

Abstract
Clostridium acetobutylicum is a strict anaerobic, endospore-forming bacterium, which is used for the production of the high energy biofuel butanol in metabolic engineering. The life cycle of C. acetobutylicum can be divided into two phases, with acetic and butyric acids being produced in the exponential phase (acidogenesis) and butanol formed in the stationary phase (solventogenesis). During the transitional phase from acidogenesis to solventogenesis and latter stationary phase, concentration peaks of the metabolic intermediates butyryl phosphate and acetyl phosphate are observed. As an acyl group donor, acyl-phosphate chemically acylates protein substrates. However, the regulatory mechanism of lysine acetylation and butyrylation involved in the phenotype and solventogenesis of C. acetobutylicum remains unknown. In our study, we conducted quantitative analysis of protein acetylome and butyrylome to explore the dynamic change of lysine acetylation and butyrylation in the exponential phase, transitional phase, and stationary phase of C. acetobutylicum. Total 458 lysine acetylation sites and 1078 lysine butyrylation sites were identified in 254 and 373 substrates, respectively. Bioinformatics analysis uncovered the similarities and differences between the two acylation modifications in C. acetobutylicum. Mutation analysis of butyrate kinase and the central transcriptional factor Spo0A was performed to characterize the unique role of lysine butyrylation in the metabolic pathway and sporulation process of C. acetobutylicum. Moreover, quantitative proteomic assays were performed to reveal the relationship between protein features (e.g. gene expression level and lysine acylation level) and metabolites in the three growth stages. This study expanded our knowledge of lysine acetylation and butyrylation in Clostridia and constituted a resource for functional studies on lysine acylation in bacteria.
Keywords: Acetylation*, Bacteria, Enzyme Regulation*, Mass Spectrometry, Post-translational modifications*, Quantification, acylation regulation, endospore-forming Clostridium, metabolic shift, quantitative acetylome, quantitative butyrylome
Nε-lysine acetylation is a reversible, dynamic and conserved post-translational modification (PTM), which plays pivotal roles in cellular physiological and pathological functions in both eukaryotes and prokaryotes (1, 2). Recently, several studies have proved that the side chain of lysine can be modified by diverse acylation reactions (3), which include propionylation (4), butyrylation (4), crotonylation (5, 6), succinylation (7), malonylation (8, 9), glutarylation (10), 2-hydroxyisobutyrylation (11) and β-hydroxybutyrylation (12). Recent breakthroughs in high-resolution mass spectrometry (MS)-based proteomics have paved the way for uncovering protein acylome mapping in various organisms (13), especially in the microbial kingdom. Several LysAcy proteomic landscapes of different bacteria were explored, which revealed various roles of lysine acylation, such as in determining enzymatic activity (14, 15), cellular motility (16, 17) and protein stability (18). However, the regulatory mechanisms of lysine acylation in the Gram-positive Clostridia remain to be elucidated.
Clostridium acetobutylicum is a rod-shaped, strict anaerobic, endospore-forming bacterium, which is used to produce several important biofuels (19), especially butanol, which has a higher energy content and lower volatility than ethanol (20). In fermentation processes based on carbohydrates as substrates, the complete life cycle of C. acetobutylicum can be divided into two phases with obvious phenotypes: acidogenic stage (acidogenesis) and soventogenic stage (solventogenesis) (21). Acetate and butyrate, which are formed in the acidogenic stage, decrease the pH of the bacterial culture. Then, the bacterium switches its metabolism to the solventogenesis phase to relieve the acid burden and biosynthesize butanol, acetone and ethanol as solvent products at a ratio of 6:3:1, which is called acetone-butanol-ethanol (ABE)1 fermentation (21). In addition, the concentrations of two key metabolic intermediates acetyl phosphate (AcP) and butyryl phosphate (BuP), which are derived from acetyl-CoA and butyryl-CoA, respectively, changes during the life span of C. acetobutylicum (22). During the transitional phase from acidogenesis to solventogenesis, BuP functions as phospho-donor to its central transcriptional factor Spo0A, triggering the downstream expression of various metabolic enzymes and regulators essential for the solvent fermentation and spore maturation processes (e.g. adhE1, adc, ctfA/B, σE, σF, and σG) (23–25). Moreover, other studies reported that AcP could chemically acetylate lysine residues through a non-enzymatic mechanism and its increasing concentration was associated with elevated protein acetylation levels in Escherichia coli (26). Based on the structural similarity of AcP, we hypothesized that BuP could act as a butyryl donor in the butyrylation of protein substrates. Because AcP and BuP are considered as the major metabolite intermediates formed during the life span of C. acetobutylicum, lysine acetylation and butyrylation were speculated to regulate the phenotype and solvent production in C. acetobutylicum. Up to date, current knowledge of the physiological and metabolic changes of this bacterium is based only on transcriptome, proteome and metabolome analysis (22, 23, 27–30), the regulatory mechanism of C. acetobutylicum at the lysine acylation level is unknown.
In this study, we conducted quantitative protein acetylome and butyrylome analysis to explore the dynamic changes of lysine acetylation and butyrylation during the exponential phase, transitional phase and stationary phase of the C. acetobutylicum life cycle. Our study identified 458 lysine acetylation sites and 1078 lysine butyrylation sites in 254 and 373 substrates, respectively. Bioinformatics analysis uncovered the similarities and differences between the two acylation modifications and provided comprehensive insights into the unique role of lysine butyrylation in the regulation of the C. acetobutylicum life span. Moreover, in vitro enzymatic activity assay and electrophoretic mobility shift assay (EMSA) were conducted to characterize the effect of lysine butyrylation on the metabolic pathway and sporulation process. In addition, quantitative proteomic assays were performed to explore the intricate relationship among protein abundance, lysine acylation level, and cellular metabolite concentrations during the three growth stages. Our findings revealed a broad scope of acetylated and butyrylated substrates and unveiled the biological role of lysine butyrylation in both physical and fermentation pathways.
EXPERIMENTAL PROCEDURES
Cell Culture of C. acetobutylicum Strain
Clostridium acetobutylicum strain was cultured anaerobically at 37 °C as previously described (22, 31). Briefly, the culture medium contained the following materials (g/L): (NH4)2SO4, 2.0; KH2PO4, 0.5; K2HPO4, 1.0; FeSO4·7H2O, 0.015; MgSO4·7H2O, 0.1; CaCl2, 0.01; MnSO4·H2O, 0.01; CoCl2, 0.002; FeSO4·7H2O, 0.002; Na2SeO3, 0.00025; tryptone, 2.0; yeast extract, 1.0; glucose, 50. After heat shock at 90 °C for 5 min, the cells were cultured in an anaerobic box for 24 h. The cells were then collected in its exponential phase, transitional phase and stationary phase according to the growth curves. Three biological replicates were analyzed.
Protein Extraction of C. acetobutylicum Strain
Bacterial cells were lysed by sonication for 5 min in the extraction buffer containing 8 m urea in PBS buffer supplemented with protease inhibitor mixture (Calbiochem, Darmstadt, Germany) and 20 mm nicotinamide. After incubation on ice for 15 min, the cell debris was removed by centrifugation at 20000 g for 15 min. Protein concentration at each growth stage was determined using the protein bicinchoninic acid (BCA) assay kit (Beyotime Biotechnology, China).
Western Blot Analysis
For each growth stage, whole cell lysates (10 μg) were separated by SDS-PAGE and transferred to a nitrocellulose membrane for 90 min at 100 V. After blocking in 5% BSA-PBST (5% bovine serum albumin in PBS containing 0.1% Tween-20) at 25 °C for 1.5 h, the membrane was incubated overnight with pan anti-acetyl-lysine and pan anti-butyryl-lysine antibodies at 4 °C. After washing with PBST, the membrane was treated with horseradish peroxidase (HRP)-conjugated anti rabbit IgG (1:5000) in PBST at 25 °C for 1 h with gentle shaking. The membrane was washed with PBST four times for 5 min each, and then imaged using an ImageQuant LAS 4000 system (GE Healthcare, UK) after chemiluminescent HRP substrate treatment (Millipore, Temecula, CA).
In Solution Tryptic Digestion
For quantitative analysis of lysine acetylome and butyrylome, protein lysates were prepared as described in a previous study (32). Protein extracts (6 mg) were precipitated with 20% (v/v) trichloroacetic acid and centrifuged at 4000 × g for 20 min. After two washes with cold acetone, the pellet was suspended in 100 mm NH4HCO3 (pH 8.0). Sequencing-grade trypsin (Promega, Madison, WI) at a trypsin-to-protein ratio of 1:100 (w/w) was added and the samples were incubated at 37 °C for 12 h. The resulting peptides were reduced using 5 mm dithiothreitol (DTT) at 55 °C for 30 min and, then, alkylated with 15 mm iodoacetamide (IAA) in the dark at room temperature for 0.5 h. Incubation for 0.5 h at room temperature with cysteine at final concentration of 30 mm was performed to stop the alkylation reaction. Finally, additional trypsin at a trypsin-to-protein ratio of 1:100 (w/w) was added to complete the digestion cycle at 37 °C for further 4 h. The digested peptides were desalted using Sep-Pak tC18 6 cc (1 g) cartridges (Waters, Milford, MA).
Stable Isotope Dimethyl Labeling
The digested peptides were treated according to the stable isotope dimethyl labeling method based on a previously reported protocol with slight modifications (33). Briefly, the peptides were dissolved in 600 μl of triethylammonium bicarbonate buffer (100 mm). Then, 30 μl of 20% (w/w) CH2O, CD2O, and 13CD2O were added to obtain light (L)-, middle (M)-, and heavy (H)-labeled samples, respectively. Next, 30 μl of 3 m NaBH3CN was added to the l- and M-labeled samples, respectively, whereas 30 μl of 3 m NaBD3CN was added to the H-labeled sample. Specifically, the peptides obtained from the exponential, transitional, and stationary phases were labeled as “H,” “M,” and “H,” respectively. The mixtures were incubated at room temperature for 1 h and the procedure was repeated twice. After checking the labeling efficiency for each sample, 30 μl of 20% ammonia and 20 μl of TFA were added to the mixture to stop the reaction and acidify the samples. The labeled samples were mixed and desalted using Sep-Pak tC18 6 cc (1 g) Cartridges (Waters).
Affinity Enrichment of Lysine Acetylation (Kac) or Lysine Butyrylation (Kbu) Peptides
Before enrichment with anti-acetyl-lysine antibodies, 600 μg of the mixture was separated as the “unenriched” sample and used for protein level normalization. For antibody enrichment, the peptides were dissolved in NETN buffer (100 mm NaCl, 1 mm EDTA, 50 mm Tris-HCl, 0.5% NP-40, pH 8.0) and reacted overnight with 10 μl of pre-washed Kac/Kbu antibody beads (PTM Biolabs, Hangzhou, China) at 4 °C with gentle shaking. After washing the beads four times with NETN buffer, twice with ETN buffer (50 mm Tris-HCl, 100 mm NaCl, 17 mm EDTA, pH 8.0), and twice with water, 0.1% TFA was added to elute the peptides. The modified peptides were dried in a SpeedVac concentrator and desalted using C18 ZipTips (Millipore) prior to nano-HPLC-MS analysis.
Nano-HPLC-MS/MS Analysis
The acetylated or butyrylated peptides were dissolved in 4 μl of solvent A (0.1% (v/v) formic acid and 2% acetonitrile in water) and loaded onto a manually packed 19 cm reverse-phase C18 column (10 cm in length × 75 μm in inner diameter; C18 resin with 3 μm particle size; 90 Å pore diameter; Dikma Technologies Inc., Lake Forest, CA) coupled to an EASY-nLC 1000 system (Thermo Fisher Scientific, Waltham, MA). The peptides were eluted by 90 min gradient with 8–35% solvent B (0.1%) formic acid and 10% water in ACN) for 54 min, 35–45% solvent B for 10 min, 45–80% solvent B for 3 min, and 80% solvent B for 3 min at a flow rate of 300 nL/min. The samples were ionized and sprayed into an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) via a nanospray ion source in a positive mode. The peptides within the range of m/z 350–1400 were analyzed with a resolution of 120000 at m/z 200, and the automatic gain control (AGC) target was set to 5E5 for Orbitrap. The electrospray voltage was 2.2 kV and the isolation window (m/z) was set to 1. Top-speed mode was used for MS/MS acquisition with a 3 s cycle time. Ions with charge states 2+ to 6+ were fragmented with higher-energy collisional dissociation (HCD) at a normalized collision energy (NCE) of 32%. The fragmented ions were analyzed in an ion trap with 30 s dynamic exclusion duration. Three technical replicates for all the quantitative analysis of lysine acylome and lysine butyrylome were examined. The peptides used for protein level normalization (“unenriched” sample) was analyzed by Q Exactive mass spectrometer with a 60 min gradient including 8–32% solvent B for 51 min, 32–48% solvent B for 5 min, 48–80% solvent B for 1 min, and 80% B for 3 min at a flow rate of 300 nL/min. The mass spectrometric analysis was conducted in a data-dependent mode. The top 15 ions were isolated and subjected to higher-energy collisional dissociation (HCD) with a normalized collision energy (NCE) of 28%. The charge exclusion was set to 1+ and more than 5+, and the dynamic exclusion was set to 60 s. Two technical replicates were conducted for “unenriched” sample used for protein level normalization.
Quantitative Proteomic Analysis of Protein Profiling in C. acetobutylicum by Stable Isotope Dimethyl Labeling
For quantitative proteomic analysis, proteins extracted from cultures in the three growth stages (200 μg) were reduced using DTT and alkylated using IAA as described above. After treatment with cysteine, the samples underwent tryptic digestion overnight at a trypsin-to-protein ratio of 1:100 (w/w) at 37 °C, followed by an additional digestion process (a trypsin-to-protein ratio of 1:100) at 37 °C for 4 h. The samples were desalted and labeled with dimethyl labeling reagents: samples from the exponential phase were labeled using the “L” labeling agents CH2O and NaBH3CN, samples from the transitional phase were labeled using the “M” labeling agents CD2O and NaBH3CN, and samples from the stationary phase were labeled using the “H” labeling agents 13CD2O and NaBD3CN. The labeled samples were mixed and analyzed with nano-HPLC-MS/MS in three technical replicates.
Experimental Design and Statistical Rationale
The overview of the mass spectrometry experiment: C. acetobutylicum cells were collected in the exponential phase, the transitional phase and the stationary phase for quantitative acetylome/butyrylome and quantitative proteomic analysis. Samples from each growth stage were labeled using stable isotope dimethyl reagents. Samples from the exponential phase were labeled using “L” labeling agents, samples from the transitional phase were labeled using “M” labeling agents, and samples from the stationary phase were labeled using “H” labeling agents. Then, the samples used for quantitative acetylome/butyrylome analysis were enriched with anti-acetyl-lysine antibodies. All samples were analyzed with nano-HPLC-MS/MS. Three technical replicates were used for both quantitative acylome and quantitative proteome analyses. Two technical replicates of unenriched samples were used for acylome protein level normalization.
The MS raw data were processed with the MaxQuant software (version 1.5.1.2) (34) and the UniProt Clostridium acetobutylicum database (3847 sequences, 2014, http://www.uniprot.org/proteomes/?query=Clostridium+acetobutylicum&sort=score) was used. The digestion enzyme was set to trypsin/P, and maximal missed cleavage was set to 2. Precursor error tolerance was set to 20 ppm. Fragment ion was set to 0.02 Da for Q Exactive and 0.5 Da for Orbitrap Fusion mass spectrometers, respectively. Carbamidomethyl (C) was specified as the fixed modification and variable modifications were oxidation (M), acetylation (protein N-term) and acetylation/butyrylation (K). False discovery rate (FDR) thresholds for protein, peptide, and modification site were set to 0.01. In the quantification process, lysine acetylation/butyrylation site identifications with localization probability lower than 0.75 and those from reverse or contaminant protein sequences were removed. All the raw data from the three technical replicates were analyzed simultaneously using the MaxQuant software. The parameters specified in the experimental Design Template txt file were then uploaded in the MaxQuant software to combine the data from all the replicates. Acetylation/butyrylation sites identified in at least two replicates were retained and manual check of the acylated peptides was performed to ensure the reliability of the results. The M/l- or H/l-labeled acylated peptides ratios obtained from the combination of the data from the three technical replicates were auto-generated by the MaxQuant software. The ratios of acetylated/butyrylated peptides were then normalized based on the “unenriched” sample of total protein quantitation. The normalized acylation ratios of the acylated peptides were used for further analysis.
For proteomic analysis, the MS raw data were processed with MaxQuant software (version 1.5.1.2). The digestion enzyme was set to trypsin/P, and maximal missed cleavage was set to 2. Precursor error tolerance was set to 20 ppm. Fragment ion was set to 0.5 Da for Orbitrap Fusion mass spectrometers. Carbamidomethyl (C) was specified as the fixed modification and variable modifications were oxidation (M), acetylation (Protein N-term). False discovery rate (FDR) thresholds for the protein, peptide, and modification site were specified to 0.01. Proteins identified in all the three technical replicates were used to calculate the protein ratio. Statistical analysis was conducted using the two-tailed t test. All the quantitative proteomic analysis data were used to generate the volcano plots. Quantitative proteomic data with p value < 0.05 and ratio >1.5 were used for further analysis.
All raw data and the results folder produced by the MaxQuant software were uploaded in the iProX database.
Flanking Sequence Analysis
The iceLogo software (version 1.2) was used to analyze the amino acid preference of the flanking sequence of Kac and Kbu sites (35). The acylated sites surrounded by 15 neighboring residues on both sides were chosen as the positive set. Moreover, the Uniprot 'C. acetobutylicum' database was used as the negative control. Finally, a p value < 0.05 was considered statistically significant.
Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis of the Acylated Proteins of C. acetobutylicum
The STRING database (https://string-db.org/) was used for the KEGG enrichment analysis of the acylated sites in C. acetobutylicum. Total genome information was used as the background. An FDR of 0.05 was set as the cutoff and the remaining proteins were used for further analysis.
Cloning and Mutagenesis of Butyrate Kinase (CA_C3075) and Spo0A (CA_C2071)
The list of primers used for PCR and mutagenesis was supplied in supplemental Table S8. The buk and spo0A genes were amplified using the primers buk_F, buk_R, spo0A_F, and spo0A_R with the genomic DNA of C. acetobutylicum as the template (supplemental Table S8). After digestion with EcoRI/XhoI and NdeI/BamHI, the gene were cloned into the pET28a and pET19b plasmid, which generated pET28a-buk and pET19b-spo0A, respectively. After verified by sequencing, point mutations were generated using the QuckChange mutagenesis kit and the primers described in supplemental Table S8. E. coli BL21 (DE3) cells transformed with wild-type and mutant pET28a-buk were cultured in Luria-Bertani (LB) medium at 37 °C and induced overnight with 0.4 mm isopropyl-β-d-thiogalactoside at 20 °C. E. coli Rosetta (DE3) cells transformed with wild-type and mutant pET19b-spo0A were cultured overnight in terrific broth (TB) medium in an orbital shaker (200 rpm, 37 °C) as seed culture. Then, 5 ml of the seed culture was transferred into a 500-ml flask containing 50 ml of TB medium and incubated at 20 °C for 24 h. Cells were collected by centrifugation and sonicated in PBS. After sonication, cells were centrifuged at 8000 × g for 20 min, and the supernatant was purified using a Ni-NTA column (Merck). The column was washed with 10 ml of W1 buffer (50 mm NaH2PO4, 300 mm NaCl, 10 mm imidazole, pH 8.0) and 10 ml of W2 buffer (50 mm NaH2PO4, 300 mm NaCl, 20 mm imidazole, pH 8.0). The target proteins were eluted with the EB buffer (50 mm NaH2PO4, 300 mm NaCl, 250 mm imidazole, pH 8.0) and dialyzed against buffer C (37 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, 5% glycerol, pH 7.9). Protein concentration was measured using the BCA assay and purity was verified by SDS-PAGE.
In Vitro Butyrate Kinase Assays
The activity of wild-type butyrate kinase and its three mutants (Buk_K167Q, Buk_K239Q and Buk_K287Q) was measured according to a previously described method (36, 37). Enzymatic analysis was conducted in 50 mm Tris-HCl buffer (pH = 7.6), containing 750 mm sodium butyrate, 10 mm ATP, and 9.3 mm MgCl2. Butyrate kinase activity was assayed in the butyryl-phosphate forming direction and absorbance at A540 nm was monitored.
Electrophoretic Mobility Shift Assay (EMSA) of Spo0A
EMSA probes of Pspo0A (promoter region of CA_C2071) and Psol (promoter region of CA_P0162–0164) were amplified by PCR using the primers listed in supplemental Table S8. PCR products were labeled with biotin using a biotin labeled universal primer (supplemental Table S8).
EMSA was carried out using the chemiluminescent EMSA kit (Beyotime Biotechnology), as previously described (38). Biotin-labeled DNA probes were incubated with a gradient concentration of Spo0A at 25 °C for 20 min. For control groups, an unlabeled specific probe (200-fold) or nonspecific competitor DNA (200-fold, sonicated salmon sperm DNA) was used. Samples were separated using 6% nondenaturing PAGE gels in ice-cold 0.5×Tris borate-EDTA at 160 V. Bands were visualized using the BeyoECL Plus Western blotting detection system (Beyotime Biotechnology).
RESULTS
The Dynamic Change of Lysine Acetylation and Butyrylation in C. acetobutylicum
Previous studies showed that, in the physiological growth cycle of C. acetobutylicum, AcP and BuP have one and two concentration peaks, respectively (22). The first BuP concentration peak was identified during the transitional phase, which coincides with the onset of solvent production, whereas the second peak appears when butyric acid is reused during the solventogenesis phase. On the other hand, AcP levels mostly increased during the stationary phase. Because acyl-phosphate chemically acylates protein substrates because of its high-energy bonds, dynamic changes in AcP or BuP concentration through the bacteria growth stages could affect the acetylation/butyrylation levels of functional proteins or metabolic enzymes and, therefore, regulate the physiological status or the ABE fermentation process of C. acetobutylicum.
To explore these dynamic changes of protein acylation levels in C. acetobutylicum, bacterial cells were collected in the exponential, transitional, and stationary phases. Western blot analysis using pan anti-acetyl-lysine or anti-butyryl-lysine antibodies showed high elevation of lysine butyrylation in the transitional phase compared with that in the exponential phase, whereas a slight increase in lysine acetylation was observed (Fig. 1A). In addition, compared with the acylation level during the transitional phase, global protein acetylation and butyrylation levels did not change during the stationary phase. To further systematically evaluate acetylated and butyrylated substrates and their dynamic changes, we conducted a quantitative analysis of lysine acetylome and butyrylome through stable isotope dimethyl labeling during the three growth stages (supplemental Fig. S1). After tryptic digestion, peptides isolated from the exponential phase were labeled using “L” dimethyl labeling reagents (i.e. CH2O and NaBH3CN), peptides isolated from the transitional phase were labeled using “M” dimethyl labeling reagents (i.e. CD2O and NaBH3CN), and peptides isolated from the stationary phase were labeled using “H” labeling reagents (i.e. 13CD2O and NaBD3CN). The digested peptides were then enriched using pan-anti-acetyl-lysine/butyryl-lysine antibodies and analyzed by high-resolution Orbitrap MS. The MaxQuant software was used to analyze the data, and subsequent manual verification was conducted to verify lysine acetylation/butyrylation substrates for further analysis based on previous criteria (39). In this experiment, we identified 458 Kac sites in 254 proteins and 1078 Kbu sites in 373 proteins (supplemental Table S1–S3). To our knowledge, this was the first bacterial butyrylome description provided to date.
Fig. 1.

Quantitative analysis of lysine acetylome and butyrylome in three growth phases in C. acetobutylicum strain. A, Western blot analysis of the global lysing acetyllysine and butyryllysine level in protein lysates from C. acetobutylicum strain in the exponential phase (EP), transitional phase (TP), and stationary phase (SP). Coomassie blue staining was used as the loading control. B, Scatter plot of the quantifiable lysine acetylation sites identified in the EP and TP of the C. acetobutylicum strain. Log2 of the normalized TP/EP (M/L) ratios of the quantifiable sites were used for X axis and log10 of total intensities for each acetylated peptide for Y axis. Blue dots represent the sites of normalized M/L ratio less than 0.67; Red dots represent the sites of normalized M/L ratio between 0.67 and 1.5; Green dots represent the sites of normalized M/L ratio higher than 1.5. The percentage of acetylated sites in different M/L ratio ranges were presented in the pie chart. C, Scatter plot of the quantifiable lysine acetylation sites identified in the TP and SP of the C. acetobutylicum strain. Log2 of the normalized SP/TP (H/M) ratios of the quantifiable sites were used for X axis and log10 of total intensities for each acetylated peptide for Y axis. Blue dots represent the sites of normalized H/M ratio less than 0.67; Red dots represent the sites of normalized H/M ratio between 0.67 and 1.5; Green dots represent the sites of normalized H/M ratio higher than 1.5. The percentage of acetylated sites in different H/M ratio ranges were presented in the pie chart. D, Scatter plot of the quantifiable lysine butyrylation sites identified in the EP and TP of the C. acetobutylicum strain. Annotation is same in Fig. 1B. E, Scatter plot of the quantifiable lysine butyrylation sites identified in the TP and SP of the C. acetobutylicum strain. Annotation is same in Fig. 1C.
Among the 458 lysine acetylation sites and 1078 lysine butyrylation sites identified, 336 Kac sites and 877 Kbu sites were quantifiable. For lysine butyrylome analysis, nearly half (48%) of the modified sites bear elevated butyrylation levels (normalized M/L ratio ≥ 1.5) in the transitional phase, whereas no global change of lysine butyrylation level was observed in the stationary phases. Moreover, global acetylation levels of whole cell lysates remained constant, showing only a slight increase during the transitional phase (normalized M/L ratio for 65.8% of the Kac peptides was between 0.67 and 1.5; normalized H/M ratio for 87.2% of the Kac peptides was between 0.67 and 1.5) (Fig. 1B–1E). Interestingly, this quantitative acetylome and butyrylome study agreed with the Western blot analysis regarding the appearance of the first BuP concentration peak. However, the second BuP concentration peak and single AcP concentration peak midway through solventogenesis, which correspond to butyrate and acetate reutilization, respectively, did not result in elevated acetylation or butyrylation levels during the stationary phase.
Next, we examined the distribution of lysine acetylation and butyrylation sites in C. acetobutylicum. The average number of acetylation and butyrylation sites occurring in an individual protein was ∼1.8 and 2.9, respectively. Nearly 39% (99) of the acetylated proteins and 53% (196) of the butyrylated proteins bear no less than two modified sites (Fig. 2A). Moreover, 200 proteins and 212 sites from C. acetobutylicum shared lysine acetylation and butyrylation (Fig. 2B). To further understand this finding, the metabolic pathways in which the 200 shared acylated substrates were enriched (supplemental Table S4). Eight major subclasses were categorized according to the KEGG pathway analysis, which included ribosome, butanoate metabolism, d-glutamine and d-glutamate metabolism, fatty acid metabolism, RNA degradation, purine metabolism, glycolysis/gluconeogenesis and pyruvate metabolism (Fig. 2C). Butanoate metabolism is a specific phenotype of C. acetobutylicum and enrichment of this pathway was unusually found in analysis of other microbial acylomes, which indicated that lysine acylation might play an important role in the biosynthesis and consumption process of butyrate and butanol.
Fig. 2.
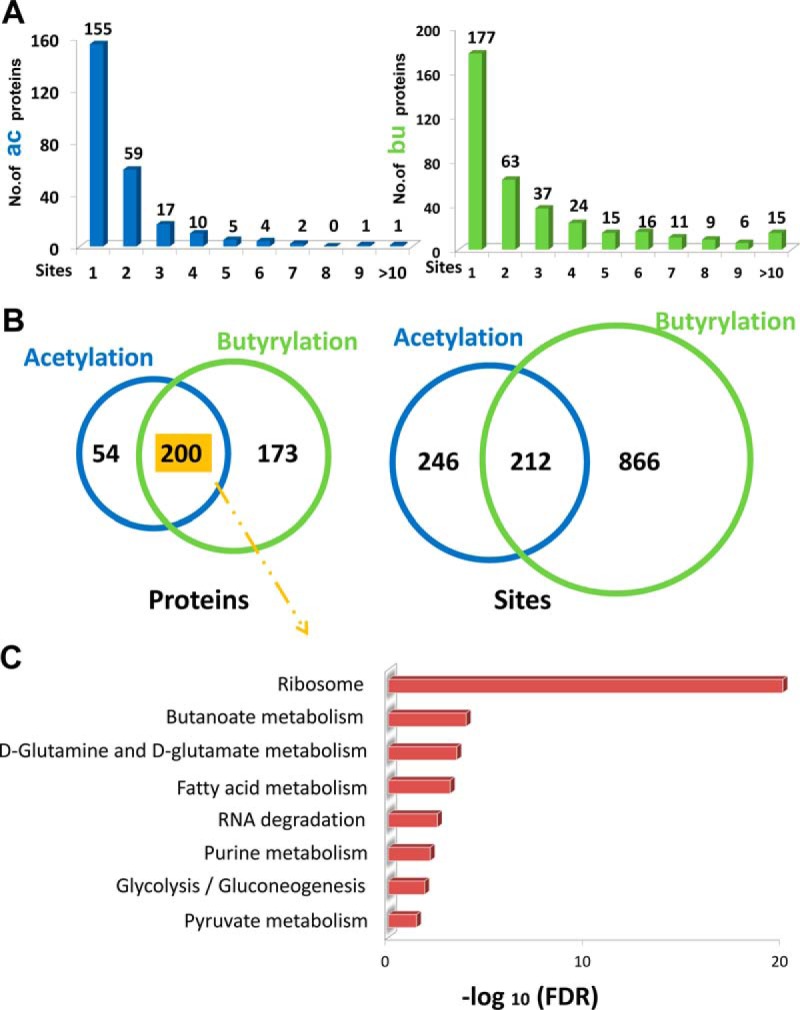
Landscape of the acylated proteins/sites in C. acetobutylicum strain. A, The number of acetylation/butyrylation sites per protein. B, The Venn diagram showing the number of overlapping acylated proteins or sites identified in our study. C, The KEGG analysis of the shared acetylated and butyrylated proteins in C. acetobutylicum strain.
Then, we analyzed the differences between the two acylation modifications and explored their implications in C. acetobutylicum. First, the iceLogo software was used to analyze the flanking sequences adjacent to the Kac and Kbu sites to evaluate the amino acid bias of the modified lysine. The results revealed that tyrosine and leucine residues were significantly over-represented at the +1 and −1 positions of the acetylated sites (supplemental Fig. S2A). Moreover, glutamic acid significantly occurred in proximity of butyrylated sites (supplemental Fig. S2B), which was similar to the lysine propionylation pattern in E. coli (40). Since lysine butyrylation differs from lysine acetylation in substrate pattern and acylation level (i.e. the butyrylation level is higher than the acetylation level during the transitional phase), it might be the modification responsible for regulating the metabolic and physiological properties of C. acetobutylicum.
The Two Lysine Acylations Involved in the Metabolic Pathways of C. acetobutylicum
Enrichment analysis against the KEGG pathway was conducted using the acetylated and butyrylated proteins with an elevated acylation level during the transitional phase (i.e., normalized M/L ratio ≥ 1.5, including 27.7% of the acetylated and 47.8% of the butyrylated sites shown in Fig. 1B and 1D). Our results showed that multiple metabolic pathways, including ribosome, biosynthesis of amino acids, purine metabolism, glycolysis/gluconeogenesis, amino acid degradation, butanoate metabolism, pentose phosphate pathway, fatty acid metabolism, and pyruvate metabolism, were highly enriched for both lysine acylation modifications. However, pathways for aminoacyl-tRNA biosynthesis, oxidative phosphorylation, tricarboxylic acid cycle and bacterial chemotaxis were only enriched for lysine butyrylation (Fig. 3A and supplemental Table S5), which suggested the potential regulatory function of protein butyrylation in these pathways. Several studies have proved that proteins with multiple acylated sites were related to ribosomal function (32, 41–43). However, according to our results, the most highly acylated protein, which bear 22 acetylated sites (6 of them with normalized M/L ratio ≥ 1.5) and 31 butyrylated sites (19 of them with normalized M/L ratio ≥ 1.5) was pyruvate-flavodoxin oxidoreductase, an essential enzyme that catalyzes the formation of acetyl-CoA through catabolization of glucose and is involved in pyruvate metabolism (Fig. 3B) (21). In the catalytic reaction, pyruvate transfers its electrons to flavodoxin and reacts with CoA to form acetyl-CoA, and the iron-sulfur binding sites in that enzyme are important for its activity. In pyruvate-flavodoxin oxidoreductase, several acetylated/butyrylated lysine sites surrounded by metal binding sites were found, such as the acetylated sites K766 and K793 and butyrylated sites K724, K761, K766, K793, and K797 (supplemental Fig. S3). Sequence alignment with its homologous enzyme from Desulfovibrio africanus was performed and the results showed that K761 and K797 were conserved in the Desulfovibrio species. Since both K761 and K797 were only butyrylated and their butyrylation level was elevated during the transitional phase (normalized M/L ratio > 1.5), the result suggested that lysine butyrylation influences the enzymatic activity of pyruvate-flavodoxin oxidoreductase and regulates the metabolic flux of acetyl-CoA and its derivatives during different growth stages.
Fig. 3.

The enrichment analysis of the protein with elevated acylation level in C. acetobutylicum strain. A, The KEGG analysis of acetylation sites and butyrylation sites with elevated acylation level in the transitional phase, including ribosome, biosynthesis of amino acids, purine metabolism, glycolysis/gluconeogenesis, amino acid degradation, butanoate metabolism, pentose phosphate pathway, fatty acid metabolism, pyruvate metabolism, aminoacyl-tRNA biosynthesis, oxidative phosphorylation, TCA cycle and bacterial chemotaxis. B, The quantitative acetylation and butyrylation sites in the Pyruvate-flavodoxin oxidoreductase of C. acetobutylicum strain.
Lysine Butyrylation Potentially Impacts the Activity of Butyrate Kinase
The typical biphasic metabolism of solventogenic Clostridia is initiated in the acidogenic phase, followed by acid re-assimilation and solvent production. Destruction of essential proton gradient across the membrane at low pH promotes the assimilation of acetate and butyrate to their corresponding CoA derivatives catalyzed by acetoacetyl-CoA/acyl-CoA transferase, with acetoacetyl-CoA as the CoA donor. Moreover, the acid formation pathway is inhibited and the bioalcohol production pathways is initiated (21). As shown in Fig. 4A, 4B and supplemental Fig. S4, almost all enzymes involved in the acid and solvent production pathways were acetylated or butyrylated, including acetate kinase (ack), phosphate acetyltransferase (pta), butyrate kinase (buk), and phosphate butyryltransferase (ptb) in the acidogenesis phase; acetoacetate decarboxylase (adc), NADH-dependent butanol dehydrogenase (bdhA), aldehyde-alcohol dehydrogenase 1 (adhe 1) and aldehyde-alcohol dehydrogenase 2 (adhe 2) in the solventogenesis phase; acetyl-CoA acetyltransferase (thlA), 3-hydroxybutyryl-CoA dehydrogenase (hbd), 3-hydroxybutyryl-CoA dehydratase (crt) and acyl-CoA dehydrogenase (bcd) in both phases. The acylation status of the four enzymes modified in the acidogenesis phase (ack, pta, ptb, buk) was analyzed. The results showed that the acylation level of acetylated and butyrylated sites did not change in pta and ack, both increased in ptb, and only the butyrylation level increased in buk.
Fig. 4.

The acylated states of the enzymes in the acid/solvent pathway. A, Diagram showing the metabolic enzymes involved in acidogenesis or solventogenesis. Enzymes in acidogenesis were shown in green; enzymes in solventogenesis were shown in blue; Enzymes in both the two phases were shown in yellow. B, The quantitative acylation level of pta, ack, ptb and buk in the acid formation pathway and adc, bdhA, adhe1 and adhe2 in the bioalcohol formation pathway.
Butyrate kinase, which catalyzes the conversion of butyryl-CoA to butyrate through butyryl phosphate, is an essential enzyme in the butyrate generation pathway (Fig. 5A). Interestingly, four lysine sites of buk (K51, K167, K239, and K287) showed butyrylation level higher in the transitional phase (normalized M/L ratio > 1.5) than that in the exponential phase (Fig. 5B). Sequence alignment analysis of butyrate kinase in several bacteria showed that K239 was highly conserved, a little lower for K51, whereas little or no conservative for K167 and K287 (Fig. 5C and supplemental Fig. S5). Therefore, it was suggested that K239 might be important for the enzymatic activity of buk. Hence, these four butyrylated lysine residues were mutated into glutamine, which mimicked the neutral charge of lysine butyrylation, to explore the potential effect of butyrylation on the enzymatic activity. Photometric activity assay showed that K239Q mutation decreased the activity of buk by 30%, which indicated that K239 might be critical for buk catalytic activity. On the other hand, no obvious changes were observed in the K167Q and K287Q mutants (Fig. 5D). Notably, the mutant K51Q expressed as an inclusion body. The results suggested that lysine butyrylation might impact the activity of buk at the initiation of solvent production. Together, these data suggested a new role of lysine butyrylation on butyrate metabolism in C. acetobutylicum, and how lysine butyrylation regulates other import enzymes, especially enzymes in butanol fermentation pathways would be explored in our further study.
Fig. 5.

Mutagenesis and enzymatic activity analysis of butyrate kinase (buk). A, The annotation of butyrate kinase in the butyrate biosynthesis pathway. B, The quantitative butyrylation sites in buk of C. acetobutylicum strain. C, Sequence alignment analysis the K239 of buk, from Exiguobacterium sibiricum (E. sibiricum), Bacillus cereus (B. cereus), Bacillus anthracis (B. anthracis), Bacillus subtilis (B. subtilis), Elusimicrobium minutum (E. minutum), Thermotoga maritima (T. maritima), Clostridium perfringens (C. perfringens) and C. acetobutylicum. D, Enzymatic activities of wild-type and K167Q, K239Q and K287Q mutant of buk. Purity of the wt and mutated proteins of buk shown by SDS-PAGE gel.
Lysine Butyrylation Potentially Regulates Bacterial Chemotaxis and Sporulation Process of C. acetobutylicum
The evolution of C. acetobutylicum from vegetative cells to active “clostridia stage” displays a series of morphological or phenotypic changes that are tightly associated with the bacterium biphasic metabolism. A previous transcriptome analysis showed that vegetative cells highly express chemotaxis and motility genes during the exponential phase, to convert to their clostridia forms in the stationary phase, triggering the expressions of genes related with sporulation and solventogenesis (Fig. 6A) (25). Therefore, whether elevated butyrylation level affected the physiological phenotypic changes and sporulation/differentiation program of C. acetobutylicum was investigated. In our experiment, several proteins functional to bacterial chemotaxis and flagellar assembly were butyrylated, including methyl-accepting chemotaxis proteins (CA_C0909, CA_C3352, CA_C3688), chemotaxis signal transduction protein CheW, chemotaxis signal receiving protein CheY, chemotaxis histidine kinase CheA, chemotaxis protein CheR, flagellar motor switch protein G, flagellar switch protein FliM and chemotaxis protein MotB (Fig. 6B). Among them, CheY is located at the center of the bacterial chemotaxis pathway. CheY transfers sensory signals from the chemoreceptors to the flagellar switch protein FliM, thereby inducing the flagellar motor to rotate clockwise (44). In addition, the homologous residue of the butyrylated K103 of C. acetobutylicum CheY was found to be acetylated in E. coli, which was reported to negatively regulate the clockwise flagellar rotation function by disturbing the salt bridge with the phosphorylation site (45, 46). Thus, the elevated butyrylation level of K103 in CheY (normalized H/M ratio = 1.5) suggested its potential role in decreasing this activity. In addition, CheA and FliM, which directly interact with CheY, also bear butyrylated sites (K534 of CheA and K49 of FliM) with obviously elevated levels (normalized M/L ratio > 1.5). Since butyrylation neutralizes the positive charge of the lysine amino group and further extends the hydrocarbon chain (i.e. enhances hydrophobicity) and increases lysine side chain dimension beyond that of Kac, we inferred that hydrogen bonds and electrostatic interactions among chemotaxis proteins would be disturbed by butyrylation, which might impact the mobility of this bacterium during the stationary phase.
Fig. 6.
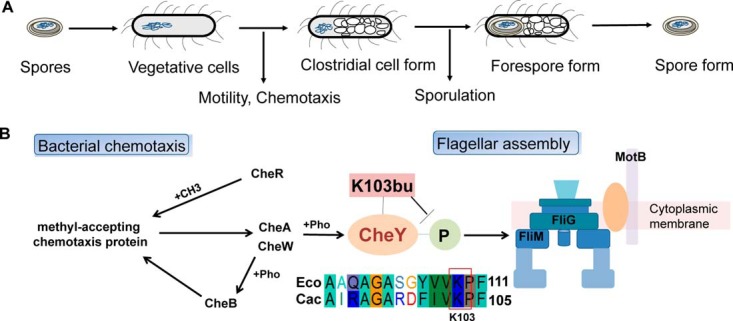
Lysine butyrylation in the bacterial chemotaxis and flagellar assembly. A, The endospore-forming process in the C. acetobutylicum strain. The phenotypes of C. acetobutylicum strain in different growth stages were shown. B, The butyrylated protein involved in the bacterial chemotaxis and flagellar assembly, including methyl-accepting chemotaxis proteins, chemotaxis signal transduction protein CheW, chemotaxis signal receiving protein CheY, chemotaxis histidine kinase CheA, chemotaxis protein CheR, flagellar motor switch protein G, flagellar switch protein FliM and chemotaxis protein MotB. Sequence alignment of CheY from E. coli and C. acetobutylicum.
Furthermore, the core transcription factor Spo0A, which promotes spore maturation and solvent formation through aspartyl phosphorylation, was butyrylated at K45 and K217 (Fig. 7A). In the sequence alignment analysis of Spo0A in four species from Bacillus to Clostridium, K217 (located in the H-T-H DNA-binding domain) was highly conserved, whereas K45 was only conserved in the Clostridium genus (Fig. 7B). Therefore, whether K217 butyrylation regulated the activity of Spo0A, especially DNA-binding ability, was investigated. K217 and K45 were mutated into glutamine to explore the effect of butyrylation on these two residues. The EMSA assay showed that wild-type Spo0A directly bound the spo0A promoter at low concentration, and the K45Q mutant had similar DNA-binding affinity, whereas little or no binding ability was observed for K217Q (Fig. 7C). Previous studies proved that K-R mutations of the transcription factor recovered the ability to bind target DNA in both higher eukaryotes and prokaryotes (47, 48), Similarly, our results demonstrated that the K217R mutant had DNA-binding affinity like that of the wild-type protein (Fig. 7C). To further describe the effect of butyrylation on Spo0A, wild-type Spo0A and its K217R mutant were chemically butyrylated with butyryl-CoA. The EMSA assay proved that the wild-type protein lowered the binding affinity to its own promoter, whereas no effect was detected in the K217R mutant (Fig. 7D). According to the computational prediction of Spo0A-binding sites, the TA-box (TGTCGAA) is in the downstream region of the ribosome binding site, suggesting a potentially negative auto-regulation mechanism. Therefore, K217 butyrylation likely relieved the auto-inhibition mechanism and promoted the expression of Spo0A during the stationary phase. In addition, whether lysine butyrylation could regulate the binding between Spo0A and the butanol formation-related sol operon, which contains target genes of Spo0A, such as adhE (encoding a bifunctional butyraldehyde/butanol dehydrogenase), ctfA, and ctfB (encoding the two subunits of CoA transferase) (49, 50) was also explored. EMSA indicated that high Spo0A concentration was essential to the interaction with the sol operon, and butyrylation did not change the binding affinity (Fig. 7E). Together, these results suggested that butyrylation on K217 might regulate Spo0A auto-expression but possibly not influence the expression of solvent-related genes.
Fig. 7.

Regulation of Spo0A by lysine butyrylation. A, The two butyrylation sites identified in the transcription factor Spo0A of C. acetobutylicum strain. B, Sequence alignment of Spo0A, from Bacillus anthracis (Ban), Bacillus subtilis (Bsu), Clostridium pasteurianum (Cpas) and Clostridium acetobutylicum (Cac). C, DNA-binding abilities of Spo0A and derivatives by EMSA. The amount of His-Spo0A used (10−7 × g) is shown at the top of each lane. From left to right panel: wild type Spo0A, Spo0A K45Q, Spo0A K217Q and Spo0A K217R. D, DNA-binding abilities of Bu-Spo0A and Bu-Spo0A K217R. The amount of His-Spo0A used (10−7 × g) is shown at the top of each lane. Left panel: The Bu-Spo0A K217R was incubated with probes. Right panel: Bu-Spo0A was incubated with probes. E, DNA-binding abilities of wt and Bu-wt. The amount of His-Spo0A used (10−7 × g) is shown at the top of each lane. Left panel: The wt was incubated with probes. Right panel: Bu-wt was incubated with probes.
Quantitative Proteomic Analysis of C. acetobutylicum in the Three Growth Stages
Genome-scale transcriptional assay and cellular AcP/BuP concentration measurement during the exponential, transitional, and stationary phases of C. acetobutylicum have been previously performed (25). To further investigate the correlation between protein expression levels, intermediate metabolite concentrations, and lysine acylation status in the three phases, quantitative proteomics assay was performed (supplemental Table S6). First, gene expression at the transcriptional and translational levels was compared. According to the KEGG analysis, proteins with high expression levels (p < 0.05, H/L ratio > 1.5) in the exponential phase were enriched in bacterial chemotaxis, flagellar assembly, and pyrimidine metabolism processes (supplemental Fig. S6A and supplemental Table S7A, S7B), which was similar to the reported transcriptional data. In addition, gene expression of ribosomal proteins was downregulated in the transitional phase and then up-regulated in the stationary phase (supplemental Fig. S6A, S6B and supplemental Table S7A, S7B), which confirmed previous studies. These results showed that the time points chosen in this study coincided with the three growth stages of C. acetobutylicum. However, our quantitative acylome data showed that global lysine acetylation and butyrylation levels did not change in the stationary phase, although AcP and BuP concentration peaks were observed (supplemental Fig. S7A). Therefore, the intricate mechanism involved in maintaining the global acylation status in the stationary phase was determined.
Previous studies reported that CobB functioned as the major lysine deacetylase conserved in bacteria, and it also catalyzed the depropionylation and desuccinylation reactions because of the structural similarity of acyl residues (32, 40). Therefore, we suggested that, in C. acetobutylicum, CaCobB (CA_C0284) could catalyze the deacetylation/debutyrylation reactions and regulate protein acylation levels in the growth cycle of C. acetobutylicum. Our quantitative proteomics assay showed that the gene expression level of CaCobB remained constant in the three growth phases.
Moreover, in the deacylation reaction catalyzed by CobB, NAD+ acts as the acyl group receptor, and the NAD+/NADH ratio influences the activity of CobB. In the transitional phase of C. acetobutylicum, formation of acetate and butyrate resulted in significant hydrogen increase, followed by accumulation of NADH and inhibition of CaCobB activity. However, when C. acetobutylicum entered the stationary phase, high expression of alcohol dehydrogenase or aldehyde dehydrogenase catalyzed the formation of NAD+ as their major reaction product, which raised the NAD+/NADH ratio and promoted CaCobB activity. Therefore, we hypothesized that two factors (concentration of acyl-phosphate and NAD+ to NADH ratio) co-determined the protein acylation levels in the different growth phases (supplemental Fig. S7B). In the exponential and transitional phases, the substrate acylation levels mainly depend on the concentration of acyl donors because the deacetylase activity of CaCobB is low. Consequently, the increase of the acetylation or butyrylation levels depends on the increase of acyl-phosphate concentration. Additionally, during the stationary phase, NADH is used as the substrate for solvent production, whereas its product NAD+ acts as the co-factors in the deacetylation/debutyrylation reaction. Hence, lysine acylation levels depend on both acyl-phosphate concentration and the deacylase activity of CaCobB, which could regulate the acylation level in the stationary phase.
DISCUSSION
Recently, proteomic advances have uncovered the diversity, frequency, and regulatory function of lysine acetylation in several microorganisms, which include E. coli (26, 51, 52), Salmonella enterica (15), Vibrio parahemolyticus (53), Thermus thermophiles (54), Cyanobacterium Synechococcus (55), Saccharopolyspora erythraea (56), Streptomyces roseosporus (57), Mycoplasma pneumonia (58) and Mycobacterium tuberculosis (59, 60). In addition to acetylation, the side chain of lysine can be modified by several other acyl moieties. In bacteria, other protein acylomes were confirmed, such as the propionylome of T. thermophiles and E. coli, succinylome of E. coli (32), M. tuberculosis (61) and V. parahemolyticus (62), malonylome of E. coli (63), S. erythraea (42), Bacillus amyloliquefaciens (64) and Synechocystis sp. (41).
In this study, quantitative acetylome and butyrylome of the ancient phylum Firmicute C. acetobutylicum, which is famous for its ABE fermentation pathway, were investigated. Collectively, 458 lysine acetylation sites and 1078 butyrylation sites were identified in 254 and 373 substrates, respectively, which is the first systematic description of lysine butyrylome in bacteria. Moreover, the amino acid bias of modified lysine residues was analyzed, and the differences in enriched KEGG pathways of the two acylomes were explored. The results showed that lysine butyrylation has specific substrate patterns and pathway enrichments different from those of acetylation. Additionally, lysine butyrylation was proved to impact the activity of the essential metabolic enzyme buk and to influence the auto-regulated gene expression of the sporulation regulator Spo0A. These results indicated that BuP-dependent lysine butyrylation is responsible for regulating the phenotypic changes and solvent production in C. acetobutylicum. Several studies proved that various signal stimulations lead to different protein acylation levels. A previous study reported that lysine butyrylation on histone H4 competes with acetylation in eukaryotic spermatogenesis (65). Moreover in E. coli, high glucose culture conditions increased global protein acetylation and succinylation but decreased the propionylation level of whole cell lysates (32, 40). Therefore, the complex distribution of intermediate metabolites in the different growth phases of C. acetobutylicum, such as AcP/BuP, acetyl/butyryl-CoA, and NAD+/NADH, determines the acylation state of protein substrates and affects their functions. Like other research groups, we proved that BuP is the major regulatory molecule acting as both phospho and butyryl donor for different proteins. For example, the transcription factor Spo0A was phosphorylated at D58 and butyrylated at K217, which co-activated Spo0A at the gene expression level and initiated the process of endospore and solvent formation in C. acetobutylicum.
At present, metabolic engineering tools and other strategies have been used to improve butanol production in C. acetobutylicum (21). Promotion of butanol formation was successfully achieved by lowering the reaction redox potential through the addition of glycerol and methyl viologen (66, 67). In the past 15 years, the development of genetic manipulation and metabolic engineering approaches facilitated the design of biobutanol-producing strains, with the major goal of elevating the metabolic flux toward the desired product by reducing byproduct formation. However, engineering C. acetobutylicum by either eliminating the acid forming pathways (i.e. buk-negative and pta-negative mutants) or over-expressing solvent-catalyzed enzymes (i.e. adhE1 overexpression mutant) was ineffective in increasing the yield of butanol. Due to the complexity of the regulatory mechanism involved in the physiological and solvent production processes of C. acetobutylicum, traditional metabolic engineering used in that strain acquired limited success (21). In our study, we found the Spo0A butyrylation could relieve auto-inhibition but had no effect on the sol operon. In addition, we found that high levels of NAD+ formed in the stationary phase might promote the activity of CaCobB and then maintained the global level of protein acylation, which avoided negatively affecting the activity of enzymes in solventogenesis. Therefore, optimization and manipulation of engineered pathways at the lysine acylation level would be a potential new strategy for generating a superior butanol producing C. acetobutylicum strain.
In addition, Clostridia include several orders, families, and genera, including species beneficial to human and animal microbiota, and human pathogens (68). For instance, C. sporogenes and C. novyi, could be used as anticancer drug delivery vehicles, whereas other species such as C. tetani and C. botulinum, which produced tetanospasmin and botulinum toxin, respectively, threaten human lives. Since these pathogenic bacteria are like C. acetobutylicum in their anaerobic and spore-forming traits, the full understanding of the regulatory mechanism of Clostridia will promote our knowledge of the infection ability and toxicity of pathogens.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited at iProX with the data set identifier IPX0001122000 (URL http://www.iprox.org/page/PDV014.html?projectId=IPX0001122000).
Supplementary Material
Footnotes
* This study was supported by grants from the National Natural Science Foundation of China (31730004, 21335003, and 21575089 to Bang-Ce Ye), Natural Science Foundation of China (31670066 and 91753203 to Minjia Tan), Innovation Project of Instrument and Equipment Function Development of the Chinese Academy of Sciences (2060499 to Minjia Tan), and China Postdoctoral Science Foundation (2017M621567 to Jun-Yu Xu).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- ABE
- acetone-butanol-ethanol
- ack
- acetate kinase
- AcP
- acetyl phosphate
- adc
- acetoacetate decarboxylase
- adhe1
- aldehyde-alcohol dehydrogenase 1
- adhe2
- aldehyde-alcohol dehydrogenase 2
- AGC
- automatic gain control
- bcd
- acyl-CoA dehydrogenase
- bdhA
- NADH-dependent butanol dehydrogenase
- buk
- butyrate kinase
- BuP
- butyryl phosphate
- CheA
- chemotaxis histidine kinase
- CheR
- chemotaxis protein
- CheW
- chemotaxis signal transduction protein
- CheY
- chemotaxis signal receiving protein
- crt
- 3-hydroxybutyryl-CoA dehydratase
- EMSA
- electrophoretic mobility shift assay
- EP
- exponential phase
- FDR
- false discovery rate
- FliM
- flagellar switch protein
- hbd
- 3-hydroxybutyryl-CoA dehydrogenase
- HCD
- higher-energy collisional dissociation
- HRP
- horseradish peroxidase
- Kac
- lysine acetylation
- Kbu
- lysine butyrylation
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- LB
- Luria-Bertani broth
- MotB
- chemotaxis protein
- NCE
- normalized collision energy
- thlA
- acetyl-CoA acetyltransferase
- TP
- transitional phase.
REFERENCES
- 1. Pietrocola F., Galluzzi L., Pedro J. M. B., Madeo F., and Kroemer G. (2015) Acetyl coenzyme A: a central metabolite and second messenger. Cell Metabolism 21, 805–821 [DOI] [PubMed] [Google Scholar]
- 2. Menzies K. J., Zhang H., Katsyuba E., and Auwerx J. (2015) Protein acetylation in metabolism - metabolites and cofactors. Nat. Rev. Endocrinol. 12, 43–60 [DOI] [PubMed] [Google Scholar]
- 3. Sabari B. R., Di Z., Allis C. D., and Zhao Y. (2017) Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18, 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen Y., Sprung R., Tang Y., Ball H. L., Sangras B., Kim S. C., Falck J. R., Peng J., Gu W., and Zhao Y. (2007) Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics 6, 812–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tan M., Luo H., Lee S., Jin F., Yang J. S., Montellier E., Buchou T., Cheng Z., Rousseaux S., and Rajagopal N. (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sabari B. R., Tang Z., Huang H., Yonggonzalez V., Molina H., Kong H. E., Dai L., Shimada M., Cross J. R., and Zhao Y. (2015) Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 58, 203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Z., Tan M., Xie Z., Dai L., Chen Y., and Zhao Y. (2011) Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., and Yang K. (2011) The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics 10, M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., and Choi B. H. (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tan M., Peng C., Anderson K. A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., and Zhang Y. (2014) Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metabolism 19, 605–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dai L., Peng C., Montellier E., Lu Z., Chen Y., Ishii H., Debernardi A., Buchou T., Rousseaux S., and Jin F. (2014) Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 10, 365–370 [DOI] [PubMed] [Google Scholar]
- 12. Xie Z., Zhang D., Chung D., Tang Z., Huang H., Dai L., Qi S., Li J., Colak G., and Chen Y. (2016) Metabolic regulation of gene expression by histone lysine β-hydroxybutyrylation. Mol. Cell 62, 194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choudhary C., Weinert B. T., Nishida Y., Verdin E., and Mann M. (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 [DOI] [PubMed] [Google Scholar]
- 14. Starai V. J., Celic I., Cole R. N., Boeke J. D., and Escalantesemerena J. C. (2002) Sir2-Dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 [DOI] [PubMed] [Google Scholar]
- 15. Wang Q., Zhang Y., Yang C., Xiong H., Lin Y., Yao J., Li H., Xie L., Zhao W., and Yao Y. (2010) Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thao S., Chen C. S., Zhu H., and Escalantesemerena J. C. (2010) Nε-lysine acetylation of a bacterial transcription factor inhibits Its DNA-binding activity. Plos One 5, e15123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li R., Gu J., Chen Y., Xiao C., Wang L., Zhang Z., Bi L., Wei H., Wang X. P., and Deng J. (2010) CobB regulates Escherichia coli chemotaxis by deacetylating the response regulator CheY. Mol. Microbiol. 76, 1162–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liang W., Malhotra A., and Deutscher M. P. (2011) Acetylation regulates the stability of a bacterial protein: growth stage-dependent modification of RNase R. Mol. Cell 44, 160–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paredes C. J., Alsaker K. V., and Papoutsakis E. T. (2005) A comparative genomic view of clostridial sporulation and physiology. Nat. Rev. Microbiol. 3, 969–978 [DOI] [PubMed] [Google Scholar]
- 20. Tracy B. P. (2012) Improving butanol fermentation to enter the advanced biofuel market. Mbio 3, e00518–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lutkeeversloh T., and Bahl H. (2011) Metabolic engineering of Clostridium acetobutylicum: recent advances to improve butanol production. Curr. Opinion Biotechnol. 22, 634–647 [DOI] [PubMed] [Google Scholar]
- 22. Zhao Y., Tomas C., Rudolph F. B., Papoutsakis E. T., and Bennett G. N. (2005) Intracellular butyryl phosphate and acetyl phosphate concentrations in Clostridium acetobutylicum and their implications for solvent formation. Appl. Environ.l Microbiol. 71, 530–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakotte S., Schaffer S., and Zickner B. (2002) Transcriptional regulation of solventogenesis in Clostridium acetobutylicum. J. Mol. Microb. Biotech. 4, 295–300 [PubMed] [Google Scholar]
- 24. Harris L. M., Welker N. E., and Papoutsakis E. T. (2002) Northern, morphological, and fermentation analysis of spo0A inactivation and overexpression in Clostridium acetobutylicum ATCC 824. J. Bacteriol. 184, 3586–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones S. W., Paredes C. J., Tracy B. P., Cheng N., Sillers R., Senger R. S., and Papoutsakis E. T. (2008) The transcriptional program underlying the physiology of Clostridial sporulation. Genome Biol. 9, 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Weinert B. T., Iesmantavicius V., Wagner S. A., Scholz C., Gummesson B., Beli P., Nystrom T., and Choudhary C. (2013) Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell 51, 265–272 [DOI] [PubMed] [Google Scholar]
- 27. Venkataramanan K. P., Min L., Hou S., Jones S. W., Ralston M. T., Lee K. H., and Papoutsakis E. T. (2015) Complex and extensive post-transcriptional regulation revealed by integrative proteomic and transcriptomic analysis of metabolite stress response in Clostridium acetobutylicum. Biotechnol. Biofuels 8, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Millat T., Janssen H., Bahl H., Fischer R., and Wolkenhauer O. (2013) Integrative modelling of pH-dependent enzyme activity and transcriptomic regulation of the acetone–butanol–ethanol fermentation of Clostridium acetobutylicum in continuous culture. Microb. Biotechnol. 6, 526–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jang Y., Han M. J., Lee J., Im J. A., Lee Y. H., Papoutsakis E. T., Bennett G. N., and Lee S. Y. (2014) Proteomic analyses of the phase transition from acidogenesis to solventogenesis using solventogenic and non-solventogenic Clostridium acetobutylicum strains. Appl. Microbiol. Biotechnol. 98, 5105–5115 [DOI] [PubMed] [Google Scholar]
- 30. Grimmler C., Janssen H., Krauβe D., Fischer R., Bahl H., Durre P., Liebl W., and Ehrenreich A. (2011) Genome-wide gene expression analysis of the switch between acidogenesis and solventogenesis in continuous cultures of Clostridium acetobutylicum. J. Mol. Microb. Biotech. 20, 1–15 [DOI] [PubMed] [Google Scholar]
- 31. Hartmanis M. G. N., and Gatenbeck S. (1984) Intermediary metabolism in Clostridium acetobutylicum: Levels of enzymes involved in the formation of acetate and butyrate. Appl. Environm. Microbiol. 47, 1277–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Colak G., Xie Z., Zhu A. Y., Dai L., Lu Z., Zhang Y., Wan X., Chen Y., Cha Y. H., and Lin H. (2013) Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol. Cell. Proteomics 12, 3509–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boersema P. J., Raijmakers R., Lemeer S., Mohammed S., and Heck A. J. R. (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat. Protocols 4, 484–494 [DOI] [PubMed] [Google Scholar]
- 34. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 35. Colaert N., Helsens K., Martens L., Vandekerckhove J., and Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Meth. 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 36. Huang K., Huang S., Rudolph F. B., and Bennett G. N. (2000) Identification and characterization of a second butyrate kinase from Clostridium acetobutylicum ATCC 824. J. Mol. Microb. Biotech. 2, 33–38 [PubMed] [Google Scholar]
- 37. Cary J. W., Petersen D. J., Papoutsakis E. T., and Bennett G. N. (1988) Cloning and expression of Clostridium acetobutylicum phosphotransbutyrylase and butyrate kinase genes in Escherichia coli. J. Bacteriol. 170, 4613–4618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liao C., Yao L., and Ye B. (2014) Three genes encoding citrate synthases in Saccharopolyspora erythraea are regulated by the global nutrient-sensing regulators GlnR, DasR, and CRP. Mol. Microbiol. 94, 1065–1084 [DOI] [PubMed] [Google Scholar]
- 39. Chen Y., Kwon S. W., Kim S. C., and Zhao Y. (2005) Integrated approach for manual evaluation of peptides identified by searching protein sequence databases with tandem mass spectra. J. Proteome Res. 4, 998–1005 [DOI] [PubMed] [Google Scholar]
- 40. Sun M., Xu J., Wu Z., Zhai L., Liu C., Cheng Z., Xu G., Tao S., Ye B., and Zhao Y. (2016) Characterization of protein lysine propionylation in Escherichia coli: global profiling, dynamic change, and enzymatic regulation. J. Proteome Res. 15, 4696–4708 [DOI] [PubMed] [Google Scholar]
- 41. Ma Y., Yang M., Lin X., Liu X., Huang H., and Feng G. (2017) Malonylome analysis reveals the involvement of lysine malonylation in metabolism and photosynthesis in cyanobacteria. J. Proteome Res. 16, 2030–2043 [DOI] [PubMed] [Google Scholar]
- 42. Xu J., Xu Z., Zhou Y., and Ye B. (2016) Lysine malonylome may affect the central metabolism and erythromycin biosynthesis pathway in Saccharopolyspora erythraea. J. Proteome Res. 15, 1685–1701 [DOI] [PubMed] [Google Scholar]
- 43. Xie L., Liu W., Li Q., Chen S., Xu M., Huang Q., Zeng J., Zhou M., and Xie J. (2015) First succinyl-proteome profiling of extensively drug-resistant Mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. J. Proteome Res. 14, 107–119 [DOI] [PubMed] [Google Scholar]
- 44. Wadhams G. H., and Armitage J. P. (2004) Making sense of it all: bacterial chemotaxis. Nat. Rev. Mol. Cell Biol. 5, 1024–1037 [DOI] [PubMed] [Google Scholar]
- 45. Lukat G. S., Lee B. H., Mottonen J., Stock A. M., and Stock J. B. (1991) Roles of the highly conserved aspartate and lysine residues in the response regulator of bacterial chemotaxis. J. Biol. Chem. 266, 8348–8354 [PubMed] [Google Scholar]
- 46. Liarzi O., Barak R., Bronner V., Dines M., Sagi Y., Shainskaya A., and Eisenbach M. (2010) Acetylation represses the binding of CheY to its target proteins. Mol. Microbiol. 76, 932–943 [DOI] [PubMed] [Google Scholar]
- 47. Bararia D., Kwok H. S., Welner R. S., Numata A., Sarosi M. B., Yang H., Wee S., Tschuri S., Ray D., and Weigert O. (2016) Acetylation of C/EBPα inhibits its granulopoietic function. Nat. Commun. 7, 10968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ren J., Sang Y., Tan Y., Tao J., Ni J., Liu S., Fan X., Zhao W., Lu J., and Wu W. (2016) Acetylation of lysine 201 inhibits the DNA-binding ability of PhoP to regulate salmonella virulence. Plos Pathog. 12, e1005458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Durre P. (2008) Fermentative butanol production: bulk chemical and biofuel. Ann. NY Acad. Sci. 1125, 353–362 [DOI] [PubMed] [Google Scholar]
- 50. Thormann K. M., Feustel L., Lorenz K., Nakotte S., and Durre P. (2002) Control of butanol formation in Clostridium acetobutylicum by transcriptional activation. J. Bacteriol. 184, 1966–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang J., Sprung R., Pei J., Tan X., Kim S., Zhu H., Liu C., Grishin N. V., and Zhao Y. (2009) Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia Coli. Mol. Cell. Proteomics 8, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang K., Zheng S., Yang J. S., Chen Y., and Cheng Z. (2013) Comprehensive profiling of protein lysine acetylation in Escherichia coli. J. Proteome Res. 12, 844–851 [DOI] [PubMed] [Google Scholar]
- 53. Pan J., Ye Z., Cheng Z., Peng X., Wen L., and Zhao F. (2014) Systematic analysis of the lysine acetylome in Vibrio parahemolyticus. J. Proteome Res. 13, 3294–3302 [DOI] [PubMed] [Google Scholar]
- 54. Meng Q., Liu P., Wang J., Wang Y., Hou L., Gu W., and Wang W. (2016) Systematic analysis of the lysine acetylome of the pathogenic bacterium Spiroplasma eriocheiris reveals acetylated proteins related to metabolism and helical structure. J. Proteomics 148, 159–169 [DOI] [PubMed] [Google Scholar]
- 55. Chen Z., Zhang G., Yang M., Li T., Ge F., and Zhao J. (2017) Lysine acetylome analysis reveals photosystem II manganese-stabilizing protein acetylation is involved in negative regulation of oxygen evolution in model Cyanobacterium Synechococcus sp. PCC 7002. Mol. Cell. Proteomics 16, 1297–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Huang D., Li Z., You D., Zhou Y., and Ye B. (2015) Lysine acetylproteome analysis suggests its roles in primary and secondary metabolism in Saccharopolyspora erythraea. Appl. Microbiol. Biotechnol. 99, 1399–1413 [DOI] [PubMed] [Google Scholar]
- 57. Liao G., Xie L., Li X., Cheng Z., and Xie J. (2014) Unexpected extensive lysine acetylation in the trump-card antibiotic producer Streptomyces roseosporus revealed by proteome-wide profiling. J. Proteomics 106, 260–269 [DOI] [PubMed] [Google Scholar]
- 58. Van Noort V., Seebacher J., Bader S., Mohammed S., Vonkova I., Betts M. J., Kuhner S., Kumar R., Maier T., and Oflaherty M. (2012) Cross-talk between phosphorylation and lysine acetylation in a genome-reduced bacterium. Mol. Syst. Biol. 8, 571–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu F., Yang M., Wang X. P., Yang S., Gu J., Zhou J., Zhang X., Deng J., and Ge F. (2014) Acetylome Analysis Reveals Diverse Functions of Lysine Acetylation in Mycobacterium tuberculosis. Mol. Cell. Proteomics 13, 3352–3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xie L., Wang X., Zeng J., Zhou M., Duan X., Li Q., Zhang Z., Luo H., Pang L., and Li W. (2015) Proteome-wide lysine acetylation profiling of the human pathogen Mycobacterium tuberculosis. Int. J. Biochem. Cell Biol. 59, 193–202 [DOI] [PubMed] [Google Scholar]
- 61. Yang M., Wang Y., Chen Y., Cheng Z., Gu J., Deng J., Bi L., Chen C., Mo R., and Wang X. P. (2015) Succinylome analysis reveals the involvement of lysine succinylation in metabolism in pathogenic Mycobacterium tuberculosis. Mol. Cell. Proteomics 14, 796–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pan J., Chen R., Li C., Li W., and Ye Z. (2015) Global analysis of protein lysine succinylation profiles and their overlap with lysine acetylation in the marine bacterium Vibrio parahemolyticus. J. Proteome Res. 14, 4309–4318 [DOI] [PubMed] [Google Scholar]
- 63. Qian L., Nie L., Chen M., Liu P., Zhu J., Zhai L., Tao S., Cheng Z., Zhao Y., and Tan M. (2016) Global profiling of protein lysine malonylation in Escherichia coli reveals its role in energy metabolism. J. Proteome Res. 15, 2060–2071 [DOI] [PubMed] [Google Scholar]
- 64. Fan B., Li Y. L., Li L., Peng X. J., Bu C., Wu X. Q., and Borriss R. (2017) Malonylome analysis of rhizobacterium Bacillus amyloliquefaciens FZB42 reveals involvement of lysine malonylation in polyketide synthesis and plant-bacteria interactions. J. Proteomics 154, 1–12 [DOI] [PubMed] [Google Scholar]
- 65. Goudarzi A., Zhang D., Huang H., Barral S., Kwon O. K., Qi S., Tang Z., Buchou T., Vitte A., and He T. (2016) Dynamic competing histone H4 k5k8 acetylation and butyrylation are hallmarks of highly active gene promoters. Mol. Cell 62, 169–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Girbal L., Croux C., Vasconcelos I., and Soucaille P. (1995) Regulation of metabolic shifts in Clostridium acetobutylicum ATCC 824. FEMS Microbiol. Rev. 17, 287–297 [Google Scholar]
- 67. Girbal L., and Soucaille P. (1998) Regulation of solvent production in Clostridium acetobutylicum. Trends Biotechnol. 16, 11–16 [Google Scholar]
- 68. Alhinai M. A., Jones S. W., and Papoutsakis E. T. (2015) The Clostridium sporulation programs: diversity and preservation of endospore differentiation. Microbiol. Mol. Biol. R 79, 19–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited at iProX with the data set identifier IPX0001122000 (URL http://www.iprox.org/page/PDV014.html?projectId=IPX0001122000).