

Abstract
Endogenous metabolism, environmental exposure, and cancer chemotherapy can lead to alkylation of DNA. It has been well documented that, among the different DNA alkylation products, minor-groove O2-alkylthymidine (O2-alkyldT) lesions are inefficiently repaired. In the present study, we examined how seven O2-alkyldT lesions, with the alkyl group being a Me, Et, nPr, iPr, nBu, iBu, or sBu, are recognized by the DNA replication machinery in human cells. We found that the replication bypass efficiencies of these lesions decrease with increasing length of the alkyl chain, and that these lesions induce substantial frequencies of T→A and T→G mutations. Replication experiments using isogenic cells deficient in specific translesion synthesis (TLS) DNA polymerases revealed that the absence of polymerase η or polymerase ζ, but not polymerase κ or polymerase ι, significantly decreased both the bypass efficiencies and the mutation frequencies for those O2-alkyldT lesions carrying a straight-chain alkyl group. Moreover, the mutagenic properties of the O2-alkyldT lesions were influenced by the length and topology of the alkyl chain and by TLS polymerases. Together, our results provide important new knowledge about the cytotoxic and mutagenic properties of O2-alkyldT lesions, and illustrate the roles of TLS polymerases in replicative bypass of these lesions in human cells.
Keywords: DNA replication, DNA damage, DNA polymerase, mutagenesis, mutagenesis mechanism, alkylating agent, DNA alkylation, O2-alkylthymidine, translesion synthesis, translesion synthesis polymerase, DNA adduct, polymerase
Introduction
DNA is intrinsically unstable, where it may undergo spontaneous deamination or depurination under physiological conditions (1). In addition, endogenous metabolic processes and environmental exposure can give rise to covalent modifications of DNA (2). The resulting DNA lesions, if left unrepaired, may compromise genomic integrity by impeding DNA replication and transcription and eliciting mutations in these processes, which may ultimately lead to the development of cancer and other human diseases (3).
Alkylation is a common type of DNA damage, and it also constitutes the major mechanism of action for some widely prescribed anti-cancer drugs (4–6). Alkylating agents can react with DNA directly or following metabolic activation, which results in the conjugation of DNA with varying sizes of alkyl groups. For instance, a number of anticancer drugs, including dacarbazine, procarbazine, streptozotocin, and temozolomide, can give rise to methylation of DNA (7). In addition, metabolites of some tobacco-derived N-nitrosamines can lead to the conjugation of bulky pyridyloxobutyl (POB)2 and pyridylhydroxybutyl groups with nucleobases and the phosphate backbone of DNA (8–10).
Among the plethora of alkylated DNA damage products, minor-groove O2-alkylthymidine (O2-alkyldT) lesions are known to be poorly repaired. In this respect, the POB and pyridylhydroxybutyl derivatives of guanine and thymine could be detected in various tissues of rats exposed with tobacco-derived N-nitrosamines (11–14). Nevertheless, O2-POBdT could accumulate at markedly higher levels than O6-POBdG, and substantial frequencies of mutations at A:T base pairs could be detected in genomic DNA of Chinese hamster ovary cells treated with NNK (11, 14). Likewise, tissues of rats treated with DNA ethylating agents displayed much higher levels of O2-EtdT than O6-EtdG (15–17), and lymphocyte DNA of smokers exhibited elevated levels of O2-EtdT relative to that of nonsmokers (18). Together, these findings suggest that the minor-groove O2-alkyldT lesions are recalcitrant to repair (15–17); hence, it is important to examine how these lesions are recognized by DNA replication machinery.
Some replication experiments have been conducted for the O2-alkyldT lesions. Zhai et al. (19, 20) showed that O2-alkyldT lesions strongly compromised the efficiency and fidelity of DNA replication in Escherichia coli cells. Additionally, the three SOS-induced DNA polymerases exert distinct effects on replicative bypass of these lesions, where depletion of Pol V, but not Pol II or Pol IV, elicits significant diminutions in bypass efficiencies of these lesions in E. coli cells. Moreover, both Pol IV and Pol V are essential for the misinsertion of dCMP opposite these lesions, whereas misincorporation of dTMP opposite the lesions requires only Pol V. A recent study by Basu and co-workers (21) showed that O2-MedT and O2-POBdT were strong impediments to DNA replication and were highly mutagenic in HEK293T cells. Replication experiments were also performed in the same cells following knockdown of individual translesion synthesis (TLS) DNA polymerases (21). However, sometimes unequivocal conclusions about the involvement of a specific TLS polymerase in bypassing a given lesion could not be reached because of the inability of the siRNA technology in depleting completely these polymerases (21, 22).
In the present study, we aim to achieve a comprehensive understanding about the recognition of minor-groove O2-alkyldT lesions by DNA replication machinery in human cells. We introduced, into double-stranded plasmids, seven O2-alkyldT lesions with varying sizes and structures of the alkyl group (Fig. 1) and investigated how these lesions impede DNA replication and induce mutations in HEK293T cells. We also assessed the roles of TLS DNA polymerases in bypassing these lesions by conducting replication experiments in the isogenic HEK293T cells where individual TLS polymerases were genetically ablated by the CRISPR/Cas9 method (23).
Figure 1.
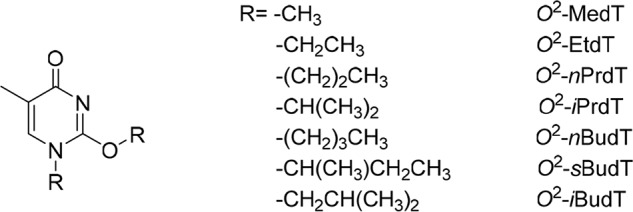
The structures of the O2-alkyldT lesions examined in the present study.
Results
The primary objectives of the present study were to assess comprehensively the extents to which the O2-alkyldT lesions with varying sizes and structures of the alkyl group perturb the efficiencies and fidelities of DNA replication in cultured human cells, and to establish the roles of TLS DNA polymerases in replication past these lesions.
We first constructed double-stranded shuttle vectors containing a site-specifically inserted O2-alkyldT lesion, as well as the corresponding nonlesion control vector with an unmodified dT at the lesion site. The lesion-carrying or the undamaged control vectors were mixed individually with the damage-free competitor vector at fixed molar ratios and cotransfected into human cells. The competitor vector, which contains three additional nucleotides relative to the control or lesion-harboring vector, serves as the internal reference for determining the degrees to which the O2-alkyldT lesions block DNA replication in human cells. The progeny genomes were extracted from human cells at 24 h following the transfection, and the residual unreplicated plasmids were removed by treatment with DpnI and exonuclease III. The progeny plasmids were then amplified by PCR using a pair of primers spanning the original lesion site. In this respect, one of the primers (P1) contains a G at the 3′ terminus corresponding to the C/C mismatch site (Fig. 2b), which permits the selective amplification of the progeny genomes arising from the replication of the bottom, lesion-situated strand of the plasmids under suitable conditions (24). The P1 primer also carries a C/A mismatch three bases from its 3′-end, which improves the specificity of strand-specific PCR, as described elsewhere (25). The ensuing PCR products were digested with appropriate restriction enzymes, i.e. NcoI and SfaNI (Fig. 3a), and the digestion products were subjected to LC-MS/MS and PAGE analyses (Fig. 3, b and c and Figs. S1–S5).
Figure 2.
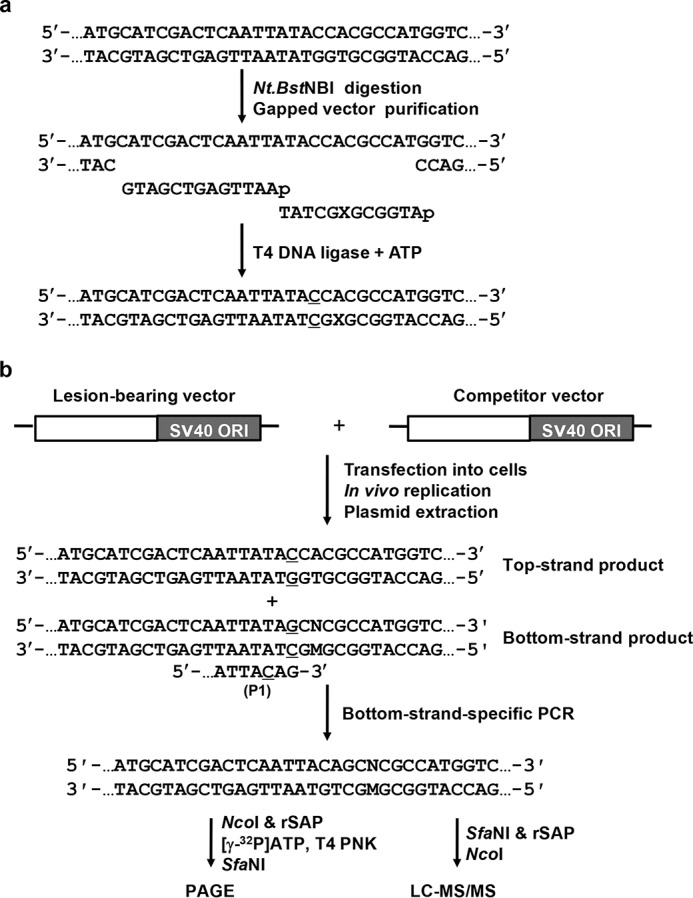
a and b, schematic diagrams outlining the procedures for the preparation of the lesion-bearing plasmid (a) and the single-stranded PCR-competitive replication and adduct bypass (SSPCR-CRAB) assay (b) (22). SV40 ORI and X indicate SV40 replication origin and the site where the O2-alkyldT lesions are situated, respectively. The C/C mismatch site is underlined. P1 represents one of the PCR primers, 5′-GCTAGCGGATGCATCGACTCAATTACAG-3′, which contains a G at the 3′ terminus corresponding to the C/C mismatch site of the lesion-bearing genome. The P1 primer also carries a C/A mismatch three bases away from its 3′-end to improve PCR specificity (the 3′ portion of the P1 primer is shown with the mismatched C being underlined). M and N designate the nucleotide incorporated at the lesion site during DNA replication and the paired nucleotide of M in the complementary strand, respectively.
Figure 3.
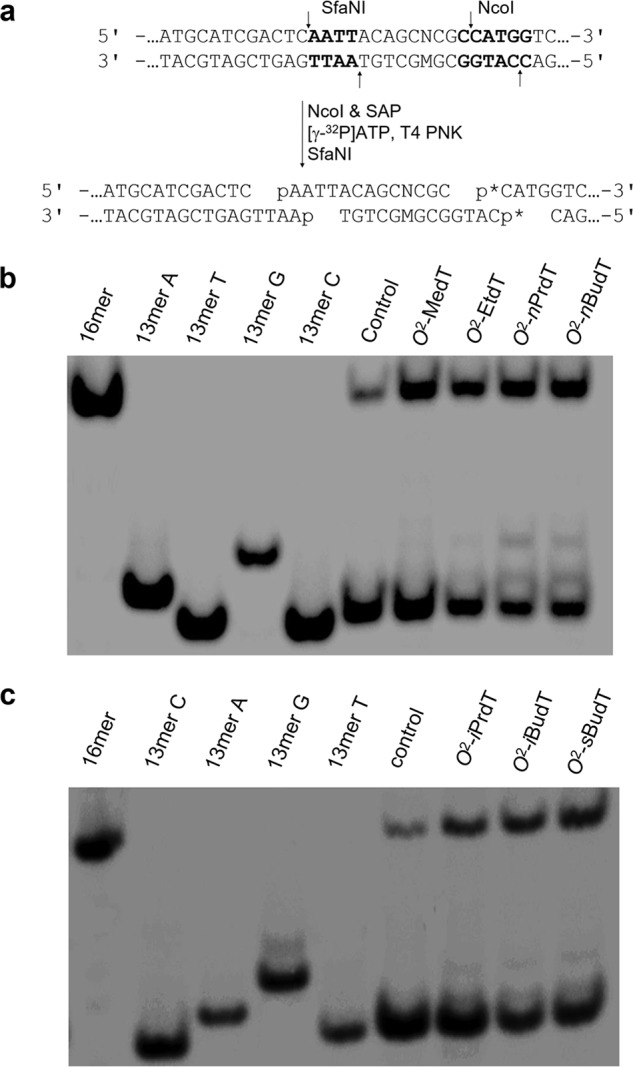
Restriction digestion and post-labeling method for determining the bypass efficiencies and mutation frequencies of the O2-alkyldT lesions in HEK293T cells. a, restriction digestion with NcoI and SfaNI and post-labeling assay (p* indicates a 32P-labeled phosphate group). The recognition sequences for the restriction enzymes are highlighted in bold, and restriction cleavage sites are designated with arrows. b and c, representative gel images showing the NcoI/SfaNI-produced restriction fragments of interest. The restriction fragment arising from the competitor vector, i.e. d(CATGGCGATATGCTGT), is designated as 16mer; 13mer C, 13mer A, 13mer G, and 13mer T indicate the standard synthetic ODNs d(CATGGCGNGCTGT), where N is C, A, G and T, respectively.
Our results showed that, in WT HEK293T cells, the bypass efficiencies were 60, 43, 37, and 29% for O2-MedT, O2-EtdT, O2-nPrdT, and O2-nBudT, respectively. Hence, the bypass efficiencies decrease with the chain length for those O2-alkyldT lesions bearing a straight-chain alkyl group. The bypass efficiencies were lower for those O2-alkyldT lesions harboring a branched-chain alkyl group, as reflected by the bypass efficiencies of 34, 14, and 14% for O2-iPrdT, O2-iBudT, and O2-sBudT, respectively (Fig. 4a). These results are reminiscent of previous findings made for the replicative bypass of these lesions in E. coli cells (20).
Figure 4.
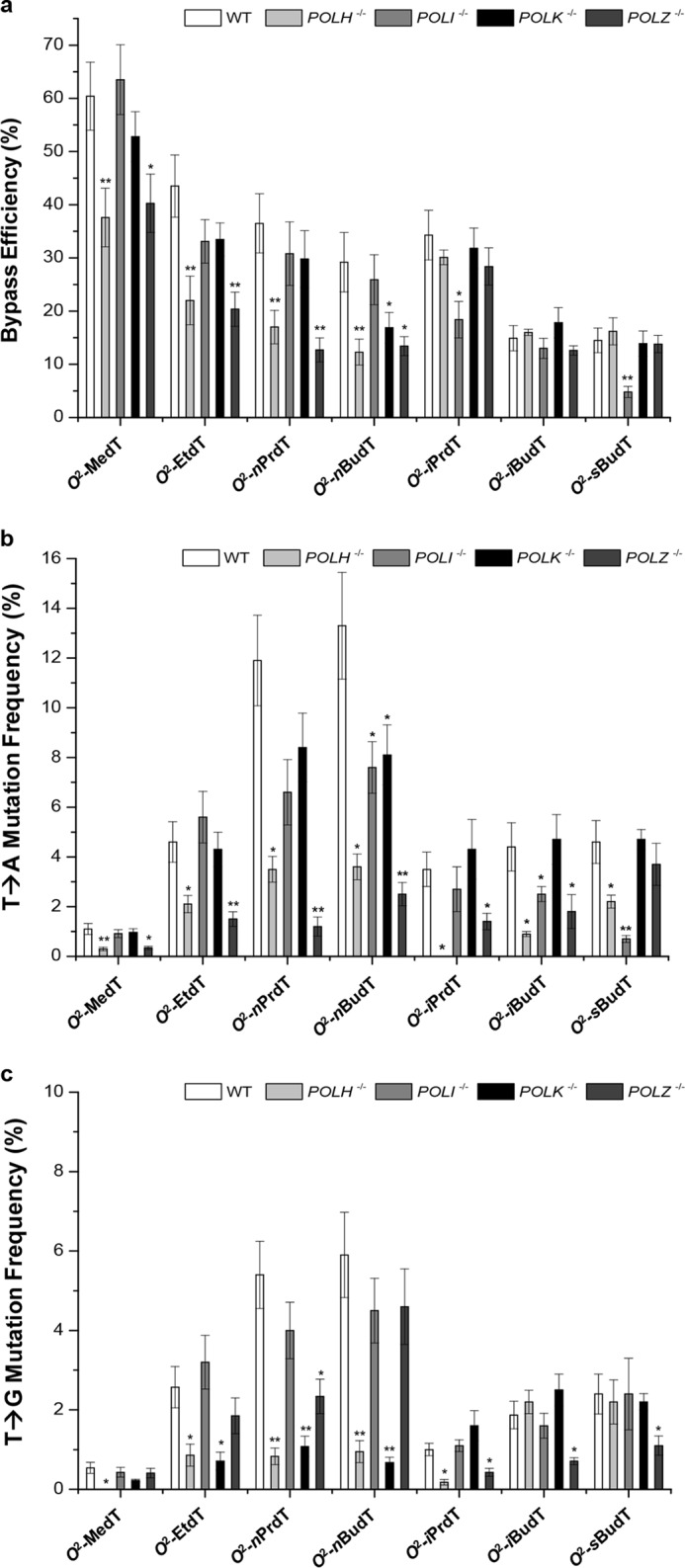
The bypass efficiencies and mutation frequencies of the O2-alkyldT lesions in HEK293T cells and the isogenic cells where the TLS polymerases were individually depleted by CRISPR/Cas9. a–c, shown are the bypass efficiencies (a) and the frequencies for the T→A (b) and T→G (c) mutations observed for the O2-alkyldT lesions. The data represent the mean and S.D. of results from three independent replication experiments. **, 0.001 < p < 0.01; *, 0.01 < p < 0.05. The p values were calculated using two-tailed, unpaired t test, and the values referred to the comparisons between WT and TLS polymerase-deficient HEK293T cells.
We next asked how the replicative bypass of the O2-alkyldT lesions in mammalian cells is affected by depletion of TLS DNA polymerases by conducting replication experiments with the use of isogenic HEK293T cells depleted of individual TLS polymerases. Our results showed that in general the depletion of Pol ι or Pol κ did not appreciably impact the bypass efficiencies of the O2-alkyldT lesions except O2-iPrdT and O2-sBudT, for which loss of Pol ι led to significant decreases in bypass efficiencies (Fig. 4a). On the other hand, individual depletion of Pol η or Pol ζ resulted in significant decreases (by ∼16–30%) in bypass efficiencies for all the O2-alkyldT lesions carrying a straight-chain alkyl group (Fig. 4a), supporting that Pol η and Pol ζ play crucial roles in bypassing these O2-alkyldT lesions. In contrast, individual depletion of Pol η or Pol ζ did not appreciably perturb the replicative bypass for the three O2-alkyldT lesions possessing a branched-chain alkyl functionality (Fig. 4a).
To determine the mutagenic properties of the O2-alkyldT lesions, we identified the mutant products by employing LC-MS/MS and PAGE analyses of restriction fragments of PCR products from progeny genomes. In the LC-MS/MS method, we monitored the fragmentations of the [M–3H]3− ions of d(AATTACAGCMCGC), with “M” designating the nucleotides inserted opposite the initial damage site. Our LC-MS/MS data revealed that all seven O2-alkyldT lesions exhibit similar miscoding properties, where T→A and T→G transversions, but not T→C transition, were observed (representative LC-MS and MS/MS data are shown in Figs. S3–S5). With the use of 30% native PAGE, we were able to resolve 5′-32P–labeled d(p*CATGGCGTGCTGT) (nonmutagenic product, 13-mer T) from the corresponding products carrying a T→A or T→G mutation, i.e. d(p*CATGGCGAGCTGT) (13-mer A) and d(p*CATGGCGGGCTGT) (13-mer G) (Fig. 3, b and c and Figs. S1 and S2). However, the nonmutagenic product comigrated with the corresponding product with a T→C mutation at the lesion site, i.e. d(p*CATGGCGCGCTGT) (13-mer C) (Fig. 3, b and c and Figs. S1 and S2). Because of the lack of T→C mutation arising from replication past the O2-alkyldT lesions, we determined the frequencies of the lesion-induced mutations by using native PAGE analysis.
Similar to what was observed previously in E. coli cells (20), the major type of mutation induced by the O2-alkyldT lesions in HEK293T cells was T→A transversion, which was detected at frequencies of 1–13% and was accompanied with lower frequencies (0.5–6%) of T→G substitution (Fig. 4, b and c). In addition, the frequencies for T→A and T→G mutations for the O2-alkyldT lesions with straight-chain alkyl group rise with the increasing length of the alkyl group (Fig. 4, b and c). These elevations in mutation frequencies were associated with concomitant decreases in bypass efficiencies for the lesions (see above). Thus, the increases in chain length of the alkyl group adducted to the O2-position of thymine led to greater diminutions in both the efficiency and fidelity of replication across the O2-alkyldT lesions in human cells.
We next examined the mutagenic properties of the O2-alkyldT lesions in HEK293T cells that are deficient in Pol η, Pol ι, Pol κ, or Pol ζ and compared the results with what we obtained for the parental HEK293T cells. Our results revealed that Pol η and Pol ζ played an important role in the misincorporation of dTMP opposite the O2-alkyldT lesions regardless of the alkyl chain being straight or branched, as reflected by the finding that the depletion of either polymerase resulted in a significant drop in T→A mutation (Fig. 4b). The only exception was that the loss of Pol ζ did not alter the frequency of T→A mutation for O2-sBudT (Fig. 4b). Likewise, genetic ablation of Pol η led to substantial diminutions in T→G mutation for all O2-alkyldT lesions except O2-iBudT and O2-sBudT (Fig. 4c), indicating a role of Pol η in the misincorporation of dCMP opposite these O2-alkyldT lesions. Moreover, for those lesions bearing straight-chain alkyl groups, depletion of Pol κ led to marked reductions in T→G mutation (Fig. 4c), whereas Pol ζ functions in dCMP misincorporation opposite those O2-alkyldT lesions with branched-chain alkyl groups (Fig. 4c). On the other hand, Pol ι played an important role in dTMP misinsertion opposite the sites of those O2-alkyldT lesions carrying a large alkyl group (i.e. O2-nBudT, O2-iBudT, and O2-sBudT) (Fig. 4b). Cumulatively, these results demonstrated that the effects of depletion of TLS DNA polymerases on the mutagenic properties of the O2-alkyldT lesions are highly dependent on the chemical structures (i.e. chain length and chain topology) of the alkyl group being conjugated with the O2-position of thymidine, and are further modulated by TLS DNA polymerases.
Discussion
In this study, we assessed comprehensively the cytotoxic and mutagenic properties of O2-alkyldT lesions in HEK293T cells and the isogenic cells where the individual TLS polymerases were depleted by the CRISPR/Cas9 genome editing method. Our results revealed that, similar to what we observed in E. coli cells (20), the O2-alkyldT lesions inhibit strongly DNA replication in mammalian cells, with the blockage effect increasing with the size and branching of the alkyl groups (Fig. 4a). This result underscores the increased difficulty experienced by the DNA polymerases in accommodating those O2-alkyldT lesions with longer and bulkier alkyl chain into their active sites, which confers reduced efficiencies in nucleotide incorporation at or near the lesion site. In this context, it is worth noting that the differential rate of repair of the O2-alkyldT lesions may also contribute in part to the differences in the observed replication bypass efficiencies. In addition, we found that the bypass efficiencies for all the O2-alkyldT lesions carrying a straight chain alkyl group were significantly reduced in cells depleted of Pol η or Pol ζ (Fig. 4a), suggesting that these two TLS polymerases play important roles in bypassing these lesions in vivo. This is reminiscent of our previous finding that Pol V, the E. coli ortholog of human Pol η, constitutes the major TLS polymerase for bypassing these lesions (20), and is consistent with results obtained from in vitro replication experiments showing that human Pol η was capable of bypassing readily these lesions in template DNA (26). In addition, Pol ζ is known to participate in extension step of the TLS after nucleotide incorporation opposite the lesion site (27). Together, our results suggest that Pol η and Pol ζ may cooperate in bypassing the O2-alkyldT lesions with a straight-chain alkyl group, with Pol η and Pol ζ being involved in the insertion and extension steps of the lesion bypass, respectively. It will be important to assess, in the future, how replication across these lesions in human cells are affected by simultaneous depletion of both Pol η and Pol ζ, and biochemically how Pol η and Pol ζ may function together in bypassing the O2-alkyldT lesions in vitro.
Our results demonstrated that the O2-alkyldT lesions primarily directed the misincorporations of pyrimidine nucleotides (i.e. dTMP and dCMP), and replicative bypass of these lesions yields T→A and T→G mutations. This finding is in line with the notion that the incorporation of an alkyl group to the O2-position of thymine may render the nucleobase unfavorable in pairing with any of the four canonical nucleobases (20). This observation is also in agreement with the finding that T→A and T→G transversions constitute the two main types of mutations induced by these lesions in E. coli (20). Different from what we found in E. coli cells, we did not observe T→C mutation for any of the O2-alkyldT lesions in WT HEK293T cells or the isogenic cells depleted of any of the TLS polymerases (Fig. S3–S5), which could be attributed to the differential recognition of these lesions by replication machineries of human and E. coli cells.
By conducting replication experiments in cells depleted of TLS polymerases, we showed that misincorporation of dTMP opposite O2-alkyldT and perhaps the subsequent extension beyond the lesion site require both Pol η and Pol ζ (Fig. 4, b and c), suggesting that Pol η and Pol ζ are the major polymerases responsible for dTMP misincorporation and subsequent extension beyond the O2-alkyldT lesions in human cells. In keeping with the previous finding that both Pol IV and Pol V were required for inducing T→G mutation (20), their orthologs in human cells, i.e. Pol κ and Pol η, were the major polymerases contributing to the induction of T→G mutation for the straight-chain O2-alkyldT lesions (Fig. 4c). On the other hand, Pol ζ was the only TLS polymerase found to be required for the T→G mutation induced by the three branched-chain O2-alkyldT lesions (Fig. 4c), although it remains unclear which polymerase might be involved in the dCMP misincorporation opposite these lesions.
Taken together, our systematic shuttle vector-based study on a group of structurally defined O2-alkyldT lesions provided important new insights into the impact of this under-investigated group of DNA lesions on the efficiency and accuracy of DNA replication in mammalian cells. The findings made in the present study unveiled that, in mammalian cells, the cytotoxic and mutagenic properties of these DNA lesions depend on the size and branching of the alkyl group and are further modulated by TLS DNA polymerases. Moreover, the significant inhibitory effects of the O2-alkyldT lesions on DNA replication, their strong mutagenic potentials, and their resistance to repair suggest that the O2-alkyldT lesions constitute a family of biologically important DNA lesions.
Experimental procedures
All chemicals, unless otherwise specified, were from Sigma-Aldrich or EMD Millipore. HFIP was obtained from TCI America (Portland, OR). [γ-32P]ATP was purchased from PerkinElmer, and all other enzymes were obtained from New England Biolabs (NEB) (Ipswich, MA). All unmodified oligodeoxyribonucleotides (ODNs) were obtained from Integrated DNA Technologies (Coralville, IA). The 12-mer ODNs harboring a site-specifically incorporated O2-alkyldT were synthesized previously (20). The identities and purities of all the lesion-harboring ODNs were confirmed by LC-MS and tandem MS (MS/MS) analyses prior to their insertion into double-stranded plasmids. HEK293T cells with POLH, POLI, POLK and POLZ genes, which encode DNA polymerases η, ι, κ, and ζ, respectively, being individually depleted by the CRISPR/Cas9 genomic editing method were described previously (23).
Construction of lesion-containing and lesion-free plasmids
The lesion-containing and lesion-free genomes were prepared according to the previously reported procedures (22, 23). The parent vector was subsequently digested with Nt.BstNBI, followed with removing the 25-mer ODN by annealing with a 25-mer complementary ODN in large excess to generate a gapped vector. The gapped vector was subsequently purified from the mixture by centrifugation using 100 kDa-cutoff ultracentrifugal filter units (EMD Millipore). The gap in the vector was filled with a 5′-phosphorylated 13-mer lesion-free ODN (5′-AATTGAGTCGATG-3′) and a 5′-phosphorylated 12-mer lesion-containing or lesion-free ODN (5′-ATGGCGXGCTAT-3′, where X = O2-alkyldT or dT) by using T4 DNA ligase in the presence of ATP (Fig. 2a). The successfully ligated, supercoiled plasmid was isolated from the ligation mixture by using agarose gel electrophoresis. The amounts of constructed lesion-containing vectors were normalized against that of the lesion-free competitor vector following published procedures (22, 23).
Cellular DNA replication and plasmid isolation
The lesion-bearing and the corresponding nonlesion control plasmids were individually premixed with the competitor genome at a molar ratio of 19:1 for all replication experiments. The HEK293T cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Invitrogen) and 100 units/ml penicillin and incubated at 37 °C in 5% CO2 atmosphere. The HEK293T cells and CRISPR/Cas9 genome-engineered cells (1 × 105) were seeded in a 24-well plate and cultured for 24 h before they were transfected with the aforementioned plasmid mixtures by using Lipofectamine 2000 following the manufacturer's recommended procedures, where a total of 300 ng of lesion and competitor or control and competitor genome mixture along with 500 ng of carrier plasmid were employed for each transfection. The cells were harvested at 24 h following the transfection, and the progeny genomes were isolated using Qiagen Spin kit (Qiagen, Valencia, CA) with minor modifications (28). The residual unreplicated plasmids were further digested with DpnI, followed by removing the resulting linear DNA with exonuclease III, as described elsewhere (29–31). Along this line, there were 25 DpnI recognition sites in the parental plasmid; therefore, digestion at any one of these sites would result in the degradation of the entire plasmid by exonuclease III and prevent the following PCR amplification of the parental vector.
PCR and PAGE analyses
The progeny genomes resulting from cellular replication were amplified by PCR with the use of GoTaq Hot Start DNA polymerase (Promega, Madison, WI). The two primers were 5′-GCTAGCGGATGCATCGACTCAATTACAG-3′ and 5′-GCTGATTATGATCTAGAGTTGCGGCCGC-3′, and the PCR amplifications started at 95 °C for 2 min, followed by 30 cycles at 95 °C for 30 s, 64 °C for 30 s, and 72 °C for 1 min, and a final extension at 72 °C for 5 min. The PCR products were purified using Cycle Pure Kit (Omega, Norcross, GA) and stored at −20 °C until use.
For PAGE analysis, a portion of the PCR products were treated with 5 units NcoI and 1 unit shrimp alkaline phosphatase at 37 °C in 10 μl of NEB buffer 3 for 1 h, and the shrimp alkaline phosphatase was subsequently deactivated by heating at 80 °C for 20 min. The above mixture was then treated with 5 units of T4 polynucleotide kinase in 15-μl NEB buffer 3 containing 5 mm DTT and ATP (10 pmol cold, premixed with 1.66 pmol [γ-32P]ATP). The reaction was continued at 37 °C for 30 min, followed by heating at 65 °C for 20 min to deactivate the polynucleotide kinase. To the reaction mixture was subsequently added 2 units of SfaNI in 5 μl NEB buffer 3, and the solution was incubated at 37 °C for 1.5 h, followed by quenching with 20 μl of formamide gel–loading buffer containing xylene cyanol FF and bromphenol blue dyes. The mixture was loaded onto 30% polyacrylamide gel (acrylamide/bisacrylamide, 19:1), and the gel band intensities were quantified by phosphorimager analysis.
The effects of DNA lesions on replication efficiency and fidelity were characterized by bypass efficiency and mutation frequency, respectively. The bypass efficiency was calculated as (lesion signal/competitor signal)/(nonlesion control signal/competitor signal) × 100%. The mutation frequency was determined from the percentage of the amount of mutagenic product among the total amounts of products formed from replication of the lesion-containing genome (32–34).
Identification of mutagenic products using LC-MS/MS
The PCR products were digested with 30 units SfaNI restriction endonuclease and 15 units shrimp alkaline phosphatase in 150 μl NEB buffer 3 at 37 °C for 2 h, followed by deactivation of the phosphatase at 80 °C for 20 min. To the mixture was subsequently added 50 units NcoI restriction endonuclease in 5 μl NEB buffer 3, and the solution was incubated at 37 °C for another 2 h. The resulting solution was extracted once with phenol/chloroform/isoamyl alcohol (25:24:1, v/v). To the aqueous layer were subsequently added 2.5 volumes of 100% ethanol and 0.1 volume of 3.0 m sodium acetate, and the solution was incubated at −20 °C overnight to precipitate the DNA. The DNA pellet was then dissolved in water for LC-MS/MS analysis. An Agilent Zorbax SB-C18 column (0.5 × 250 mm, 5 μm in particle size) was employed, and the gradient for LC-MS/MS analysis was 5 min of 5–20% methanol followed by 50 min of 20–45% methanol in 400 mm HFIP. The temperature for the ion-transport tube was maintained at 300 °C. The mass spectrometer was set up for monitoring the fragmentation of the [M–3H]3− ions of the 13-mer d(AATTACAGCMCGC), where “M” represents A, T, C or G. The fragment ions detected in MS/MS were manually assigned.
Author contributions
J. W. and Y. W. data curation; J. W. and Y. W. formal analysis; J. W. and P. W. investigation; J. W. and C. Y. methodology; J. W. and L. L. writing-original draft; P. W., L. L., and C. Y. resources; P. W. and Y. W. writing-review and editing; Y. W. conceptualization; Y. W. supervision; Y. W. funding acquisition; Y. W. project administration.
Supplementary Material
This work was supported by the National Institutes of Health Grant R01 ES025121 (to Y. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5.
- POB
- pyridyloxobutyl
- O2-alkyldT
- O2-alkylthymidine
- Pol
- polymerase
- NNK
- 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- TLS
- translesion synthesis
- ODN
- oligodeoxyribonucleotide
- NEB
- New England Biolabs
- HFIP
- 1,1,1,3,3,3-hexafluoro-2-propanol
References
- 1. Lindahl T. (1993) Instability and decay of the primary structure of DNA. Nature 362, 709–715 10.1038/362709a0 [DOI] [PubMed] [Google Scholar]
- 2. Liu S., and Wang Y. (2015) Mass spectrometry for the assessment of the occurrence and biological consequences of DNA adducts. Chem. Soc. Rev. 44, 7829–7854 10.1039/C5CS00316D [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., and Ellenberger T. (2006) DNA Repair and Mutagenesis, ASM Press, Washington, D.C. [Google Scholar]
- 4. Singer B., and Grunberger D. (1983) Molecular Biology of Mutagens and Carcinogens, Springer Science & Business Media, New York, NY [Google Scholar]
- 5. Sedgwick B., Bates P. A., Paik J., Jacobs S. C., and Lindahl T. (2007) Repair of alkylated DNA: Recent advances. DNA Repair 6, 429–442 10.1016/j.dnarep.2006.10.005 [DOI] [PubMed] [Google Scholar]
- 6. Helleday T., Petermann E., Lundin C., Hodgson B., and Sharma R. A. (2008) DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 8, 193–204 10.1038/nrc2342 [DOI] [PubMed] [Google Scholar]
- 7. Gerson S. L. (2004) MGMT: Its role in cancer aetiology and cancer therapeutics. Nat. Rev. Cancer 4, 296–307 10.1038/nrc1319 [DOI] [PubMed] [Google Scholar]
- 8. Wang L., Spratt T. E., Liu X.-K., Hecht S. S., Pegg A. E., and Peterson L. A. (1997) Pyridyloxobutyl adduct O6-[4-oxo-4-(3-pyridyl) butyl] guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O6-alkylguanine-DNA alkyltransferase. Chem. Res. Toxicol. 10, 562–567 10.1021/tx9602067 [DOI] [PubMed] [Google Scholar]
- 9. Wang M., Cheng G., Sturla S. J., Shi Y., McIntee E. J., Villalta P. W., Upadhyaya P., and Hecht S. S. (2003) Identification of adducts formed by pyridyloxobutylation of deoxyguanosine and DNA by 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone, a chemically activated form of tobacco specific carcinogens. Chem. Res. Toxicol. 16, 616–626 10.1021/tx034003b [DOI] [PubMed] [Google Scholar]
- 10. Upadhyaya P., Sturla S. J., Tretyakova N., Ziegel R., Villalta P. W., Wang M., and Hecht S. S. (2003) Identification of adducts produced by the reaction of 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanol with deoxyguanosine and DNA. Chem. Res. Toxicol. 16, 180–190 10.1021/tx0256376 [DOI] [PubMed] [Google Scholar]
- 11. Lao Y., Yu N., Kassie F., Villalta P. W., and Hecht S. S. (2007) Analysis of pyridyloxobutyl DNA adducts in F344 rats chronically treated with (R)-and (S)-N′-nitrosonornicotine. Chem. Res. Toxicol. 20, 246–256 10.1021/tx060208j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lao Y., Yu N., Kassie F., Villalta P. W., and Hecht S. S. (2007) Formation and accumulation of pyridyloxobutyl DNA adducts in F344 rats chronically treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 20, 235–245 10.1021/tx060207r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Upadhyaya P., Kalscheuer S., Hochalter J. B., Villalta P. W., and Hecht S. S. (2008) Quantitation of pyridylhydroxybutyl-DNA adducts in liver and lung of F-344 rats treated with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and enantiomers of its metabolite 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol. Chem. Res. Toxicol. 21, 1468–1476 10.1021/tx8001109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li L., Perdigao J., Pegg A. E., Lao Y. B., Hecht S. S., Lindgren B. R., Reardon J. T., Sancar A., Wattenberg E. V., and Peterson L. A. (2009) The influence of repair pathways on the cytotoxicity and mutagenicity induced by the pyridyloxobutylation pathway of tobacco specific nitrosamines. Chem. Res. Toxicol. 22, 1464–1472 10.1021/tx9001572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Den Engelse L., De Graaf A., De Brij R.-J., and Menkveld G. J. (1987) O2-and O4-ethylthymine and the ethylphosphotriester dTp (Et) dT are highly persistent DNA modifications in slowly dividing tissues of the ethylnitrosourea-treated rat. Carcinogenesis 8, 751–757 10.1093/carcin/8.6.751 [DOI] [PubMed] [Google Scholar]
- 16. Brent T. P., Dolan M. E., Fraenkel-Conrat H., Hall J., Karran P., Laval L., Margison G. P., Montesano R., Pegg A. E., and Potter P. M. (1988) Repair of O-alkylpyrimidines in mammalian cells: a present consensus. Proc. Natl. Acad. Sci. U.S.A. 85, 1759–1762 10.1073/pnas.85.6.1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bronstein S. M., Skopek T. R., and Swenberg J. A. (1992) Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells. Cancer Res. 52, 2008–2011 [PubMed] [Google Scholar]
- 18. Chen H.-J. C., Wang Y.-C., and Lin W.-P. (2012) Analysis of ethylated thymidine adducts in human leukocyte DNA by stable isotope dilution nanoflow liquid chromatography—nanospray ionization tandem mass spectrometry. Anal. Chem. 84, 2521–2527 10.1021/ac203405y [DOI] [PubMed] [Google Scholar]
- 19. Zhai Q., Wang P., and Wang Y. (2014) Cytotoxic and mutagenic properties of regioisomeric O2-, N3- and O4-ethylthymidines in bacterial cells. Carcinogenesis 35, 2002–2006 10.1093/carcin/bgu085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhai Q., Wang P., Cai Q., and Wang Y. (2014) Syntheses and characterizations of the in vivo replicative bypass and mutagenic properties of the minor-groove O2-alkylthymidine lesions. Nucleic Acids Res. 42, 10529–10537 10.1093/nar/gku748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weerasooriya S., Jasti V. P., Bose A., Spratt T. E., and Basu A. K. (2015) Roles of translesion synthesis DNA polymerases in the potent mutagenicity of tobacco-specific nitrosamine-derived O2-alkylthymidines in human cells. DNA Repair 35, 63–70 10.1016/j.dnarep.2015.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. You C., Swanson A. L., Dai X., Yuan B., Wang J., and Wang Y. (2013) Translesion synthesis of 8, 5′-cyclopurine-2′-deoxynucleosides by DNA polymerases η, ι, and ζ. J. Biol. Chem. 288, 28548–28556 10.1074/jbc.M113.480459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu J., Li L., Wang P. C., You C. J., Williams N. L., and Wang Y. S. (2016) Translesion synthesis of O4-alkylthymidine lesions in human cells. Nucleic Acids Res. 44, 9256–9265 10.1093/nar/gkw662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Delaney J. C., and Essigmann J. M. (2006) Assays for determining lesion bypass efficiency and mutagenicity of site-specific DNA lesions in vivo. Methods Enzymol. 408, 1–15 10.1016/S0076-6879(06)08001-3 [DOI] [PubMed] [Google Scholar]
- 25. Newton C. R., Graham A., Heptinstall L. E., Powell S. J., Summers C., Kalsheker N., Smith J. C., and Markham A. F. (1989) Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res. 17, 2503–2516 10.1093/nar/17.7.2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams N. L., Wang P., and Wang Y. (2016) Replicative bypass of O2-alkylthymidine lesions in vitro. Chem. Res. Toxicol. 29, 1755–1761 10.1021/acs.chemrestox.6b00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shachar S., Ziv O., Avkin S., Adar S., Wittschieben J., Reißner T., Chaney S., Friedberg E. C., Wang Z., Carell T., Geacintov N., and Livneh Z. (2009) Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 28, 383–393 10.1038/emboj.2008.281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ziegler K., Bui T., Frisque R. J., Grandinetti A., and Nerurkar V. R. (2004) A rapid in vitro polyomavirus DNA replication assay. J. Virol. Methods 122, 123–127 10.1016/j.jviromet.2004.08.012 [DOI] [PubMed] [Google Scholar]
- 29. Burns J. A., Dreij K., Cartularo L., and Scicchitano D. A. (2010) O6-Methylguanine induces altered proteins at the level of transcription in human cells. Nucleic Acids Res. 38, 8178–8187 10.1093/nar/gkq706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanchez J. A., Marek D., and Wangh L. J. (1992) The efficiency and timing of plasmid DNA replication in Xenopus eggs: Correlations to the extent of prior chromatin assembly. J. Cell Sci. 103, 907–918 [DOI] [PubMed] [Google Scholar]
- 31. Taylor E. R., and Morgan I. M. (2003) A novel technique with enhanced detection and quantitation of HPV-16 E1- and E2-mediated DNA replication. Virology 315, 103–109 10.1016/S0042-6822(03)00588-9 [DOI] [PubMed] [Google Scholar]
- 32. You C., Dai X., Yuan B., Wang J., Wang J., Brooks P. J., Niedernhofer L. J., and Wang Y. (2012) A quantitative assay for assessing the effects of DNA lesions on transcription. Nat. Chem. Biol. 8, 817–822 10.1038/nchembio.1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. You C., Dai X., Yuan B., and Wang Y. (2012) Effects of 6-thioguanine and S6-methylthioguanine on transcription in vitro and in human cells. J. Biol. Chem. 287, 40915–40923 10.1074/jbc.M112.418681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delaney J. C., and Essigmann J. M. (2004) Mutagenesis, genotoxicity, and repair of 1-methyladenine, 3-alkylcytosines, 1-methylguanine, and 3-methylthymine in alkB Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 101, 14051–14056 10.1073/pnas.0403489101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.