cell numbers in aorta by counting α-SMA–positive cells and by setting the cell number in Ctrl group as 1. *, p < 0.05 compared with the Ctrl group, n = 10. Error bars indicate S.D.
Discussion
The present studies have demonstrated that Meox1 plays an important role in regulating TGF-β–induced SMC differentiation. Meox1 is induced along with the expression of early SMC markers. Knockdown of Meox1 inhibits SMC marker gene expression whereas forced expression of Meox1 enhances the marker expression. The essential roles of Meox1 in TGF-β–induced SMC marker mRNA expression and promoter activities suggest that Meox1 regulates SMC differentiation by controlling SMC gene transcription. Most importantly, Meox1 is required for SMC differentiation during the embryonic development in vivo. It appears that Meox1 is involved in Smad3 regulation of SMC genes because knockdown of Meox1 diminishes Smad3-mediated SMC gene promoter activities. Mechanistically, Meox1 promotes PPM1A degradation or reduces its expression, which permits Smad3 to preserve its phosphorylation status in the nuclei of 10T1/2 cells upon TGF-β treatment, and thus allows Smad3 to activate SMC gene transcription, leading to the initiation of SMC differentiation (Fig. 8).
Figure 8.

A schematic mechanism by which Meox1 regulates SMC differentiation. Upon TGF-β stimulation, Smad3 is phosphorylated and shuttled into nucleus where Smad3 binds to Smad-binding element (SBE) in SMC marker gene promoters to initiate SMC marker transcription and subsequent SMC differentiation. As a mild activator for SMC genes, Smad3 facilitates SMC differentiation in early stage of TGF-β treatment through a temperate and precise regulation. In the late stages, nuclear Smad3 is exported to cytoplasm because of the dephosphorylation by PPM1A. In the initial stage of SMC differentiation, Meox1 confines PPM1A level through promoting its degradation, which enhances nuclear Smad3 retention and thus safeguards SMC marker gene transcription.
TGF-β/Smad signaling plays a critical role in SMC differentiation (22, 23). In fact, Smad proteins are involved in progenitor-specific regulation of SMC differentiation, i.e. Smad2 is important for neural crest cell differentiation to SMC whereas Smad3 is essential for the SMC differentiation from mesenchymal progenitors (24). Although PPM1A dephosphorylates and promotes nuclear export of both the TGF-β–activated Smad2 and Smad3 (19), we only focus on the effect of PPM1A on Smad3 phosphorylation because Smad2 appears not to be involved in TGF-β–induced SMC differentiation from mesenchymal progenitors (25). Smad3 activity may be regulated in multiple levels such as expression, phosphorylation, nuclear translocation, and interactions with other transcription factor or Smad-binding elements on the target gene promoters. Our results suggest that Meox1 regulates Smad3 phosphorylation status in the nuclei rather than participating in the TGF-β receptor–mediated Smad3 phosphorylation in the cytoplasm, largely because of the nuclear location of Meox1. Meox1 appears to be a novel suppressor for PPM1A expression because knockdown of Meox1 promotes the expression of PPM1A, even in the basal state prior to TGF-β stimulation. In addition, Meox1 promotes PPM1A degradation because knockdown of Meox1 causes an enhanced accumulation of PPM1A when the protein synthesis is blocked.
In addition to serving as a PPM1A inhibitor, Meox1 is likely to also function as a transcription factor directly regulating SMC gene promoter. This notion is supported by the fact that Meox1 alone is able to enhance the activities of exogenously introduced SMC promoters in quiescent SMCs where Smad3 is mainly located in the cytoplasm. These results are consistent with a previous report showing that Meox1 may be involved in gene transcription because it can specifically bind the DNA sequences recognized by Hoxa2 on its target genes (26). A future identification of the Meox1-binding element on SMC marker promoters and the in vivo function may provide more detailed insights into the regulatory mechanism governing Meox1 function in SMC differentiation.
A recent study has shown that Meox1 up-regulated by PDGF-BB inhibits α-SMA and calponin expression in cultured SMCs (35), which is opposite to the role of Meox1 identified in this study. The discrepancy could be because of the different environments and cellular contexts involved in these two studies. TGF-β–treated progenitors mimic the development program whereas PDGF-BB–treated SMCs imitate the injury-induced vascular pathology, which may lead to completely different intracellular responses and thus may produce opposite outcome even to the same gene. Indeed, TGF-β–induced Meox1 promotes Smad3 activity via inhibiting PPM1A, resulting in the increased SMC marker expression. However, PDGF-BB–induced Meox1 activates FAK-ERK1/2 signaling and induces autophagy, causing down-regulation of SMC markers (35). These results suggest that the functional outcome of a gene may not only depend on its target, but also is influenced largely by the intracellular environment especially its associated factors.
Taken together, our studies have identified Meox1 as a novel regulator for TGF-β–induced SMC differentiation from mesenchymal progenitors. Meox1 regulates SMC marker gene expression by preserving Smad3 phosphorylation in cell nuclei through promoting PPM1A degradation and possibly also by blocking PPM1A expression, which prevents Smad3 dephosphorylation. Meox1 may also serve as a transcription factor directly regulating SMC gene promoter activation. Because Meox1 is involved in early somite development and somite is one of the progenitors for vascular SMCs, Meox1-mediated SMC differentiation is likely to be important for embryonic vasculature development.
Experimental procedures
Cell culture and transfection
C3H10T1/2 (10T1/2) cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 5% l-glutamine. Cells were starved in serum-free medium for 48 h followed by incubating with TGF-β (5 ng/ml) for various times as needed. For transfection of plasmid DNA, cells were plated on 12-well plates or 6-cm dishes with 70–80% confluence. 24 h later, transfection was carried out using Lipofectamine LTX reagents (Life Technologies) according to the manufacturer's instruction.
Construction of adenoviral vectors
Mouse Meox1 cDNA was inserted into the XhoI site of pShuttle-IRES-hrGFP-1 (Agilent Technologies) and was confirmed by sequencing. For Meox1 shRNA adenoviral vector, dsDNAs coding Meox1 shRNAs were cloned into pRNAT-H1.1/Adeno shuttle vector (GenScript). Recombinant adenoviral vector was produced in AD-1 competent cells (Agilent Technologies) according to the manufacturer's instruction (27). shRNA target sequences for mouse Meox1 were 5′-GGA CTG AGC GAA TCT TCA ACG AGC AGC AT-3′ (top strand) and 5′-ATG CTG CTC GTT GAA GAT TCG CTC AGT CC-3′ (bottom strand).
Viral inoculation of mouse embryo
C57BL/6J mice were housed under conventional conditions in the animal care facilities and received humane care in compliance with the Principles of Laboratory Animal Care formulated by the National Society for Medical Research and the Guide for the Care and Use of Laboratory Animals. Animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Georgia. 3-month-old mice were mated to produce embryos for viral inoculation. Females were checked to see whether there was a vaginal plug every morning. The day that the plug was found was considered as E0.5. Pregnant mice with E12.5 embryos were intraplacentally administered 3 μl 5.3 × 105 TCID50/ml adenovirus (where TCID50 is tissue culture infective dose) expressing Meox1 shRNA into E12.5 embryos; scramble shRNA–expressing adenovirus was injected as control. To improve retention of the pregnancy, the viral injection of the two embryos next to the ovaries and the upper vagina was avoided. Injected embryos were placed back in pregnant dams and allowed to develop to the full term (28, 29). The descending aorta of postnatal mice were collected day 1 after the mice were born.
Quantitative reverse transcription PCR (qPCR)
Total RNA was extracted from cells using TRIzol Reagent (Life Technologies) followed by a reverse transcription using iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer's instructions. qPCR was performed using Mx3005P qPCR machine with SYBR Green Master Mix (Agilent Technologies). Mouse Meox1 primers used were 5′-GGA AGG AGA GGA CAG CCT TC-3′ (forward) and 5′-CCC TTC ACA CGT TTC CAC TT-3′ (reverse). Smooth muscle marker primers were described previously (30).
Immunofluorescent staining
10T1/2 cells were seeded on sterile coverslips and transfected with Meox1 or control (Ctrl) shRNA in complete medium for 1 day and then starved for 2 days before TGF-β treatment for 30 min and 2 h. Cells were washed with PBS three times before fixing in 4% paraformaldehyde for 5 min. For immunostaining of aorta tissues, 10 μm fresh frozen sections were air-dried for 30 min at room temperature followed by 10 min of fixation with 4% paraformaldehyde. The fixed cells or tissue sections were washed by PBS three times followed by incubation with PBS containing 0.1% Triton X-100 for 10 min. After washing with PBS three times, 5% goat serum in PBS was used to block cells for 30 min. Anti-Meox1 (Abcam, ab105349), anti-Smad3 (Cell Signaling Technology, 9523s), anti-α-SMA (Sigma-Aldrich, A2547), or anti-calponin (Abcam, ab46794) primary antibodies were diluted at 1:50 or 1:100 and the cells incubated at 4 °C overnight. Cells and sections were then incubated with FITC- or TRITC-conjugated secondary antibodies (1:150 and 1:50, respectively) for 30 min followed by PBS washing for three times. Stained cells and sections were observed and imaged with Nikon Eclipse 90i microscope, and images were captured with Nikon 12.7MP digital Sight DS-Ri1 color camera as described previously (31).
Western blotting
10T1/2 cells were cultured in DMEM and treated with or without TGF-β or other factors as needed. Total proteins were extracted as described previously (32). Nuclear and cytoplasmic proteins were extracted using nuclear extraction kit (EMD Millipore) according to the manufacturer's instructions. Protein concentration was measured using BCA Protein Assay reagent (Thermo Scientific). Protein lysates were resolved by SDS-PAGE and transferred to PVDF membrane (Bio-Rad) or nitrocellulose membrane (Bio-Rad) that was blocked with 5% nonfat dry milk or 5% BSA separately. Membranes were then incubated with the following primary antibodies in blocking buffers at room temperature for 2–3 h or in 4 °C overnight: anti-phospho-Smad3 (Cell Signaling Technology, 9520s), anti-PPM1A (Thermo Scientific, PA5–29275), anti-αSMA (Sigma-Aldrich, A2547), anti-SM22α (Abcam, ab10135), anti-calponin (Abcam, ab46794), anti-α-tubulin (Sigma-Aldrich, T9026), and anti-GAPDH (Sigma-Aldrich, G8795). Then, HRP-conjugated or immunofluorescent secondary antibodies were incubated with the membranes for 1 h. The protein expression levels were detected with enhanced chemiluminescence (EMD Millipore) or scanned by Odyssey fluorescence scanner (LI-COR Biosciences) (33).
Promoter-reporter luciferase assay
α-SMA or SM22α luciferase promoter constructs were transfected into 10T1/2 cells with or without other plasmids using Lipofectamine LTX (Life Technologies) as described previously (34). 24 h after the transfection, 10T1/2 cells were starved for 2 days before TGF-β treatment for 8 h. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega).
Statistical analysis
All values are expressed as mean ± S.E. Data were evaluated with a two-tailed, unpaired, Student's t test or compared by one-way analysis of variance (ANOVA) followed by Fisher exact test. A p value <0.05 was considered statistically significant.
Author contributions
K. D., X. G., W. C., A. C. H., and S.-Y. C. data curation; K. D., W. C., A. C. H., and S.-Y. C. formal analysis; K. D. validation; K. D. and X. G. investigation; K. D., A. C. H., Q. S., J.-F. C., and S.-Y. C. methodology; K. D. writing-original draft; K. D., A. C. H., J.-F. C., and S.-Y. C. writing-review and editing; A. C. H., J.-F. C., and S.-Y. C. conceptualization; A. C. H., J.-F. C., and S.-Y. C. supervision; A. C. H., J.-F. C., and S.-Y. C. funding acquisition; A. C. H., J.-F. C., and S.-Y. C. project administration.
This work was supported by National Institutes of Health Grants HL119053, HL123302, and HL135854 (to S. Y. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- SMC
- smooth muscle cell
- Ctrl
- control
- TRITC
- tetramethylrhodamine isothiocyanate.
References
- 1. Owens G. K., Kumar M. S., and Wamhoff B. R. (2004) Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801 10.1152/physrev.00041.2003 [DOI] [PubMed] [Google Scholar]
- 2. Majesky M. W. (2007) Developmental basis of vascular smooth muscle diversity. Arterioscler. Thromb. Vasc. Biol. 27, 1248–1258 10.1161/ATVBAHA.107.141069 [DOI] [PubMed] [Google Scholar]
- 3. Majesky M. W., Dong X. R., Regan J. N., and Hoglund V. J. (2011) Vascular smooth muscle progenitor cells: Building and repairing blood vessels. Circ. Res. 108, 365–377 10.1161/CIRCRESAHA.110.223800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferreira L. S., Gerecht S., Shieh H. F., Watson N., Rupnick M. A., Dallabrida S. M., Vunjak-Novakovic G., and Langer R. (2007) Vascular progenitor cells isolated from human embryonic stem cells give rise to endothelial and smooth muscle–like cells and form vascular networks in vivo. Circ. Res. 101, 286–294 10.1161/CIRCRESAHA.107.150201 [DOI] [PubMed] [Google Scholar]
- 5. Manabe I., and Owens G. K. (2001) Recruitment of serum response factor and hyperacetylation of histones at smooth muscle–specific regulatory regions during differentiation of a novel P19-derived in vitro smooth muscle differentiation system. Circ. Res. 88, 1127–1134 10.1161/hh1101.091339 [DOI] [PubMed] [Google Scholar]
- 6. Hirschi K. K., Rohovsky S. A., and D'Amore P. A. (1998) PDGF, TGF-beta, and heterotypic cell-cell interactions mediate endothelial cell-induced recruitment of 10T1/2 cells and their differentiation to a smooth muscle fate. J. Cell Biol. 141, 805–814 10.1083/jcb.141.3.805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Candia A. F., Hu J., Crosby J., Lalley P. A., Noden D., Nadeau J. H., and Wright C. V. (1992) Mox-1 and Mox-2 define a novel homeobox gene subfamily and are differentially expressed during early mesodermal patterning in mouse embryos. Development 116, 1123–1136 [DOI] [PubMed] [Google Scholar]
- 8. Mankoo B. S., Skuntz S., Harrigan I., Grigorieva E., Candia A., Wright C. V., Arnheiter H., and Pachnis V. (2003) The concerted action of Meox homeobox genes is required upstream of genetic pathways essential for the formation, patterning and differentiation of somites. Development 130, 4655–4664 10.1242/dev.00687 [DOI] [PubMed] [Google Scholar]
- 9. Douville J. M., Cheung D. Y., Herbert K. L., Moffatt T., and Wigle J. T. (2011) Mechanisms of MEOX1 and MEOX2 regulation of the cyclin dependent kinase inhibitors p21 and p16 in vascular endothelial cells. PLoS One 6, e29099 10.1371/journal.pone.0029099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wasteson P., Johansson B. R., Jukkola T., Breuer S., Akyürek L. M., Partanen J., and Lindahl P. (2008) Developmental origin of smooth muscle cells in the descending aorta in mice. Development 135, 1823–1832 10.1242/dev.020958 [DOI] [PubMed] [Google Scholar]
- 11. Nguyen P. D., Hollway G. E., Sonntag C., Miles L. B., Hall T. E., Berger S., Fernandez K. J., Gurevich D. B., Cole N. J., Alaei S., Ramialison M., Sutherland R. L., Polo J. M., Lieschke G. J., and Currie P. D. (2014) Haematopoietic stem cell induction by somite-derived endothelial cells controlled by meox1. Nature 512, 314–318 10.1038/nature13678 [DOI] [PubMed] [Google Scholar]
- 12. Mack C. P., Somlyo A. V., Hautmann M., Somlyo A. P., and Owens G. K. (2001) Smooth muscle differentiation marker gene expression is regulated by RhoA-mediated actin polymerization. J. Biol. Chem. 276, 341–347 10.1074/jbc.M005505200 [DOI] [PubMed] [Google Scholar]
- 13. Miano J. M. (2003) Serum response factor: Toggling between disparate programs of gene expression. J. Mol. Cell Cardiol. 35, 577–593 10.1016/S0022-2828(03)00110-X [DOI] [PubMed] [Google Scholar]
- 14. Mack C. P., and Hinson J. S. (2005) Regulation of smooth muscle differentiation by the myocardin family of serum response factor co-factors. J. Thromb. Haemost. 3, 1976–1984 10.1111/j.1538-7836.2005.01316.x [DOI] [PubMed] [Google Scholar]
- 15. Xie W. B., Li Z., Miano J. M., Long X., and Chen S. Y. (2011) Smad3-mediated myocardin silencing: A novel mechanism governing the initiation of smooth muscle differentiation. J. Biol. Chem. 286, 15050–15057 10.1074/jbc.M110.202747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi X., DiRenzo D., Guo L. W., Franco S. R., Wang B., Seedial S., and Kent K. C. (2014) TGF-β/Smad3 stimulates stem cell/developmental gene expression and vascular smooth muscle cell de-differentiation. PLoS One 9, e93995 10.1371/journal.pone.0093995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hill C. S. (2009) Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 19, 36–46 10.1038/cr.2008.325 [DOI] [PubMed] [Google Scholar]
- 18. Massagué J., and Wotton D. (2000) Transcriptional control by the TGF-β/Smad signaling system. EMBO J. 19, 1745–1754 10.1093/emboj/19.8.1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lin X., Duan X., Liang Y. Y., Su Y., Wrighton K. H., Long J., Hu M., Davis C. M., Wang J., Brunicardi F. C., Shi Y., Chen Y. G., Meng A., and Feng X. H. (2006) PPM1A functions as a Smad phosphatase to terminate TGFβ signaling. Cell 125, 915–928 10.1016/j.cell.2006.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mumby M. C., and Walter G. (1993) Protein serine/threonine phosphatases: Structure, regulation, and functions in cell growth. Physiol. Rev. 73, 673–699 10.1152/physrev.1993.73.4.673 [DOI] [PubMed] [Google Scholar]
- 21. Shi Y. (2009) Serine/threonine phosphatases: Mechanism through structure. Cell 139, 468–484 10.1016/j.cell.2009.10.006 [DOI] [PubMed] [Google Scholar]
- 22. Chen S., Crawford M., Day R. M., Briones V. R., Leader J. E., Jose P. A., and Lechleider R. J. (2006) RhoA modulates Smad signaling during transforming growth factor-β–induced smooth muscle differentiation. J. Biol. Chem. 281, 1765–1770 10.1074/jbc.M507771200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kurpinski K., Lam H., Chu J., Wang A., Kim A., Tsay E., Agrawal S., Schaffer D. V., and Li S. (2010) Transforming growth factor-β and notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells 28, 734–742 10.1002/stem.319 [DOI] [PubMed] [Google Scholar]
- 24. Xie W. B., Li Z., Shi N., Guo X., Tang J., Ju W., Han J., Liu T., Bottinger E. P., Chai Y., Jose P. A., and Chen S. Y. (2013) Smad2 and myocardin-related transcription factor B cooperatively regulate vascular smooth muscle differentiation from neural crest cells. Circ. Res. 113, e76–e86 10.1161/CIRCRESAHA.113.301921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen S., Kulik M., and Lechleider R. J. (2003) Smad proteins regulate transcriptional induction of the SM22α gene by TGF-β. Nucleic Acids Res. 31, 1302–1310 10.1093/nar/gkg224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kirilenko P., He G., Mankoo B. S., Mallo M., Jones R., and Bobola N. (2011) Transient activation of meox1 is an early component of the gene regulatory network downstream of hoxa2. Mol. Cell Biol. 31, 1301–1308 10.1128/MCB.00705-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo J., Deng Z. L., Luo X., Tang N., Song W. X., Chen J., Sharff K. A., Luu H. H., Haydon R. C., Kinzler K. W., Vogelstein B., and He T. C. (2007) A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protoc. 2, 1236–1247 10.1038/nprot.2007.135 [DOI] [PubMed] [Google Scholar]
- 28. Shao Q., Herrlinger S., Yang S. L., Lai F., Moore J. M., Brindley M. A., and Chen J. F. (2016) Zika virus infection disrupts neurovascular development and results in postnatal microcephaly with brain damage. Development 143, 4127–4136 10.1242/dev.143768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buckley S. M., Waddington S. N., Jezzard S., Bergau A., Themis M., MacVinish L. J., Cuthbert A. W., Colledge W. H., and Coutelle C. (2008) Intra-amniotic delivery of CFTR-expressing adenovirus does not reverse cystic fibrosis phenotype in inbred CFTR-knockout mice. Mol. Ther. 16, 819–824 10.1038/mt.2008.26 [DOI] [PubMed] [Google Scholar]
- 30. Chen S., and Lechleider R. J. (2004) Transforming growth factor-β–induced differentiation of smooth muscle from a neural crest stem cell line. Circ. Res. 94, 1195–1202 10.1161/01.RES.0000126897.41658.81 [DOI] [PubMed] [Google Scholar]
- 31. Shi N., Xie W. B., and Chen S. Y. (2012) Cell division cycle 7 is a novel regulator of transforming growth factor-β–induced smooth muscle cell differentiation. J. Biol. Chem. 287, 6860–6867 10.1074/jbc.M111.306209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shi N., and Chen S. Y. (2013) Cell division cycle 7 mediates transforming growth factor-β–induced smooth muscle maturation through activation of myocardin gene transcription. J. Biol. Chem. 288, 34336–34342 10.1074/jbc.M113.498238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shirai T., Nazarewicz R. R., Wallis B. B., Yanes R. E., Watanabe R., Hilhorst M., Tian L., Harrison D. G., Giacomini J. C., Assimes T. L., Goronzy J. J., and Weyand C. M. (2016) The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 213, 337–354 10.1084/jem.20150900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li F., Luo Z., Huang W., Lu Q., Wilcox C. S., Jose P. A., and Chen S. (2007) Response gene to complement 32, a novel regulator for transforming growth factor-β–induced smooth muscle differentiation of neural crest cells. J. Biol. Chem. 282, 10133–10137 10.1074/jbc.C600225200 [DOI] [PubMed] [Google Scholar]
- 35. Wu B., Zhang L., Zhu Y. H., Zhang Y. E., Zheng F., Yang J. Y., Guo L. Y., Li X. Y., Wang L., Tang J. M., Chen S. Y., and Wang J. N. (2018) Mesoderm/mesenchyme homeobox gene l promotes vascular smooth muscle cell phenotypic modulation and vascular remodeling. Int. J. Cardiol. 251, 82–89 10.1016/j.ijcard.2017.10.098 [DOI] [PubMed] [Google Scholar]