

Abstract
Yersinia pseudotuberculosis is a Gram-negative bacterium and zoonotic pathogen responsible for a wide range of diseases, ranging from mild diarrhea, enterocolitis, lymphatic adenitis to persistent local inflammation. The Y. pseudotuberculosis invasin D (InvD) molecule belongs to the invasin (InvA)-type autotransporter proteins, but its structure and function remain unknown. In this study, we present the first crystal structure of InvD, analyzed its expression and function in a murine infection model, and identified its target molecule in the host. We found that InvD is induced at 37 °C and expressed in vivo 2–4 days after infection, indicating that InvD is a virulence factor. During infection, InvD was expressed in all parts of the intestinal tract, but not in deeper lymphoid tissues. The crystal structure of the C-terminal adhesion domain of InvD revealed a distinct Ig-related fold that, apart from the canonical β-sheets, comprises various modifications of and insertions into the Ig-core structure. We identified the Fab fragment of host-derived IgG/IgA antibodies as the target of the adhesion domain. Phage display panning and flow cytometry data further revealed that InvD exhibits a preferential binding specificity toward antibodies with VH3/VK1 variable domains and that it is specifically recruited to a subset of B cells. This finding suggests that InvD modulates Ig functions in the intestine and affects direct interactions with a subset of cell surface-exposed B-cell receptors. In summary, our results provide extensive insights into the structure of InvD and its specific interaction with the target molecule in the host.
Keywords: adhesin, protein chemistry, X-ray crystallography, phage display, small-angle X-ray scattering (SAXS), Fab-binding, invasin, InvD, Yersinia pseudotuberculosis
Introduction
Yersinia pseudotuberculosis is a Gram-negative bacterium belonging to the family of Enterobacteriaceae. This zoonotic pathogen is responsible for a wide range of diseases ranging from mild diarrhea, enterocolitis, lymphatic adenitis to sequelae such as reactive arthritis and iritis (1). Transmission of Y. pseudotuberculosis occurs via the oral–fecal route. Once in the intestine, the bacteria have to penetrate through the epithelial cell layer to colonize and invade their hosts. For this purpose Y. pseudotuberculosis utilizes a variety of multifunctional adhesins (2). Bacterial adhesins have in common that they target specific host cell receptors or components of the extracellular matrix. Therefore, adhesins mediate bacterial attachment to mammalian cells and activate host cell signaling cascades, leading to bacterial uptake and efficient dissemination to Peyer's patches, mesenteric lymph nodes (mLNs),4 liver, and spleen. Enteropathogenic yersiniae express three important adhesins, belonging to different protein classes: YadA, Ail, and invasin (InvA) (3). Yersinia adhesin A (YadA) from enteropathogenic Yersinia species is one of the best-characterized members of the family of homotrimeric autotransporters, which mediates tight adhesion to the eukaryotic host cell and efficient injection of effector proteins/virulence factors (4–8). The protein termed “attachment and invasion locus” (Ail) belongs to the family of outer membrane proteins and promotes cell attachment and invasion. Furthermore, Ail is required for full virulence of Y. pestis and the delivery of pathogenicity factors into host cells (9–12).
The invasin subfamily comprises adhesins of the inverse autotransporter group also referred to as the type Ve secretion system (13, 14). InvA represents the prototype of the large bacterial invasin subfamily of adhesins (2, 3, 13). InvA is the major adhesion factor of Y. pseudotuberculosis and is sufficient to promote tight binding to cells by exploiting β1 integrins as cellular receptors (15). The interaction between InvA and β1 integrins activates actin rearrangement resulting in the internalization of bacteria (16, 17). In addition to InvA, four additional invasins InvB (lfp), InvC, InvD, and InvE have been identified in Y. pseudotuberculosis (18–20).
InvB was reported to support colonization of the host lymphatic tissues and organs (18, 19). InvC promoted adhesion to intestinal cells, but its loss did significantly affect survival of infected mice (18, 19). We recently reported the structure of InvE (20), but further details regarding the specific function of InvE as well as of InvD remain elusive. All five invasins share a common architecture. They consist of the following: (i) an N-terminal β-barrel–like domain, which is responsible for anchoring invasins in the bacterial outer membrane; (ii) repetitive immunoglobulin-like (Ig-like) domains, which vary significantly in number among all the invasins; and (iii) typically a C-terminal C-type lectin-like domain, often described as the capping or adhesion domain, which provides specificity for interaction with host-derived factors, as seen for the interaction between InvA and β1 integrins.
The C-terminal domains of InvA, InvB, and InvC share a sequence similarity of 33–42% (sequence identity 16–24%) and are thus likely to share a common fold. In contrast, the InvD C-terminal domain is not related to any of the other four invasins and only shares a sequence similarity of 17% or less (identity 11% or less) (20). In this study, we gained insight into the structure and function of InvD. We present the crystal structure of the InvD adhesion domain along with two Ig-like domains at 2.6 Å resolution. The InvD adhesion domain adopts a novel fold, raising the question of the identity of its host-derived target. We show that InvD is predominantly induced at 37 °C and expressed within the intestinal tract during infection. Moreover, we identify immunoglobulins (Igs) with VH3/VK1 variable domains as its host-derived target molecule. In summary, after structural determination of InvA (21) and InvE (20), our work presents structural and functional details for a novel member of the Yersinia invasin family and provides insights into the potential role of InvD during infection.
Results
InvD domain architecture
Sequence analysis of InvD using a combination of BLAST, Pfam (ID: A0A0H3B1G5), and Signal-BLAST identifies a putative signal peptide between residues 32 and 57, sharing 46% sequence identity (73% similarity) with the signal peptide (residues 20–45) in the “putative attaching and effacing protein homolog” (UNIPROT:P36943-EAEH_ECOLI). This N-terminal sequence stretch is followed by a region (residues 146–421) assigned as domain of unknown function (DUF3442), predicted to form a β-barrel domain structure required for anchoring the protein in the bacterial outer membrane. The putative β-barrel domain is followed by 13 bacterial Ig-like (BIg) domains (Fig. 1A). Of these, the first 12 BIg-domains share high sequence identity in the range of 56–97% (72–100% similarity), whereas BIg13 is significantly different from BIg1–12 (sequence identity 22–28%; similarity 40–47%) (Fig. S1). BIg1–13 are capped by a predicted C-terminal domain (residues 1829–1976) with no similarity to known proteins, which, according to the canonical domain architecture, we assumed to harbor the adhesion or host–interaction domain. To gain insight into the structure and function of the putative adhesion domain (hereafter referred to as the AD), we tested three expression constructs: InvD1640 that comprises BIg12/13-AD (residues 1640–1976); InvD1737 including BIg13–AD (residues 1737–1976); and InvD1838 covering only the AD (residues 1838–1976) (Fig. 1A). Of these, InvD1640 and InvD1737 resulted in soluble, monodisperse protein samples expressed in Escherichia coli and were purified to homogeneity by affinity chromatography followed by size-exclusion chromatography, whereas the AD alone was not soluble.
Figure 1.

Domain architecture, structure, and stability of Y. pseudotuberculosis InvD. A, domain architecture of InvD with the signal peptide (SP, black), the β-barrel domain (gray), bacterial Ig-like domains (BIg1–12 in blue, BIg13 in green), and the C-terminal adhesion domain (orange). The same color scheme is consistently used in subsequent figures, unless otherwise stated. The nomenclature and boundaries of C-terminal constructs used for the initial protein expression and solubility analysis are depicted below the schematic domain representation of InvD. B, domain organization of InvD1640 with boundaries depicted below the schematic domain representation of the InvD1640 construct. C, cartoon model of the crystal structure of InvD1640. The structure of InvD1640 comprises BIg12 (blue), BIg13 (green), and the C-terminal AD (orange). D, rigid-body fitting of the crystal structure into the ab initio-determined small-angle X-ray scattering envelope. E, fit of the rigid-body model (red) with the experimental scattering (blue). The quality of the fit is expressed in terms of χ value. F and G, stability of InvD against proteases. InvD1737 (BIg13–AD) (F) and InvD1640 (BIg12/13-AD) (G) were treated with 12 different proteases (BR, bromelain; TR, trypsin; EG-C, endoproteinase Glu-C; PA, papain; TH, thermolysin; CL, clostripain; AE, actinase; PK, proteinase-K; EL, elastase; SU, subtilisin; α-C, α-chymotrypsin; PE, pepsin). Apart from treatment with proteinase-K or subtilisin, InvD1737 (BIg13–AD) is highly stable with no major degradation detectable, and InvD1640 (BIg12/BIg13–AD) shows high stability with only minor degradation. Control samples (not treated with proteases) incubated under the same condition are shown on the right-hand side.
Structure of InvD
Of the two InvD constructs expressed and purified, only the construct InvD1640, comprising BIg12/13-AD, yielded crystals of sufficient quality for structural determination. InvD1640 crystallized in space group P43 with two molecules in the asymmetric unit, and the structure was determined by a combination of molecular replacement and single-wavelength anomalous diffraction (Table 1). The overall structure of the construct is rod-like, with a length of 12.9 nm, and it reveals three domains: two BIg domains (BIg12, residues 1640–1736; BIg13, residues 1737–1837) and the C-terminal AD (residues 1838–1976) (Fig. 1, B and C). The two molecules in the asymmetric unit are highly similar, and they superimpose with an r.m.s.d. of 0.923 Å. The separate superimposition of BIg13/AD or BIg12 results in a lower r.m.s.d. of 0.814 and 0.761 Å, respectively, indicative of a minor structural flexibility between BIg12 and BIg13. Structure description hereafter will be focusing on one of the two molecules in the asymmetric unit. We have additionally confirmed the overall rod-shaped architecture by small-angle X-ray scattering. The χ value of the fit to the scattering data using InvD1640 structure as model equals 1.09, and the model is in very good agreement with the calculated small-angle X-ray scattering envelope (Fig. 1, D and E). In the crystal structure, small differences between the structure and the small-angle X-ray scattering envelope are seen in the orientation of BIg12, indicating that the junction between BIg12 and BIg13 allows for minor flexibility, in line with the small differences seen between the structures of the two molecules in the asymmetric unit. In summary, the low r.m.s.d., in comparing the two molecules in the asymmetric unit in conjunction with the very good agreement with the small-angle X-ray scattering data and the high-protease stability (Fig. 1, F and G), suggests that the three domains form a chain with only a low degree of conformational flexibility between BIg13 and AD.
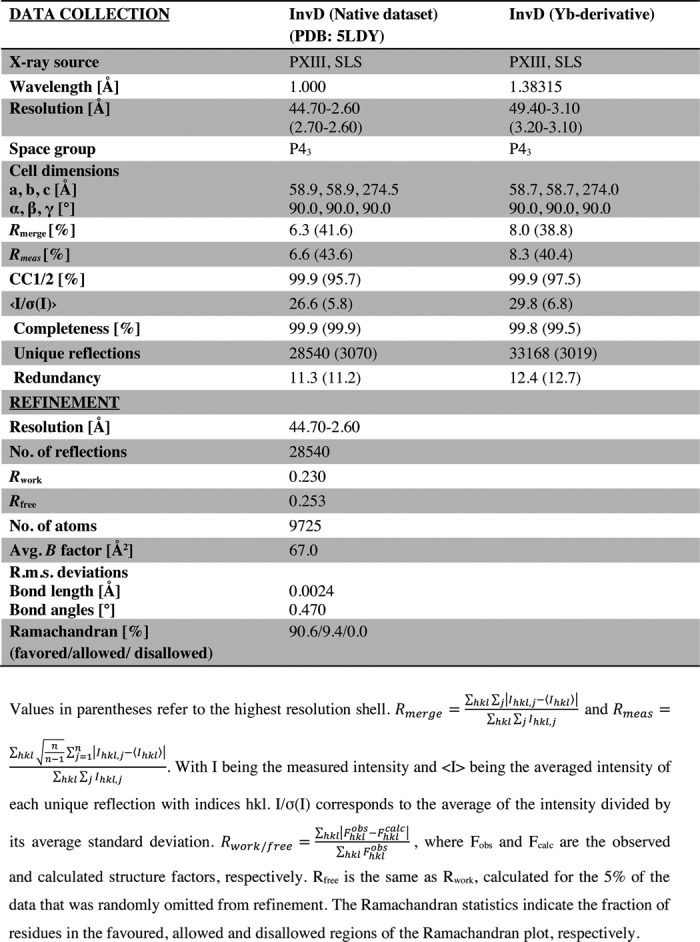
Table 1.
Data collection and refinement statistics
BIg12 and BIg13 domains
Both the BIg12 and BIg13 adopt a β-sandwich fold resembling eukaryotic members of the immunoglobulin superfamily. Immunoglobulin superfamily domains have been previously classified into V, C1, C2, I1, and I2 sets according to the strand arrangement (22, 23). The first sheet in BIg12 and BIg13 comprises strands ABED, and the second comprises strands A′GFC (Fig. 2A, B, and E). Despite the lower sequence identity between BIg12 and BIg13 (compared with BIg1–12), their structures are highly similar and superimpose with an r.m.s.d. of 1.58 Å (93 aligned residues, residues 1640GNLS…ITLA1736, 1737PGAL…VTIT1837) (Fig. 2C). The kink in strand A/A′ and crossover to the opposite sheet (forming strand A′), the presence of strand D, as well as the lack of strand C″ are reminiscent of the I1 set with ABED and A′GFCC′. Even though BIg12 and BIg13 lack strand C′, which has also been observed previously in other I1 set domains (24), we propose to classify both BIg12 and BIg13 as the I1 set. The contact area between BIg12 and BIg13 comprises an interface of 199 Å2 with a total of 13 interacting residues (seven on BIg12 and six on BIg13). Although these interactions partially stabilize the relative orientation of BIg12–BIg13 and rod-like arrangement, the rather small interface is likely to allow for a minor degree of flexibility in solution, as seen in the small-angle X-ray scattering data (Fig. 1, D and E) and the minor differences between the domain arrangements of the two molecules in the crystal structure.
Figure 2.

Architecture of InvD1640 domains. A, topology diagram of BIg12/13 and AD of InvD1640 with the β-strand nomenclature according to the immunoglobulin superfamily fold. β-Strands in BIg12/13 as well as β-strands shown with a red border in the AD correspond to the canonical β-strands in the immunoglobulin superfamily. Noncanonical modifications and insertions of secondary structure elements into the immunoglobulin superfamily core structure of the AD are shown with an orange border. B, overlay of surface and cartoon representation of the X-ray crystal structure of InvD1640. In the AD, β-strands representing the canonical immunoglobulin superfamily fold are shown in red, whereas noncanonical modifications/insertions are shown in orange. C, superposition of BIg12 and BIg13 shown in two orientations. D, superposition of BIg13 and the AD shown in two orientations. E, cartoon representation of InvD–AD with β-strands labeled as shown in A. F, structural comparison of InvD–AD with other structurally similar proteins. Superposition of InvD–AD (orange) was with following proteins (in pink): adnectin (PDB code 4OV6:G) (r.m.s.d. 4.3 Å); PsaA (PDB code 4F8O:A) (r.m.s.d. 3.7 Å); and SafA pilin (PDB code-2CNY:A) (r.m.s.d. 3.7 Å). Structural comparison was done by using the PDBeFold program (http://www.ebi.ac.uk/msd-srv/ssm; please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.). Numbers in parentheses correspond to the percentage of sequence identity between the two superposed proteins.
Adhesion domain
The C-terminal adhesion domain (AD) comprises residues 1838–1976 with a molecular mass of 14.8 kDa. It is mainly composed of β-strands with a single α-helix at the tip of the rod-shaped protein. Interestingly, the AD shares 37 and 27% sequence identity to BIg12 and BIg13, respectively. This is also reflected in the high degree of structural similarity of the two BIg domains with the core structure of the adhesion domain (Fig. 2D). The overall topology of the AD core is reminiscent of an immunoglobulin fold with the two canonical β-sheets, the four-stranded anti-parallel GFCC′-sheet and the three-stranded ABE-sheet (Fig. 2, A, B, and E). Based on this topology, the core of the adhesion domain can be classified as C2 set, due to the lack of strand A/A′ subdivision, lack of strand D, combined with the presence of strand C′. However, apart from the canonical strands present in the immunoglobulin superfamily fold, various modifications of and insertions into the core structure can be observed: (i) an elongated linker region is present between strand A and B; (ii) a short helix is inserted between strands E and F that also includes a disulfide bridge formed between Cys-1859 and Cys-1939; (iii) two short antiparallel β-strands (I/I′) are inserted connecting strands B and C and enlarge the interaction surface with BIg13; moreover, (iv) an additional insertion between strands C and C′ composed of two antiparallel β-strands (C′*/C*) is present, which in combination with (v), the extended strand F, form a third antiparallel three-stranded β-sheet (C′*C*F), sharing strand F with the canonical GFCC′ sheet.
Compared with BIg12–BIg13, the BIg13–AD interdomain interface spans a significantly larger area of 317 Å2 attributed to 19 interacting residues in total (9 on BIg13 and 10 on AD). These interactions, the high-protease stability of the BIg13–AD construct (Fig. 1F) and the fit to the small-angle X-ray scattering envelope, suggest that these two domains, BIg13 and AD, form a rigid unit with low-conformational flexibility.
Structure and sequence similarity to the adhesion domain of InvD
Despite the structural homology of the AD core to the immunoglobulin superfamily, taking the insertions into account, the AD adopts a distinct fold. A search for structural similarity to InvD–AD (including the insertions) results only in weak structural similarity (r.m.s.d. >3 Å) with proteins of various functions. These include the following: (i) Adnectin, a monobody with specificity for different proteins (25); (ii) PsaA from Yersinia pestis, which has been shown to agglutinate red blood cells, binds to carbohydrates and lipids, and also inhibits phagocytosis by macrophages by binding to the Fc part of IgG (26, 27); and (iii) SafA pilus, which is involved in host cell attachment and biofilm formation (28) (Fig. 2, E and F, and Fig. S2). Analysis based on the primary sequence, however, reveals four structurally uncharacterized Yersinia proteins in the database, which share more than 65% sequence identity (similarity 76–97%) with the AD of InvD (Fig. 3A). Of these, three belong to different strains of the Y. pseudotuberculosis species, and one belongs to the species Yersinia wautersii. We mapped the sequence conservation on the structure of InvD–AD to identify potential functional patches with a high conservation rate (Fig. 3, B and C), and we identified two major, highly conserved patches on the surface. The first one is a large surface generated by the C′CFG sheet extending to the opposite sheet on strand A and the short antiparallel β-strands inserted between strands B and C. The second patch is located at the tip of the AD and comprises residues of the α-helix and the C*/C′* β-turn. The two highly conserved patches are separated by a deep cleft in the protein with lower conservation, extending from strands C*/C′* to strands E, B, and partially A (Fig. 3, B and C).
Figure 3.

Sequence conservation of the InvD adhesion domain. A, alignment of InvD related proteins (red, 100% conserved amino acids; orange, 80%; yellow, 60%). Proteins belonging to different strains of Y. pseudotuberculosis: CNI94919.1, strain IP32881; CFV23641.1, strain OK6088 and AIN12719.1, strain ATCC6904. CRG49100.1 belongs to the species Y. wautersii (alignment generated with GeneDoc (76)). B and C, conserved regions on the structure of InvD AD as cartoon representation (B) and surface representation (C). Color scheme as in A. The cleft is indicated with a dashed line.
Expression pattern of InvD differs significantly from InvA
To gain more information about the contribution of InvD to virulence, we first analyzed the expression pattern of InvD in response to temperature and growth phase using an invD-luxCDABE reporter fusion strain. The invD-luxCDABE was maximally expressed at 37 °C throughout the growth curve. At 25 °C expression was generally lower (Fig. 4A). This is in strong contrast to InvA, which is more strongly expressed at 25 °C to prime the bacteria for uptake during the early stages of the infection (Fig. 4B) (29). This suggests that InvD is required at later time points of the infection.
Figure 4.

Expression analysis of InvD in vitro and in vivo. A and B, expression of InvD and InvA under in vitro conditions. Overnight culture of Y. pseudotuberculosis YPIII pRG05 (PinvD::luxCDABE) was diluted 1:50 and grown at 25 or 37 °C in LB medium. Light emission of the luxCDABE reporter and optical density were measured. The dashed line represents the detection limit. C, in vivo expression analysis of an invD-luxCDABE fusion shows expression of invD during infection. BALB/c mice were orally infected with 9 × 108 CFU of Y. pseudotuberculosis YPIII pFU189 (empty vector) and YPIII pRG05 (PinvD::luxCDABE). Mice were anesthetized and ventrally imaged with the in vivo imaging system camera at given time points. D, expression of invD in murine intestines visualized with GFP reporter fusion. BALB/c mice were infected orally with 3 × 108 CFU of Y. pseudotuberculosis YPIII pFU228/pRG01 (PgapA::dsred, PinvD::gfpmut3.1). Mice were sacrificed 3 days post-infection, and the small intestine, colon, mLNs, and spleen were isolated. Cryosections (thickness, 7 μm) were prepared and imaged with a fluorescence microscope. Bacteria were detected by the DsRed2 reporter protein. Cell nuclei were stained with DAPI. White bars indicate 10 μm.
InvD expression in the murine infection model
To gain more information about the expression and role of InvD for the infection, BALB/c mice (Janvier, Saint Berthevin Cedex, France) were orally infected with 9 × 108 colony-forming units (CFU) of Y. pseudotuberculosis YPIII harboring an invD-luxCDABE reporter construct. Bioluminescence was detected in anesthetized mice using the in vivo imaging system. Barely any signal was found in mice infected with Y. pseudotuberculosis expressing/harboring a promoterless luciferase gene as negative control. In contrast, light emission was observed in the intestinal tract of mice infected with bacteria harboring the invD-luxCDABE. No activity or only a very low luciferase activity could be measured before and during the early time points of the infection (after 6 h, 1 day), but a substantial bioluminescent signal was detectable at days 2–4 after infection (Fig. 4C), indicating that the invD gene is expressed during the acute phase of infection.
To assess invD expression within different tissues at single cell level, BALB/c mice were infected orally with 3 × 108 CFU of Y. pseudotuberculosis YPIII containing a constitutive gapA-dsred2 and an invD-gfpmut3.1 construct. Microscopic analyses of organ cryosections were performed 3 days post-infection, and bacteria were detected and visualized via the DsRed2 signal. Expression of the invD-gfp fusion was found in all parts of the intestinal tract, but not in deeper lymphoid tissues, spleen, and mLNs (Fig. 4D).
Impact of InvD on colonization of the intestinal tract
To address whether InvD plays a role for the colonization of the intestinal tract, we performed a competitive infection experiment to detect even minor differences in bacterial numbers. For the competitive experiment, mice were orally infected with 2 × 108 CFU in an equal mixture of WT YPIII and mutant strain (YP197). The small intestine and colon were isolated 6 h and 1 and 3 days post-infection to determine the bacterial loads within the organs. We found that less mutant bacteria were found within both compartments of the intestinal tract, suggesting that InvD provides an advantage for the WT strain in the intestine (Fig. 5).
Figure 5.
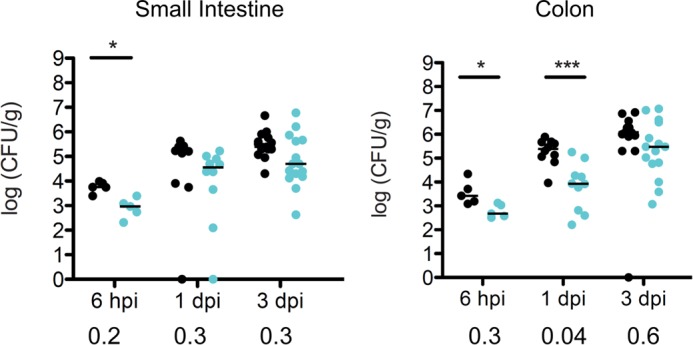
Colonization of the intestinal tract of mice infected with WT or ΔinvD strain of Y. pseudotuberculosis. Intestinal colonization of the invD deletion mutant (YP197) is impaired in a competitive infection with the WT Y. pseudotuberculosis strain YPIII. 2 × 108 CFU in an equal mixture of WT (black circles) and invD mutant (cyan circles) bacteria were used to orally infect mice for the competitive infection experiment. 6 h and 1 and 3 days post-infection, mice were sacrificed, and the small intestine and colon were isolated. The spots indicate the organ-specific amount of viable bacteria within one mouse; lines illustrate the medians. The Mann-Whitney test was performed to specify statistically significant differences between the groups of WT and mutant strain, indicated by asterisks: * p < 0.05; *** p < 0.001. Stated numbers below the diagrams represent the competitive index determined for the individual time points.
InvD does not bind to carbohydrates or lipids and does not mediate attachment to human epithelial type 2 (HEp-2) cells
As InvD is expressed during the infection in the intestine, we aimed to identify the host-derived target and gain insight into the specific function of InvD on the molecular level. We thus focused on a systematic analysis and identification of the target molecule using recombinant InvD. Various adhesins harbor an adhesion domain with a lectin-like fold to bind glycans (30). Despite the lack of structural similarity to lectin-binding domains, the adhesion domain of InvD might provide a glycan-binding domain of a different fold or a platform for binding to specific lipids as seen in adhesin PsaA of Y. pestis, which binds to galactose and phosphocholine (27). However, screening for interaction of InvD1640 with glycans using the mammalian glycan array (within the Consortium for Functional Glycomics) or with lipids using membrane lipid strips did not show any significant binding (data not shown). We next reasoned that a host protein represents the target of InvD, in analogy to InvA. Usually, the adhesion domain in the invasin family represents the domain that targets a specific host molecule and mediates interaction with the host cell during infection (18, 19). Because of the low structural similarity of the adhesion domain of InvD to known proteins, we expected the adhesion domain to bind a novel, yet unidentified molecule. To identify whether InvD targets a receptor on the host cell surface to promote adhesion and eventually invasion, similar to InvA, we analyzed the attachment efficiency of InvD1640-coated latex beads to HEp-2 cells. Although beads coated with the extracellular part of InvA (hereafter referred to as InvA500, comprising residues 500–985) were readily attached, beads coated with InvD1640 or the negative control bovine serum albumin (BSA) showed no efficient cell attachment (Fig. 6). We therefore exclude that InvD targets a molecule, which is commonly exposed on the cellular surface of HEp-2 cells.
Figure 6.

Attachment of InvD-coated latex beads to HEp-2 cells. A, latex beads (green) coated with InvA500 (positive control, corresponding to the extracellular part of InvA encompassing residues Pro-500–Ile-985 with four BIg domains and the adhesion domain), InvD1640 or BSA (negative control) were incubated with HEp-2 cells (HEp-2, ATCC® CCL-23TM). B, attached beads per cell were quantified after removal of unattached beads. Numbers in parentheses correspond to # beads/# of total cells.
InvD targets soluble Igs
To extend the search for the potential target of InvD in the host, we performed pulldown assays using 3×FLAG-tagged InvD1640 with homogenized extract of the Peyer's patches and intestine (small intestine, caecum, and colon) of uninfected mice. A 3×FLAG-tagged ΔAD-InvD construct was used as a negative control, which lacks the adhesion domain AD and only comprises the BIg12 and BIg13 domains of InvD (residues 1640–1839). Upon analysis by SDS-PAGE, we observed a 25-kDa band co-purifying with InvD1640 in pulldowns using both the extract from mouse intestine (Fig. 7A) and Peyer's patches (Fig. 7B). A less prominent 50-kDa band co-purifying with InvD1640 was visible in the pulldown with extract from Peyer's patches (Fig. 7B). Both the 25- and 50-kDa band were not co-purified with the negative control ΔAD-InvD, indicating that the interaction with the co-purified proteins was mediated by and dependent on the presence of the adhesion domain of InvD. As these represented the potential host target molecule(s) of InvD, the 25- and 50-kDa proteins were subjected to analysis by MS. The co-purified 25- and 50-kDa proteins were identified as the immunoglobulin light and heavy chain of antibodies, respectively, giving rise to the notion that Igs represent the host–target molecule of InvD (Fig. 7, A and B). We used microscale thermophoresis to validate the interaction between InvD and Igs isolated from pooled human or mouse serum, thus comprising a large variety of antibodies with combinations of diverse heavy and light chain subtypes. Cy5-labeled InvD1640, ΔAD-InvD, and InvA500 were titrated with increasing concentrations of human IgG (Fig. 7C), mouse IgG, and human IgA (Fig. S3A and B). Because obtaining higher concentrations of the titrated Igs was limited by their solubility, microscale thermophoresis data did not reach saturation. In consequence, the affinity constant (Kd) could not be precisely quantified, but the data allowed for an estimation of the approximate Kd value. The obtained results confirmed the binding of InvD with IgG/IgA with an estimated Kd in the range of ∼3–10 μm. In addition, no binding was observed when labeled controls ΔAD-InvD or InvA500 were titrated with human/mouse IgG or human IgA, confirming an AD-dependent, specific interaction of InvD (Fig. 7C and Fig. S3, A and B). To identify the domain of the immunoglobulin to which InvD binds, we next investigated whether InvD1640 binding to Igs was mediated by the Fc or Fab fragment. Purified IgG-Fc and IgG-F(ab′)2 were separately used for interaction assays with InvD1640 by microscale thermophoresis, demonstrating that InvD1640 selectively binds to the F(ab′)2 but not the Fc part of IgG (Fig. S3C).
Figure 7.

InvD interacts with Igs. A and B, homogenized extract of mouse intestines (A) and mouse Peyer's patches (B) mixed with 3×FLAG-tagged InvD1640 or ΔAD-InvD was subjected to FLAG pulldown assay. When using InvD1640 as bait, 25-kDa (in A and B) and 50-kDa (in B) bands were co-purified that were identified as the immunoglobulin light and heavy chain of antibodies. These bands were, however, not co-purified with the negative control ΔAD–InvD, indicating that the interaction is mediated by the adhesion domain of InvD. C, analysis of the interaction of InvD1640 with human IgG by microscale thermophoresis. InvA500 or ΔAD-InvD was used as negative control. Data shown are the average of three (InvD1640; ΔAD-InvD) or two (InvA500) technical repeats.
InvD specifically binds to a subset of murine B cells
Based on our observation that InvD1640 binds Igs, we further analyzed binding of 3×FLAG-tagged InvD1640 to ex vivo isolated murine B-cell populations via flow cytometry. Initially, B cells were identified, using multicolor flow cytometry, as single viable CD19+B220+ encompassing all B cells with a clonotypic B-cell antigen receptor (Fig. 8A) (31). To exclude unspecific binding of InvD1640 to the cell surface of splenic cells in the single-cell suspension, we utilized 3×FLAG-tagged ΔAD-InvD as control and could validate that the binding of InvD1640 relied on the adhesion domain as samples subjected to ΔAD-InvD showed a lower signal in frequency and intensity (Fig. 8A and Fig. S4). Importantly, the isotype control for the anti-FLAG antibody did not bind unspecifically to B cells previously subjected to 3×FLAG-tagged InvD1640. As over 99% of the CD19+ B cells of the murine spleen are B220+, CD19+ was sufficient to identify B cells in conjunction with additional immunological cell types (Fig. 8A). As expected, only B cells were enriched for cells bound by InvD, whereas total T cells (CD3+), γδT cells (γδTCR+), monocytes (CD11b+), NK cells (CD49b+), and dendritic cells (CD11c+) did not show any binding of InvD1640 (Fig. 8B and Fig. S4), indicating that InvD binds specifically to the Fab fragment of the cell-surface–exposed B-cell receptors.
Figure 8.

InvD binds predominantly to B cells. A, InvD1640 binds to 0.58% of the B cell population, whereas ΔAD-InvD or the isotype control show a strongly reduced binding to 0.03 and 0.02% of the B cell population, respectively. Single cell suspensions from spleens were analyzed using flow cytometry. Previous to B220+ gating, B cells were gated as alive cells, excluded for duplets, and gated on CD19+ cells. Numbers indicate frequency of parental population. Anti-FLAG allophycocyanin antibody was used to detect the 3×FLAG-tagged InvD1640. Exemplary dot plots depict frequencies and fluorescence intensity of 3×FLAG-tagged InvD1640 or 3×FLAG-tagged ΔAD-InvD on living B cells using anti-FLAG-antibody conjugated to allophycocyanin. Isotype controls were performed using 3×FLAG-tagged InvD1640 and subsequently subjecting the cells to allophycocyanin-conjugated anti-FLAG-antibody isotype control antibody (InvD + Iso). Exemplary data of five independent experiments. B, scatterplot summarizes frequencies of InvD+ cells of the respective cell population. Pooled data of two independent experiments (n = 2–3 mice per experiment) are shown. B cells, CD19+; T cells, CD3+; γδT cells, CD19−γδTCR+; NK cells, CD49b+; monocytes, CD19−γδTCR−CD11b+CD11c−; dendritic cells, CD19−γδTCR−CD11bmedCD11c+.
Characterization of Fab-binding specificity
To gain a detailed insight into the interaction of InvD with Fab on the molecular level, we performed phage display panning. Two scFv naive universal libraries (HAL10 and HAL9 + HAL10) were employed (32). Three rounds of panning were performed on immobilized strep-tagged InvD1640 followed by enzyme-linked immunosorbent assay (ELISA) with soluble scFv. To ensure that the interaction between scFvs and InvD1640 is via the adhesion domain, ELISA was also performed between strep-tagged ΔAD-InvD and soluble scFvs. A 20–60-fold higher absorbance was observed with InvD1640 as compared with ΔAD-InvD in ELISA, confirming that interaction with the scFvs isolated by panning is mediated by the adhesion domain of InvD (Fig. S5, A and B). Phagemid DNA of 57 and 59 randomly selected individual scFvs after the 3rd round of panning with HAL10 (κ library) and HAL9 + HAL10 library (combination of κ and λ library), respectively, were sequenced. Sequence analysis demonstrates that scFv isolated by panning on InvD are highly enriched in the VK1 light-chain subfamily in combination with the VH3 heavy-chain subfamily. Although this combination only represents ∼16 and 6% of the unselected HAL10 and HAL9 + 10 library, 86 and 59% of the clones isolated after panning correspond to VH3/VK1, respectively (Fig. 9, A and B). In contrast to the panning with InvD, in a general antigen panning, antibodies with V-genes from different V families (internal data from M. Hust) will be enriched, and more than 90% of the individual binders contain a λ chain. Therefore, the high percentage of isolated VH3/VK1 antibodies is indicative of an InvD-mediated recognition of VH3/VK1 family antibodies, instead of being a classical antibody-mediated recognition of InvD as antigen. Furthermore, sequence alignment of VH and VL Complementarity Determining Regions indicates broad diversity of Complementarity Determining Regions H3 (Fig. S5C). Complementarity Determining Regions H3 are largely responsible for antigen specificity as these represent unique loops that arise from the hypervariable nature of the VDJ region (33).
Figure 9.

InvD targets VH3/VK1 subfamily antibodies. A, heat map representation of the percentage abundance of VH-VK gene pairs in HAL10 (κ) library composed of 456 different clones (left) and VH-VK gene pairs obtained from the 3rd round of panning against InvD1640 using the HAL10 library (based on sequence analysis of 57 clones) (right). B, heat map representation of the percentage abundance of VH-VK/VL gene pairs in HAL9 + 10 (λ + κ) library composed of 1282 different clones (left) and (right) VH-VK/VL gene pairs obtained from the 3rd round of panning against InvD1640 using the HAL9 + 10 library (based on sequence analysis of 59 clones). C, pulldown-based interaction analysis of InvD1640 and scFv-Fc antibodies with different heavy/light chain subtype combinations. Three VH3/VK1 antibodies and three non-VH3/VK1 antibodies were randomly selected from a selection of 3000 antibodies that were previously identified in panning against target proteins unrelated to InvD. (Antibodies used and their respective targets are as follows: SH83-A7, anti-CD71 (92); SH863-B6, anti-PAK5 (M. Hust, unpublished data.); SUW61-II-A10, anti-CD33 (91); CHN24-2-B7, anti-VEEV (93); TM43-F8, anti-OmpD (94); SH565-II-D6, anti-CD71 (M. Hust, unpublished data.). D, interaction of InvD1640/ΔAD-InvD with VH3/VK1 (SH83-A7) or VH1/VL2 (CHN24–2-B7) subfamily scFv-Fc antibodies analyzed by microscale thermophoresis. Data shown are the average of two technical repeats.
To further validate that this interaction represents a specific InvD-mediated recognition of VH3/VK1 family antibodies, we used scFv-Fc antibodies (Yumabs) selected in other projects by phage display from different human naive HAL antibody gene libraries against targets not related to InvD. Three VH3/VK1 as well as three non-VH3/VK1 antibodies were randomly chosen from a selection of more than 3000 antibodies according to the V-gene combination. Interaction in the pulldown was only seen for the VH3/VK1 family when InvD1640 was used as bait, whereas no interaction was detected when using the negative control ΔAD-InvD, which lacks the adhesion domain (Fig. 9C). The in vitro pulldown assay using these recombinant scFv-Fc antibodies thus confirms the specificity toward the VH3/VK1 subfamily combination and dependence on the presence of the adhesion domain. Compared with the estimated Kd of ∼3–10 μm using IgG/IgA mixtures of different VH/VL subfamilies, the estimated affinity to the recombinant VH3/VK1 scFv-Fc is in the range of 0.7 μm (Fig. 9D).
In summary, we convincingly demonstrate that InvD interacts with the Fab region of IgG/IgA via the C-terminal adhesion domain and that the interaction is targeting the VH3/VK1 subfamily combination of antibodies in a highly specific manner.
Discussion
Y. pseudotuberculosis produces different adhesins, which target certain host factors to promote host cell attachment and invasion, as well as to overcome the host immune system and colonize specific niches. Y. pseudotuberculosis possesses five different invasins (InvA, InvB, InvC, InvD, and InvE). So far, details on the regulation/expression, structure, and function of InvD have been elusive. In this study, we provide the first structural data as well as in vitro and in vivo functional analysis for the recently identified InvD from Y. pseudotuberculosis strain YPIII.
Invasins share a common architecture and consist of an N-terminal β-barrel-like domain that anchors the protein in the bacterial outer membrane, a varying number of repetitive Ig-like domains, and the C-terminal adhesion domain that commonly mediates interaction with host-derived factors. With the outer membrane anchor located at the N terminus, the C-terminal adhesion domain is placed distal to the bacterial surface, oriented toward the specific host-derived target. The central BIg domains thus act as linker/tether arm to span the distance between the bacterial surface and the host-derived target molecule, and they are likely to pre-align the C-terminal adhesion domain for binding to its host–target molecule. But despite their similarities in the overall architecture, invasins differ significantly in their function and the host–target molecule. Here, we present the structure of InvD1640 comprising bacterial Ig-like domains BIg12/13 and the C-terminal adhesion domain. In full-length InvD, BIg1–12 domains of InvD are highly repetitive sequences except for the BIg13 domain, which links BIg1–12 and the adhesion domain. BIg1–12 adopt an I set IgSF fold, which is often seen in extended molecules with varying number of repetitive arrangements of related BIg domains. The high sequence similarity within the central bacterial Ig-like domains of InvD appears to be a common motif, as it is reminiscent of InvE, where the central bacterial Ig-like domains BIg1–20 share a high sequence identity between 56 and 98% (20). In contrast to the sequence similarities within BIg1–12 of InvD, the difference in the last BIg13 domain of InvD suggests a special role of this domain in bacterial pathogenicity along with the C-terminal adhesion domain. This is also seen in the case of InvA where the D4 domain interacts with the head/adhesion domain (D5) to form a superdomain necessary to bind β1 integrins (21), as well as in the case of InvE, where BIg21 appears to form a functional superdomain with the C-terminal adhesion domain (20). Based on the high stability and the structural data, we thus conclude that BIg13 and AD are also likely to form a functional superdomain in InvD.
However, although InvD shares the presence of the N-terminal β-barrel-like domain and the central bacterial Ig-like domains with other members of the invasin family, the C-terminal adhesion domain of InvD differs significantly in sequence and fold from the adhesion domains of the other invasins. Despite a low sequence identity, the structures of the C-terminal adhesion domains of InvA and InvE reveal a C-type lectin-like fold, in common with the presence of a C-terminal C-type lectin-like domain in the adhesin protein Intimin from enteropathogenic E. coli/enterohemorrhagic E. coli (20, 21, 34). The C-type lectin-like domain is however not specific for bacterial adhesins, but it is also present in many other proteins involved in various processes, such as signaling and cell–cell recognition (35). However, the C-terminal adhesion domain of InvD shares no similarity with the C-type lectin-like domain of InvA/InvE or Intimin and instead adopts a β-sandwich core resembling that of an immunoglobulin superfamily fold with extensive variations and insertions. Ig-folds are not necessarily uncommon in bacterial adhesins, as these are commonly found in the fimbrial adhesins, where the noncovalently linked structural subunits of the fimbria and the one subunit involved in adhesion fold into an incomplete Ig-like domain that is completed by interaction with the subsequent subunit (22). As described previously, the central BIg domains of the invasins are known to adopt an Ig-like fold. However, to our knowledge this is the first example of an adhesin of the nonfimbrial invasin/Intimin family, where the distal C-terminal adhesion domain adopts a fold related to the immunoglobulin superfamily. The similarities to the immunoglobulin superfamily in structure and topology further suggest that Yersinia has evolved the C-terminal adhesion domain of InvD from its BIg-like domain to carry out a specific function.
To gain information about the function of this novel invasin, we characterized the expression in vitro and in vivo as well as the influence of InvD on the colonization of mice infected with WT or an invD knockout mutant strain. InvD is maximally expressed at 37 °C in vitro and in the intestinal tract during the acute stage of the infection. In a competitive infection experiment, we also observed that an invD mutant is less efficient in the colonization of intestinal tissues. We further demonstrate that InvD1640 is highly resistant to proteolytic degradation, which further strengthens our in vivo data indicating that InvD is potentially involved in the infection process such as in efficient colonization of the host intestinal tract and/or in host immune evasion. We identified the host target molecule of InvD and conclusively show by in vitro pulldown and panning data a highly specific role for InvD in selectively binding antibodies, such as IgA/IgG, via the Fab region with nanomolar to low micromolar affinity and that this interaction is highly selective for VH3/VK1 antibodies. Notably, in a normal antigen panning, antibodies with V-genes from different V families (internal data from M. Hust) will be enriched and more than 90% of the individual binders have a λ chain. In addition, independent of the panning, we have used additional VH3/VK1 scFv-Fc antibodies (Yumabs) to confirm the specific interaction of InvD with VH3/VK1 antibodies (Fig. 9, C and D). This high selectivity for VH3/VK1 in panning and the interaction with three different VH3/VK1 scFv-Fc antibodies thus strongly suggest that this interaction does not represent a classical antigen–antibody interaction, but rather a specific recognition of the VH3/VK1 family of antibodies by InvD. Although the majority of antibodies isolated by panning with InvD corresponds to the VH3/VK1 subtype, interestingly, the panning libraries also included additional antibodies of the VH3/VK1 subtype, which have however not been targeted by InvD. This is not uncommon as also for gp120 from HIV-1 (HIV) and staphylococcal protein A (SpA), it has been observed that not all antibodies of the specific antibody subfamily, but rather a restricted set of VH3, are targeted by gp120 and SpA (36, 37). In the case of InvD, this suggests the presence of slight differences in the amino acid composition within the VH3/VK1 subfamily that are likely to influence the interaction of InvD with the Fab region. Nonetheless, the VH3 heavy chain and the VK1 light chain are the most commonly observed families in the human germ line repertoire (38–40). Despite the presence of some VH3/VK1 nonbinders, InvD thus appears to selectively bind the most common heavy and light chain. The Fab region, which is targeted by InvD, is found on antibodies such as IgA and IgG, which are commonly expressed during bacterial infection. The highly specific targeting of antibodies with the most common heavy/light chain combination might thus suggest that InvD is utilized for defense against the host immune system to contribute to bacterial pathogenicity by targeting one of the most abundant populations of host-defensive antibodies (41).
Two major functions of pathogenic proteins binding to antibodies are commonly known. The first function is to interact with the Fc region of Igs and act as an inhibitor of phagocytosis (function I), such as SpA and staphylococcal binder of IgG (Sbi) from Staphylococcus aureus. Interaction of SpA and Sbi with the Fc region of IgG sterically blocks the binding of Fc receptors on macrophages to the immunoglobulin, thereby preventing recognition by the immune system and bacterial uptake by phagocytic cells present at the site of infection (42–46). This group of bacterial Fc-binding proteins also includes the E. coli immunoglobulin-binding (Eib) proteins, consisting of six homologous proteins (EibA, -C, -D, -E, -F, and -G), which have been shown to increase the resistance of the bacteria to the human serum complement system (47, 48). In regard to the potential function of InvD in the context of antibody binding, we can exclude a function as phagocytosis inhibitor or complement resistance factor, as seen for SpA, Sbi, or Eib (function I. described above), because InvD clearly binds to the Fab but not to the Fc part. Furthermore, a function to neutralize highly specific anti-pathogen antibodies is unlikely, as recognition by the immune system or anti-Yersinia antibodies would not be restricted to a specific variable region and light chain–heavy chain combination.
The second known function of pathogenic proteins binding to antibodies (function II) is to act as B-cell or immunoglobulin superantigens. These proteins bind to Igs and/or the B-cell surface via the exposed antibody segments of the B-cell receptor, thereby modulating and/or impairing immunoglobulin and B-cell–mediated immune response (function II). The group of immunoglobulin-binding proteins classified as “immunoglobulin superantigens” includes the previously mentioned gp120, SpA, as well as protein L from Peptostreptococcus magnus (PpL) (49, 50). Prime examples of bacterial proteins acting as B-cell superantigens include the following: the Moraxella catarrhalis IgD-binding protein (MID) (51), targeting the CH1 region in the Fab; the protein SpA from S. aureus, which apart from its previously mentioned function as an Fc-binding protein also targets the VH3 subfamily of Fab (52, 53); and PpL, which binds with a broader specificity range to VK1, VK3, and VK4 subfamilies (54, 55). Viral “B-cell superantigens” include gp120 (56) that targets the VH3 subfamily of Fab. The reported ability of these superantigens to bind to Igs in the serum as well as to the B-cell receptor triggers one or more of a variety of different responses in the host cell. These responses range from B-cell proliferation, as seen for MID (51); B-cell activation and subsequent induction of B-cell apoptosis, as seen for HIV gp120 (56); overactivation of the classical complement pathway by SpA (57); to stimulation of proinflammatory cytokine release, as reported for SpA, protein L, and HIV gp120 (58, 59). Notably, the overall estimated affinity of ∼700 nm for the InvD–scFv interaction is similar to the previously reported affinities of 400–5000 nm for the bacterial SpA–Fab interaction (60) and 130 nm for the bacterial PpL–Fab interaction (55). However, InvD only binds to ∼0.5% of the B-cell population (Fig. 8B), whereas for canonical B-cell superantigens, a population of bound B cells between 3 and 8% has been reported (41), with the exception of PpL, which has a broad specificity and has been shown to bind >50% of the B-cell population (61). However, in these cases the B-cell superantigen shows specificity for only the light or the heavy chain (55, 62). It is possible that the dual specificity for the heavy and light chain (VH3/VK1) is responsible for the comparably small population of InvD-bound B cells and does not necessarily exclude InvD from the group of potential superantigens.
However, expression of InvD in the small intestine and the colon, but not in the deeper colonized lymphatic tissues, and its influence on the colonization of the intestinal compartments suggest a function in the intestine during the acute phase of the infection. Secretory IgAs (SIgAs) are the most abundant antibodies in the intestinal lumen, and they play an important role in limiting access of commensals and pathogenic microorganisms to the mucosal barrier. By this process, which is also referred to as “immune exclusion,” SIgA prevents colonization and invasion, thus contributing to luminal compartmentalization and pathogen clearance by peristalsis (63, 64). In addition to this host defense mechanism, the recent report of IgA-mediated cross-linking and elimination of the bacteria (65) from the intestine indicates an alternative function of InvD in neutralizing immunoglobulin-mediated defense mechanisms, for example by preventing IgA-mediated “immune exclusion” and/or clumping via secretory IgA in the intestine. SIgA also exhibits the ability to selectively adhere to M cells, allowing for limited transport of SIgA across the epithelial layer (66–69). Interestingly, Shigella flexneri, a pathogen that is unable to enter the epithelium, was detected in Peyer's patches and mLNs after coating with specific SIgA antibodies (68). Thus, InvD-mediated binding of IgA could potentially also provide a mechanism to enhance translocation of Yersinia across the epithelial barrier to promote infection.
Conclusions
In summary, we present the structure of the Yersinia surface protein InvD revealing a C-terminal adhesion domain that is related to an immunoglobulin-fold but that contains extensive modifications and insertions. InvD is thus the first member of the invasin family of adhesins with an adhesion domain that adopts an Ig-related fold. We further conclusively show that the C-terminal adhesion domain of InvD specifically interacts with the Fab region of IgA/IgG antibodies. Expression in the intestine and preferential binding to antibodies with VH3/VK1 variable domains indicate that InvD neutralizes the function of intestinal antibodies. As InvD also attaches specifically to a subset of B cells, this suggests that InvD might in addition target surface-exposed B-cell receptors, similar to bacterial proteins that act as superantigens. Although these hypotheses require further validation in the future, the results presented in this work demonstrate that a subclass of invasin-type outer membrane proteins has gained additional functions, which are not related to epithelial cell adhesion and invasion. A following more detailed analysis of InvD–IgA/IgG interactions in the context of a Yersinia infection will reveal how this process contributes to pathogenesis.
Experimental procedures
The strains and primers used in this study are listed in supporting Table S1.
Bacterial strains, media, and growth conditions
Overnight cultures of E. coli were grown at 37 °C, and Yersinia strains were grown at 25 °C in LB (Luria-Bertani) broth. The antibiotics used for bacterial selection were ampicillin (100 μg/ml), chloramphenicol (30 μg/ml), and kanamycin (50 μg/ml). No differences between the in vitro growth characteristics of the Y. pseudotuberculosis ΔinvD mutant and those of the WT strain were observed.
Cloning
The DNA fragments encoding invD constructs were amplified by PCR from the genomic DNA of Y. pseudotuberculosis YPIII (Swiss-Prot accession no. A0A0H3B1G5) using KOD Hot Start DNA polymerase (Novagen). The PCR product was digested with NotI-HF (New England Biolabs) and PstI (New England Biolabs), purified using QIAquick PCR purification kit (Qiagen), cloned into a modified pCOLA-Duet vector containing an N-terminal His6 tag and TEV cleavage site (MGHHHHHHVDENLYFQGGGR), an N-terminal strep affinity tag with TEV cleavage site (MASWSHPQFEKVDENLYFQGGGR), or an N-terminal His6, TEV-protease cleavage site followed by a 3×FLAG tag (HHHHHHVDENLYFQGGGRGDYKDHDGDYKDHDIDYKDDDDKGGR).
Protein expression and purification
The protein was expressed in E. coli strain Rosetta2 (DE3) (Invitrogen). A 50-ml preculture was grown overnight in LB medium supplemented with 30 μg/ml kanamycin and 34 μg/ml chloramphenicol in an incubator at 37 °C and 130 rpm. For large-scale production, Terrific Broth (TB) medium was inoculated with the overnight culture and was grown at 130 rpm, 37 °C. At an OD600 (optical density at 600 nm) of 0.7, the expression was induced by addition of 100 μm isopropyl β-d-1-thiogalactopyranoside (IPTG) (Carbosynth-EI05931). Upon addition of IPTG, the temperature was shifted to 20 °C, and cultures were incubated for 21 h in the incubator at 130 rpm. Afterward, cells were harvested by centrifugation at ∼4690 × g (5000 rpm, Fiberlite F9-4 × 1000y) for 15 min at 4 °C. To purify the His6-tagged proteins, cell pellets were resuspended in 300 ml of lysis buffer (50 mm Tris-HCl, pH 8.0, 200 mm NaCl, 5 mm imidazole, pH 8.0, 5 mm β-mercaptoethanol (BME), 5 mm MgCl2, 1 mm phenylmethanesulfonyl fluoride, DNase (40 μl at 1 mg/ml per liter of culture)) and lysed by homogenization at 16,000 p.s.i. (Emulsi-Flex C3, Avestin). Lysate was cleared by centrifugation at ∼34,780 × g (15500 rpm, SA-600) at 4 °C for 50 min. Cleared lysate was loaded onto a 2.5-ml nickel-Sepharose gravity column, which was pre-equilibrated with 50 ml of buffer containing 50 mm Tris-HCl, pH 8.0, 200 mm NaCl, 5 mm BME, 5 mm imidazole, pH 8.0. Nickel-Sepharose resin (GE Healthcare) was allowed to incubate with the protein for 1 h at 4 °C with gentle shaking. The nickel-Sepharose column was washed with wash buffer containing 50 mm Tris-HCl, pH 8.0, 200 mm NaCl, 5 mm BME, 15 mm imidazole, and protein was eluted in 50 mm Tris-HCl, pH 8.0, 200 mm NaCl, 5 mm BME, 100 mm imidazole. To purify the strep-tagged proteins, the same procedure was used to lyse the cells with the exception that the lysis buffer did not contain imidazole. Lysate was cleared as discussed before and was loaded onto 4-ml Strep-Tactin-Sepharose beads (iba-2-1201-025) gravity column equilibrated with 50 mm Tris-HCl, pH 8.0, 200 mm NaCl. After incubation of the lysate with the beads, the column was washed with wash buffer containing 50 mm Tris-HCl, pH 8.0, 200 mm NaCl, and protein was eluted with wash buffer containing 3 mm d-desthiobiotin (iba-2-1000-005). The elution pool was concentrated using a Vivaspin 10,000 molecular weight cutoff concentrator and loaded onto a HiLoad 16/60 Superdex 200 column (GE Healthcare), which was pre-equilibrated with buffer containing 20 mm HEPES, pH 7.4, 200 mm NaCl, and 5 mm DTT. Purified protein was concentrated to 25 mg/ml for crystallization. For all the binding assays, buffer without DTT was used.
Crystallization and structure elucidation
His6-tagged InvD1640 was crystallized in 1.9 m ammonium dihydrogen phosphate, 0.1 m Tris-HCl, pH 8.0. The crystals were grown by vapor diffusion in hanging drops at 20 °C by mixing 0.5 μl of concentrated protein with 0.5 μl of the crystallization solution. Crystals were cryoprotected in a solution with a final concentration of 25% v/v PEG 400, 1.9 m ammonium dihydrogen phosphate, and 0.1 m Tris-HCl, pH 8.0.
The native data set (collected at a wavelength of 1 Å) was collected at the Swiss Light Source X06DA/PXIII beamline on a DECTRIS PILATUS 2 M-F detector at 100 K. Data processing was carried out with XDS (70). Crystals of the native protein diffracted to 2.6 Å resolution. Phaser was used to phase the data by molecular replacement (71). Individual Ig-like domains of InvA (PDB entry 1CWV) (21), which shares ∼40% sequence identity with InvD Ig-like domain, were used as a search model, and a partial solution with two Ig-like domains in the asymmetric unit was found, which did not result in an electron density map/phase information with a quality that was sufficient for manual completion of the model. Further attempts to use the integrin binding domain of InvA (PDB entry 1CWV) (21) as a search model did not improve the quality of the electron density. To solve the phase problem, heavy-atom derivatives were prepared by soaking the crystals for 1 min in 1.9 m ammonium dihydrogen phosphate, 0.1 m Tris-HCl, pH 8.0, 25% PEG400, 100 mm Yb-HPDO3A (lanthanide phasing kit, Jena Bioscience), followed by flash-freezing in liquid nitrogen. The heavy atom data set (collected at a wavelength of 1.38315 Å) was collected at the Swiss Light Source at X06DA/PXIII beamline on a DECTRIS PILATUS 2 M-F detector at 100 K. The heavy atom-soaked crystals diffracted to 3.3 Å resolution. Phaser-EP was employed to calculate the phases (72). Single-wavelength anomalous diffraction alone was not sufficient to calculate the phases. We then used a combination of molecular replacement and single-wavelength anomalous diffraction for phasing. The model obtained after molecular replacement and single-wavelength anomalous diffraction was subsequently used as the template to phase the native dataset by a molecular replacement approach. Coot was used for model building (73). The final model was refined to 2.6 Å resolution using TLS refinement in phenix.refine (74) taking into account the twin law (h,-k,-l). The atomic coordinates and structure factors have been deposited in the Protein Data Bank with the PDB code 5LDY. Structure figures were prepared using PyMOL. Surface areas of interfaces were calculated using the PISA server (75). Alignments are generated with GeneDoc (76).
Proteolysis
InvD1737 and InvD1640 at a concentration of 1 mg/ml were treated with the respective proteases at a concentration of 0.01 mg/ml and incubated for 20 h at room temperature. Reaction was stopped by addition of SDS-PAGE sample buffer and incubation at 95 °C for 10 min.
Small-angle X-ray scattering data collection
Small-angle X-ray scattering data were measured for InvD1640 on the P12 beamline at PETRAIII EMBL Hamburg with an X-ray wavelength of 1.0332 Å. InvD1640 was prepared in a buffer consisting of 20 mm HEPES, pH 7.4, 200 mm NaCl, and 5 mm DTT at a protein concentration of 3.5 mg/ml. For buffer-scattering subtraction, identical buffer sample was prepared. All the data were processed using tools in the ATSAS software package (77). The net protein-scattering data were generated by subtracting scattering of buffer from the scattering of protein using PRIMUS (78). The distance distribution function was calculated by GNOM (79). 18 independent ab initio models were built using DAMMIF (80). These models were then averaged with DAMAVER package (81) to yield the small-angle X-ray scattering envelope. Fitting of the crystal structure with the experimental small-angle X-ray scattering curve was performed using CRYSOL (82). UCSF Chimera (83) was used to fit the X-ray structure of InvD1640 in the small-angle X-ray scattering envelope.
Coating of latex beads for adhesion assay
A 30-μl suspension of amine-modified polystyrene, fluorescent yellow-green latex beads (L1030; Sigma) were added to 1 ml of PBS (1× PBS: 140 mm NaCl, 3 mm KCl, 12 mm Na2HPO4, 2 mm KH2PO4, pH 7.4). After one washing step with PBS, beads were suspended in 1 ml of coupling buffer (0.2 m NaHCO3, pH 8.6, 0.5 m NaCl) in the absence of light. After centrifugation at ∼20,850 × g (12,000 rpm, SA-600) for 5 min, beads were resuspended in 100 μl of coupling buffer, and 100 μl of 0.6–1 mg/ml InvA500, BSA, or InvD1640 were added to it and incubated at 37 °C for 1 h. Then 500 μl of coupling buffer was added and sonicated for 10 s. To the coated beads 500 μl of 30 mg/ml BSA in coupling buffer was added, followed by 1 h of incubation at 37 °C, followed by washing with 1× PBS, 0.2 mg/ml BSA. Coated beads were resuspended in 1 ml of 1× PBS, 0.2 mg/ml BSA and stored at 4 °C until use.
Cell adhesion assay
5 × 104 HEp-2 (ATCC® CCL-23TM) cells were seeded and grown overnight in individual wells of 12-well cell culture plates. Cells were washed three times with PBS and incubated in binding buffer (RPMI 1640 medium supplemented with 20 mm HEPES, pH 7.0, and 0.4% BSA) before the addition of latex beads coated with InvD1640, BSA or InvA500. 2 × 106 beads were added to the cells, and the cell culture plate was centrifuged for 5 min at ∼169 × g (1000 rpm, A-4–81 with MTP/flex buckets). Cells were incubated for 1 h at 25 °C to check for adhesion. After 1 h of incubation, cells were washed three times with PBS and fixed with 2% paraformaldehyde (Sigma P6184). 10 min postincubation at room temperature, cells were washed once with PBS, and fixed cells were stained with 4′,6-diamidino-2-phenylindole (DAPI; Roth 6335.1) to visualize the nuclei of the cells. After another 4-min incubation at room temperature, cells were again washed with PBS, and after addition of 200 μl of PBS, cells were visualized with a Zeiss Axiovert II fluorescence microscope.
Lipid-binding assay
To assess lipid binding of InvD1640, a protein–lipid assay was performed using Membrane Lipid Strips (P-6002; Echelon Biosciences) according to the manufacturer's recommendations. Briefly, membranes were blocked in 3% (w/v) fatty acid-free BSA (Sigma A4503) in TBST (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 0.1% (v/v) Tween 20) for 1 h at room temperature in the dark followed by 1-h incubation with 3×FLAG-tagged InvD1640 (1.0 μg/ml in TBST) at room temperature with gentle agitation. After washing the membranes three times over a 1-min period in TBST, they were incubated for 1 h with 0.8 μg/ml rabbit anti-FLAG polyclonal antibody (Sigma F7425). Membranes were washed as before and incubated for 1 h with anti-rabbit-HRP conjugate (1:10,000 dilution; Jackson Immuno Research 111-035-045) followed by washing as done before and detection by Echelon's K-TMBP.
Reporter gene fusion plasmids
The upstream region of invD (448 nucleotides) was amplified using the primers III790/791 and the genomic DNA of Y. pseudotuberculosis YPIII as a template. The fragment was ligated into the gfpmut3.1-containing plasmid pFU58 (for pRG01) or into the luxCDABE-containing pFU54 (for pRG03) after restriction digestion with BamHI and SalI. Clones were selected on carbenicillin-containing agar plates and sequenced. For the construction of pRG05, the restriction fragment of pFU189 with AvrII and SacI, containing the origin of replication, was ligated into pRG03.
Construction of ΔinvD strain (YP197)
Mutagenesis plasmids were constructed for the replacement of a gene of interest by a kanamycin-resistance cassette. The resistance gene was amplified by PCR using the primers I661 and I662 and the plasmid pKD4 as template. For homologous recombination, flanking upstream and downstream regions of the target genes of about 500 bp were amplified using genomic DNA of the Y. pseudotuberculosis strain YPIII as template. Primers used for the amplification contained 20 nucleotides, homologous to the start/end of the kanamycin-resistance gene at their 5′-end (antisense primer) or 3′-end (sense primer). Primers used to generate the up- and downstream fragments for invD were III792/797. A fusion PCR was performed with the up- and downstream PCR products of the target gene and the kanamycin-resistance gene fragment as template. Primers used for this reaction were sense and antisense primers of the flanking regions, harboring SacI restriction sites. After digestion with SacI, the fusion product was cloned into the suicide vector pAKH3 resulting in plasmid pRG09. Clones were selected on kanamycin containing LB agar plates and further tested by sequencing. For the construction of the invD mutant strains, the invD::kan encoding plasmid pRG09 was transformed into E. coli S17 λpir, conjugated into Y. pseudotuberculosis strain YPIII, and strains with chromosomal integration of the pRG09 plasmid were selected by plating on Yersinia selective agar plates supplemented with kanamycin and carbenicillin as described previously (84). Positive clones were plated on LB agar containing 10% sucrose, and fast-growing variants were selected. Subsequently, clones were tested by plating on antibiotic-containing agar plates and by PCR using the test primers for invD III793/795. For removal of the kanamycin-resistance cassette, the mutant was transformed with the temperature-sensitive plasmid pCP20, encoding the FLP recombinase. Subsequently, the transformants were inoculated overnight at 37 °C in brain heart infusion medium to remove the kanR gene at the FLP recognition sites. The resulting clones were grown at 37 °C to exponential phase and then shifted for 1 h at 42 °C to provoke loss of the temperature-sensitive plasmid as described previously (85). Afterward, bacteria were plated on agar plates, and the resulting clones were tested for growth on kanamycin and carbenicillin and via PCR with the test primers.
Luciferase expression assay
For the determination of gene expression by luciferase expression assays, Y. pseudotuberculosis was transformed with the corresponding plasmids, grown at 25 or 37 °C overnight, and diluted 1:50. Subsequently, cultures were grown at 25 and 37 °C, and every hour the OD600 and bioluminescence were measured with the Varioskan Flash plate reader (Thermo Fisher Scientific) using the ScanIt software (Thermo Fisher Scientific). Background level of the luciferase activity was subtracted, and the relative luminescence units/OD600 were calculated.
Ethics statement
All animal work was performed in strict accordance with the German Recommendations of the Society for Laboratory Animal Science and the European Health Recommendations of the Federation of Laboratory Animal Science Associations. The animal protocol was approved by the “Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit” animal licensing committee permission no. 33.9.43502-04-55/09 and 33.9-42502-04-12/0907. Animals were handled with appropriate care and welfare, and all efforts were made to minimize suffering.
Mouse infection
For all experiments, female BALB/c mice between 6 and 8 weeks old were purchased from Janvier (Saint Berthevin Cedex, France) and infected via the oral route with a ball-tipped feeding needle. Bacteria were grown to stationary phase in LB liquid medium at 25 °C, washed, and diluted in PBS. Expression analysis of invD during an infection was assessed using the in vivo imaging system. Mice were orally infected with 9 × 108 CFU of Y. pseudotuberculosis harboring a plasmid-encoded invD-luxCDABE reporter fusion (pRG05) or the empty vector (pFU189). Three independent clones with pRG05 were tested using three mice each. Mice were anesthetized with isoflurane and ventrally imaged using the in vivo imaging Lumina system (Xenogen). Cryosections (thickness, 7 μm) of different organs were prepared for the expression analysis of invD at a single cell level. Three mice were orally infected with 3 × 108 CFU of Y. pseudotuberculosis harboring a plasmid-encoded gapA-dsred2 reporter fusion (pFU228) for the detection of all bacteria and an invD-gfpmut3.1 reporter fusion (pRG01). Three days after infection, mice were sacrificed by CO2 asphyxiation. The small intestine, colon, mLNs, and spleen were frozen on dry ice in Tissue-Tek OCT freezing medium (Sakura Finetek). Sections of 6–8 μm were prepared (Zeiss cryostat Hyrax C50), mounted on SuperFrost Plus slides (Thermo Fisher Scientific). Sections were fixed for 10 min in ice-cold acetone and washed twice with PBS. Cell nuclei were stained with DAPI by using a drop of Roti-Mount FluorCare DAPI (Roth), and slides were subsequently equipped with a coverslip. For visualization, a fluorescence microscope (Zeiss Axiovert II) and the AxioVision software were used (Zeiss).
To analyze the influence of InvD on the colonization of the intestinal tract, two groups of 3–7 mice were orally infected with 2 × 108 CFU in an equal mixture of WT and mutant strain. At 6 h and 1 and 3 days post-infection, mice were euthanized by CO2 asphyxiation, and small intestine and colon were dissected. The organs were rinsed with PBS and incubated for 30 min with gentamicin-containing PBS (50 μg/ml) to kill all extracellular bacteria. Afterward, organs were rinsed with PBS again to remove the gentamycin. All organs were weighted and homogenized in sterile PBS at 25,000 rpm for 30 s using the Polytron PT 2100 homogenizer (Kinematica). Colonization rates of Y. pseudotuberculosis strains were determined by plating serial dilutions of the homogenates on Yersinia selective agar plates with and without antibiotics. CFU/g of tissue are given. The levels of statistical significance for differences were determined by the Mann-Whitney test.
Pulldown experiments
For FLAG-tagged pulldown experiments, InvD1640 (Gly-1640–Asn-1976) and ΔAD-InvD (Gly-1640–Gly-1839) were cloned into a modified pCOLA-Duet vector containing an N-terminal His6, TEV-protease cleavage site, and 3×FLAG site. Expression and purification conditions were the same as for InvD construct with a His6 tag. To prepare tissue lysates, organs were removed from uninfected BALB/c mice and immediately homogenized in cold PBS supplemented with one protease inhibitor tablet (S8820 Sigma). To the homogenate, 200 mm octyl-d-glucopyranoside (Fluka-75081) was added, and the homogenate was further incubated at 4 °C for 1 h with rotation. Postincubation, the homogenate extract was centrifuged at ∼9300 × g (10,000 rpm, F-45-30-11) for 10 min at 4 °C. Supernatant was then used immediately for pulldown experiments (15).
For FLAG-tagged pulldown experiments, 20 μg of 3×FLAG-tagged InvD1640 or 3×FLAG-tagged ΔAD-InvD were incubated with 30 μl of anti-FLAG M2 magnetic beads (Sigma M8823) in TBS for 1 h at 4 °C. Supernatant was discarded, and beads were then incubated with 600 μl of supernatant obtained from homogenized extract from mouse organs for 16 h at 4 °C. The beads were washed three times with 150 μl of TBS buffer. Proteins were eluted in three packed gel volumes of 0.1 m glycine HCl, pH 3.0, collecting 50 μl of eluate in a vial containing 5 μl of 1 m Tris, pH 8.0. The eluted fractions were analyzed by 15% SDS-PAGE and stained by blue silver stain (86).
For strep-tagged pulldown experiments, 20 μg of strep-tagged InvD or ΔAD-InvD were incubated with 20 μl of Strep-Tactin® beads (Strep-Tactin® macroprep beads-iba-2–1505-025) in PBS for 1 h at 4 °C. Supernatant was discarded, and beads were then incubated with 6 μg of scFv-Fc fragments for 1 h at 4 °C. The beads were washed three times with PBS and boiled in 20 μl of SDS loading buffer. Samples were analyzed on 15% SDS-PAGE and stained by Coomassie Blue (R-250) staining solution.
Mass spectrometry
For the identification of the protein bands, the band of interest was excised from the blue silver-stained SDS gel and subjected to MS (MALDI) analysis performed by the mass spectrometry platform at the HZI using an Ultraflex MALDI TOF/TOF (Bruker Daltonics).
Protein labeling for microscale thermophoresis
The accessible primary amine residues of InvD1640, ΔAD-InvD, or InvA500 were covalently labeled with cyanine 5 NHS ester (GE Healthcare) in PBS, using a 3-fold excess of dye. The dye excess was removed via a buffer exchange column (illustra NAP-5 columns GE Healthcare). The protein/dye ratio was determined from the absorption at 280 and 647 nm using an extinction coefficient of Cy5 of 250,000 m−1 cm−1.
Kd determination by microscale thermophoresis
Binding affinities of InvD1640, ΔAD-InvD, or InvA500 to immunoglobulin were determined in PBS at 25 °C by microscale thermophoresis (87) on a NanoTemper Monolith NT.115 (20% LED and 20–60% power; 30 s laser-on time for microscale thermophoresis power 20/40% and 20 s for microscale thermophoresis power 60%; 5 s laser-off time) in standard treated capillaries. Cy-5-labeled InvD was used at a concentration of 50 nm. Equal amounts of labeled protein were titrated with the following proteins: purified human IgG (Life Technologies, Inc., catalog no. 02-7102); mouse IgG (Life Technologies, Inc., catalog no. 02-6502); human IgA (Sigma catalog no. I4036); human IgG Fc (0855911-MP Biomedicals); human IgG F(ab′)2 fragment (catalog no. 009-000-006 Dianova); or scFv-Fc fragments. Fitting was done using the MO.Affinity Analysis (×64) software (NanoTemperTechnologies) in the Thermophoresis + T-jump mode or DYNAFIT (88), as indicated.
Flow cytometry
For spleen, single cell suspensions were obtained by meshing organs through a 100-μm sieve and rinsing with PBS containing 0.2% w/v BSA. From spleens, erythrocytes were eliminated by resuspending splenocytes in 1 ml of erythrocyte/lysis buffer per spleen (0.01 m KHCO3, 0.155 m NH4Cl, 0.1 mm EDTA, adjusted to pH 7.5; Sigma) for 3 min and neutralized by adding 10-fold PBS containing 0.2% w/v BSA.
Fluorochrome-conjugated anti-CD3 (17A2), anti-CD19 (6D5), anti-CD11b (M1/70), anti-CD11c (N418), anti-CD45R(B220, RA3–6B2), anti-CD49b (DX5), anti-γδTCR (eBioGL3), and anti-FLAG (L5, DYKDDDDK tag epitope) were purchased from eBioscience (San Diego) and Biolegend (San Diego). Dead cells were identified using live/dead fixable blue dead cell stain kit (Thermo Fisher Scientific L23105). To exclude unspecific binding of antibodies, samples were incubated for 15 min with anti-CD16/CD32 (BioXCell catalog no. CUS-HB-197) and rat IgG (Jackson ImmunoResearch catalog no. 012-000-003). 3×FLAG-tagged InvD1640 and ΔAD-InvD were used at 100 μg/ml, and staining was performed together with the antibodies staining for 15 min at 4 °C in PBS containing 0.2% (w/v) BSA and 2 mm Ca2+ (Roth catalog no. CN93.1). Flow cytometry was performed using LSR Fortessa flow cytometer with Diva software (BD Biosciences), and data were analyzed with FlowJo software (TreeStar, Ashland, OR).
scFv phage library panning
HAL9 and HAL10 scFv libraries with a size of 1.04 × 1010 and 4.45 × 109, respectively, were used for panning (32). Panning and production of soluble scFv were performed according to Ref. 89. 5 μg of strep-tagged InvD1640 was coated on a microtiter plate (U96 PP 0,5 ml, Greiner, Frickenhausen, Germany) and incubated with the HAL10 library/HAL9 + HAL10 library. After washing, bound phages were eluted with trypsin. The eluted phages were used to infect an E. coli culture, which was then co-infected with the helper phage and grown overnight. Phage particles were precipitated in PEG/NaCl and then used for the next round of panning. This process was repeated for three rounds, and plasmids of isolated clones were purified and inserts sequenced.
DNA sequencing
scFv clones from the third round of panning were randomly picked and inoculated in 2× TY-GA (2× TY + 100 mm glucose + 100 μg/ml ampicillin) media and incubated overnight at 37 °C, 130 rpm. Plasmids were extracted from Qiagen Miniprep kit and sequenced with primer 5′-GGCTCGTATGTTGTGTGG-3′, and afterward sequences were analyzed with VBASE2 (90).
ELISA
200 ng of InvD 1640 or ΔAD-InvD was coated onto a 96-well microtiter plate in PBS. After overnight incubation at 4 °C, plates were blocked with 2% MPBST for 1 h followed by three washings with PBST. 100 μl of scFv-containing supernatant were added to the wells and incubated for 1.5 h at room temperature. Wells were washed three times with PBST and incubated with clone 9E10 mouse anti-Myc antibody (1:50 dilution in 2% MPBST) (Yumab, Braunschweig, Germany) for 1.5 h at room temperature. Plates were washed again as described earlier. For detection, goat anti-mouse IgG-HRP-coupled antibody (1:1000 dilution in 2% MPBST) (Sigma catalog no. A0168) was added to the wells and incubated for 45 min at room temperature followed by addition of 3,3′,5,5′-tetramethylbenzidine. Absorbance was measured at 450 nm (620 nm reference) in an ELISA plate reader (Tecan, Crailsheim, Germany). As a control, four wells were also coated with lysozyme. For detection of the background signal, four wells with streptavidin and E. coli supernatant were incubated only with the components of the detection system.
For the production of soluble scFv, clones were picked from the third round of panning and inoculated in 2× YT-GA media and incubated overnight at 30 °C at 250 rpm. Phosphate-buffered 2× YT-GA was inoculated with the overnight culture and grown for 2 h at 37 °C and 800 rpm. Cells were harvested, and the pellet was suspended in 2× YT supplemented with 100 μg/ml ampicillin, 100 mm sucrose, and 50 μm IPTG and incubated at 30 °C and 800 rpm overnight. Bacteria were pelleted and supernatant was used for ELISA.
scFv-Fc expression and purification
scFv-Fc fragments of different variable heavy and light chain subtypes were cloned, expressed, and purified as described in Ref. 91.
Author contributions
P. S., R. G., J. P., J. H., M. H., P. D., and A. S. conceptualization; P. S., R. G., J. P., S. H., J. H., M. H., P. D., and A. S. formal analysis; P. S., R. G., J. P., S. H., J. H., M. H., P. D., and A. S. validation; P. S., R. G., J. P., and S. H. investigation; P. S., R. G., J. P., and A. S. visualization; P. S., R. G., J. P., P. D., and A. S. writing-original draft; J. H., M. H., P. D., and A. S. supervision; P. D. and A. S. funding acquisition.
Supplementary Material
Acknowledgments
We are grateful to Petra Büsing, Stefan Schmelz, Carina Schmühl, Julius Rabl, and Maria Ebel for expert assistance and to the members of the contributing labs for useful discussions. We acknowledge the Consortium for Functional Glycomics for performing the glycan array screening (supported by National Institutes of Health Grants GM62116 and GM098791). We are grateful to the staff of beamlines P12 at PETRAIII EMBL Hamburg, Germany, and X06DA/PXIII Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland, for assistance with data collection and processing.
This work was supported in part by the Helmholtz Association Young Investigator Grant VH-NG-727 (to A. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Table S1, Figs. S1–S5, and supporting references.
The atomic coordinates and structure factors (code 5LDY) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- mLN
- mesenteric lymph node
- AD
- adhesion domain
- Ail
- attachment and invasion locus
- BIg
- bacterial immunoglobulin-like
- CFU
- colony-forming unit
- Eib
- E. coli immunoglobulin-binding
- HEp-2
- human epithelial type 2
- InvD
- invasinD
- MID
- Moraxella catarrhalis IgD-binding protein
- PpL
- protein L from Peptostreptococcus magnus
- r.m.s.d.
- root mean square deviation
- Sbi
- staphylococcal binder of IgG
- scFv
- single chain fragment variable
- SIgA
- secretory IgA
- SpA
- protein A from Staphylococcus aureus
- YadA
- Yersinia adhesin A
- TEV
- tobacco etch virus
- BME
- β-mercaptoethanol
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- PDB
- Protein Data Bank
- Ig
- immunoglobulin.
References
- 1. Dube P. (2009) Interaction of Yersinia with the gut: mechanisms of pathogenesis and immune evasion. Curr. Top. Microbiol. Immunol. 337, 61–91 [DOI] [PubMed] [Google Scholar]
- 2. Chauhan N., Wrobel A., Skurnik M., and Leo J. C. (2016) Yersinia adhesins: an arsenal for infection. Proteomics Clin. Appl. 10, 949–963 10.1002/prca.201600012 [DOI] [PubMed] [Google Scholar]
- 3. Mikula K. M., Kolodziejczyk R., and Goldman A. (2012) Yersinia infection tools–characterization of structure and function of adhesins. Front. Cell. Infect. Microbiol. 2, 169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bliska J. B., Copass M. C., and Falkow S. (1993) The Yersinia pseudotuberculosis adhesin YadA mediates intimate bacterial attachment to and entry into HEp-2 cells. Infect. Immun. 61, 3914–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eitel J., Heise T., Thiesen U., and Dersch P. (2005) Cell invasion and IL-8 production pathways initiated by YadA of Yersinia pseudotuberculosis require common signalling molecules (FAK, c-SRC, Ras) and distinct cell factors. Cell Microbiol. 7, 63–77 [DOI] [PubMed] [Google Scholar]
- 6. Flügel A., Schulze-Koops H., Heesemann J., Kühn K., Sorokin L., Burkhardt H., von der Mark K., and Emmrich F. (1994) Interaction of enteropathogenic Yersinia enterocolitica with complex basement membranes and the extracellular matrix proteins collagen type IV, laminin-1 and -2, and nidogen/entactin. J. Biol. Chem. 269, 29732–29738 [PubMed] [Google Scholar]
- 7. Schulze-Koops H., Burkhardt H., Heesemann J., Kirsch T., Swoboda B., Bull C., Goodman S., and Emmrich F. (1993) Outer membrane protein YadA of enteropathogenic yersiniae mediates specific binding to cellular but not plasma fibronectin. Infect. Immun. 61, 2513–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tertti R., Skurnik M., Vartio T., and Kuusela P. (1992) Adhesion protein YadA of Yersinia species mediates binding of bacteria to fibronectin. Infect. Immun. 60, 3021–3024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bartra S. S., Ding Y., Miya Fujimoto L. M., Ring J. G., Jain V., Ram S., Marassi F. M., and Plano G. V. (2015) Yersinia pestis uses the Ail outer membrane protein to recruit vitronectin. Microbiology 161, 2174–2183 10.1099/mic.0.000179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsang T. M., Annis D. S., Kronshage M., Fenno J. T., Usselman L. D., Mosher D. F., and Krukonis E. S. (2012) Ail protein binds ninth type III fibronectin repeat (9FNIII) within central 120-kDa region of fibronectin to facilitate cell binding by Yersinia pestis. J. Biol. Chem. 287, 16759–16767 10.1074/jbc.M112.358978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsang T. M., Felek S., and Krukonis E. S. (2010) Ail binding to fibronectin facilitates Yersinia pestis binding to host cells and Yop delivery. Infect. Immun. 78, 3358–3368 10.1128/IAI.00238-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Felek S., and Krukonis E. S. (2009) The Yersinia pestis Ail protein mediates binding and Yop delivery to host cells required for plague virulence. Infect Immun. 77, 825–836 10.1128/IAI.00913-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leo J. C., Oberhettinger P., Schütz M., and Linke D. (2015) The inverse autotransporter family: intimin, invasin and related proteins. Int. J. Med. Microbiol. 305, 276–282 [DOI] [PubMed] [Google Scholar]
- 14. Fan E., Chauhan N., Gupta Udatha D. B., Leo J. C., and Linke D. (2016) Type V secretion systems in bacteria. Microbiol. Spectr. 4, 10.1128/microbiolspec.VMBF-0009-2015 [DOI] [PubMed] [Google Scholar]
- 15. Isberg R. R., and Leong J. M. (1990) Multiple beta 1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell 60, 861–871 10.1016/0092-8674(90)90099-Z [DOI] [PubMed] [Google Scholar]
- 16. Alrutz M. A., Srivastava A., Wong K. W., D'Souza-Schorey C., Tang M., Ch'Ng L. E., Snapper S. B., and Isberg R. R. (2001) Efficient uptake of Yersinia pseudotuberculosis via integrin receptors involves a Rac1-Arp 2/3 pathway that bypasses N-WASP function. Mol. Microbiol. 42, 689–703 [DOI] [PubMed] [Google Scholar]
- 17. Alrutz M. A., and Isberg R. R. (1998) Involvement of focal adhesion kinase in invasin-mediated uptake. Proc. Natl. Acad. Sci. U.S.A. 95, 13658–13663 10.1073/pnas.95.23.13658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pisano F., Kochut A., Uliczka F., Geyer R., Stolz T., Thiermann T., Rohde M., and Dersch P. (2012) In vivo-induced InvA-like autotransporters Ifp and InvC of Yersinia pseudotuberculosis promote interactions with intestinal epithelial cells and contribute to virulence. Infect. Immun. 80, 1050–1064 10.1128/IAI.05715-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Strong P. C., Hinchliffe S. J., Patrick H., Atkinson S., Champion O. L., and Wren B. W. (2011) Identification and characterisation of a novel adhesin Ifp in Yersinia pseudotuberculosis. BMC Microbiol. 11, 85 10.1186/1471-2180-11-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sadana P., Mönnich M., Unverzagt C., and Scrima A. (2017) Structure of the Y. pseudotuberculosis adhesin InvasinE. Protein Sci. 26, 1182–1195 10.1002/pro.3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hamburger Z. A., Brown M. S., Isberg R. R., and Bjorkman P. J. (1999) Crystal structure of invasin: a bacterial integrin-binding protein. Science 286, 291–295 10.1126/science.286.5438.291 [DOI] [PubMed] [Google Scholar]
- 22. Bodelón G., Palomino C., and Fernández L. Á. (2013) Immunoglobulin domains in Escherichia coli and other enterobacteria: from pathogenesis to applications in antibody technologies. FEMS Microbiol. Rev. 37, 204–250 10.1111/j.1574-6976.2012.00347.x [DOI] [PubMed] [Google Scholar]
- 23. Casasnovas J. M., Stehle T., Liu J. H., Wang J. H., and Springer T. (1998) A dimeric crystal structure for the N-terminal two domains of intercellular adhesion molecule-1. Proc. Natl. Acad. Sci. U.S.A. 95, 4134–4139 10.1073/pnas.95.8.4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tan K., Casasnovas J. M., Liu J. H., Briskin M. J., Springer T. A., and Wang J. H. (1998) The structure of immunoglobulin superfamily domains 1 and 2 of MAdCAM-1 reveals novel features important for integrin recognition. Structure 6, 793–801 10.1016/S0969-2126(98)00080-X [DOI] [PubMed] [Google Scholar]
- 25. Lipovsek D. (2011) Adnectins: engineered target-binding protein therapeutics. Protein Eng. Des. Sel. 24, 3–9 10.1093/protein/gzq097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zav'yalov V. P., Abramov V. M., Cherepanov P. G., Spirina G. V., Chernovskaya T. V., Vasiliev A. M., and Zav'yalova G. A. (1996) pH 6 antigen (PsaA protein) of Yersinia pestis, a novel bacterial Fc-receptor. FEMS Immunol. Med. Microbiol. 14, 53–57 10.1111/j.1574-695X.1996.tb00267.x [DOI] [PubMed] [Google Scholar]
- 27. Bao R., Nair M. K., Tang W., Esser L., Sadhukhan A., Holland R. L., Xia D., and Schifferli D. M. (2013) Structural basis for the specific recognition of dual receptors by the homopolymeric pH 6 antigen (Psa) fimbriae of Yersinia pestis. Proc. Natl. Acad. Sci. U.S.A. 110, 1065–1070 10.1073/pnas.1212431110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Remaut H., Rose R. J., Hannan T. J., Hultgren S. J., Radford S. E., Ashcroft A. E., and Waksman G. (2006) Donor-strand exchange in chaperone-assisted pilus assembly proceeds through a concerted β strand displacement mechanism. Mol. Cell 22, 831–842 10.1016/j.molcel.2006.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Isberg R. R., Swain A., and Falkow S. (1988) Analysis of expression and thermoregulation of the Yersinia pseudotuberculosis inv gene with hybrid proteins. Infect. Immun. 56, 2133–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Esko J. D., and Sharon N. (2009) in Essentials of Glycobiology (Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., and Etzler M. E., eds) 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York: http://www.ncbi.nlm.nih.gov/books/NBK1907/ [PubMed] [Google Scholar]
- 31. Allman D., and Pillai S. (2008) Peripheral B cell subsets. Curr. Opin. Immunol. 20, 149–157 10.1016/j.coi.2008.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kügler J., Wilke S., Meier D., Tomszak F., Frenzel A., Schirrmann T., Dübel S., Garritsen H., Hock B., Toleikis L., Schütte M., and Hust M. (2015) Generation and analysis of the improved human HAL9/10 antibody phage display libraries. BMC Biotechnol. 15, 10 10.1186/s12896-015-0125-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu J. L., and Davis M. M. (2000) Diversity in the CDR3 region of V(H) is sufficient for most antibody specificities. Immunity 13, 37–45 10.1016/S1074-7613(00)00006-6 [DOI] [PubMed] [Google Scholar]
- 34. Luo Y., Frey E. A., Pfuetzner R. A., Creagh A. L., Knoechel D. G., Haynes C. A., Finlay B. B., and Strynadka N. C. (2000) Crystal structure of enteropathogenic Escherichia coli intimin-receptor complex. Nature 405, 1073–1077 10.1038/35016618 [DOI] [PubMed] [Google Scholar]
- 35. Zelensky A. N., and Gready J. E. (2005) The C-type lectin-like domain superfamily. FEBS J. 272, 6179–6217 10.1111/j.1742-4658.2005.05031.x [DOI] [PubMed] [Google Scholar]
- 36. Neshat M. N., Goodglick L., Lim K., and Braun J. (2000) Mapping the B cell superantigen binding site for HIV-1 gp120 on a V(H)3 Ig. Int. Immunol. 12, 305–312 10.1093/intimm/12.3.305 [DOI] [PubMed] [Google Scholar]
- 37. Karray S., Juompan L., Maroun R. C., Isenberg D., Silverman G. J., and Zouali M. (1998) Structural basis of the gp120 superantigen-binding site on human immunoglobulins. J. Immunol. 161, 6681–6688 [PubMed] [Google Scholar]
- 38. Guigou V., Cuisinier A. M., Tonnelle C., Moinier D., Fougereau M., and Fumoux F. (1990) Human immunoglobulin VH and VK repertoire revealed by in situ hybridization. Mol. Immunol. 27, 935–940 10.1016/0161-5890(90)90161-R [DOI] [PubMed] [Google Scholar]
- 39. Dudgeon K., Rouet R., Kokmeijer I., Schofield P., Stolp J., Langley D., Stock D., and Christ D. (2012) General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. U.S.A. 109, 10879–10884 10.1073/pnas.1202866109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu G., Kim D., Park B. K., Park S., Ha J.-H., Kim T. H., Gautam A., Kim J. N., Lee S. I., Park H.-B., Kim Y.-S., Kwon H.-J., and Lee Y. (2016) Anti-metastatic effect of the TM4SF5-specific peptide vaccine and humanized monoclonal antibody on colon cancer in a mouse lung metastasis model. Oncotarget 7, 79170–79186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Silverman G. J., and Goodyear C. S. (2006) Confounding B-cell defenses: lessons from a staphylococcal superantigen. Nat. Rev. Immunol. 6, 465–475 10.1038/nri1853 [DOI] [PubMed] [Google Scholar]
- 42. Sulica A., Medesan C., Laky M., Onică D., Sjöquist J., and Ghetie V. (1979) Effect of protein A of Staphylococcus aureus on the binding of monomeric and polymeric IgG to Fc receptor-bearing cells. Immunology 38, 173–179 [PMC free article] [PubMed] [Google Scholar]
- 43. Deisenhofer J. (1981) Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry 20, 2361–2370 10.1021/bi00512a001 [DOI] [PubMed] [Google Scholar]
- 44. Zhang L., Jacobsson K., Vasi J., Lindberg M., and Frykberg L. (1998) A second IgG-binding protein in Staphylococcus aureus. Microbiology 144, 985–991 10.1099/00221287-144-4-985 [DOI] [PubMed] [Google Scholar]
- 45. Smith E. J., Visai L., Kerrigan S. W., Speziale P., and Foster T. J. (2011) The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect. Immun. 79, 3801–3809 10.1128/IAI.05075-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smith E. J., Corrigan R. M., van der Sluis T., Gründling A., Speziale P., Geoghegan J. A., and Foster T. J. (2012) The immune evasion protein Sbi of Staphylococcus aureus occurs both extracellularly and anchored to the cell envelope by binding lipoteichoic acid. Mol. Microbiol. 83, 789–804 10.1111/j.1365-2958.2011.07966.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Leo J. C., and Goldman A. (2009) The immunoglobulin-binding Eib proteins from Escherichia coli are receptors for IgG Fc. Mol. Immunol. 46, 1860–1866 10.1016/j.molimm.2009.02.024 [DOI] [PubMed] [Google Scholar]
- 48. Sandt C. H., and Hill C. W. (2001) Nonimmune binding of human immunoglobulin A (IgA) and IgG Fc by distinct sequence segments of the EibF cell surface protein of Escherichia coli. Infect. Immun. 69, 7293–7303 10.1128/IAI.69.12.7293-7203.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marone G., Rossi F. W., Detoraki A., Granata F., Marone G., Genovese A., and Spadaro G. (2007) Role of superallergens in allergic disorders. Chem. Immunol. Allergy 93, 195–213 [DOI] [PubMed] [Google Scholar]
- 50. Marone G., Rossi F. W., Bova M., Detoraki A., Liccardo B., and Petraroli A. (2004) Superallergens: a novel mechanism of IgE-mediated activation of human basophils and mast cells. Clin. Exp. Allergy Rev. 4, 64–75 10.1111/j.1472-9725.2004.00036.x [DOI] [Google Scholar]
- 51. Gjörloff Wingren A., Hadzic R., Forsgren A., and Riesbeck K. (2002) The novel IgD binding protein from Moraxella catarrhalis induces human B lymphocyte activation and Ig secretion in the presence of Th2 cytokines. J. Immunol. 168, 5582–5588 10.4049/jimmunol.168.11.5582 [DOI] [PubMed] [Google Scholar]
- 52. Sasso E. H., Silverman G. J., and Mannik M. (1989) Human IgM molecules that bind staphylococcal protein A contain VHIII H chains. J. Immunol. 142, 2778–2783 [PubMed] [Google Scholar]
- 53. Sasano M., Burton D. R., and Silverman G. J. (1993) Molecular selection of human antibodies with an unconventional bacterial B cell antigen. J. Immunol. 151, 5822–5839 [PubMed] [Google Scholar]
- 54. Björck L. (1988) Protein L. A novel bacterial cell wall protein with affinity for Ig L chains. J. Immunol. 140, 1194–1197 [PubMed] [Google Scholar]
- 55. Beckingham J. A., Bottomley S. P., Hinton R., Sutton B. J., and Gore M. G. (1999) Interactions between a single immunoglobulin-binding domain of protein L from Peptostreptococcus magnus and a human κ light chain. Biochem. J. 340, 193–199 [PMC free article] [PubMed] [Google Scholar]
- 56. Townsley-Fuchs J., Neshat M. S., Margolin D. H., Braun J., and Goodglick L. (1997) HIV-1 gp120: a novel viral B cell superantigen. Int. Rev. Immunol. 14, 325–338 10.3109/08830189709116523 [DOI] [PubMed] [Google Scholar]
- 57. Kozlowski L. M., Soulika A. M., Silverman G. J., Lambris J. D., and Levinson A. I. (1996) Complement activation by a B cell superantigen. J. Immunol. 157, 1200–1206 [PubMed] [Google Scholar]
- 58. Genovese A., Bouvet J. P., Florio G., Lamparter-Schummert B., Björck L., and Marone G. (2000) Bacterial immunoglobulin superantigen proteins A and L activate human heart mast cells by interacting with immunoglobulin E. Infect. Immun. 68, 5517–5524 10.1128/IAI.68.10.5517-5524.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Patella V., Florio G., Petraroli A., and Marone G. (2000) HIV-1 gp120 induces IL-4 and IL-13 release from human Fc epsilon RI+ cells through interaction with the VH3 region of IgE. J. Immunol. 164, 589–595 10.4049/jimmunol.164.2.589 [DOI] [PubMed] [Google Scholar]
- 60. Graille M., Stura E. A, Corper A. L., Sutton B. J., Taussig M. J., Charbonnier J. B., and Silverman G. J. (2000) Crystal structure of a Staphylococcus aureus protein A domain complexed with the Fab fragment of a human IgM antibody: structural basis for recognition of B-cell receptors and superantigen activity. Proc. Natl. Acad. Sci. U.S.A. 97, 5399–5404 10.1073/pnas.97.10.5399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goodyear C. S., Narita M., and Silverman G. J. (2004) In vivo VL-targeted activation-induced apoptotic supraclonal deletion by a microbial B cell toxin. J. Immunol. 172, 2870–2877 10.4049/jimmunol.172.5.2870 [DOI] [PubMed] [Google Scholar]
- 62. Axcrona K., Björckf L., and Leanderson T. (1995) Multiple ligand interactions for bacterial immunoglobulin-binding proteins on human and murine cells of the hematopoetic lineage. Scand. J. Immunol. 42, 359–367 10.1111/j.1365-3083.1995.tb03668.x [DOI] [PubMed] [Google Scholar]
- 63. Cerutti A., and Rescigno M. (2008) The biology of intestinal immunoglobulin A responses. Immunity 28, 740–750 10.1016/j.immuni.2008.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Corthésy B. (2013) Multi-faceted functions of secretory IgA at mucosal surfaces. Front. Immunol. 4, 185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moor K., Diard M., Sellin M. E., Felmy B., Wotzka S. Y., Toska A., Bakkeren E., Arnoldini M., Bansept F., Co A. D., Völler T., Minola A., Fernandez-Rodriguez B., Agatic G., Barbieri S., et al. (2017) High-avidity IgA protects the intestine by enchaining growing bacteria. Nature 544, 498–502 10.1038/nature22058 [DOI] [PubMed] [Google Scholar]
- 66. Mantis N. J., Cheung M. C., Chintalacharuvu K. R., Rey J., Corthésy B., and Neutra M. R. (2002) Selective adherence of IgA to murine Peyer's patch M cells: evidence for a novel IgA receptor. J. Immunol. 169, 1844–1851 10.4049/jimmunol.169.4.1844 [DOI] [PubMed] [Google Scholar]
- 67. Favre L., Spertini F., and Corthésy B. (2005) Secretory IgA possesses intrinsic modulatory properties stimulating mucosal and systemic immune responses. J. Immunol. 175, 2793–2800 10.4049/jimmunol.175.5.2793 [DOI] [PubMed] [Google Scholar]
- 68. Kadaoui K. A., and Corthésy B. (2007) Secretory IgA mediates bacterial translocation to dendritic cells in mouse Peyer's patches with restriction to mucosal compartment. J. Immunol. 179, 7751–7757 10.4049/jimmunol.179.11.7751 [DOI] [PubMed] [Google Scholar]
- 69. Matysiak-Budnik T., Moura I. C., Arcos-Fajardo M., Lebreton C., Ménard S., Candalh C., Ben-Khalifa K., Dugave C., Tamouza H., van Niel G., Bouhnik Y., Lamarque D., Chaussade S., Malamut G., Cellier C., et al. (2008) Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J. Exp. Med. 205, 143–154 10.1084/jem.20071204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kabsch W. (2010) XDS. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Read R. J., and McCoy A. J. (2011) Using SAD data in Phaser. Acta Crystallogr. D Biol. Crystallogr. 67, 338–344 10.1107/S0907444910051371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S090744491200130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 76. Nicholas K. B., Nicholas H. B. Jr., and Deerfield D. W. II. (1997) GeneDoc: analysis and visualization of genetic variation. EMBNEW. NEWS 4, 14 [Google Scholar]
- 77. Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D., Konarev P. V., and Svergun D. I. (2012) New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 10.1107/S0021889812007662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 10.1107/S0021889803012779 [DOI] [Google Scholar]
- 79. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 10.1107/S0021889892001663 [DOI] [Google Scholar]
- 80. Franke D., and Svergun D. I. (2009) DAMMIF, a program for rapid ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346 10.1107/S0021889809000338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Beaucage B. Y. (1995) Approximations leading to unified exponential/power-law approach to small-angle scattering. J. Appl. Crystallogr. 28, 717–728 10.1107/S0021889895005292 [DOI] [Google Scholar]
- 82. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL–a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 10.1107/S0021889895007047 [DOI] [Google Scholar]
- 83. Yang Z., Lasker K., Schneidman-Duhovny D., Webb B., Huang C. C., Pettersen E. F., Goddard T. D., Meng E. C., Sali A., and Ferrin T. E. (2012) UCSF Chimera, MODELLER, and IMP: an integrated modeling system. J. Struct. Biol. 179, 269–278 10.1016/j.jsb.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nagel G., Lahrz A., and Dersch P. (2001) Environmental control of invasin expression in Yersinia pseudotuberculosis is mediated by regulation of RovA, a transcriptional activator of the SlyA/Hor family. Mol. Microbiol. 41, 1249–1269 10.1046/j.1365-2958.2001.02522.x [DOI] [PubMed] [Google Scholar]
- 85. Datsenko K. A., and Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Candiano G., Bruschi M., Musante L., Santucci L., Ghiggeri G. M., Carnemolla B., Orecchia P., Zardi L., and Righetti P. G. (2004) Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25, 1327–1333 10.1002/elps.200305844 [DOI] [PubMed] [Google Scholar]
- 87. Wienken C. J., Baaske P., Rothbauer U., Braun D., and Duhr S. (2010) Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1, 100 10.1038/ncomms1093 [DOI] [PubMed] [Google Scholar]
- 88. Kuzmic P. (1996) Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273 10.1006/abio.1996.0238 [DOI] [PubMed] [Google Scholar]
- 89. Frenzel A., Kügler J., Wilke S., Schirrmann T., and Hust M. (2014) Construction of human antibody gene libraries and selection of antibodies by phage display. Methods Mol. Biol. 1060, 215–243 10.1007/978-1-62703-586-6_12 [DOI] [PubMed] [Google Scholar]
- 90. Retter I., Althaus H. H., Münch R., and Müller W. (2005) VBASE2, an integrative V gene database. Nucleic Acids Res. 33, D671–D674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jäger V., Büssow K., Wagner A., Weber S., Hust M., Frenzel A., and Schirrmann T. (2013) High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotechnol. 13, 52 10.1186/1472-6750-13-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hust M., Meyer T., Voedisch B., Rülker T., Thie H., El-Ghezal A., Kirsch M. I., Schütte M., Helmsing S., Meier D., Schirrmann T., and Dübel S. (2011) A human scFv antibody generation pipeline for proteome research. J. Biotechnol. 152, 159–170 10.1016/j.jbiotec.2010.09.945 [DOI] [PubMed] [Google Scholar]
- 93. Kirsch M. I., Hülseweh B., Nacke C., Rülker T., Schirrmann T., Marschall H.-J., Hust M., and Dübel S. (2008) Development of human antibody fragments using antibody phage display for the detection and diagnosis of Venezuelan equine encephalitis virus (VEEV). BMC Biotechnol. 8, 66 10.1186/1472-6750-8-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Meyer T., Stratmann-Selke J., Meens J., Schirrmann T., Gerlach G. F., Frank R., Dübel S., Strutzberg-Minder K., and Hust M. (2011) Isolation of scFv fragments specific to OmpD of Salmonella typhimurium. Vet. Microbiol. 147, 162–169 10.1016/j.vetmic.2010.06.023 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.