

Abstract
Prostate cancer is a common cause of cancer-related death in men. E6AP (E6-Associated Protein), an E3 ubiquitin ligase and a transcription cofactor, is elevated in a subset of prostate cancer patients. Genetic manipulations of E6AP in prostate cancer cells expose a role of E6AP in promoting growth and survival of prostate cancer cells in vitro and in vivo. However, the effect of E6AP on prostate cancer cells is broad and it cannot be explained fully by previously identified tumor suppressor targets of E6AP, promyelocytic leukemia protein and p27. To explore additional players that are regulated downstream of E6AP, we combined a transcriptomic and proteomic approach. We identified and quantified 16,130 transcripts and 7,209 proteins in castration resistant prostate cancer cell line, DU145. A total of 2,763 transcripts and 308 proteins were significantly altered on knockdown of E6AP. Pathway analyses supported the known phenotypic effects of E6AP knockdown in prostate cancer cells and in parallel exposed novel potential links of E6AP with cancer metabolism, DNA damage repair and immune response. Changes in expression of the top candidates were confirmed using real-time polymerase chain reaction. Of these, clusterin, a stress-induced chaperone protein, commonly deregulated in prostate cancer, was pursued further. Knockdown of E6AP resulted in increased clusterin transcript and protein levels in vitro and in vivo. Concomitant knockdown of E6AP and clusterin supported the contribution of clusterin to the phenotype induced by E6AP. Overall, results from this study provide insight into the potential biological pathways controlled by E6AP in prostate cancer cells and identifies clusterin as a novel target of E6AP.
Keywords: Prostate cancer, Transcription*, SILAC, Ubiquitinases, Protein Identification*, Clusterin, E6AP, Proteomics, Transcriptomics
Prostate cancer (PC)1 is one of the leading causes of cancer-related mortality in men, with one in five men newly diagnosed with prostate cancer present locally advanced or metastatic form of the disease (1). Treatment options for both metastatic and castration resistant (CRPC) forms of PC are still limited. Clinical trials with proteasomal inhibitor Bortezomib alone or in combination with docetaxel for treatment of CRPC have shown promising results (as reviewed in (2)). However, lack of specificity of Bortezomib leads to patients suffering from significant side-effects (3, 4). Inhibition of specific proteasome degradation pathways by defining key E3 ligases that promote PC can lead to the development of more effective PC therapies.
E6-associated protein (E6AP; UBE3A) is the founding member of the HECT (Homologous to E6AP Carboxyl Terminus) domain E3 ligases (5). E6AP was initially identified in the context of high risk human papillomavirus (HPV)-related cancers, where E6AP in association with viral oncoprotein E6 ubiquitinates and degrades the tumor suppressor p53 (6). Recent work from our group and others reveal roles of E6AP in HPV-independent cancers, including lung and blood cancer (7, 8). E6AP functions as an E3 ligase and as a transcriptional cofactor (9). The functions of E6AP, mostly attributed to its ligase activity include cellular growth, cell cycle, apoptosis, cellular senescence, cellular stress response and proteasomal regulation (10–16). Independent of its E3 ligase activity, E6AP also co-activates transcription by the steroid hormone receptors and by E2F1 (7, 9, 17).
E6AP plays an important part in prostate gland development and growth. E6AP-null mice have reduced wet weight of prostate glands. Consistently, mice over-expressing E6AP develop enlarged prostate glands and show prostatic intraepithelial neoplasia (18, 19). Moreover, knockdown of E6AP in PC cells attenuates cellular growth in vitro and in vivo (20), further supporting an oncogenic function of E6AP in PC. We have recently demonstrated that this is partially explained by the effect of E6AP on the key tumor suppressors, promyelocytic leukemia protein (PML; (20)) and p27 (17). This in vitro and in vivo work is further supported by tissue microarray data demonstrating that patients with localized PC expressing high levels of E6AP and low levels of PML have the shortest clinical relapse-free survival and this correlation is a significant predictor of prostate cancer-associated death (21). Additionally, a subset of PC patients with elevated Gleason's score express high levels of E6AP and low levels of p27 (17), suggesting that high E6AP/low p27 correlation is associated with late stage PC. However, there are subsets of patients that do not present an inverse correlation between E6AP/PML (62%; (21)) or E6AP/p27 (68%; (17)) protein levels. As E6AP regulates a plethora of functions in PC cells, we hypothesize that other targets of E6AP are likely to contribute to its oncogenic activities. This study aims to identify novel targets of E6AP by using transcriptomic and proteomic approaches. Pathway analyses exposed novel links of E6AP with cancer metabolism, DNA damage repair and immune response. Moreover, we identify clusterin, a stress-induced chaperon protein, as a novel tumor suppressor whose expression is regulated by E6AP.
EXPERIMENTAL PROCEDURES
Cell Culture
DU145, PC3 and HEK293T cells were maintained in DMEM (Thermo Fisher, Melbourne, Victoria, Australia) containing 10% fetal calf serum (FCS; Sigma-Aldrich, Sydney, New South Wales, Australia) and 0.1% penicillin/streptomycin (Sigma-Aldrich). All cell lines were acquired from American Type Culture Collection (ATCC, Melbourne, Victoria, Australia). Cell counts were performed using the Coulter cell counter (Beckman, Sydney, New South Wales, Australia).
For SILAC labeling, DU145 cells were cultured in DMEM supplemented with 10% heat-inactivated dialyzed FCS (Thermo Fisher), 0.1% penicillin/streptomycin, 4 mm l-glutamine (Thermo Fisher), 84 mg/L l-arginine and 146 mg/L l-lysine. DU145 shControl cells were cultured in 'light' SILAC media containing naturally abundant l-arginine (12C6, 14N4; Sigma-Aldrich) and l-lysine (12C6, 14N2; Sigma-Aldrich). DU145 shE6AP cells were cultured in 'heavy' SILAC media containing heavy isotope l-arginine (13C6, 15N4; Cambridge Isotope Laboratories Inc, MA) and l-lysine (13C6, 15N2; Cambridge Isotope Laboratories Inc). Incorporation of SILAC label and arginine-to-proline conversion in the cells was verified by mass spectrometry prior to experimentation.
Plasmid and Lentivirus Generation
The sequences for lentiviruses expressing shRNA against E6AP (shE6AP) and its control (shControl) and methodology for viral production and infection have previously been described (20). The sequence of lentivirus expressing shRNA against clusterin (shCLU) was F 5′-TCCCGCTCAGCAACCTAGAAGAATTCAAGAGATTCTTCTAGG TTGCTGAGCTTTTTC-3′ and R 5′-TCGAGAAAAAGCTCAGCAACCTAGAAGAATCTCTTGAATTCTTCTAGGTTGCTGAGC-3′ and a control against clusterin (shControl-CLU) was F 5′-TCCCGCTCAGCTGACTAGAAGAATTCAAGAGATTCTTC-TAGTCAGCTGAGCTTTTTC-3′ and R 5′-TCGAGAAAAAGCTCAGCTGACTAGAA GAATCTCTTGAATTCTTCTAGTCAGCTGAGC-3′. Knockdown of shRNA was induced with 0.2 μg/ml doxycycline (dox; Sigma-Aldrich) in DU145 and 0.05 μg/ml dox in PC3 cells. HA-E6AP and HA-E6AP-C820A plasmids were kindly provided by Zafar Nawaz, Baylor College, Houston, TX. Clusterin plasmid was a kind gift from Saverio Bettuzzi, University of Parma, Parma, Italy (22).
Experimental Design and Statistical Rationale
PC cell line DU145 was chosen for the discovery-based approach as the phenotypic effects of E6AP knockdown both in vitro and in vivo have been explored in DU145 cells in our laboratory (17, 20). Triplicate samples of DU145 cells, transduced with shRNA against E6AP (shE6AP) or control (shControl), were treated with 0.2 μg/ml dox for 2.5 days to induce knockdown of E6AP. We chose this time point because it maximizes E6AP knockdown before any observed growth inhibition (20). The changes in the global transcriptome and proteome were assessed using RNA-seq and SILAC-based proteomics. Data was normalized and mRNA and protein identification and quantitation performed. GO cellular component and Reactome pathway enrichment analysis was performed on significantly (p value < 0.05) altered (≥/≤ 1.5-fold change) transcripts and proteins before validation (Fig. 1).
Fig. 1.
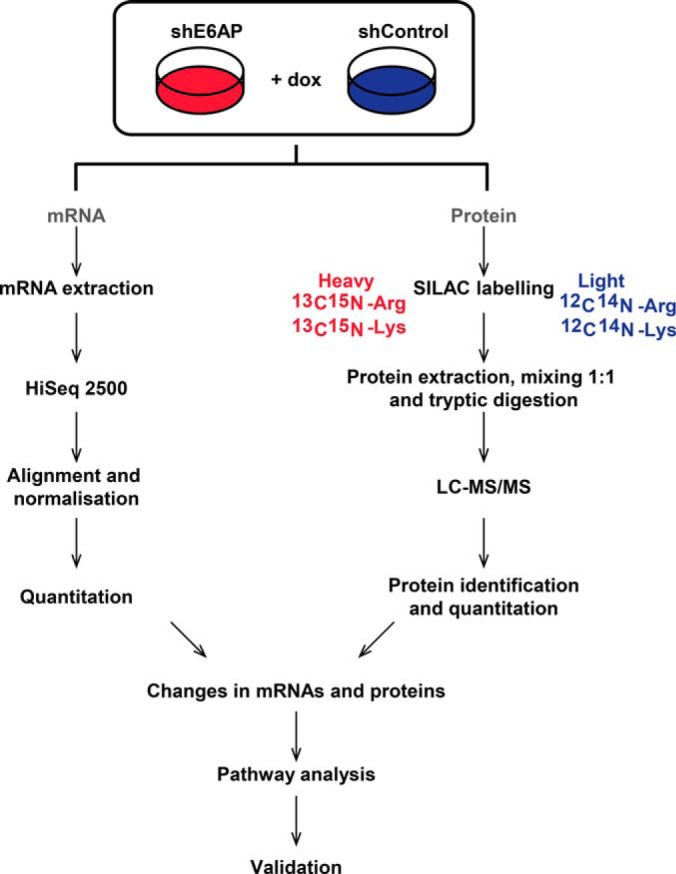
Experimental design for omics approach. DU145 cells transduced with shE6AP and shControl were treated with 0.2 μg/ml dox for 2.5 days. Changes in mRNA and protein expression between triplicate samples of DU145 shE6AP + dox and shControl + dox cells were assessed by Hi Seq 2500 and SILAC-based LC-MS/MS analysis. Significantly altered transcripts and proteins were subjected to pathway analysis. Top regulated candidates altered at both transcript and protein levels were validated using qRT-PCR.
Protein Preparation for Mass Spectrometry
Proteins were extracted from cell pellets with 1% sodium deoxycholate (SDC) in 50 mm HEPES pH 8.0. The lysates were boiled at 95 °C for 5 min before probe sonication. Equal amount of protein (250 μg) from SILAC-labeled DU145 shE6AP and DU145 shControl cells was mixed. Proteins were reduced with 10 mm tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and alkylated with 40 mm chloroacetamide (CAA) by boiling at 95 °C for 5 min. Lysates were digested with sequencing grade modified trypsin (Promega, Madison, WI) with an enzyme-to-substrate ratio of 1:100 overnight at 37 °C. The trypsin was inactivated by acidification with formic acid (FA; Sigma-Aldrich) and SDC was removed from the tryptic digest by two extractions with ethyl acetate (Sigma-Aldrich). Peptides were reverse-phase fractionated as previously described (23). The lyophilized peptides were resuspended in 2% ACN/0.1% FA, aided by sonication (Ultrasonic Bath XUBA3, Grant Instruments Ltd, Cambridge, UK) before MS analysis.
LC-MS/MS
Data-dependent acquisition was performed on a QExactiveTM Plus Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to an UltiMate® 3000 Ultra High Performance Liquid Chromatography (Thermo Fisher Scientific) equipped with an Acclaim PepMap RSLC column (75 μm × 50 cm, nanoViper, C18, 2 μm, 100 Å Thermo Fisher Scientific) using a 155 min gradient. The precursor MS scan, which covered a range of 375–1600 m/z at a resolution of 70,000, was followed by up to 10 subsequent MS/MS scans measured at a resolution of 17,500. Only the most intense multiple charged precursors were selected for higher energy collision induced dissociation fragmentation. The automatic gain control targets were set to 1E6 for the MS scans and 5E4 for the MS/MS scans. Dynamic exclusion was applied for 20 s.
Peptide and Protein Identification
MaxQuant (version 1.5.2.8; (24)) was used for peptide and protein identification using the human proteome database downloaded from Uniprot (UP000005640) in February 2015. The number of entries in the database searched in the MS search parameters were 89,824. The parameters for MaxQuant searches were as follows: precursor mass tolerance was set to 20 ppm (parts per million) for the first search and 4.5 ppm for the main search. Carbamidomethylation of cysteines was entered as a fixed modification. Arg10 and Lys8 were used to specify heavy labeled amino acids, a maximum labeling of three amino acids was allowed and minimum peptide length was set to 7. Oxidation of methionines and acetylation of protein N termini were considered as variable modifications. Enzyme digestion was set to trypsin with a maximum number of two missed cleavages. Using the target-decoy approach, the false discovery rate (FDR) for peptide, modification site and protein identifications was set to 1%. Match between runs was performed with a 2 min retention time window. Only unmodified and razor peptides were used for quantification with the option to re-quantify. Mass spectrometry data was deposited to PRIDE using the ProteomeXchange consortium guidelines (25) with the dataset identifier PXD008743 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD008743).
The ProteinGroups file obtained from MaxQuant was further analyzed with Perseus (version 1.5.2.6; (26)). Proteins that were identified as “only-by-site”, reverse hits and potential contaminants were removed. Outlier significance score for protein subsets was generated by intensity binning, called significance B, with a p value < 0.05 (24). Based on this statistical test, proteins could be significant in 1, 2 or 3 replicates. Proteins meeting all the following three criteria in at least 2 replicates: significance, percentage of peptide quantitative variation of < 40% (as determined by MaxQuant in the ProteinGroups file) and ≥/≤ 1.5-fold difference were considered significant.
Next Generation RNA Sequencing
RNA was extracted using TRIzol (Thermo Fisher) according to the manufacturer's instructions. RNA quality was determined using Agilent Bioanalyser 2100 Expert and samples with RNA integrity number > 9 were used for next generation sequencing. One μg of RNA was used for preparation of the library using TruSeq RNA library preparation kit version 2 (Illumina Inc.) as per manufacturers' instructions. Samples were run on HiSeq 2500 Flowcell System (Illumina) and generated ∼65 million paired-end 50 bp reads per sample.
Reads were aligned using Bowtie2 Align (version 2.2.3) and TopHat (version 2.0.13). The data was normalized using voom (27) and differential expression analysis was performed on normalized data using LIMMA (version 3.1; (28)). The transcripts, shortlisted based on p value < 0.05 and at least 1.5-fold differential expression, were considered significant.
Bioinformatics Analysis
Bioinformatics analyses were performed on Enrichr (29, 30). Gene names of significantly different transcripts and proteins were uploaded on Enrichr. Gene Ontology (GO) enrichment analysis for cellular component 2017b and Reactome 2016 biological pathways were generated using Fisher's exact test (p value < 0.05).
Quantitative Real-time PCR
RNA extraction, cDNA synthesis and quantitative real-time PCR were performed as previously described (17). Primers used are listed in supplemental Table S1. All experiments were performed in technical triplicates in three independent biological samples. Gene expression was calculated using ΔΔCt ± standard deviation and normalized to human housekeeping gene RPL37A. Statistical analysis was performed using GraphPad Prism 7 (GraphPad Software, Inc., La Jolla, CA). The unpaired student's t test was used for comparison of ΔΔCt and p values < 0.05 was considered significant.
Immunoblotting
Immunoblotting was performed as previously described (20). Primary antibodies against E6AP (E6AP-330, Sigma-Aldrich), clusterin (41D, Millipore, Melbourne, Victoria, Australia), CDK6 (C21, Santa Cruz, Dallas, TX), HSP60 (H-300, Santa Cruz), HA (3F10, Roche, Melbourne, Victoria, Australia) and GFP (B2, Santa Cruz) were used. Secondary antibodies used were goat anti-mouse IgG, anti-rabbit IgG (Thermo Fisher) or anti-rat IgG (Dako, Santa Clara, CA).
Immunofluorescence
Immunofluorescence was performed as previously described (17). Antibody against MDC1 (Sigma-Aldrich) and anti-mouse Alexa Fluor 568 (Thermo Fisher) were used. Cells were imaged on Olympus BX-51 (Olympus, Melbourne, Victoria, Australia) and quantitation of nuclear staining was performed on at least 600 cells using Metamorph® (version7.10.1.161; Molecular Devices, San Jose, CA). The experiment was performed on three biological replicates with technical duplicates.
Flow Cytometry
Flow cytometry was performed as previously described (31), except non-permeabilized cells were resuspended in 2% FCS/0.1% NaN3/PBS−/− and stained with antibody against SUSD2 (Novus Biological, Melbourne, Victoria, Australia) and anti-mouse Alexa Fluor 568. Flow cytometry analysis was assessed by BD FACSCantoII (BD Biosciences, Franklin Lakes, NJ) cytometer and gated to exclude cellular debris. Percentage of cells double positive for GFP and Alexa Fluor 568 were calculated. The experiment was performed on two biologically independent samples with technical triplicates.
Xenografts Study
DU145 xenografts experiments were performed as explained in (20). The animal experiments were performed with the ethics approval of the Peter MacCallum Institute Animal Experimentation Committee.
Statistical Analysis
Statistical analysis for cell culture studies was performed using GraphPad Prism 7. All experiments were performed in technical replicates in at least two biological replicates. Data is presented as mean ± standard deviation (S.D.) for technical replicates. Data is presented as mean ± standard error of mean (S.E.) for biological replicates. Unpaired student t test or analysis of variance (ANOVA) was performed to determine significance and was represented as * for p value < 0.05, ** for p value < 0.01, *** for p value < 0.001 or **** for p value < 0.0001.
RESULTS
Exploring the Transcripts Altered on E6AP Knockdown in PC Cells
We recently demonstrated that E6AP plays an oncogenic role in PC (17, 20, 21). Here we aimed to identify novel downstream effectors of E6AP that mediate its oncogenic functions in PC. PC cell line DU145 was chosen for this discovery-based approach as the phenotypic effects of E6AP knockdown both in vitro and in vivo have been explored in DU145 cells in our laboratory (17, 20). DU145 cells, transduced with shRNA against E6AP (shE6AP) or control (shControl), were treated with 0.2 μg/ml dox for 2.5 days to induce knockdown of E6AP. We chose this time point because it maximizes E6AP knockdown before any observed growth inhibition (20). Because E6AP acts as a transcriptional cofactor (32), we explored the transcripts altered on E6AP knockdown in PC cells by RNA-seq. This led to the identification and quantification of 16,130 transcripts across triplicate samples of E6AP knockdown and control cells (supplemental Table S2). Principle component analysis revealed that replicates of the two conditions are well separated in the first dimension, suggesting that a distinct transcriptional signature was associated with cells when the expression of E6AP was knocked down (supplemental Fig. S1A). A histogram of the total number of transcripts against the log2 of fold change demonstrated a binomial distribution (supplemental Fig. S1B). The change in mRNA expression induced following knockdown of E6AP was reproducible between replicates, supported by a 4% median coefficient of variation between the samples (supplemental Fig. S1C). Of the total transcripts identified, 2763 transcripts were significantly (p value < 0.05) altered by at least 1.5-fold, with increased expression of 1139 transcripts and decreased expression of 1624 transcripts (supplemental Table S3). Expectedly, the expression of E6AP mRNA was reduced by 4-fold (Fig. 2A). Transcripts significantly altered on E6AP knockdown were enriched in GO cellular components including axonal initial segment, clathrin-coated phagocytic vesicle membrane, clathrin coat of endocytic vesicle and node of Ranvier (supplemental Fig. S1D). Reactome pathways associated with up-regulated transcripts were G1/S-specific transcription, E2F mediated regulation of DNA replication, RNA polymerase I promoter opening and DNA methylation (Fig. 2B). The biological pathways linked with down-regulated transcripts were mostly related to metabolism: metabolism of lipids and lipoproteins, peroxisomal lipid metabolism, β-oxidation of very long fatty acids and nephrin interactions (Fig. 2C).
Fig. 2.

Transcripts altered on E6AP knockdown in DU145 cells. Triplicate samples of DU145 shE6AP and shControl cells were treated with 0.2 μg/ml dox for 2.5 days before extraction of mRNA and RNA sequencing. A, Volcano plot visualizing transcriptomic alterations where the observed log2 fold changes of each transcript are plotted against their -log10 p values. The horizontal dashed line corresponds to p value < 0.05 and the vertical dashed line corresponds to a 1.5-fold decrease or increase. B, Significant biological pathways (–log10 p value; orange bars) and percentage of transcripts (blue dots) associated with significantly up-regulated transcripts on knockdown of E6AP. C, Significant biological pathways (–log10 p value; orange bars) and percentage of transcripts (blue dots) associated with significantly down-regulated transcripts following knockdown of E6AP.
Exploring the Proteins Altered on E6AP Knockdown in PC Cells
E6AP is an established E3 ubiquitin ligase. Multiple E6AP targets have been identified in the context of viral-independent cancers (7, 8), however only a limited number of targets have been described to modulate the effect of E6AP in prostate tumorigenesis (17, 20). Analysis of proteins in DU145 cells led to identification and quantification of 7209 proteins (supplemental Table S4). These proteins were identified by two or more unique peptides and were quantified with more than one shE6AP/shControl ratio between the triplicate samples. A strong Pearson's correlation (average of 0.75; supplemental Fig. S2A) and a less than 20% median coefficient of variation (supplemental Fig. S2B) between three independent biological replicates demonstrated that the replicates produce similar results and the identified proteins showed a normal distribution (supplemental Fig. S2C). Of the total identified proteins, 309 were significantly altered with up-regulation of 138 proteins and down-regulation of 171 proteins on E6AP knockdown (supplemental Table S5). Expectedly, E6AP abundance was reduced by 4-fold (Fig. 3A). GO analysis of proteins significantly altered in DU145 cells following E6AP knockdown revealed enrichment in the focal adhesion, actin cytoskeleton, mitochondrial pyruvate dehydrogenase complex and mitochondrial endopeptidase Clp complex (supplemental Fig. S2D). Endosomal/vacuolar pathway, antigen processing-cross presentation, ER-phagosome pathway and trafficking and processing of endosomal TLR, pathways associated with the immune system, were associated with significantly up-regulated proteins on E6AP knockdown (Fig. 3B). The biological pathways linked with proteins down-regulated following E6AP knockdown were metabolism, tRNA aminoacylation, metabolism of amino acids and derivatives and cytosolic tRNA aminoacylation (Fig. 3C). The pathways associated with aminoacylation indicate a role of E6AP in protein metabolism, particularly, with translation of proteins (33), which has been shown to be relevant in the context of prostate cancer pathogenesis (34).
Fig. 3.

Proteins altered on E6AP knockdown in DU145 cells. The expression of E6AP was knocked down in triplicate SILAC-labeled DU145 shE6AP and shControl cells for 2.5 days. This was followed by extraction of proteins, mixing of proteins in 1: 1 ratio, tryptic digestion of the proteins to peptides and fractionation of peptides before analysis on LC-MS/MS. A, Volcano plot visualizing proteomic alterations where the observed log2 fold changes of each transcript are plotted against their -log10 p values. The horizontal dashed line corresponds to p value < 0.05 and the vertical dashed line corresponds to a 1.5-fold decrease or increase. B, Significant biological pathways (–log10 p value; orange bars) and percentage of proteins (blue dots) associated with significantly up-regulated proteins on knockdown of E6AP. C, Significant biological pathways (–log10 p value; orange bars) and percentage of proteins (blue dots) associated with significantly down-regulated proteins following knockdown of E6AP.
Biological Pathways Shared Between Significantly Altered Transcripts and Proteins on E6AP Knockdown
Subsequently, we compared significant biological pathways associated with only up-regulated or only down-regulated transcripts and proteins following E6AP knockdown. Of all the statistically significant biological pathways associated with the up-regulated transcripts and proteins, 6 biological pathways were common between them following E6AP knockdown (supplemental Fig. S3A). Notably, the pathways “cell cycle, mitotic” and “cell cycle” were associated with up-regulated transcripts and proteins. Similarly, of all the significant biological pathways associated with the down-regulated transcripts and proteins, 13 biological pathways were common between them following E6AP knockdown (supplemental Fig. S3B). Interestingly, most of the pathways associated with transcripts and proteins down-regulated on E6AP knockdown were related to various metabolic pathways including, but not limited to, fatty acids, lipids, lipoproteins and vitamins. This analysis suggests a correlation between E6AP and metabolism of prostate cancer cells.
Integration of the Transcriptomics and Proteomics Screens
To gain further insight into the levels at which E6AP affects its candidate targets, we compared the results of the significantly altered transcripts and proteins following E6AP knockdown in DU145 cells (Fig. 4A). Most of the identified transcripts were altered only at the mRNA level on E6AP knockdown in DU145 cells with up-regulation of 1080 transcripts and down-regulation of 1541 transcripts. Conversely, 79 and 88 proteins were observed to be only post-transcriptionally up- and down- regulated respectively, without any accompanying changes of their mRNA transcript levels. A total of 142 SILAC-labeled proteins were also significantly changed at their mRNA level in DU145 cells on E6AP knockdown. The change in expression of these transcripts was consistently in the same direction as the proteins (59 up-regulated and 83 down-regulated) on knockdown of E6AP (supplemental Fig. S4) and this positive correlation was further supported by a Spearman's correlation of 0.83 (p value < 0.0001; Fig. 4B). The most predominant pathways associated with these proteins were interferon gamma signaling, interferon signaling, cytokine signaling in immune system and formation of transcription-coupled nuclear excision repair (TC-NER) pre-incision complex (Fig. 4C). The identification of these pathways raises a novel potential link of E6AP to the immune system and DNA damage repair. The pathway signature of candidates that are altered at transcript and protein level was surprisingly distinct from the biological pathways altered by E6AP candidates changing only transcriptionally (supplemental Fig. S5A) or only post-transcriptionally (supplemental Fig. S5B).
Fig. 4.

Integration of the transcriptomics and proteomics screens. A, Venn diagram comparing significantly altered transcripts and proteins on E6AP knockdown. B, Correlation plot between candidates altered at both transcript and protein level on knockdown of E6AP. C, Significant biological pathways (–log10 p value; orange bars) and percentage of proteins (blue dots) associated with candidates altered at transcript and protein level following knockdown of E6AP.
Validation of Selected Candidates Altered on Knockdown of E6AP
To validate the results of the screen, we selected seven top-regulated candidates that were altered on knockdown of E6AP (Fig. 5A). These included cell growth regulators such as, clusterin (CLU), cyclin dependent kinase 6 (CDK6), sushi domain-containing protein 2 (SUSD2) and SWI/SNF complex subunit, SMARCC1; enzymes such as glycogen phosphorylase, liver form (PYGL) and 2′,5′-phosphodiesterase 12 (PDE12); as well as the DNA damage repair protein, mediator of DNA damage checkpoint protein 1 (MDC1). The expression of candidates that were altered at both mRNA and protein level following E6AP knockdown was confirmed using qRT-PCR. Validating the RNA-seq data, we observed increased expression of CLU, CDK6 and SUSD2 and decreased expression of SMARCC1, PYGL and PDE12 on E6AP knockdown (Fig. 5B–5G). The protein abundance of clusterin, CDK6 and SUSD2 was increased on knockdown of E6AP, corroborating the results of qRT-PCR and SILAC data (supplemental Fig. S6). In addition, a reduction in MDC1 protein expression, as identified by SILAC, was confirmed using immunofluorescence (Fig. 5H and supplemental Fig. S6). Taken together, these results validate selected candidates from the transcriptomic and proteomic screens.
Fig. 5.

Validation of E6AP candidates. A, Table depicting fold change (FC) and p value of top-regulated candidates from the transcriptomic and proteomic screens on knockdown of E6AP in DU145 cells. ∧ Sig B represents the Significance B criteria was used to determine significance of proteins (24). B–H, Three independent samples of DU145 shE6AP and shControl cells were treated with 0.2 μg/ml dox for 3 days. CLU (B), CDK6 (C), SUSD2 (D), SMARCC1 (E), PYGL (F) and PDE12 (G) mRNA level was normalized to RLP37A and mRNA expression in shE6AP cells was expressed relative to shControl. Data represents mean ± standard deviation of technical triplicates of one biological replicate. p value was determined using unpaired Student's t test with ** p value < 0.01; *** p value < 0.001; **** p value < 0.0001. H, Representative immunofluorescence staining of MDC1 protein in DU145 cells on E6AP knockdown.
Clusterin Expression is Elevated on E6AP Knockdown in Vitro and in Vivo
Consistent with results in Fig. 5B and (supplemental Fig. S6A), clusterin expression was increased at both mRNA and protein level on E6AP knockdown using a different shRNA targeting E6AP, discarding the possibility that the observed changes are off-target effects of the short hairpin used in the screen (supplemental Fig. S7). To study whether the inverse correlation between E6AP and clusterin was restricted to DU145 cells or a more general characteristic of PC cells, we analyzed the effect of down-regulation of E6AP on clusterin levels in the castration resistant and metastatic PC cell line, PC3. E6AP knockdown in PC3 cells resulted in a significant increase in the abundance of clusterin mRNA (Fig. 6A) and protein level (Fig. 6B). In addition, we evaluated the effect of E6AP knockdown on clusterin mRNA and protein levels in an in vivo setting, using DU145 xenograft model previously described (20). qRT-PCR and immunoblotting of tumors collected at the ethical end point (1500 mm3) demonstrated that knockdown of E6AP increased clusterin mRNA (Fig. 6C) and protein (Fig. 6D) expression compared with control tumors. Of note, the different molecular masses of clusterin protein represent different stages of clusterin glycosylation (35). Taken together, our data strongly demonstrates that down-regulation of E6AP is associated with increased levels of clusterin mRNA and protein in PC in vitro as well as in vivo.
Fig. 6.

Increased clusterin mRNA and protein expression on knockdown of E6AP in vitro and in vivo. qRT-PCR (A) and immunoblotting (B) of clusterin mRNA and protein levels in PC3 shE6AP and Control (parental) cells treated with 0.05 μg/ml dox for 3 days. mRNA data representative of mean ± S.D. of technical triplicates of one of three independent experiments. Immunoblot represents three independent experiments. DU145 shE6AP and shControl cells were injected subcutaneously in NSG mice and treated with dox as detailed under “Experimental Procedures.” End point tumor samples were analyzed for clusterin mRNA and protein levels by qRT-PCR (C) and immunoblotting (D). mRNA data represents mean ± S.E. of three independent mice. For all qRT-PCR, CLU mRNA expression was normalized to housekeeping gene hRLP37A and data is expressed relative to shControl or Control. p value is calculated by unpaired Student t test * p value < 0.05; **** p value < 0.0001.
E6AP Regulates Clusterin also at the Post-transcriptional Level
To test the possibility that E6AP also regulates clusterin post-transcriptionally, we measured the effect of E6AP on the steady-state levels of clusterin. To achieve this, we examined the expression of clusterin in presence of a HA-tagged wild-type (WT) form of E6AP (E6AP-HA) or a catalytically inactive mutant E6AP-C820A-HA. Forty-eight hours post transfection, cells were harvested and total protein lysates were subjected to immunoblotting. As shown in Fig. 7A, transfection of increasing amounts of E6AP-HA resulted in a reduction of clusterin protein levels. GFP expression was used to control for the transfection efficiency. Consistently, there was an increase in clusterin levels with increasing concentrations of E6AP-C820A-HA. This suggested that the overexpressed catalytically mutant E6AP had a dominant negative effect on the endogenous E6AP. Overall, these results indicate that the catalytic activity of E6AP is required for the reduction in clusterin protein levels.
Fig. 7.

E6AP regulates clusterin post-transcriptionally. A, HEK293T cells were transfected with various concentrations of HA-E6AP or HA-E6AP-C820A and constant concentrations of clusterin (0.01 μg) and GFP (0.05 μg). Forty-eight hours post-transfection, proteins were extracted. Left, immunoblot shows expression of E6AP and clusterin, with their expression normalized to GFP loading control. Right, densitometric analysis of clusterin expression normalized to GFP. Graph represents mean ± S.E. of four independent experiments. B, DU145 shE6AP#2 cells were treated with (+ dox) or without (− dox) 0.2 μg/ml dox for 2.5 days. Subsequently, cells were treated with the indicated concentration of cycloheximide (CHX) over time. Left, immunoblot shows expression of E6AP, clusterin and loading control HSP60. Right, densitometric analysis of clusterin abundance normalized to HSP60. Data is representative of three independent experiments.
Subsequently, we determined whether the increase in clusterin protein on knockdown of E6AP was because of reduced protein degradation of clusterin. For this purpose, we examined clusterin protein stability by measuring the effect of E6AP on the half-life of clusterin in DU145 cells. The half-life of clusterin was prolonged (∼90 min) on knockdown of E6AP compared with a half-life of clusterin of ∼50 min in the presence of E6AP (Fig. 7B). In contrast, the half-life of clusterin remained unchanged in control cells (supplemental Fig. S8), demonstrating dependence on E6AP. Overall, these results demonstrate that knockdown of E6AP prolongs the half-life of clusterin protein.
Concomitant Knockdown of Clusterin and E6AP Partially Restores Cell Growth
To determine the contribution of clusterin to the effect of E6AP on PC cell proliferation, we concomitantly knocked down clusterin along with E6AP. To achieve this, PC3 shE6AP or shControl cells were transduced with a short hairpin against clusterin (shCLU) or a control for clusterin (shControl-CLU) in the dox-inducible expression system described previously. The experiment was performed for 6 days as a significant effect on cell growth following knockdown of E6AP was observed at this time point (20). Following treatment of cells with dox for 6 days, simultaneous reduction in E6AP and clusterin protein levels was observed (Fig. 8A). Knockdown of clusterin in PC3 cells increased cell growth. As we previously demonstrated (17), there was a 51% reduction in cell numbers following knockdown of E6AP in PC3 cells. Notably, a concomitant knockdown of E6AP and clusterin partially restored cell growth by 23% (Fig. 8B), supporting the contribution of clusterin to the effect of E6AP on the proliferation of PC cells.
Fig. 8.

Double knockdown of E6AP and clusterin partially rescues PC cell growth. PC3 shE6AP or Control (parental) cells were transduced with shRNA against clusterin (shCLU) or a control for clusterin (shControl-CLU) in a dox-inducible lentivirus-mediated expression system. Cells were treated with 0.05 μg/ml dox for 6 days. A, Immunoblot shows expression of proteins E6AP, clusterin and HSP60 as the loading control. B, Cell counts were performed on PC3 cells following knockdown of E6AP and/or clusterin expression. Graph represents mean ± S.E. for two biological replicates with technical triplicates. Statistical analysis was performed using one-way ANOVA with * p value < 0.05; **** p value < 0.0001.
DISCUSSION
Although E6AP has historically been studied in the context of HPV E6-associated cancers, the functional significance of E6AP in promoting cancer in an E6-independent manner recently attracted attention. In the current study, we identify novel E6AP-regulated transcripts and proteins by performing a combined transcriptomics and proteomics approach. Our results support the recent work undertaken in our laboratory, particularly on how E6AP expression interplays with cell cycle regulators beyond the known link with p27 (17). Moreover, pathway analyses corroborate the phenotypic effect of E6AP knockdown observed in DU145 cells, namely, decreased cell growth, increased cell death and increased sensitivity to DNA damage (20).
To date, limited numbers of transcriptional targets of E6AP have been identified. We observe a greater number of E6AP-regulated transcripts compared with E6AP-regulated proteins in the current study. There are several possible explanations for this. Firstly, E6AP co-activates top layer transcription factors, such as E2F1 (7, 17), SP1, PR, ER, and GR (9), resulting in a potentially broad transcriptional cascade via activation of secondary response genes (36). Secondly, RNA sequencing is much more sensitive compared with the limited dynamic range of current mass spectrometers, contributing to the disparity in number of transcripts versus proteins identified (37). Thirdly, cellular mRNA and protein abundances are controlled by transcription rate, mRNA stability and mRNA splicing in addition to translation rate and protein degradation (38, 39). This suggests that variation in protein abundances may be partially predicted by mRNA expression (40). Lastly, marginal changes in mRNA expression may lead to substantial changes in protein expression over time (41), suggesting that the time at which changes at mRNA versus protein expression are measured can be important.
To filter out the most dominant changes related to E6AP knockdown, we carried out our analysis on candidates that were significantly changed at both mRNA and protein level. The modes of regulation of these candidates by E6AP could be either as (1) a transcription cofactor, as ∼25–30% of the variation in protein abundance can be explained by mRNA expression (38); (2) an E3 ligase for a transcription factor such as C/EBPα (42, 43), thereby indirectly affecting the regulation of candidates downstream of C/EBPα; or (3) a transcription cofactor and an E3 ligase as in the case of p27, AR and amplified in breast cancer 1 (11, 17, 19, 44). The perturbation of the steady state level of a candidate by E6AP as both a transcription cofactor and an E3 ligase in part reflects the different kinetics of the two distinct functions of E6AP. Therefore, it is conceivable that the distinct biological pathways associated with candidates altered only at the transcriptional level versus those altered only post-transcriptionally could be attributed to these different functions of E6AP as a transcription cofactor and an E3 ligase.
Consistent with previous work undertaken in our laboratory (15, 20), pathway analyses revealed a functional link between E6AP and DNA damage response. The current study builds on this link between E6AP and DNA damage response by associating it with a candidate target, MDC1. MDC1, in conjunction with the MRE complex, is recruited to damaged DNA to induce homologous DNA repair and is essential for intra-S and G2/M cell cycle checkpoints (45, 46). There is evidence that MDC1 also plays a role in non-homologous end joining repair (NHEJ) (47) and nucleotide excision repair pathways (48, 49). Recent work by Wang et al. demonstrated that MDC1 co-actives AR transcription, underlining the importance of MDC1 in PC (50). This raises several questions including how E6AP regulates MDC1, which DNA damage repair pathways are altered by E6AP and whether there is a possible therapeutic opportunity with E6AP inhibitors (51) and PARP inhibitors (52) for the treatment of PC.
Jensen et al. previously demonstrated a role of E6AP in Drosophila metabolism; however, no candidate was associated with this link (53). Our study is the first to show a potential link between E6AP and cellular metabolism in the context of cancer. PYGL, the liver form of glycogen phosphorylase, is important for glycogen breakdown and is associated with tumor progression (54). Functionally, knockdown of PYGL leads to glycogen accumulation, reduction in cell growth, increased reactive oxygen species and premature senescence under hypoxic conditions (54). Similarly, an inverse correlation between glycogen accumulation and cell growth was also observed in PC cell lines in the presence of androgen (55). These results raise the possibility that modulation of PYGL by E6AP may explain the phenotypic effects of E6AP knockdown in PC cells.
Our results demonstrate a novel putative link between E6AP and cytokine signaling in the immune system. Interferon signaling suppresses PC cell growth (56). Indeed, overexpression of mitochondrial nuclease, PDE12 prevents interferon induced cell death in PC cells (57) and suppression of PDE12 imparts resistance to and spread of viral infection (57, 58). Blocking PDE12 using inhibitors would promote the anti-proliferative and antiviral response induced by interferon (57, 58). Therefore, reduction in PDE12 expression on E6AP knockdown may act as a feedback loop in promoting interferon induced cell death.
Regulation of cell cycle proteins by E6AP is extensively studied (14, 59–61). However, to date p27 has been the only E6AP target identified in PC (17). Our study further expands on this by identifying novel cell cycle targets of E6AP, including CDK6, SUSD2 and SMARCC1. CDK6 is responsible for G1/S transition in the cell cycle (62). Several studies demonstrated a tumor suppressive function of CDK6 (63–65). Independent of its kinase activity, CDK6 regulates transcription of p16, leading to cell cycle inhibition (62). This poses an interesting possibility that E6AP may modulate p16 expression via CDK6, as an alternative pathway to E2F1 described previously (7). Alternatively, E6AP may mediate CDK6-dependent transcription of AR in an androgen-dependent context (66). SUSD2 is a type I membrane protein containing somatomedin B, adhesion-associated domain present in MUC4 and other proteins (AMOP), von Willebrand factor type D, and Sushi domains, important for cell-cell and cell-matrix adhesion (67). SUSD2 expression is decreased in lung and kidney (67). Furthermore, the role of SUSD2 in reducing cell proliferation and colony formation and promoting apoptosis is well documented (67), highlighting the potential modulation of cell growth by E6AP in PC cells via SUSD2. SMARCC1 is a core component of the SWI/SNF chromatin remodeling complex that induces replicative senescence in colon and ovarian cell lines (68). SMARCC1 is deregulated in PC (69, 70), however, the correlation between SMARCC1 expression and PC progression is not consistent between the two studies and these studies do not define the functional significance of SMARCC1 deregulation in PC. Our laboratory has recently demonstrated that E6AP knockdown induces cellular senescence in PC xenografts models (20), suggesting a potential role of SMARCC1 in modulating cellular senescence in PC cells.
Clusterin, a stress-induced chaperon protein (71), is involved in cell signaling (72), apoptosis (73), cellular migration (74), metastasis (75), DNA repair (76, 77), lipid accumulation (78), and lipid transport (79). Clusterin was selected as a candidate to study further as it ranked as one of the top regulated candidates of E6AP and some of its functions are related to the biology of E6AP. Moreover, several lines of evidence support the role of clusterin as a tumor suppressor in PC (22, 35, 80–82). Clusterin transcript and protein expression are decreased in epithelial cells of low- and high-grade PC samples from patients that underwent androgen deprivation therapy followed by radical prostatectomy (83, 84). Moreover, significant reduction in clusterin transcript expression was correlated with negative prognosis (defined by increased PSA levels following surgery, distant metastasis and/or lymph node involvement) in these PC patients (84). Bettuzzi et al. demonstrated that 67% of CLU homozygous KO mouse developed PINs and 34% develop well-differentiated carcinomas (85). Clusterin overexpression in immortalized human prostate epithelial cells and PC cell lines reduced colony formation, cellular proliferation, induced cell cycle arrest and increased cell death (22, 81, 82). Consistent with these studies, our results demonstrate a tumor suppressive role of clusterin in PC3 cells. Further, concomitant knockdown of clusterin together with E6AP partially restores the effect of E6AP on the proliferation of PC3 cells. These results support the contribution of clusterin to the promotion of PC cell proliferation by E6AP (Fig. 9).
Fig. 9.
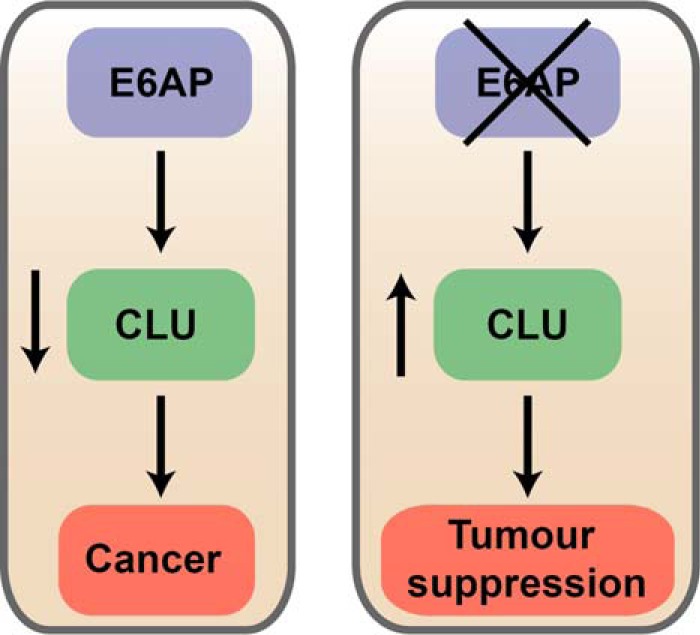
Diagram depicting our proposed model of how loss of E6AP can restore clusterin and lead to tumor suppression in PC. High levels of E6AP lead to prostate cancer progression and loss of clusterin promotes prostate cancer formation. Therefore, reduction of E6AP levels in prostate cancer would restore clusterin expression, leading to tumor suppression.
Clusterin is regulated by various transcription factors, including, AR, cFOS, cMYC, Activator Protein 1 (AP1), ERG1, heat shock factor (HSF)1-HSF2 hetercomplex and HIF-1α (86–90). The mechanism by which E6AP transcriptionally regulates clusterin remains to be investigated. Clusterin is a poly-ubiquitinated protein that is degraded by the ubiquitin proteasome system (80). Until now, endoplasmic reticulum-associated ubiquitin ligase Hrd1 is the only ubiquitin ligase that has been shown to bind and ubiquitinate clusterin (71). Our results suggest that E6AP may be an E3 ubiquitin ligase of clusterin.
In conclusion, this study enhances our understanding of the biology of E6AP in prostate cancer. The combined transcriptomic and proteomic discovery approach exposed novel biological pathways and candidates that are affected by modulating E6AP expression in PC cells. Some of the candidates identified have not been linked to E6AP previously, expanding the pool of tumor suppressors that are regulated by E6AP, including clusterin.
DATA AVAILABILITY
Mass spectrometry data was deposited to PRIDE using the ProteomeXchange consortium guidelines (25) with the dataset identifier PXD008743 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD008743). The RNA-seq data is accessible through GEO Series accession number GSE107245 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?accGSE107245).
Supplementary Material
Footnotes
* This work was supported by Cancer Therapeutics CRC (Graduate Student Scholarship), the Australian Government Training Program (Graduate Student Scholarship), the Prostate Cancer Foundation (PCF) (Creativity Award), the Victorian Cancer Agency (VCA) (Richard Pratt Fellowship in Prostate Cancer Research and Grant ID 10851514), and the Peter Mac Foundation.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- PC
- prostate cancer
- CRPC
- castration resistant PC
- E6AP
- E6-Associated Protein
- PML
- Promyelocytic Leukaemia
- SILAC
- stable isotope labelling with amino acids in culture
- qRT-PCR
- quantitative real-time polymerase chain reaction
- HECT
- Homologous to E6AP Carboxyl Terminus
- HPV
- human papillomavirus
- DMEM
- Dulbecco's Modified Eagle Medium
- FCS
- fetal calf serum
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- FDR
- false discovery rate
- shE6AP
- short hairpin against E6AP
- shControl
- short hairpin control
- dox
- doxycycline
- GO
- Gene Ontology
- CLU
- clusterin
- SDC
- sodium deoxycholate
- TCEP
- tris(2-carboxyethyl)phosphine hydrochloride
- CAA
- chloroacetamide
- FA
- formic acid
- SD
- standard deviation
- SEM
- standard error of mean
- ANOVA
- analysis of variance
- dox
- doxycycline
- GFP
- green fluorescent protein
- SUSD2
- sushi domain-containing protein 2
- CDK6
- cyclin-dependent kinase 6
- SMARCC1
- SWI/SNF complex subunit SMARCC1
- PYGL
- glycogen phosphorylase, liver form
- PDE12
- 2′,5′-phosphodiesterase 12
- ppm
- parts per million
- HSP60
- heat shock protein 60
- HA
- haemagglutinin
- MDC1
- mediator of DNA damage checkpoint protein 1
- AP1
- Activator Protein 1
- HSF
- heat shock factor
- ATCC
- American Type Culture Collection
- ER
- endoplasmic reticulum
- TLR
- toll-like receptor
- TC-NER
- transcription-coupled nuclear excision repair
- AR
- androgen receptor
- PR
- progesterone receptor
- ER
- estrogen receptor
- C/EBPα
- CCAAT/Enhancer Binding Protein Alpha.
REFERENCES
- 1. Tilki D., and Evans C. P. (2014) The changing landscape of advanced and castration resistant prostate cancer: latest science and revised definitions. Canadian J. Urol. 21, 7–13 [PubMed] [Google Scholar]
- 2. Voutsadakis I. A., and Papandreou C. N. (2012) The ubiquitin-proteasome system in prostate cancer and its transition to castration resistance. Urol. Oncol. 30, 752–761 [DOI] [PubMed] [Google Scholar]
- 3. Kraft A. S., Garrett-Mayer E., Wahlquist A. E., Golshayan A., Chen C. S., Butler W., Bearden J., and Lilly M. (2011) Combination therapy of recurrent prostate cancer with the proteasome inhibitor bortezomib plus hormone blockade. Cancer Biol. Therapy 12, 119–124 [DOI] [PubMed] [Google Scholar]
- 4. Papandreou C. N., and Logothetis C. J. (2004) Bortezomib as a potential treatment for prostate cancer. Cancer Res. 64, 5036–5043 [DOI] [PubMed] [Google Scholar]
- 5. Matentzoglu K., and Scheffner M. (2008) Ubiquitin ligase E6-AP and its role in human disease. Biochem. Soc. Trans. 36, 797–801 [DOI] [PubMed] [Google Scholar]
- 6. Scheffner M., Huibregtse J. M., Vierstra R. D., and Howley P. M. (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505 [DOI] [PubMed] [Google Scholar]
- 7. Gamell C., Gulati T., Levav-Cohen Y., Young R. J., Do H., Pilling P., Takano E., Watkins N., Fox S. B., Russell P., Ginsberg D., Monahan B. J., Wright G., Dobrovic A., Haupt S., Solomon B., and Haupt Y. (2017) Reduced abundance of the E3 ubiquitin ligase E6AP contributes to decreased expression of the INK4/ARF locus in non-small cell lung cancer. Sci. Signal. 10 [DOI] [PubMed] [Google Scholar]
- 8. Wolyniec K., Shortt J., de Stanchina E., Levav-Cohen Y., Alsheich-Bartok O., Louria-Hayon I., Corneille V., Kumar B., Woods S. J., Opat S., Johnstone R. W., Scott C. L., Segal D., Pandolfi P. P., Fox S., Strasser A., Jiang Y. H., Lowe S. W., Haupt S., and Haupt Y. (2012) E6AP ubiquitin ligase regulates PML-induced senescence in Myc-driven lymphomagenesis. Blood 120, 822–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nawaz Z., Lonard D. M., Smith C. L., Lev-Lehman E., Tsai S. Y., Tsai M. J., and O'Malley B. W. (1999) The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol. 19, 1182–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee S. Y., Ramirez J., Franco M., Lectez B., Gonzalez M., Barrio R., and Mayor U. (2014) Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell. Mol. Life Sci. 71, 2747–2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mishra A., Godavarthi S. K., and Jana N. R. (2009) UBE3A/E6-AP regulates cell proliferation by promoting proteasomal degradation of p27. Neurobiol. Dis. 36, 26–34 [DOI] [PubMed] [Google Scholar]
- 12. Levav-Cohen Y., Wolyniec K., Alsheich-Bartok O., Chan A. L., Woods S. J., Jiang Y. H., Haupt S., and Haupt Y. (2012) E6AP is required for replicative and oncogene-induced senescence in mouse embryo fibroblasts. Oncogene 31, 2199–2209 [DOI] [PubMed] [Google Scholar]
- 13. Tomaic V., and Banks L. (2015) Angelman syndrome-associated ubiquitin ligase UBE3A/E6AP mutants interfere with the proteolytic activity of the proteasome. Cell Death Dis. 6, E1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuhne C., and Banks L. (1998) E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J. Biol. Chem. 273, 34302–34309 [DOI] [PubMed] [Google Scholar]
- 15. Louria-Hayon I., Alsheich-Bartok O., Levav-Cohen Y., Silberman I., Berger M., Grossman T., Matentzoglu K., Jiang Y. H., Muller S., Scheffner M., Haupt S., and Haupt Y. (2009) E6AP promotes the degradation of the PML tumor suppressor. Cell Death Differentiation 16, 1156–1166 [DOI] [PubMed] [Google Scholar]
- 16. Wolyniec K., Levav-Cohen Y., Jiang Y. H., Haupt S., and Haupt Y. (2013) The E6AP E3 ubiquitin ligase regulates the cellular response to oxidative stress. Oncogene 32, 3510–3519 [DOI] [PubMed] [Google Scholar]
- 17. Raghu D., Jan Paul P., Gulati T., Deb S., Khoo C., Russo A., Gallo E., Blandino G., Chan A. L., Takano E., Sandhu S. K., Fox S. B., Williams S., Haupt S., Gamell C., and Haupt Y. (2017) E6AP promotes prostate cancer by reducing p27 expression. Oncotarget 8, 42939–42948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Srinivasan S., and Nawaz Z. (2011) E3 ubiquitin protein ligase, E6-associated protein (E6-AP) regulates PI3K-Akt signaling and prostate cell growth. Biochim. Biophys. Acta 1809, 119–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khan O. Y., Fu G., Ismail A., Srinivasan S., Cao X., Tu Y., Lu S., and Nawaz Z. (2006) Multifunction steroid receptor coactivator, E6-associated protein, is involved in development of the prostate gland. Mol. Endocrinol. 20, 544–559 [DOI] [PubMed] [Google Scholar]
- 20. Paul P. J., Raghu D., Chan A. L., Gulati T., Lambeth L., Takano E., Herold M. J., Hagekyriakou J., Vessella R. L., Fedele C., Shackleton M., Williams E. D., Fox S., Williams S., Haupt S., Gamell C., and Haupt Y. (2016) Restoration of tumor suppression in prostate cancer by targeting the E3 ligase E6AP. Oncogene 35, 6235–6245 [DOI] [PubMed] [Google Scholar]
- 21. Birch S. E., Kench J. G., Takano E., Chan P., Chan A. L., Chiam K., Veillard A. S., Stricker P., Haupt S., Haupt Y., Horvath L., and Fox S. B. (2014) Expression of E6AP and PML predicts for prostate cancer progression and cancer-specific death. Ann. Oncol. 25, 2392–2397 [DOI] [PubMed] [Google Scholar]
- 22. Scaltriti M., Santamaria A., Paciucci R., and Bettuzzi S. (2004) Intracellular clusterin induces G2-M phase arrest and cell death in PC-3 prostate cancer cells1. Cancer Res. 64, 6174–6182 [DOI] [PubMed] [Google Scholar]
- 23. Udeshi N. D., Svinkina T., Mertins P., Kuhn E., Mani D. R., Qiao J. W., and Carr S. A. (2013) Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol. Cell. Proteomics 12, 825–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 25. Vizcaino J. A., Csordas A., del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M. Y., Geiger T., Mann M., and Cox J. (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 [DOI] [PubMed] [Google Scholar]
- 27. Law C. W., Chen Y., Shi W., and Smyth G. K. (2014) voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., and Smyth G. K. (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen E. Y., Tan C. M., Kou Y., Duan Q., Wang Z., Meirelles G. V., Clark N. R., and Ma'ayan A. (2013) Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuleshov M. V., Jones M. R., Rouillard A. D., Fernandez N. F., Duan Q., Wang Z., Koplev S., Jenkins S. L., Jagodnik K. M., Lachmann A., McDermott M. G., Monteiro C. D., Gundersen G. W., and Ma'ayan A. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Watson A. P., Evans R. L., and Egland K. A. (2013) Multiple functions of sushi domain containing 2 (SUSD2) in breast tumorigenesis. Mol. Cancer Res. 11, 74–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ramamoorthy S., and Nawaz Z. (2008) E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nuclear Receptor Signal. 6, e006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Park S. G., Schimmel P., and Kim S. (2008) Aminoacyl tRNA synthetases and their connections to disease. Proc. Natl. Acad. Sci. U.S.A. 105, 11043–11049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vellaichamy A., Sreekumar A., Strahler J. R., Rajendiran T., Yu J., Varambally S., Li Y., Omenn G. S., Chinnaiyan A. M., and Nesvizhskii A. I. (2009) Proteomic interrogation of androgen action in prostate cancer cells reveals roles of aminoacyl tRNA synthetases. PloS one 4, e7075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rizzi F., and Bettuzzi S. (2010) The clusterin paradigm in prostate and breast carcinogenesis. Endocrine-related Cancer 17, R1–R17 [DOI] [PubMed] [Google Scholar]
- 36. Jothi R., Balaji S., Wuster A., Grochow J. A., Gsponer J., Przytycka T. M., Aravind L., and Babu M. M. (2009) Genomic analysis reveals a tight link between transcription factor dynamics and regulatory network architecture. Mol. Syst. Biol. 5, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Billing A. M., Ben Hamidane H., Dib S. S., Cotton R. J., Bhagwat A. M., Kumar P., Hayat S., Yousri N. A., Goswami N., Suhre K., Rafii A., and Graumann J. (2016) Comprehensive transcriptomic and proteomic characterization of human mesenchymal stem cells reveals source specific cellular markers. Sci. Rep. 6, 21507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vogel C., Abreu Rde S., Ko D., Le S. Y., Shapiro B. A., Burns S. C., Sandhu D., Boutz D. R., Marcotte E. M., and Penalva L. O. (2010) Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol. 6, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Y., Gonzalez-Porta M., Santos S., Brazma A., Marioni J. C., Aebersold R., Venkitaraman A. R., and Wickramasinghe V. O. (2017) Impact of Alternative Splicing on the Human Proteome. Cell Rep. 20, 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vogel C., and Marcotte E. M. (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Drabovich A. P., Pavlou M. P., Schiza C., and Diamandis E. P. (2016) Dynamics of protein expression reveals primary targets and secondary messengers of estrogen receptor alpha signaling in MCF-7 breast cancer cells. Mol. Cell. Proteomics 15, 2093–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pal P., Lochab S., Kanaujiya J. K., Kapoor I., Sanyal S., Behre G., and Trivedi A. K. (2013) E6AP, an E3 ubiquitin ligase negatively regulates granulopoiesis by targeting transcription factor C/EBPalpha for ubiquitin-mediated proteasome degradation. Cell Death Disease 4, e590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pal P., Lochab S., Kanaujiya J. K., Kapoor I., Sanyal S., Behre G., and Trivedi A. K. (2013) E3 ubiquitin ligase E6AP negatively regulates adipogenesis by downregulating proadipogenic factor C/EBPalpha. PloS one 8, e65330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mani A., Oh A. S., Bowden E. T., Lahusen T., Lorick K. L., Weissman A. M., Schlegel R., Wellstein A., and Riegel A. T. (2006) E6AP mediates regulated proteasomal degradation of the nuclear receptor coactivator amplified in breast cancer 1 in immortalized cells. Cancer Res. 66, 8680–8686 [DOI] [PubMed] [Google Scholar]
- 45. Goldberg M., Stucki M., Falck J., D'Amours D., Rahman D., Pappin D., Bartek J., and Jackson S. P. (2003) MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature 421, 952–956 [DOI] [PubMed] [Google Scholar]
- 46. Stewart G. S., Wang B., Bignell C. R., Taylor A. M., and Elledge S. J. (2003) MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421, 961–966 [DOI] [PubMed] [Google Scholar]
- 47. Lou Z., Chen B. P., Asaithamby A., Minter-Dykhouse K., Chen D. J., and Chen J. (2004) MDC1 regulates DNA-PK autophosphorylation in response to DNA damage. Biol. Chem. 279, 46359–46362 [DOI] [PubMed] [Google Scholar]
- 48. Wakasugi M., Sasaki T., Matsumoto M., Nagaoka M., Inoue K., Inobe M., Horibata K., Tanaka K., and Matsunaga T. (2014) Nucleotide excision repair-dependent DNA double-strand break formation and ATM signaling activation in mammalian quiescent cells. J. Biol. Chem. 289, 28730–28737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marteijn J. A., Bekker-Jensen S., Mailand N., Lans H., Schwertman P., Gourdin A. M., Dantuma N. P., Lukas J., and Vermeulen W. (2009) Nucleotide excision repair-induced H2A ubiquitination is dependent on MDC1 and RNF8 and reveals a universal DNA damage response. J. Cell Biol. 186, 835–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang C., Sun H., Zou R., Zhou T., Wang S., Sun S., Tong C., Luo H., Li Y., Li Z., Wang E., Chen Y., Cao L., Li F., and Zhao Y. (2015) MDC1 functionally identified as an androgen receptor co-activator participates in suppression of prostate cancer. Nucleic Acids Res. 43, 4893–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamagishi Y., Shoji I., Miyagawa S., Kawakami T., Katoh T., Goto Y., and Suga H. (2011) Natural product-like macrocyclic N-methyl-peptide inhibitors against a ubiquitin ligase uncovered from a ribosome-expressed de novo library. Chem. Biol. 18, 1562–1570 [DOI] [PubMed] [Google Scholar]
- 52. Mateo J., Carreira S., Sandhu S., Miranda S., Mossop H., Perez-Lopez R., Nava Rodrigues D., Robinson D., Omlin A., Tunariu N., Boysen G., Porta N., Flohr P., Gillman A., Figueiredo I., Paulding C., Seed G., Jain S., Ralph C., Protheroe A., Hussain S., Jones R., Elliott T., McGovern U., Bianchini D., Goodall J., Zafeiriou Z., Williamson C. T., Ferraldeschi R., Riisnaes R., Ebbs B., Fowler G., Roda D., Yuan W., Wu Y. M., Cao X., Brough R., Pemberton H., A'Hern R., Swain A., Kunju L. P., Eeles R., Attard G., Lord C. J., Ashworth A., Rubin M. A., Knudsen K. E., Feng F. Y., Chinnaiyan A. M., Hall E., and de Bono J. S. (2015) DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 373, 1697–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jensen L., Farook M. F., and Reiter L. T. (2013) Proteomic profiling in Drosophila reveals potential Dube3a regulation of the actin cytoskeleton and neuronal homeostasis. PloS One 8, e61952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Favaro E., Bensaad K., Chong M. G., Tennant D. A., Ferguson D. J., Snell C., Steers G., Turley H., Li J. L., Gunther U. L., Buffa F. M., McIntyre A., and Harris A. L. (2012) Glucose utilization via glycogen phosphorylase sustains proliferation and prevents premature senescence in cancer cells. Cell Metab. 16, 751–764 [DOI] [PubMed] [Google Scholar]
- 55. Schnier J. B., Nishi K., Gumerlock P. H., Gorin F. A., and Bradbury E. M. (2005) Glycogen synthesis correlates with androgen-dependent growth arrest in prostate cancer. BMC Urol. 5, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sokoloff M. H., Tso C. L., Kaboo R., Taneja S., Pang S., deKernion J. B., and Belldegrun A. S. (1996) In vitro modulation of tumor progression-associated properties of hormone refractory prostate carcinoma cell lines by cytokines. Cancer 77, 1862–1872 [DOI] [PubMed] [Google Scholar]
- 57. Kubota K., Nakahara K., Ohtsuka T., Yoshida S., Kawaguchi J., Fujita Y., Ozeki Y., Hara A., Yoshimura C., Furukawa H., Haruyama H., Ichikawa K., Yamashita M., Matsuoka T., and Iijima Y. (2004) Identification of 2′-phosphodiesterase, which plays a role in the 2–5A system regulated by interferon. J. Biol. Chem. 279, 37832–37841 [DOI] [PubMed] [Google Scholar]
- 58. Wood E. R., Bledsoe R., Chai J., Daka P., Deng H., Ding Y., Harris-Gurley S., Kryn L. H., Nartey E., Nichols J., Nolte R. T., Prabhu N., Rise C., Sheahan T., Shotwell J. B., Smith D., Tai V., Taylor J. D., Tomberlin G., Wang L., Wisely B., You S., Xia B., and Dickson H. (2015) The role of phosphodiesterase 12 (PDE12) as a negative regulator of the innate immune response and the discovery of antiviral inhibitors. J. Biol. Chem. 290, 19681–19696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Thomas M., and Banks L. (1998) Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 17, 2943–2954 [DOI] [PubMed] [Google Scholar]
- 60. Kumar S., Talis A. L., and Howley P. M. (1999) Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J. Biol. Chem. 274, 18785–18792 [DOI] [PubMed] [Google Scholar]
- 61. Zheng L., Ding H., Lu Z., Li Y., Pan Y., Ning T., and Ke Y. (2008) E3 ubiquitin ligase E6AP-mediated TSC2 turnover in the presence and absence of HPV16 E6. Genes Cells 13, 285–294 [DOI] [PubMed] [Google Scholar]
- 62. Kollmann K., Heller G., Schneckenleithner C., Warsch W., Scheicher R., Ott R. G., Schafer M., Fajmann S., Schlederer M., Schiefer A. I., Reichart U., Mayerhofer M., Hoeller C., Zochbauer-Muller S., Kerjaschki D., Bock C., Kenner L., Hoefler G., Freissmuth M., Green A. R., Moriggl R., Busslinger M., Malumbres M., and Sexl V. (2013) A kinase-independent function of CDK6 links the cell cycle to tumor angiogenesis. Cancer Cell 24, 167–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lucas J. J., Domenico J., and Gelfand E. W. (2004) Cyclin-dependent kinase 6 inhibits proliferation of human mammary epithelial cells. Mol. Cancer Res. 2, 105–114 [PubMed] [Google Scholar]
- 64. Nagasawa M., Gelfand E. W., and Lucas J. J. (2001) Accumulation of high levels of the p53 and p130 growth-suppressing proteins in cell lines stably over-expressing cyclin-dependent kinase 6 (cdk6). Oncogene 20, 2889–2899 [DOI] [PubMed] [Google Scholar]
- 65. Wang X., Sistrunk C., and Rodriguez-Puebla M. L. (2011) Unexpected reduction of skin tumorigenesis on expression of cyclin-dependent kinase 6 in mouse epidermis. Am. J. Pathol, 178, 345–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lim J. T., Mansukhani M., and Weinstein I. B. (2005) Cyclin-dependent kinase 6 associates with the androgen receptor and enhances its transcriptional activity in prostate cancer cells. Proc. Natl. Acad. Sci. U.S.A. 102, 5156–5161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cheng Y., Wang X., Wang P., Li T., Hu F., Liu Q., Yang F., Wang J., Xu T., and Han W. (2016) SUSD2 is frequently downregulated and functions as a tumor suppressor in RCC and lung cancer. Tumour Biol 37, 9919–9930 [DOI] [PubMed] [Google Scholar]
- 68. DelBove J., Rosson G., Strobeck M., Chen J., Archer T. K., Wang W., Knudsen E. S., and Weissman B. E. (2011) Identification of a core member of the SWI/SNF complex, BAF155/SMARCC1, as a human tumor suppressor gene. Epigenetics 6, 1444–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hansen R. L., Heeboll S., Ottosen P. D., Dyrskjot L., and Borre M. (2011) Smarcc1 expression: a significant predictor of disease-specific survival in patients with clinically localized prostate cancer treated with no intention to cure. Scand. J. Urol. Nephrol. 45, 91–96 [DOI] [PubMed] [Google Scholar]
- 70. Heeboll S., Borre M., Ottosen P. D., Andersen C. L., Mansilla F., Dyrskjot L., Orntoft T. F., and Torring N. (2008) SMARCC1 expression is upregulated in prostate cancer and positively correlated with tumour recurrence and dedifferentiation. Histol. Histopathol. 23, 1069–1076 [DOI] [PubMed] [Google Scholar]
- 71. Nizard P., Tetley S., Le Drean Y., Watrin T., Le Goff P., Wilson M. R., and Michel D. (2007) Stress-induced retrotranslocation of clusterin/ApoJ into the cytosol. Traffic 8, 554–565 [DOI] [PubMed] [Google Scholar]
- 72. Zoubeidi A., Ettinger S., Beraldi E., Hadaschik B., Zardan A., Klomp L. W., Nelson C. C., Rennie P. S., and Gleave M. E. (2010) Clusterin facilitates COMMD1 and I-kappaB degradation to enhance NF-kappaB activity in prostate cancer cells. Mol. Cancer Res. 8, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang H., Kim J. K., Edwards C. A., Xu Z., Taichman R., and Wang C. Y. (2005) Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 7, 909–915 [DOI] [PubMed] [Google Scholar]
- 74. Kang B. H., Shim Y. J., Tae Y. K., Song J. A., Choi B. K., Park I. S., and Min B. H. (2014) Clusterin stimulates the chemotactic migration of macrophages through a pertussis toxin sensitive G-protein-coupled receptor and Gbetagamma-dependent pathways. Biochem. Biophys. Res. Commun. 445, 645–650 [DOI] [PubMed] [Google Scholar]
- 75. Moretti R. M., Mai S., Montagnani Marelli M., Rizzi F., Bettuzzi S., and Limonta P. (2011) Molecular mechanisms of the antimetastatic activity of nuclear clusterin in prostate cancer cells. Int. J. Oncol. 39, 225–234 [DOI] [PubMed] [Google Scholar]
- 76. Pucci S., Mazzarelli P., Sesti F., Boothman D. A., and Spagnoli L. G. (2009) Interleukin-6 affects cell death escaping mechanisms acting on Bax-Ku70-Clusterin interactions in human colon cancer progression. Cell Cycle 8, 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yang C. R., Leskov K., Hosley-Eberlein K., Criswell T., Pink J. J., Kinsella T. J., and Boothman D. A. (2000) Nuclear clusterin/XIP8, an x-ray-induced Ku70-binding protein that signals cell death. Proc. Natl. Acad. Sci. U.S.A. 97, 5907–5912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Matukumalli S. R., Tangirala R., and Rao C. M. (2017) Clusterin: full-length protein and one of its chains show opposing effects on cellular lipid accumulation. Sci. Rep. 7, 41235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. de Silva H. V., Stuart W. D., Duvic C. R., Wetterau J. R., Ray M. J., Ferguson D. G., Albers H. W., Smith W. R., and Harmony J. A. (1990) A 70-kDa apolipoprotein designated ApoJ is a marker for subclasses of human plasma high density lipoproteins. J. Biol. Chem. 265, 13240–13247 [PubMed] [Google Scholar]
- 80. Rizzi F., Caccamo A. E., Belloni L., and Bettuzzi S. (2009) Clusterin is a short half-life, poly-ubiquitinated protein, which controls the fate of prostate cancer cells. J. Cell. Physiol. 219, 314–323 [DOI] [PubMed] [Google Scholar]
- 81. Bettuzzi S., Scorcioni F., Astancolle S., Davalli P., Scaltriti M., and Corti A. (2002) Clusterin (SGP-2) transient overexpression decreases proliferation rate of SV40-immortalized human prostate epithelial cells by slowing down cell cycle progression. Oncogene 21, 4328–4334 [DOI] [PubMed] [Google Scholar]
- 82. Scaltriti M., Bettuzzi S., Sharrard R. M., Caporali A., Caccamo A. E., and Maitland N. J. (2004) Clusterin overexpression in both malignant and nonmalignant prostate epithelial cells induces cell cycle arrest and apoptosis. Br. J. Cancer 91, 1842–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Scaltriti M., Brausi M., Amorosi A., Caporali A., D'Arca D., Astancolle S., Corti A., and Bettuzzi S. (2004) Clusterin (SGP-2, ApoJ) expression is downregulated in low- and high-grade human prostate cancer. Int. J. Cancer 108, 23–30 [DOI] [PubMed] [Google Scholar]
- 84. Bettuzzi S., Davalli P., Astancolle S., Carani C., Madeo B., Tampieri A., and Corti A. (2000) Tumor progression is accompanied by significant changes in the levels of expression of polyamine metabolism regulatory genes and clusterin (sulfated glycoprotein 2) in human prostate cancer specimens. Cancer Res. 60, 28–34 [PubMed] [Google Scholar]
- 85. Bettuzzi S., Davalli P., Davoli S., Chayka O., Rizzi F., Belloni L., Pellacani D., Fregni G., Astancolle S., Fassan M., Corti A., Baffa R., and Sala A. (2009) Genetic inactivation of ApoJ/clusterin: effects on prostate tumourigenesis and metastatic spread. Oncogene 28, 4344–4352 [DOI] [PubMed] [Google Scholar]
- 86. Criswell T., Beman M., Araki S., Leskov K., Cataldo E., Mayo L. D., and Boothman D. A. (2005) Delayed activation of insulin-like growth factor-1 receptor/Src/MAPK/Egr-1 signaling regulates clusterin expression, a pro-survival factor. J. Biol. Chem. 280, 14212–14221 [DOI] [PubMed] [Google Scholar]
- 87. Loison F., Debure L., Nizard P., le Goff P., Michel D., and le Drean Y. (2006) Up-regulation of the clusterin gene after proteotoxic stress: implication of HSF1-HSF2 heterocomplexes. Biochem. J. 395, 223–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cochrane D. R., Wang Z., Muramaki M., Gleave M. E., and Nelson C. C. (2007) Differential regulation of clusterin and its isoforms by androgens in prostate cells. J. Biol. Chem. 282, 2278–2287 [DOI] [PubMed] [Google Scholar]
- 89. Park J., Park S. Y., Shin E., Lee S. H., Kim Y. S., Lee D. H., Roh G. S., Kim H. J., Kang S. S., Cho G. J., Jeong B. Y., Kim H., and Choi W. S. (2014) Hypoxia inducible factor-1alpha directly regulates nuclear clusterin transcription by interacting with hypoxia response elements in the clusterin promoter. Mol. Cells 37, 178–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shiota M., Zoubeidi A., Kumano M., Beraldi E., Naito S., Nelson C. C., Sorensen P. H., and Gleave M. E. (2011) Clusterin is a critical downstream mediator of stress-induced YB-1 transactivation in prostate cancer. Mol. Cancer Res. 9, 1755–1766 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data was deposited to PRIDE using the ProteomeXchange consortium guidelines (25) with the dataset identifier PXD008743 (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD008743). The RNA-seq data is accessible through GEO Series accession number GSE107245 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?accGSE107245).