

Abstract
High grade gliomas are the most common brain tumors in adult. These tumors are characterized by a high infiltration in microglial cells and macrophages. The immunosuppressive tumor environment is known to orient immune cells toward a pro-tumoral and anti-inflammatory phenotype. Therefore, the current challenge for cancer therapy is to find a way to reorient macrophages toward an antitumoral phenotype. Previously, we demonstrated that macrophages secreted antitumoral factors when they were invalidated for the proprotein converstase 1/3 (PC1/3) and treated with LPS. However, achieving an activation of macrophages via LPS/TLR4/Myd88-dependent pathway appears yet unfeasible in cancer patients. On the contrary, the antitumor drug Paclitaxel is also known to activate the TLR4 MyD88-dependent signaling pathway and mimics LPS action. Therefore, we evaluated if PC1/3 knock-down (KD) macrophages could be activated by Paclitaxel and efficient against glioma. We report here that such a treatment of PC1/3 KD macrophages drove to the overexpression of proteins mainly involved in cytoskeleton rearrangement. In support of this finding, we found that these cells exhibited a Ca2+ increase after Paclitaxel treatment. This is indicative of a possible depolymerization of microtubules and may therefore reflect an activation of inflammatory pathways in macrophages. In such a way, we found that PC1/3 KD macrophages displayed a repression of the anti-inflammatory pathway STAT3 and secreted more pro-inflammatory cytokines. Extracellular vesicles isolated from these PC1/3 KD cells inhibited glioma growth. Finally, the supernatant collected from the coculture between glioma cells and PC1/3 KD macrophages contained more antitumoral factors. These findings unravel the potential value of a new therapeutic strategy combining Paclitaxel and PC1/3 inhibition to switch macrophages toward an antitumoral immunophenotype.
Keywords: Calcium Signaling*, Cancer therapeutics, Exosomes, Immunology*, Secretome, glioma, macrophages, Paclitaxel, proprotein convertase 1/3, proteomics
Cancer cells share eight common traits (“hallmarks”) that govern their transformation from normal cells and maintenance in the host environment: (1) they stimulate their own growth (self-sufficiency in growth signals), (2) they resist inhibitory signals that might otherwise hinder their growth and proliferation (insensitivity to antigrowth signals), (3) they resist programmed cell death (evading apoptosis) and other cell death mechanisms, (4) they can multiply indefinitely (limitless replicative potential), (5) they stimulate blood vessels growth to supply nutrients to tumors (sustained angiogenesis), (6) they invade local tissues and metastasize to distant sites (tissue invasion and metastasis), (7) they have altered metabolic pathways, and (8) finally, they evade the immune system (1). This last-mentioned feature has radically changed our views on cancer pathophysiology by pointing the crucial instructing effects exerted by cancer cells on both local and systemic immune responses. Thus, working toward new immunotherapeutic approaches for cancer is now considered as a priority task.
Among the many mechanisms that drive tumor immune evasion, tumor-associated macrophages (TAM)1 greatly contribute to the resistance to immune responses. Indeed, the local and systemic environments shaped by tumor microenvironment suppress the antitumor functions of macrophages (2, 3). Moreover, TAMs promote tumor growth. In fact, a high TAM number coupled with a low T cell number correlate with poor prognosis in cancer patients (4, 5). Furthermore, the expression of the chemokine CCL2 is associated with TAM migration to the tumor site (6) and the M-CSF factor secreted by tumor cells shapes TAM functional activity. Indeed, M-CSF not only attracts TAMs (7) but also induces a switch from a pro-inflammatory M1 into an anti-inflammatory M2 immunophenotype (8). Accordingly, elevated systemic or local levels of M-CSF are associated with poor outcomes (7, 9, 10). Taken together, these findings highlight the major roles that TAMs play in mediating tumor growth and metastasis. This vital interaction between tumor cells and TAMs implicates a key opportunity for cancer treatment intervention.
Considering the above, we recently demonstrated in proprotein convertase PC1/3 knock-out mice (11), as well as in the pulmonary resident NR8383 rat macrophage PC1/3 knock-down (PC1/3-KD) cell line (12), that PC1/3-deficient macrophages produce high levels of cytokines and chemokines through autocrine and paracrine pathways (13). Challenging these cells with endotoxin/LPS, results in a cytokine storm that impacts cell survival of cancer cells (13). In fact, PC1/3 protein regulates cytokines and chemokines secretion in macrophages. Indeed, under LPS stimulation, the TLR4 MyD88-dependent signaling pathway is enhanced in PC1/3-deficient macrophages promoting a stable pro-inflammatory activated phenotype. Secreted factors from these PC1/3-KD macrophages not only attract naïve T helper cells but also inhibit ovarian and breast cancer cell lines viability via innate immune mechanisms (13).
However, achieving an activation of TAM via the LPS/TLR4/Myd88-dependent pathway appears yet unfeasible in cancer patients. On the contrary, the sterile ligand Paclitaxel is also known to activate the TLR4 MyD88-dependent signaling pathway mimicking LPS action (14, 15). It has been widely used for the treatment of breast, ovarian, lung, and colon cancers (16). Paclitaxel is an antineoplastic agent that stabilizes microtubules against depolymerization and blocks cells at the G2/M junction of the cell cycle. Although treatment with Paclitaxel has led to improvement in the duration and quality of life for some cancer patients, most Paclitaxel-treated patients eventually develop drug resistance leading to disease progression (17). In addition to its cytostatic and cytotoxic effects, Paclitaxel has immunomodulatory properties that may partly interfere with its antitumoral effects (18). Paclitaxel stimulates tumor cells to secrete different chemo-attractant factors such as macrophage colony-stimulating factor 1 (M-CSF) that promote immune evasion. Interestingly, our previous results showed that PC1/3 inhibition in macrophages strongly impacted cytoskeleton organization (13). Therefore, because of the key roles of Paclitaxel in microtubule stabilization and tumor resistance, we aimed to study the impact of Paclitaxel on our PC1/3-KD macrophage cell model (Fig. 1A).
Fig. 1.

Experimental strategy. A, Graphical abstract resuming the conducted experiments in this study and the main results for each part. B, Table showing the number of identified proteins in (i) the supernatants of PC1/3 KD and NT macrophages stimulated during 24 h with Paclitaxel (suppl. Data 1), (ii) in the cell extracts after 24 h stimulation with Paclitaxel (suppl. Data 2) and (iii) in the supernatants of the coculture between C6 glioma spheroids and macrophages (supplemental Data S4). The number of differential proteins (p < 0.05) and unique proteins is also indicated.
We report here that such a treatment of PC1/3 KD macrophages drove to the secretion of pro-inflammatory cytokines and chemokines. In addition, proteomics analysis of whole cell extracts indicated that these macrophages display overexpression of proteins mainly involved in cytoskeleton rearrangement. In support of this finding, we found that these cells exhibited a Ca2+ increase after Paclitaxel treatment. This is indicative of a possible depolymerization of microtubules and this may therefore act on the activation of inflammatory pathways in macrophages. In such a way, we found that PC1/3 KD macrophages displayed a repression of the anti-inflammatory pathway STAT3. We also isolated extracellular vesicles from these PC1/3 KD cells and showed that their content inhibited glioma growth. Finally, analysis of the cell supernatant collected from the coculture between glioma cells and PC1/3 KD macrophages led to a more important release of antitumoral factors.
This work will contribute to our understanding on microtubule network dynamics in PC1/3-KD macrophages and Paclitaxel impact on PC1/3-KD cells in term of intracellular signaling, trafficking, and proteins profiles and antitumor activities through EVs production.
EXPERIMENTAL PROCEDURES
Experimental Design and Statistical Rationale
Shotgun proteomics experiments on PC1/3 knockdown (KD) and nontarget (NT) cells treated with Paclitaxel were conducted in biological triplicate. Spheroids studies, viability tests, Western blotting analyses, calcium imaging experiments were also conducted in biological triplicate.
Statistical Analysis
For the proteomics statistical analysis of extracted proteins or secreted media, only proteins presenting as significant by the ANOVA test were used with FDR 5%. Normalization was achieved using a Z-score with a matrix access by rows. Obtained data from Western blotting were reported as mean ± S.E. Mean values among different experimental groups were statistically compared by one-way ANOVA tests using Graph pad PRISM software or by student t test. When we compared NT control cells versus NT treated cells and between the various time points, the tests were performed on a parametric basis because the variance in the group is equal and n > 2. KD group was analyzed in the same way. By contrast, the variance between NT and KD cells is not equal. Therefore, we compared these two groups on a non-parametric basis i.e. test U Mann-Withney.
Values of p < 0.05 were considered statistically significant (*p value of < 0.05, **p value of < 0.01, ***p < 0.001). All experiments were performed as 3 independent biological assays (interassays).
Reagents
The rat alveolar macrophage NR8383 cell line (CRL-2192) was obtained from ATCC (USA). The rat C6 glioma cell line was kindly provided by Prof. Dr. Bernd Kaina (Institute of Toxicology, University Medical Center, Mainz, Germany). Paclitaxel was obtained from Sigma (Lyon, France) (C47H51NO14, MW = 853.91). The level of endotoxins of Paclitaxel solution is less than 0.1EU/ml. Antibodies against phospho-STAT3 and STAT3 were obtained from CellSignaling Technology (Leiden, The Netherlands). Antibodies against ANP32A and Factor IX were obtained from Abcam. Ham's F12K, puromycin, phosphate buffer saline (PBS), fetal bovine serum (FBS) were obtained from Invitrogen Life Technologies (Milan, Italy). LysC/Trypsin was obtained from Promega (Madison, WI). Peroxydase-conjugated secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA). Rat Cytokine Array Panel A was obtained from R&D Systems (Minneapolis, MN). The CellTiter 96 AQueous One Solution Cell Proliferation reagent assay was purchased from Promega (Southampton, UK).
Cell Culture
NR8383 PC1/3 KD and NR8383 NT shRNA cell lines were cultured in Ham's F12K medium supplemented with 15% fetal bovine serum and 12 μg/ml puromycin at 37 °C in a humidified atmosphere (5% CO2). NR8383 PC1/3 knockdown was performed using lentivirus transduction, as described previously (12). C6 cells were cultured in high-glucose Dulbecco's Modified Eagle's Medium (DMEM) and supplemented with 10% heat-inactivated fetal bovine serum, 1% l-glutamine (2 mm) and 1% gentamicin (50 units per ml), all from Sigma-Aldrich. This medium is referred to as complete DMEM (cDMEM). The FBS concentration was reduced to 5% in medium used for the coculture experiments (c-DMEM-5).
Identification of Cytokines and Chemokines Using Rat Cytokine Antibody Arrays
NR8383 KD and NT cells were plated on sterile 6-well plates to reach confluence. The cells were starved overnight with Ham's F12K medium supplemented with 2% FBS and stimulated for 24 h with 20 ng/ml IL-10 in serum-free medium or were left untreated. Then, the medium was replaced, and the cells were stimulated for 24 h with 30 μm Paclitaxel or were left untreated. Cell supernatants were collected, centrifuged at 500 × g, passed through a 0.22 μm filter to remove cells and immediately frozen in liquid nitrogen. The Rat Cytokine Array Panel A from the R&D system was used to probe cytokines in the cell supernatant of stimulated and unstimulated NR8383 cells by following the procedures recommended by the manufacturer. Briefly, the array membranes were first incubated in the blocking buffer for 1 h. In the meantime, cell supernatants were mixed with the Detection Antibody Mixture and incubated for 1 h at room temperature. The volume of cell supernatant used for this experiment was determined according to the number of cells counted after stimulation. Then, after removing the blocking buffer, the sample/antibody mixture was added to array membranes and incubated overnight at 4 °C. After incubation, the membranes were washed 3 times with the wash buffer and then incubated with the Streptavidin-HRP solution for 30 min at room temperature. The membranes were finally washed with wash buffer 3 times, and the bound antibodies were detected by chemoluminescence using the Chemi Reagent Mix. The membranes were quantified by densitometry using ImageJ software. Statistical analysis was carried out using a paired t test.
Total Protein Extractions
NR8383 KD and NT cells were plated on sterile 6-well plates to reach confluence. For Paclitaxel stimulation, the cells were starved overnight with Ham's F12K medium supplemented with 2% FBS. The cells were stimulated with 30 μm Paclitaxel in serum-free medium or left untreated. After stimulation (24 h for FASP analysis, 1 h and 3 h for Western blot analysis), cells were collected, washed once with ice-cold PBS and then lysed with RIPA buffer for total protein extraction (150 mm NaCl, 50 mm Tris, 5 mm EGTA, 2 mm EDTA, 100 mm NaF, 10 mm sodium pyrophosphate, 1% NP40, 1 mm PMSF, and 1× proteases inhibitors). Cells debris were removed by centrifugation (20,000 × g, 10 min, 4 °C), and supernatants were collected, and protein concentrations were measured using a Bio-Rad Protein Assay according to the manufacturer's instructions.
Western Blot Analysis
Total cell NT or PC1/3-KD cells extracts (40 μg) were then analyzed by Western blotting assays. First, proteins were separated by SDS-PAGE electrophoresis and then transferred onto a nitrocellulose membrane. Membranes were blocked for 1 h at room temperature in TBS-Tween 0.1% + milk 5% and incubated overnight at 4 °C with primary antibodies directed against rabbit antiphospho STAT3 (1:2000, from Cell Signaling), rabbit anti-STAT3 (1:1000, from Cell Signaling), rabbit anti-Factor IX (1:500, from Abcam) and rabbit anti-ANP32A (1:500, from Abcam). Horseradish peroxidase-coupled goat anti-mouse and goat anti-rabbit secondaries (Jackson ImmunoResearch) were used at 1:30,000 and 1:20,000 respectively. The proteins were visualized with the enhanced chemiluminescence kit (West Dura from Pierce) according to the manufacturer's instructions. ImageJ software was used to quantify the bands. Experiments were done in triplicate (n = 3).
Filter-aided Sample Preparation (FASP)
Total protein extract (0.1 mg) was used for FASP analysis as described previously. We performed FASP using Microcon devices YM-30 (Millipore, Burlington, MA) before adding trypsin (Promega) for protein digestion (40 μg/ml in 0.05 m NH4HCO3). The samples were incubated overnight at 37 °C. The digests were collected by centrifugation, and the filter device was rinsed with 50 μl of NaCl 0.5 m. Next, 5%TFA was added to the digests, and the peptides were desalted with a Millipore ZipTip device before LC-MS/MS analysis (13). The solution was then dried using the SpeedVac. Dried samples were solubilized in water/0.1% formic acid before LC MS/MS analysis. Experiments were done in triplicate (n = 3).
Proteomics Analysis of the Cell Supernatants
Cell supernatants obtained from macrophages treated or not with Paclitaxel during 24 h or from the coculture of C6 spheroids and macrophages (after 6 days) were centrifuged at 500 × g and passed through a 0.22-μm filter to remove cells and debris. The experiments were performed in biological triplicates. Four hundred microliters of the cell supernatant were collected for each condition. The volume was reduced to 100 μl in a SpeedVac. Cell supernatant digestion was performed as previously described (19). In brief, the cell supernatants were denatured with 2 m urea in 10 mm HEPES, pH 8.0 by sonication on ice. The proteins were reduced with 10 mm DTT for 40 min followed by alkylation with 55 mm iodoacetamide for 40 min in the dark. The iodoacetamide was quenched with 100 mm thiourea. The proteins were digested with 1 μg LysC/Trypsin mixture (Promega) overnight at 37 °C. The digestion was stopped with 0.5% TFA. The peptides were desalted with a Millipore ZipTip device in a final volume of 20 μl of 80% ACN elution solution. The solution was then dried using the SpeedVac. Dried samples were solubilized in water/0.1% formic acid before LC MS/MS analysis. Experiments were done in triplicate (n = 3).
LC MS/MS Analysis
Samples were separated by online reversed-phase chromatography using a Thermo Scientific Proxeon Easy-nLC system equipped with a Proxeon trap column (100 μm ID × 2 cm, Thermo Scientific, Waltham, MA) and a C18 packed-tip column (75 μm ID × 50 cm, Thermo Scientific). Peptides were separated using an increasing amount of acetonitrile (5–35% for 100 min) at a flow rate of 300 nL/min. The LC eluent was electrosprayed directly from the analytical column, and a voltage of 1.7 kV was applied via the liquid junction of the nanospray source. The chromatography system was coupled with a Thermo Scientific Q Exactive mass spectrometer programmed to acquire a data dependent Top 10 method. Survey scans were acquired at a resolution of 70,000 at m/z 400.
Data Analyses
All the MS data were processed with MaxQuant (20) (version 1.5.1.2) using the Andromeda (21) search engine. Proteins were identified by searching MS and MS/MS data against Decoy version of the complete proteome for Rattus norvegicus of the UniProt database (22) (Release June 2014, 33,675 entries) combined with 262 commonly detected contaminants. Trypsin specificity was used for the digestion mode with N-terminal acetylation and methionine oxidation selected as the variable. Carbarmidomethylation of cysteines was set as a fixed modification, and we allowed up to two missed cleavages. For MS spectra, an initial mass accuracy of 6 ppm was selected, and the MS/MS tolerance was set to 20 ppm for HCD data. For identification, the FDR at the peptide spectrum matches (PSMs) and protein level was set to 0.01. Relative, label-free quantification of proteins was performed using the MaxLFQ algorithm (23) integrated into MaxQuant with the default parameters. Analysis of the proteins identified was performed using Perseus software (http://www.perseus-framework.org/) (version 1.5.0.31). The file containing the information from identification was used with hits to the reverse database, and proteins only identified with modified peptides and potential contaminants were removed. Then, the LFQ intensity was logarithmized (log2[x]). Categorical annotation of rows was used to define different groups depending on the following: 1) the cell line (NT or KD), 2) the treatment (Control/Paclitaxel). Multiple-samples tests were performed using an ANOVA test with a FDR of 5% and preserved grouping in randomization. To determine enrichment of categorical annotations (Gene Ontology terms and KEGG pathway), a Fisher's exact test was used, taking in account the results of the ANOVA test for each group. Normalization was achieved using a Z-score with matrix access by rows. Only proteins presenting as significant by the ANOVA tests were used for statistical analysis. A hierarchical clustering was first performed using the Euclidean parameter for distance calculation and an average option for linkage in row and column trees using a maximum of 300 clusters. To quantify fold changes of proteins across samples, we used MaxLFQ. To visualize these fold changes in the context of individual protein abundances in the proteome, we projected them onto the summed peptide intensities normalized by the number of theoretically observable peptides. Functional annotation and characterization of identified proteins were obtained using PANTHER software (version 9.0, http://www.pantherdb.org) and STRING (version 9.1, http://string-db.org). The GeneMANIA Cytoscape plugin (24) was used to generate 2 distinct coexpression networks from cell extract proteomics data: (1) a ≪ Paclitaxel-treated NT cells ≫ network composed of 99,556 recognized interactions, generated from the analysis of a data subset gathering control NT cells and Paclitaxel-treated NT cells and (2) a ≪ Paclitaxel-treated KD cells ≫ network composed of 86,780 recognized interactions, generated from the analysis of a data subset gathering control KD cells and Paclitaxel-treated KD cells. A supervised clustering was then performed to identify the top 100 molecules that coregulated with the following query molecules: Tuba1c/Tuba4a. The list of 100 genes that encoded molecules was identified and was then assessed for gene set enrichment using EnrichR and the GO classification. Finally, subnetworks of genes presenting significant enrichments for specific GO terms were selected and visualized on Cytoscape. For presentation purposes, nodes were assigned equal weights and subnetworks were slightly distorted to avoid node superimposition. Fig. 1B resumed the number of intracellular and secreted proteins in the different experiments we conducted. We also indicated the number of differential proteins between conditions (p < 0.05) and unique proteins.
Cell Proliferation Measured by MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tatrazolium, inner salt] Assay
C6 cells were seeded into 96-well white plates at 70% confluence with NR8383 cell supernatants obtained after 24 h of Paclitaxel (30 μm) stimulation or no stimulation. The assay was conducted for 24, 48, 72 or 96 h. For the time point “96h+medium”, conditioned mediums were removed at 72 h and replaced with fresh ones. CellTiter 96 AQueous One Solution Cell Proliferation reagent (Promega) was added to the wells and incubated at 37 °C for 1 h protected from light. The absorbance was recorded at 490 nm using a 96-well plate reader. The results are expressed as absorbance values. Experiments were performed as four independent experiments.
Spheroid Generation and Embedding in a Collagen Matrix
C6 rat glioma cells were resupended in cDMEM at the final concentration of 12,500 cells in 200 μl. Cells (200 μl per well) were distributed in flat 96-well low attachment surface plates (Corning®). Plates were incubated at standard culture conditions for 96 h. The newly formed C6 cell spheroids were then implanted in the center of each well of a 24-well plate coated with a 2.2 mg/ml collagen mixture (one spheroid per well in 400 μl of collagen mixture per well). The collagen mixture was prepared by mixing 2 ml of PureCol® bovine collagen type I solution (3 mg/ml; Advanced BioMatrix) with 250 μl of 10X minimal essential medium (MEM) (Sigma-Aldrich, Carlsbad, CA) and 500 μl of sodium hydroxide 0.1 m. After cell spheroid embedding, the plate was incubated for 30 min at standard culture conditions to solidify the gels. Thereafter 400 μl of cDMEM was overlaid on the collagen matrix in each well. The complete system was incubated for a total of 6 days.
Coculture of Paclitaxel Pretreated Macrophages and C6 Spheroids
For coculture experiments, 800.000 NT or PC1/3 KD macrophages were seeded in a T25 flask and treated for 24 h with 30 μm Paclitaxel in cDMEM medium with 12 μg/ml puromycin or with the corresponding volume of DMSO (control, untreated macrophages). The cell supernatants were collected (cell supernatants 24 h for proteomics analysis), the cells harvested, washed in PBS, resuspended in MEM 10× and mixed with components of the collagen mixture as described above. The collagen mixture containing macrophages was distributed in 24-well plates (400 μl/100.000 macrophages per well) for embedding of C6 spheroids as described above. Supernatants of all conditions were collected at the end of the coculture, immediately frozen and stored at −80 °C for further proteome analysis (cell supernatant day 6). Experiments were done as four independent experiments (n = 4).
Coculture of Extracellular Vesicles of NR8383 Cell Line with C6 Spheroids
NT and PC1/3 KD macrophages were treated or not with 30 μm Paclitaxel for 24h. Medium was collected and EVs isolated. Differential steps of centrifugation and ultracentrifugation to pellet extracellular vesicles (EVs) were performed. Membranes and debris were discarded from the cleared medium by centrifugation for 30 min at 2000 × g at 4 °C. The supernatant was centrifuged once at 10,000 × g for 30 min, 4 °C and once ultracentrifuged at 100,000 × g (Beckman Optima TLX Ultracentrifuge) for 70 min, 4 °C. The pellet was washed in 5 ml of phosphate buffered saline (PBS) and reultracentrifuged at 100,000 × g for 120 min, 4 °C. The EVs pellet were resuspended in MEM 10X, filtrated with 0.20 μm filter and analyzed with a Nanosight NS300 instrument (Malvern, UK) to assess the quality of EVs purification and to determine the amount of exosomes in each condition. An equal amount of EVs were added to the collagen mixture as described above. The collagen mixture containing EVs was distributed in 24-well plates for embedding of C6 spheroids as described above (n = 3).
Quantification of Spheroid Size and Invaded Area
After the spheroids were embedded, cell invasion out of the spheroid was monitored by digital photography using a Leica DM IL LED Fluo inverted light microscope (Leica DFC450C camera) at room temperature, with the Leica Application Suite (LAS V4.4). Images were acquired every day (day 0 = time of embedding in collagen; picture taken immediately after embedding) using a 4×/0.10 objective. Image processing and quantification of spheroids and of invasion areas was performed using an in-house software. This in-house software considers cell density and not the (observer-dependent) limits of cell migration in the collagen matrix. The implemented algorithm uses local fluctuations of the image intensity for an automated estimation of the invasion magnitude. It is robust enough to handle micrographs of different generation methods and various qualities without the concept of an invasive front of the spheroids (25). Invasion and spheroid areas are normalized for each day to the invasion and spheroid areas measured at day 0 and expressed in protocol defined units (PDU). These normalized data are reported as relative size to day zero. Relative size of day 0 thus equals 1.
Calcium Imaging
NT and PC1/3-KD cells were plated onto glass coverslips and were loaded with 4 μm Fura-2 AM at room temperature for 45 min in the growth medium. Recordings were performed in HBSS containing 140 mm NaCl, 5 mm KCl, 2 mm MgCl2, 0.3 mm Na2HPO3, 0.4 mm KH2PO4, 4 mm NaHCO3, 5 mm glucose and 10 mm HEPES adjusted to pH 7.4 with NaOH. The cells were then washed three times in HBSS. Paclitaxel (30 μm) was added at the time of analysis. The fluorescent intensity of Fura-2 in each cell was monitored and recorded at 340 nm and 380 nm. To represent the variation in the intracellular free calcium concentration, the fluorescence intensity ratio represented by F340/F380 was used as an indicator of changes in cytosolic Ca2+ concentrations. Experiments were done in triplicate (n = 3).
RESULTS
Paclitaxel Induces Secretion of Proinflammatory Cytokines by PC1/3 KD Cells
To deepen our understanding on the intracellular impact of Paclitaxel treatment on NT and PC1/3 KD macrophages, we performed a complete proteome analyses of cell supernatants and intracellular proteins.
First, we analyzed the protein over or under-secreted by macrophages stimulated during 24 h with 30 μm Paclitaxel (Fig. 2A, supplemental Data S1). Paclitaxel treatment induced a modulation of the amount of proteins released by both cells compared with control condition (Fig. 2A). Cluster 1 corresponded to proteins over-expressed by untreated macrophages, irrespective of PC1/3 expression. In that case, 9 proteins were identified. Most of them were related to immune response and pro-tumoral activities: Gamma-interferon-inducible lysosomal thiol reductase (Ifi30), granulins isoform a precursor (Grn), Lysozyme C-1 precusor (Lyz2), Macrophage metalloelastase (MMP12), Beta-2-microglobulin (B2m), Interleukin-1 receptor antagonist protein (Il1rn), proteogycan 4 precursor (Pgr4), cathepsin D (Ctsd). We also detected the presence of Amyloid beta A4 protein N-APP Soluble, linked to tumor infiltration of macrophages. Fifty-nine proteins were identified in cluster 2 corresponding the proteins over-secreted by macrophages stimulated during 24h with Paclitaxel. The STRING analysis indicated a central huge network focused on ribonucleoproteins and translation (supplemental Fig. 1). It is known that patients treated with Paclitaxel can eventually develop drug resistance leading to disease progression (17). Of note, under Paclitaxel treatment of macrophages we found proteins that are known to protect the tumor: Rho GDP-dissociation inhibitor 1 (Arhgdia), Stress-induced-phosphoprotein 1 (Stip1), the Heat shock proteins (HSP 90-alpha and HSP-90 beta).
Fig. 2.

Paclitaxel induces secretion of pro-inflammatory cytokines by PC1/3 KD cells. NT and KD macrophages were treated with 30 μm Paclitaxel for 24 h and supernatants were collected. Proteomics analyses of factors secreted by NT and KD cells after 24 h treatment with 30 μm Paclitaxel or DMSO as control. A, MaxQuant and Perseus software were used for the statistical analysis, and a heat map was generated to show proteins that were significantly different between macrophages stimulated or not with Paclitaxel in the supernatants. B, String protein analyses of the unique proteins identified in PC1/3-KD (i) and NT cell supernatants (ii). C, Effect of PC1/3 downregulation on cytokine secretion under Paclitaxel and inhibitory conditions. The rat cytokine array assay was performed with NR8383 cell supernatants (NT/KD). The cells were untreated (control) or treated with IL-10 for 24 h without stimulation (IL-10) or with Paclitaxel for 24 h (IL-10+Paclitaxel). Blue shows NT cell supernatant, and orange indicates KD cell supernatant. The bar diagrams represent the ratio of the spot mean pixel densities/reference point pixel densities. Significant differences were analyzed using Student's t test. *p < 0.05.
In a next step, we focused on unique proteins secreted by NT and PC1/3 KD cells (Fig. 2B). We found that 25 unique proteins were detected in the supernatant of PC1/3 KD cells whereas 65 proteins were identified for NT cells (Table I). For the unique proteins identified in the supernatant of PC1/3 KD cells after 24h of Paclitaxel activation, STRING analysis indicated a single network involving ribosomal and ubiquitin proteins (RPS14, RPS16, RPS35, Eif4e, Pppr21a, Dynlrb1) (Fig. 2Ba). Again, several tumor growth factors were detected: apoptotic inhibitory 5 (Api5), angiopoietin-related protein 3 precursor (ANGPLT3), Dystroglycan precursor (Dag1).
Table I. Unique proteins from the cell supernatants of NT and PC1/3 KD macrophages obtained after 24h treatment with Paclitaxel.
| Unique identified proteins | |||
|---|---|---|---|
| NT cell supernatant - Paclitaxel | KD cell supernatant - Paclitaxel | ||
| H2afz | Rbbp4 | Matr3 | Gart |
| Taf15 | Eif3c | Eif4e | Tardbp |
| Sec24c | Cd44 | Glrx3 | Gnai2 |
| Rab11b | Eif4g1 | Api5 | Rps14 |
| Nudc | Txndc5 | Otub1 | Psma5 |
| Copg1 | Ipo5 | Pmm2 | Rps16 |
| Cth | Srp72 | Rpl35 | Dynlrb1 |
| Gk | Hmha1 | Prkcd | Fau |
| Atp6v1h | Hnrnpul2 | Fgfr1 | Cbx3 |
| Tars | Col4a3bp | Dag1 | Ppp2r1a |
| Rpl18 | Rpl38 | Angptl3 | Gsn |
| Ola1 | Fkbp15 | Igf2r | Rtcb |
| Tpmt | Hnrnpl | Ehd4 | |
| Eif3i | Nsf | ||
| Atp6v1g1 | Lima1 | ||
| Ola1 | Hnrnpm | ||
| Coro1b | Eno2 | ||
| Apeh | Arg1 | ||
| Plin3 | Acaca | ||
| Trim28 | Asl | ||
| Gpd1 | Comt | ||
| Ptma | Grb2 | ||
| Rps27l | Lars | ||
| Rps9 | Farsb | ||
| Pfkl | Rab21 | ||
| Psmc1 | Tmod3 | ||
| Nploc4 | Eif3b | ||
| Atox1 | Capza1 | ||
| Vars | Snrnp70 | ||
| Aimp2 | S100a10 | ||
| Upp1 | Stam | ||
| Pygb | Tsta3 | ||
| Rab1b | |||
For NT cells, more unique proteins were identified in their supernatant after 24h of Paclitaxel treatment (Table I). The STRING analysis (Fig. 2Bb) revealed at least 3 networks i.e. one related to vesicle transport centered on vesicle-fusing ATPase (NSF) protein, the second one is related to proteins involved in translation, the third one is based on ribonucleoproteins. Factors involved in tumor invasion were also detected: CD44, Nuclear migration protein nudC (NudC), tropmodulin-3 (Tmod3), arginase 1 (Arg1), Protein S100-A10 (S100-10).
From these proteomics analysis of macrophages' supernatants, we could establish that immunosuppressive and pro-tumoral factors were released by both cell types, even after Paclitaxel treatment. This observation may explained the resistance to this chemotherapeutic drug well described in the literature (18, 26), partly because of macrophages and the factors they released as we demonstrated here. However, the factors identified with this experiment represent only a part of the whole supernatant of macrophages. Cytokines and chemokines are also important in the tumor environment because they may recruit cytotoxic immune cells. Therefore, to assess the inflammatory phenotypes of NT and PC1/3 KD macrophages, a cytokine array experiment was conducted (Fig. 2C). This showed that PC1/3 KD cells exhibited a more pronounced inflammatory state as depicted by the release of more pro-inflammatory cytokines such as TNF-α, IL-6, CCL5, CXCL1, CXCL2 and CXCL10.
Cells were then stimulated with 30 μm Paclitaxel for 24h (Fig. 2C). This revealed that under Paclitaxel treatment, the nature of the cytokines and chemokines was modified. Indeed, TNF-α, IL-6 and CXCL10 specifically released by untreated PC1/3 KD cells were no more secreted after such a treatment. On the contrary, Paclitaxel induced a more important release of CCL3 in PC1/3 KD cells compared with its control and Paclitaxel treated NT cells. Of note, secretion of CXCL3 appeared only after stimulation of PC1/3-KD cells. IL-1β was also detected after Paclitaxel treatment of both cell lines but with higher levels in PC1/3 KD cells. Besides, CXCL1 release decreased in both cells sub-types but the level remained higher in PC1/3 KD cells. In addition, IL-1α dropped in NT cells whereas stayed unaffected in PC1/3 KD cells. Of note, the level of the anti-inflammatory cytokine IL-1ra increased strongly in NT cells whereas it was unchanged in PC1/3 KD cells. Altogether, this demonstrated that even if the nature of the cytokines and chemokines detected was modified in both cell lines after Paclitaxel treatment, PC1/3 KD cells continue to exhibit a higher pro-inflammatory state than NT cells. In brief, Paclitaxel induced a profile of secretion characterized by GRO (α, β, γ), also known as chemokines CXCL1, CXCL2, CXCL3, and interleukin 1 (α, β), which are pro-inflammatory and attractive factors for Polymorphonuclear leukocyte (PMN) cells and which are over-produced in PC1/3-KD cells. In contrast, the inhibitory and anti-inflammatory factor Il-1ra was only over-produced in NT cells under Paclitaxel stimulation.
As a next step, to mimic the anti-inflammatory tumor environment, macrophages were cultured in an inhibitory medium containing the anti-inflammatory cytokine IL-10 (Fig. 2C). Again, a treatment with IL-10 led to a modification of the cytokines and chemokines profiles. In such a way, we observed that IL-6 specifically detected in the supernatant of untreated PC1/3 KD cells were no more detected after IL-10 treatment. TNF-α level decreased in these cells but remained secreted. The levels of CCL5 and CXCL10 also diminished in PC1/3 KD cells whereas increased in NT cells to reach a similar level. Moreover, IL-10 treatment decreased the quantities of CXCL2 released by NT cells whereas no change was observed in PC1/3 KD cells. More generally, the levels of most of these secreted factors were higher in PC1/3 KD macrophages compared with NT macrophages even after a treatment with the inhibitory cytokine IL-10. When we combined IL-10 and Paclitaxel treatment, we did not observe any difference for both cell lines compared with Paclitaxel stimulation alone (Fig. 2C).
We then compared the effect of the treatment with IL-10 versus the combined stimulation with IL-10 and Paclitaxel (Fig. 2C). We again observed that CXCL10 and TNF-α were no more detected in PC1/3 KD cells like for Paclitaxel treatment alone. On the contrary, the combination of IL-10 and Paclitaxel triggered the increase of CCL3 in PC1/3 KD cells. Concerning IL-1ra, a higher level was observed in both cell lines but still higher in NT cells compared with PC1/3 KD cells. Then, CCL5 was more secreted only by PC1/3 KD cells. In conclusion, the inhibitory effect observed with the treatment of macrophages with IL-10 was abolished when combined with Paclitaxel.
IL-10 is highly secreted in the tumor microenvironment and is responsible for the immunosuppression observed in many tumors like glioma (27, 28). Here, we demonstrated that PC1/3 KD macrophages were more resistant to the anti-inflammatory effect of IL-10 and that Paclitaxel treatment is not really affected by this cytokine.
Altogether, these analyses revealed that under Paclitaxel treatment, NT and PC1/3 KD macrophages secreted pro-tumoral factor. Conversely and positively, PC1/3 KD macrophages released more pro-inflammatory cytokines and chemokines and were more resistant to an anti-inflammatory environment. Thus, at this step, it was still unclear if the use of Paclitaxel treated PC1/3 KD macrophages could be a promising antitumoral therapy. Nevertheless, we must consider that macrophages effects against tumor cells do not only rely on secreted factors. Indeed, their immune response may also involve the activities of the cells themselves such as phagocytosis. Therefore, we performed proteomics studies of the intracellular proteins.
Proteins Overexpressed by PC1/3 KD Cells Are Related to Macrophages Immune Functions
For these experiments, we analyzed the intracellular proteins of NT and PC1/3 KD macrophages. We used the ANOVA test to perform a clustering of samples grouped on the basis of PC1/3 deficiency (NT versus PC1/3 KD cells) and Paclitaxel treatment (treated versus untreated cells) (Fig. 3A, supplemental Data S2 and S3). Eight specific clusters were identified. Cluster 1 corresponds to overexpressed proteins under Paclitaxel treatment in PC1/3 KD cells and cluster 3 to overexpressed proteins under Paclitaxel in both cell lines. Cluster 2 is specific for PC1/3 KD cells regardless the treatment. Cluster 6 represents proteins overexpressed in resting PC1/3 KD cells. We investigated using systems biology analysis, the interactome and network of the overexpressed proteins in PC1/3 KD cells after Paclitaxel treatment utilizing the GeoSoftware corresponding to clusters 1 and 2 (Fig. 3B). Differential pathways were generated using the “direct interaction” algorithm to map the relationships of the identified proteins. We found that among the 50 altered proteins, 35 proteins had direct regulatory relationships, including integrins (i.e. integrin beta 2 precursor, Protein Itgax, integrin alpha-5), cytoskeleton proteins (i.e. twinfilin, filamin-A, calponin 2, emerin, moesin, Alpha-actinin-4, gelsolin, lymphocyte specific 1, coronin 1c), proteasomes (i.e. Proteasome subunit beta type-10; Proteasome activator complex subunit 2, WD repeat-containing protein) and ionic channels (the voltage-gated potassium channel subunit beta-2 or the anoctamin-6) (Fig. 3B). Among the altered proteins identified, some of them have functions directly linked to macrophages activities like Anoctamin-6 (Ano6) that contributes to both phagocytosis (29) and macrophage migration. Some specific proteins involved in the immune response were also overexpressed in PC1/3 KD macrophages, i.e. galectins (1 and 3) and CD36. Galectins have been identified as modulators of many monocyte/macrophage functions (30).
Fig. 3.

PC1/3 KD macrophages express intracellular proteins related to macrophages immune functions. A, NR8383 cells were stimulated with Paclitaxel (30 μm) or not (control) and lysed before FASP and LC-MS/MS analysis. MaxQuant and Perseus software were used for the statistical analysis, and a heat map was generated to show proteins that were significantly different between NT and PC1/3-KD NR8383 macrophages stimulated or not with Paclitaxel in the cell extracts. Eight clusters are highlighted. B, Global pathway analysis of the overexpressed proteins specifically identified in PC1/3-KD cells. The relationship between the overexpressed proteins with PCSk1/3 (pink) is also showed. C, Co-expression network analyses. i) Co-expression of Tuba1c/Tuba4a with mitochondria-related molecules in Paclitaxel-treated NT versus Paclitaxel-treated KD cells. The 100 genes which encoded molecules were the most tightly coexpressed with Tuba4a/Tuba1c were identified in Paclitaxel-treated NT versus Paclitaxel-treated KD cells. Shown are subnetworks of genes annotated by the GO term “mitochondrion” (adjusted p value for enrichment significance = 0.11e-3 in Paclitaxel-treated NT cells and 0.10e-5 in Paclitaxel-treated KD cells). ii) Co-expression of tubulins with immune-related molecules in Paclitaxel-treated NT versus Paclitaxel-treated KD cells. The 100 genes which encoded molecules were the most tightly coexpressed with Tuba4a/Tuba1c were identified in Paclitaxel-treated NT versus Paclitaxel-treated KD cells. Shown are subnetworks of immune-related genes.
To complement these unsupervised clustering bioinformatics-based analyses, we performed coexpression network enrichment study to identify molecules that- in Paclitaxel-treated NT or PC1/3 KD cells- were co-upregulated with tubulins, the primary molecular targets of Paclitaxel (31, 32) (Figs. 3Ci and 3Cii). We chose Tuba1c and Tuba4a proteins as query molecules because these 2 tubulins have shown the highest fold change among the 6 identified tubulins that were overexpressed in Paclitaxel-treated cells (i.e. Tubb4b, Tubb2b, Tubb6, Tuba4a, Tuba1c, and Tuba1b). We found that in both NT and PC1/3 KD cells, the Paclitaxel-induced upregulation of Tuba1ac and Tuba4a was accompanied by the co-upregulation of a highly significant number of mitochondria-related molecules (Fig. 3Ci). This result was in line with previous studies that demonstrated that Paclitaxel targets mitochondria (33), possibly as a direct consequence of microtubule network alterations (34). Interestingly, we also observed that Tuba1ac/Tuba4a coexpression networks comprised immune-related molecules that, for some, were distinct when comparing Paclitaxel-treated NT cells versus Paclitaxel-treated PC1/3 KD cells. Anti-inflammatory molecules such as IL1rn and Arg1 were co-upregulated with Tuba1ac/Tuba4a in Paclitaxel-treated NT cells, whereas pro-inflammatory molecules Aif1, B2m, CD44, Sod1 and Calr were co-upregulated with Tuba1ac/Tuba4a in Paclitaxel-treated PC1/3 KD (Fig. 3Cii). This reveals that Paclitaxel-treated KD cells exhibit a stronger proinflammatory state than Paclitaxel-treated NT cells. This was validated by the pro-inflammatory cytokines and chemokines profile observed in Fig. 2C.
Altogether, these proteomics studies showed that a high number of proteins involved in immune response and cytoskeleton were modulated in PC1/3 KD macrophages at basal state and even more after Paclitaxel treatment. Among these cytoskeleton proteins, several were related to Ca2+ signaling. Therefore, our hypothesis was that Ca2+ signaling pathway was modulated in PC1/3 KD macrophages, leading to cytoskeleton rearrangement.
Paclitaxel Acts as a Signalosome by Stimulating [Ca2+]c
We performed calcium imaging experiments using a fura-2AM calcium probe to evaluate the cytosolic calcium concentration ([Ca2+]c) after Paclitaxel treatment on NT and PC1/3-KD macrophages (Fig. 4). We demonstrated that Paclitaxel (30 μm) rapidly induced a pronounced elevation of [Ca2+]c in PC1/3 KD cells compared with NT cells (Fig. 4A). Quantification of these experiments is given in Fig. 4B. We also observed that other features of calcium homeostasis were affected in KD NR8383 cells. Indeed, Figs. 4B and 4C show that the resting [Ca2+]c levels were significantly increased in PC1/3-KD cells compared with NT cells in accordance with our previous publication (13). This indicates that Paclitaxel induced a higher increase of [Ca2+]c in PC1/3 KD cells compared with NT cells, which can act as a depolymerization agent for microtubules (35) and therefore may act on the activation of inflammatory pathways in macrophages. Interestingly, STAT3 is known to mediate an anti-inflammatory signaling through IL-10 activation (36) and tubulins association (37). Several studies have shown the effect of microtubule-targeted chemotherapeutic drug, like Paclitaxel, on the inhibition of STAT3 signaling (37). Paclitaxel-treated PC1/3 KD macrophages exhibits a more pronounced pro-inflammatory state and a cytoskeleton modification partly linked to an increase of [Ca2+]c. We, thus, thought that STAT3 activation may be impaired in these cells.
Fig. 4.
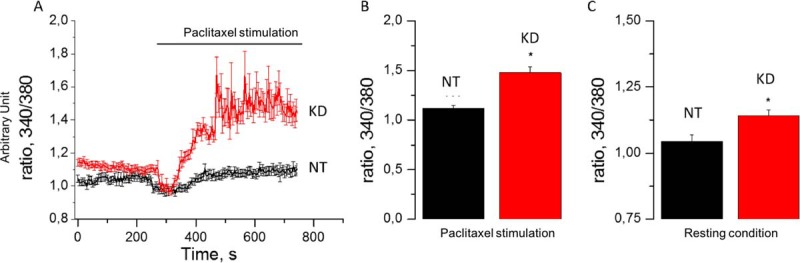
Paclitaxel stimulates [Ca2+]c production in PC1/3 KD macrophages. A, [Ca2+]c rise in NT and KD NR8383 cells in response to Paclitaxel (horizontal bar). B, Quantification of Paclitaxel mediated [Ca2+]c increase for NT and KD NR8383 cells. C, Quantification of resting [Ca2+]c for NT and KD NR8383 cells in basal condition.
STAT3 Activation is Decreased in Paclitaxel-treated KD Cells
To assess signal transducer and activator of transcription 3 (STAT3) activation, a time course of the signal transducer and activator of transcription 3 (STAT3) phosphorylation was studied (Fig. 5A and 5B). The level of the ratio of phosphorylated/total STAT3 was lower in PC1/3 KD than in NT cells at any time point and, in any condition, (Fig. 5B). Following Paclitaxel treatment, it dropped after 1 h in PC1/3-KD cells and went back to the control level at 3 h. IL-10 treatment, which is known to induce STAT3 phosphorylation, induced a similar kinetics than the one observed by Paclitaxel. Taken together, these results clearly indicate that in PC1/3 KD cells, Paclitaxel leads to the decrease of STAT3 activation as expected. Since STAT3 signaling is anti-inflammatory, this is in accordance with our proteomics and cytokine array analysis displaying the higher pro-inflammatory state of PC1/3 KD cells (Fig. 2C). As a result, STAT3 inhibition leads to a stronger activation of PC1/3 KD macrophages as depicted by the secretion of proinflammatory factors. To assess the potential of Paclitaxel-treated PC1/3 KD cells as a good antiglioma therapy, we evaluated the toxicity of their supernatants on glioma cells.
Fig. 5.

Paclitaxel inhibits STAT3 signalization in PC1/3 KD macrophages. A, Western blot analysis of phospho STAT3 and total STAT3 in NT or KD PC1/3 NR8383 macrophages after Paclitaxel stimulation (30 μm), IL10 stimulation (20 ng/ml) or not at 1 h and 3 h. B, Results are depicted through graphic representations of the quantification of phospho STAT3. The data are represented as the ratio of phosphorylated STAT3 on total STAT3 in samples for each conditions. * Significant differences between NT cells and PC1/3-KD cells (p ≤ 0.05) by t test. Experiments were performed in triplicates.
PC1/3 KD Macrophages Cell Supernatants are Cytotoxic for Glioma Cells
Tumor viability assay on the rat C6 glioma cell line was performed with the supernatant of macrophages treated with 30 μm of Paclitaxel for 24 h (Fig. 6). To test the direct effect of Paclitaxel, C6 cells were also directly cultured with medium containing or not Paclitaxel (conditions F12 Control and F12 Paclitaxel). Viability tests were performed for 24 h, 48 h, 72 h and 96 h. The time point 96 h+medium means that the medium was renewed at 72 h and the viability of cancer cells was registered 24 h later. These experiments revealed that Paclitaxel itself significantly decreased the viability of C6 glioma cells at 72 h, 96 h and 96 h+medium (Figs. 6A and 6B). On the contrary, the effects of supernatant from NT or PC1/3 KD cells treated with Paclitaxel on C6 glioma cells viability were registered as soon as 24 h and 48 h (Figs. 6A and 6B). Even though no specific differences were observed between NT cells supernatants and PC1/3 KD cells supernatants on the viability of C6 glioma cells, some interesting observations can be made. In fact, significant differences were observed between the supernatants from untreated macrophages and supernatants from Paclitaxel treated macrophages. Indeed, for NT cells these differences were observed at 48 h, 72 h, 96 h and 96 h + medium (Fig. 6A). On the contrary, for cell supernatants of PC1/3 KD cells, these differences were observed as soon as 24 h post-treatment and were observable at each time point of the assay (Fig. 6A). Compared with the situation depicted in NT cells, the supernatants from Paclitaxel treated PC1/3 KD cells not only exert a quicker antitumoral activity but also a more robust effect at 48 h and 72 h as revealed by higher significance levels (Fig. 6B). During the assay, the highest impact on C6 viability (55.08%) was also observed at 96 h, with supernatant from PC1/3 KD cells stimulated with Paclitaxel. In conclusion, these viability assays showed that in addition to the direct effect of Paclitaxel remaining in the medium and compared with NT cells, Paclitaxel treated PC1/3 KD macrophages secreted additional antitumoral factors active against glioma cells. We then decided to further explore the effect of Paclitaxel activation of the macrophages using direct cocultures of pretreated macrophages and tumor cells.
Fig. 6.

Supernatants of PC1/3 KD macrophages are cytotoxic for glioma cells. The cell viability of C6 rat glioma was determined by the CellTiter Aqueous One Solution Cell Proliferation reagent. The cells were incubated with NR8383 cell supernatants obtained at 24 h with or without Paclitaxel treatment. The assays were conducted for 24 h, 48 h, 72 h and 96 h. For “96h + medium” condition, medium was replaced with fresh medium after 72h of culture. Four experiments were performed independently. A, The results obtained are depicted through a box plot figure. Significant differences were identified using Tukey's multiple comparisons test with ****p < 0.0001; ***p < 0.001; **p < 0.01 and *p < 0.05. B, Raw p values. NS, non-significant.
Paclitaxel Triggers the Secretion of Antiglioma Factors by PC1/3-KD Cells
As a model for a direct coculture of macrophages and glioma cells, we chose to work with a 3D culture system. Spheroids generated with C6 glioma cells were cocultured for 6 days in a collagen matrix containing untreated or Paclitaxel-treated macrophages (NT or PC1/3-KD cells). The growth of the spheroids and the invasion of the matrix by cells migrating out the initial core were monitored over 6 days. As shown in Fig. 7Aa, neither the NT nor the KD cells present in the collagen impaired the growth and invasion of the spheroids, an observation in line with the reported tumor-supportive effects of macrophages. Addition of low concentrations of Paclitaxel (20 nm, 40 nm, 80 nm) in the culture of spheroids in absence of macrophages induced a decrease of spheroid growth which was concentration-dependent (Fig. 7Ab lower panel). This inhibitory effect was much less pronounced in the presence of macrophages in the collagen matrix and was Paclitaxel concentration-dependent as well (Fig. 7Ab, upper panels). Macrophages thus appeared to exert a protective effect to cancer cells against these low concentrations of Paclitaxel, as already highlighted in the literature (38). Addition however of 30 μm of Paclitaxel (i.e. the concentration used to induce the proinflammatory supernatant) directly to the cocultures led to a complete growth and invasion arrest of the spheroids, irrespective of the presence or absence of the macrophages (data not shown). Thus, with this system we could not distinguish the effect of Paclitaxel alone from the effects of Paclitaxel stimulation on macrophages. By contrast, when the macrophages (NT or PC1/3 KD cells) were pre-activated by 30 μm Paclitaxel before their embedding in collagen, thus avoiding the presence of Paclitaxel during the coculture, tumor growth and invasion were inhibited (Fig. 7Ba). Quantification of the growth and invasion rate indicated that pre-activated NT and KD cells exerted a similar inhibitory effect (Fig. 7Bb).
Fig. 7.

Paclitaxel triggers the secretion of antiglioma factors by PC1/3-KD cells cocultured with C6 glioma spheroids. A, Antitumor properties of NR8383 cell supernatants on glioma cells in a 3D coculture. a) Invasion of rat glioma cells (C6) spheroids in the absence (No Mφ) and in the presence of NR8383 non-target (NT) or PC1/3 knockdown (KD) macrophages. Representative images of the invasion of C6 spheroids in the collagen matrix at day 0, day 3 and day 6. All images were acquired with an inverted light microscope at 4× magnification. Scale bar: 500 μm. b) Invasion of rat glioma cells (C6) spheroids in the absence (No Mφ) and in the presence of NR8383 non target (NT) or PC1/3 knockdown (KD) macrophages treated or not with increasing concentrations of Paclitaxel (20 nm, 40 nm, 80 nm). Representative images of the invasion of C6 spheroids in the collagen matrix at day 0 and day 6. All images were acquired with an inverted light microscope at 4× magnification. Scale bar: 500 μm. B, Invasion of rat glioma cells (C6) spheroids in the presence of NR8383 NT or PC1/3 KD macrophages pretreated or not with 30 μm Paclitaxel for 24h. a) Representative images of the invasion of C6 spheroids in the collagen matrix at day 0, day 2, day 4 and day 6. b) Graph representing the quantification of C6 spheroids invasion into the collagen. C, Analysis of the supernatants after 6 days of coculture between macrophages and C6 spheroids. a) At day 6 after invasion, the cell supernatants were harvested and analyzed by mass spectrometry (Time 6 days). Venn Diagram of the all coculture conditions used (Paclitaxel, control DMSO) for PC1/3 KD and NT cells is presented. Western blots detecting antitumoral proteins (F9, Anp32a) in the supernatant of KD cells treated with Paclitaxel are also presented. b) A heat map was generated to show proteins that significantly differed between the supernatants of NT and PC1/3-KD NR8383 macrophages after 6 days of coculture.
We also analyzed the conditioned medium obtained after 6 days of coculture between C6 spheroids and Paclitaxel pre-activated NT or PC1/3-KD cells to identify the factors specifically or more secreted. The results showed differences between NT and PC1/3 KD cells (supplemental Data S4). Indeed, 10 unique proteins were identified in the supernatant of C6 spheroids cultured with Paclitaxel preactivated NT macrophages (Figs. 7Ca, Table II). Markers for tumor invasion were observed including the Complement component 1 Q subcomponent-binding protein (C1qbp), pro low-density lipoprotein receptor-related protein 1 precursor (LRP1 LDL), Ezrin (Ezr), sorcin (Sri) and GrpE protein homolog 1 (Grpel1) and Nucleoplasmin. In addition, after coculture, 2 proteins were over-represented in the supernatant of Paclitaxel-pretreated NT cells i.e. Mortalin (HSPA9) and SH3 domain-binding glutamic acid-rich-like protein 3 (Sh3bgrl) (Fig. 7Cb). These proteins are known to support cancer progression (39, 40) (Fig. 7Cb).
Table II. Unique proteins from the cell supernatant of NT and PC1/3 KD macrophages in co-culture with C6 spheroids obtained after 6 days of invasion into the collagen matrix (Day 6). Macrophages were preactivated with Paclitaxel or not during 24 h and then embedded into collagen with C6 spheroids. The cell supernatants from the co-culture were collected after 6 days.
| Unique identified proteins | |||
|---|---|---|---|
| NT cells Paclitaxel Time 6 days (Coculture) | NT cells Control Time 6 days (Coculture) | KD cells Paclitaxel Time 6 days (Coculture) | KD cells Control Time 6 days (Coculture) |
| Rbmxrtl | Stmn1 | Myl6 | Thbs4 |
| Grpel1 | C5 | Rac1 | Tgfbi |
| Ezr | Ak2 | Anp32a | Hba1 |
| Sri | Prdx2 | Nqo1 | Plod1 |
| Lrp1 | Plod2 | F9 | Inhba |
| C1qbp | Psma7 | Dpysl2 | |
| Npm1 | Alyref | Cand1 | |
| Fh | Atp5b | Wdr1 | |
| H1f0 | Fkbp3 | Eef1g | |
| Eif2s2 | Calu | Dstn | |
| Igf2r | |||
| Dbi | |||
| Rpl30 | |||
| Rpl10a | |||
| Pgm1 | |||
| Arhgdib | |||
| Fus | |||
| Il1rl1 | |||
| Etfb | |||
| Capg | |||
| Gm2a | |||
| Pabpc1 | |||
| Atox1 | |||
After 6 days of coculture between PC1/3 KD macrophages and C6 spheroids, 5 unique proteins were identified in the cell supernatant (Figs. 7Ca, Table II). Among them, a balance of factors produced by tumor cells was also observed, as indicated by the presence of Rac1 and Myl6. Rac1 is described as a major player in glioma invasion and progression (41) and myosin light polypeptide 6 (Myl6) is found in glioblastoma exosomes (42). However, the three other proteins were related to tumor suppression: (1) NAD(P)H dehydrogenase quinone 1 (Nqo1) known to inhibit the degradation of the tumor suppressor gene p53 and involved in the metabolism of Vitamin K (inhibitor of glioma cell growth) (43), (2) acidic leucine-rich nuclear phosphoprotein 32 family member A (ANP32A, pp32) present in macrophages exosomes during inflammation (44) and known to inhibit pancreatic cancer cells survival (45), and (3) Coagulation factor IX (F9) known to regulate carcinoma migration (46).
Even if the effect of NT and PC1/3 KD cells on C6 glioma spheroids growth was similar (Fig. 7Ba), these results suggested that in presence of PC1/3 KD cells, a resistance to Paclitaxel treatment could be less developed. As mentioned above, some of the proteins detected are found in exosomes. Previously, we demonstrated that formation of multivesicular bodies was strongly increased in PC1/3 KD cells (13, 47). Multivesicular bodies are a source of extracellular vesicles (EVs) (48). Therefore, we tested the effects of exosomes purified from the cell supernatants of untreated or Paclitaxel treated NT and PC1/3 KD cells on C6 spheroids growth and invasion.
Extracellular Vesicles Released by PC1/3 KD Cells after Paclitaxel Treatment Repress More Efficiently the Growth and Invasion of C6 Spheroids
EVs were isolated from the cell supernatants of NT and PC1/3 KD cells treated or not with 30 μm Paclitaxel for 24h, quantified with Nanosight NS300 and added in equal amount in the collagen matrix before embedding of the C6 spheroids. Spheroid's growth and cell invasion in the matrix were monitored over 3 days (Fig. 8a). After 24 h, addition of EVs, irrespective of their origin, had no effect on growth and invasion of C6 spheroids. It did exert however a cell- and treatment-dependent inhibitory effect at 48 h. EVs from control NT cells were slightly, though not significantly, more inhibitory than EVs from control KD cells. EVs from Paclitaxel-treated KD cells however showed a stronger inhibitory effect than EVs derived from Paclitaxel-treated NT cells. At 72 h, EVs from both cell lines, irrespective of their treatment, similarly and strongly inhibited growth and invasion of C6 spheroids. Expression of the data as fold change of growth/invasion after Paclitaxel treatment does support these effects and their cell- and treatment-specificity (Fig. 8b).
Fig. 8.

EVs from PC1/3 KD macrophages supernatants repress more efficiently the growth and invasion of C6 spheroids. a) NT and KD macrophages were treated with 30 μm Paclitaxel for 24h and secreted EVs were isolated by ultracentrifugation. An equal amount of EVs for each condition were diluted in the collagen matrix prior embedding with C6 glioma spheroids. Images of the invasion of C6 spheroids in the collagen matrix were taken every 24 h during 3 days. All images were acquired with an inverted light microscope at 5× magnification. Images were quantified with the in-house software as described in Material and Methods. b) Graphic representation showing ratio of C6 area invasion of Paclitaxel condition over control condition at each time point. * Significant differences between NT cells and PC1/3-KD cells (p ≤ 0.05) by t test.
DISCUSSION
Since the last decade, many studies have demonstrated that the density of TAMs is associated with a poor prognosis, suggesting macrophages as a target for clinical therapy (49–55). Macrophages can be activated through cytokines or via secondary signals supplied by either antibodies or LPS/endotoxin/TLR stimulants. Thus, new therapeutic strategies have been elaborated to counteract tumor outgrowth by either inhibiting macrophages infiltration or by promoting a macrophages pro-inflammatory phenotype (49). Strategies targeting TLR4 pathway have shown some success in antitumoral activity (56, 57). Paclitaxel, an antitumor drug, is also known to activate the TLR4 pathway and therefore could be a potential strategy to reactivate macrophages within tumors. This drug is currently used for the treatment of breast, ovarian, lung, and colon cancers (16). More recently different strategies based on liposomes or nanoparticles with Paclitaxel have also been investigated in glioma (58–61). Based on this knowledge, this work aimed to investigate the possibility to trigger macrophage mediated tumor cytotoxicity based on a TLR4 activation using Paclitaxel drug as ligand.
Previously, we have shown that PC1/3 KD macrophages were able to produce pro-inflammatory cytokines in autocrine and paracrine ways and were more pro-inflammatory compared with macrophages expressing PC1/3 (12, 13). We thus concluded that this PC1/3 enzyme controlled the activation of macrophages. In the present study, we also revealed by proteomics approaches differences between NT and PC1/3 KD cells after Paclitaxel stimulation (Fig. 1A). In PC1/3 KD macrophages proteins involved in microtubule cytoskeleton assembly, actin organization, cell-cell adhesion and cell matrix adhesion modifications are upregulated. The overexpression of cytoskeleton proteins is expected to impact the endosomal and phagosome movements (62, 63), which would also lead to a higher secretion of immune factors. This modulation of cytoskeleton organization is supported by the high increase of Ca2+ release in these cells (Fig. 4). We also confirmed the release of pro-inflammatory cytokines and chemokines by untreated and Paclitaxel-treated PC1/3 KD macrophages reflective of a higher level of activation of these cells (Fig. 2 and 5). Of note, this secretory activity was not really affected by the presence of the anti-inflammatory cytokines IL-10. Among the factors released by PC1/3 KD cells after Paclitaxel stimulation, we found growth related oncogene (GRO) (α, β, γ) and interleukin 1 (α, β). These factors are known to elicit recruitment of PMN cells. GRO (α, β, γ), also known as chemokines CXCL1, CXCL2, CXCL3, mediates varied functions such as attracting neutrophils to sites of inflammation, regulating angiogenesis, and modulating neurotransmitter release. However, in cancer, they are involved in tumor initiation, progression, and metastasis (64, 65). For example, GRO-β forms an autocrine loop that activates the Ras-Erk1/2 signaling pathway (66) which is linked to phosphorylation of STAT3 (67) signal transducer activator of transcription which is important for cell proliferation. At a first sight, this did not support that Paclitaxel treated PC1/3 KD cells may exert a stronger antitumoral activity. However, we cannot exclude that (i) combination of other molecules contained in the cell supernatant may exhibit antitumoral activity and (ii) in a coculture with tumor cells the nature of the factors secreted will vary. Therefore, we decided to evaluate the potential cytotoxic activity of the supernatant of NT and PC1/3 KD cells after Paclitaxel stimulation. This revealed that the supernatant of PC1/3 KD macrophages stimulated with Paclitaxel exerted an antitumor activity on C6 glioma cells (Fig. 6). Of note, we showed that the effect observed is higher than the direct effect of Paclitaxel alone. In 3D coculture experiments, untreated or treated with low doses of Paclitaxel, macrophages supported the growth and invasion of glioma cells (Fig. 7Ab), as expected from previous observations with murine microglia and human tumor-associated microglia/macrophages (68, 69). We showed that these supportive activities were suppressed by a pre-treatment of macrophages with Paclitaxel, which triggered an efficient antitumor response. Indeed, growth of spheroids and cell invasion in the matrix were severely decreased by macrophages pre-activated with Paclitaxel, irrespective of their PC1/3 content. However, in the supernatant of the coculture between C6 glioma cells and Paclitaxel pretreated PC1/3 KD macrophages cocultures, we found that expression of tumor supportive proteins such as Mortalin or SH3BGRL was lower. We could also identify unique proteins exhibiting antitumor properties such as Nqo1, ANP32A and Coagulation factor IX. This unique profile of secreted proteins suggests a possible scenario whereby these factors may stabilize P53, inhibit its degradation through Nqo1 and inhibit cancer cell survival and facilitate drug efficacy thanks to ANP32A production (45). It may also block the tumor migration with Coagulation factor IX in conjunction of production of immune factors (46).
We also demonstrated that several proteins identified in the supernatant of the coculture between glioma C6 spheroids and PC1/3 KD macrophages were related to exosomes. Interestingly, at 48 h, extracellular vesicles (EVs) from control NT cells had the tendency to block C6 spheroids growth and invasion (Fig. 8). Quite the opposite, EVs from Paclitaxel-treated NT cells did not exert this inhibitory effect. In this case, growth and invasion were restored to a similar level as those observed for untreated C6 cells. This suggests that EVs may contribute to the development of drug resistance observed after Paclitaxel treatment (17). On the contrary, we demonstrated that PC1/3 inhibition in macrophages abolished this phenomenon. Indeed, EVs from Paclitaxel-treated PC1/3 KD cells showed an inhibitory effect on growth and invasion of the C6 spheroids at 48 h of coculture. This effect was however limited in time. In fact, after 72 h, EVs from both NT and KD cells were equally effective in inhibiting C6 spheroids to an extent comparable to the one observed with cocultures of macrophages and spheroids. This transient effect might reflect the capacity of glioma cells to re-program macrophages in a way that is PC1/3 independent and/or that masks the inhibitory capacity of PC1/3 KD cells-derived EVs through secreted molecules.
In conclusion of this study, we showed that Paclitaxel increased the cytosolic calcium concentration [Ca2+]c, diminishes STAT3 activation and reveals a potential of the coaction of PC1/3 inhibition and Paclitaxel treatment in macrophages for glioma therapy. The invasion assays in complement to proteomics analyses of the cell supernatants of C6 spheroids cultured with Paclitaxel pre-activated NT or PC1/3-KD cells, demonstrated that PC1/3 KD cells secrete antitumor factors active toward C6 glioma but they were not sufficient to observe an effect on their invasion in a spheroid coculture assay. In this context, because PC1/3 KD cells have constitutively a higher amount of MVBs, we thought that EVs release through Paclitaxel treatment may influence glioma spheroids growth. Indeed, we showed that EVs from Paclitaxel treated PC1/3 KD cells could escape glioma resistance to treatment. To conclude, these experimental observations lay the basis for a cell therapy based on EVs released by PC1/3 KD macrophages activated with Paclitaxel.
DATA AVAILABILITY
The data sets and the Perseus result files used for analysis were deposited at the ProteomeXchange Consortium (70) (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (71) with the dataset identifier PXD004444 for cellular extracts.
Supplementary Material
Acknowledgments
We thank the labcom o'dreams (PRISM-OCR) for its contribution to this project.
Footnotes
* This research was supported by grants from the Ministère de L'Education Nationale, de L'Enseignement Supérieur et de la Recherche, ANR (IF), Région Nord-Pas de Calais ARCIR (IF), the Université de Lille (MD) SIRIC ONCOLille (IF, MD), Grant INCa-DGOS-Inserm 6041aa, the CCMIC, APVV 15 0613(DC), and INSERM.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- TAM
- Tumor Associated Macrophages
- ACN
- Acetonitrile
- Ca2+
- Calcium
- CCL
- Chemokine (CC-motif) Ligand
- CSF
- Colony Stimulating Factor
- CXCL
- Chemokine (CXC-motif) Ligand
- DTT
- Dithiothreitol
- EV
- Extracellular Vesicle
- FASP
- Filter Aided Sample Preparation
- FBS
- Fetal Bovine Serum
- FDR
- False Discovery Rate
- HCD
- Higher energy Collisional Dissociation
- IL
- interleukin
- KD
- Knock-Down
- KO
- Knock-Out
- LC
- Liquid Chromatography
- LFQ
- Label Free Quantification
- LPS
- Lipopolysaccharide
- MMP
- Matrix Metalloproteinase
- MS
- Mass Spectrometry
- NaOH
- Sodium Hydroxide
- NO
- Nitric Oxide
- NT
- Non-Target
- PBS
- Phosphate Buffer Saline
- PC
- Proprotein Convertase
- SEM
- Standard Error of the Mean
- STAT
- Signal Transducer and Activator of Transcription
- TFA
- Trifluoroacetic Acid
- TLR
- Toll Like Receptor
- TNF
- Tumor Necrosis Factor.
REFERENCES
- 1. Hanahan D., and Weinberg R. A. (2011) Hallmarks of Cancer: The Next Generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. De Palma M., and Lewis C. E. (2011) Cancer: Macrophages limit chemotherapy. Nature 472, 303–304 [DOI] [PubMed] [Google Scholar]
- 3. F. K., and K. A. (2012) The Role of tumor-associated macrophage in tumor progression. Frontiers in Bioscience - Scholar 4S, 787–798 [DOI] [PubMed] [Google Scholar]
- 4. Taskinen M., Karjalainen-Lindsberg M. L., Nyman H., Eerola L. M., and Leppä S. (2007) A high tumor-associated macrophage content predicts favorable outcome in follicular lymphoma patients treated with rituximab and cyclophosphamide- doxorubicin-vincristine-prednisone. Clin. Cancer Res. 13, 5784–5789 [DOI] [PubMed] [Google Scholar]
- 5. Shree T., et al. (2011) Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 25, 2465–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Svensson S., et al. (2015) CCL2 and CCL5 are novel therapeutic targets for estrogen-dependent breast cancer. Clin. Cancer Res. 21, 3794–3805 [DOI] [PubMed] [Google Scholar]
- 7. L. D., van O. E., de B. P., and van G. J.A. (2014) Functional relationship between tumor-associated macrophages and macrophage colony-stimulating factor as contributors to cancer progression. Front. Immunol. 5, no pagination [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mantovani A., and Locati M. (2013) Tumor-associated macrophages as a paradigm of macrophage plasticity, diversity, and polarization lessons and open questions. Arterioscler. Thromb. Vasc. Biol. 33, 1478–1483 [DOI] [PubMed] [Google Scholar]
- 9. Zhu Y., et al. (2014) CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 74, 5057–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hung J. Y., et al. (2014) Colony-stimulating factor 1 potentiates lung cancer bone metastasis. Lab. Invest. 94, 371–381 [DOI] [PubMed] [Google Scholar]
- 11. Refaie S., et al. (2012) Disruption of proprotein convertase 1/3 (PC1/3) expression in mice causes innate immune defects and uncontrolled cytokine secretion. J. Biol. Chem. 287, 14703–14717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gagnon H., et al. (2013) Proprotein convertase 1/3 (PC1/3) in the rat alveolar macrophage cell line NR8383: localization, trafficking and effects on cytokine secretion. PLoS ONE 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duhamel M., et al. (2015) Molecular consequences of proprotein convertase 1/3 (PC1/3) inhibition in macrophages for application to cancer immunotherapy: a proteomic study. Mol. Cell. Proteomics 14, 2857–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kawasaki K., Nogawa H., and Nishijima M. (2003) Identification of mouse MD-2 residues important for forming the cell surface TLR4-MD-2 complex recognized by anti-TLR4-MD-2 antibodies, and for conferring LPS and taxol responsiveness on mouse TLR4 by alanine-scanning mutagenesis. J. Immunol. 170, 413–420 [DOI] [PubMed] [Google Scholar]
- 15. Kawasaki K., et al. (2001) Involvement of TLR4/MD-2 complex in species-specific lipopolysaccharide-mimetic signal transduction by Taxol. J. Endotoxin Res. 7, 232–236 [PubMed] [Google Scholar]
- 16. Edwards S. J., Barton S., Thurgar E., and Trevor N. (2015) Topotecan, pegylated liposomal doxorubicin hydrochloride, paclitaxel, trabectedin and gemcitabine for advanced recurrent or refractory ovarian cancer: a systematic review and economic evaluation. Health Technol. Assess. 19, 1–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Orr G. A., Verdier-Pinard P., McDaid H., and Horwitz S. B. (2003) Mechanisms of Taxol resistance related to microtubules. Oncogene 22, 7280–7295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Javeed A., et al. (2009) Paclitaxel and immune system. European Journal of Pharmaceutical Sciences 38, 283–290 [DOI] [PubMed] [Google Scholar]
- 19. Meissner F., Scheltema R. A., Mollenkopf H.-J., and Mann M. (2013) Direct proteomic quantification of the secretome of activated immune cells. Science (80) 340, 475–478 [DOI] [PubMed] [Google Scholar]
- 20. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 21. Cox J., et al. (2011) Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 22. UniProt Consortium. (2012) Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71–D75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cox J., et al. (2014) Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Montojo J., et al. (2010) GeneMANIA cytoscape plugin: Fast gene function predictions on the desktop. Bioinformatics 26, 2927–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cisneros Castillo L. R., Oancea A.-D., Stüllein C., and Régnier-Vigouroux A. (2016) A Novel Computer-Assisted Approach to evaluate Multicellular Tumor Spheroid Invasion Assay. Sci. Rep. 6, 35099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeNardo D. G., et al. (2011) Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 1, 54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huettner C., Czub S., Kerkau S., Roggendorf W., and Tonn J. C. (1997) Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 17, 3217–3224 [PubMed] [Google Scholar]
- 28. Huettner C., Paulus W., and Roggendorf W. (1995) Messenger RNA expression of the immunosuppressive cytokine IL-10 in human gliomas. Am. J. Pathol. 146, 317–322 [PMC free article] [PubMed] [Google Scholar]
- 29. Ousingsawat J., et al. (2015) Anoctamin 6 mediates effects essential for innate immunity downstream of P2X7 receptors in macrophages. Nat. Commun. 6, 6245. [DOI] [PubMed] [Google Scholar]
- 30. Dhirapong A., Lleo A., Leung P., Gershwin M. E., and Liu F. T. (2009) The immunological potential of galectin-1 and -3. Autoimmunity Reviews 8, 360–363 [DOI] [PubMed] [Google Scholar]
- 31. Schiff P. B., and Horwitz S. B. (1980) Taxol stabilizes microtubules in mouse fibroblast cells. Proc. Natl. Acad. Sci. U.S.A. 77, 1561–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schiff P. B., Fant J., and Horwitz S. B. (1979) Promotion of microtubule assembly in vitro by taxol. Nature 277, 665–667 [DOI] [PubMed] [Google Scholar]
- 33. Patel N., et al. (2010) Rescue of paclitaxel sensitivity by repression of Prohibitin1 in drug-resistant cancer cells. Proc. Natl. Acad. Sci. U.S.A. 107, 2503–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shprung T., and Gozes I. (2009) A novel method for analyzing mitochondrial movement: Inhibition by paclitaxel in a pheochromocytoma cell model. J. Mol. Neurosci. 37, 254–262 [DOI] [PubMed] [Google Scholar]
- 35. O'Brien E. T., Salmon E. D., and Erickson H. P. (1997) How calcium causes microtubule depolymerization. Cell Motil. Cytoskeleton 36, 125–135 [DOI] [PubMed] [Google Scholar]
- 36. Murray P. J. (2006) STAT3-mediated anti-inflammatory signalling. Biochem. Soc. Trans. 34, 1028–1031 [DOI] [PubMed] [Google Scholar]
- 37. Walker S. R., Chaudhury M., Nelson E. A., and Frank D. A. (2010) Microtubule-targeted chemotherapeutic agents inhibit signal transducer and activator of transcription 3 (STAT3) signaling. Mol. Pharmacol. 78, 903–908 [DOI] [PubMed] [Google Scholar]
- 38. Murray S., Briasoulis E., Linardou H., Bafaloukos D., and Papadimitriou C. (2012) Taxane resistance in breast cancer: Mechanisms, predictive biomarkers and circumvention strategies. Cancer Treatment Reviews 38, 890–903 [DOI] [PubMed] [Google Scholar]
- 39. Lu W-J., et al. (2011) Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 18, 1046–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang H., et al. (2015) Dual-faced SH3BGRL: oncogenic in mice, tumor suppressive in humans. Oncogene 1–11. doi: 10.1038/onc.2015.391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chan A. Y., et al. (2005) Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene 24, 7821–7829 [DOI] [PubMed] [Google Scholar]
- 42. Skog J., et al. (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oztopçu P., Kabadere S., Mercangoz A., and Uyar R. (2004) Comparison of vitamins K1, K2 and K3 effects on growth of rat glioma and human glioblastoma multiforme cells in vitro. Acta Neurol. Belg. 104, 106–110 [PubMed] [Google Scholar]
- 44. Eldh M., et al. (2010) Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS ONE 5, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Williams T. K., et al. (2010) pp32 (ANP32A) expression inhibits pancreatic cancer cell growth and induces gemcitabine resistance by disrupting HuR binding to mRNAs. PLoS ONE 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kitano H., Mamiya A., Tomomi I., Shinichiro K., and Chiaki H. (2015) Coagulation factor IX regulates cell migration and adhesion in vitro. Cell Biol. Int. 39, 1162–1172 [DOI] [PubMed] [Google Scholar]
- 47. Duhamel M., et al. (2016) The proprotein convertase PC1/3 regulates TLR9 trafficking and the associated signaling pathways. Sci. Rep. 6, 19360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Minciacchi V. R., Freeman M. R., and Di Vizio D. (2015) Extracellular vesicles in cancer: exosomes, microvesicles and the emerging role of large oncosomes. Sem. Cell Develop. Biol. 40, 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tang X., Mo C., Wang Y., Wei D., and Xiao H. (2013) Anti-tumour strategies aiming to target tumour-associated macrophages. Immunology 138, 93–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tang X. (2013) Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett 332, 3–10 [DOI] [PubMed] [Google Scholar]
- 51. Siveen K. S., and Kuttan G. (2009) Role of macrophages in tumour progression. Immunol. Lett. 123, 97–102 [DOI] [PubMed] [Google Scholar]
- 52. Laoui D., et al. (2011) Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int. J. Dev. Biol. 55, 861–867 [DOI] [PubMed] [Google Scholar]
- 53. Jung K. Y., et al. (2015) Cancers with higher density of tumor-associated macrophages were associated with poor survival rates. J. Pathol. Transl. Med. 49, 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen P., et al. (2011) Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin. Cancer Res. 17, 7230–7239 [DOI] [PubMed] [Google Scholar]
- 55. Allavena P., and Mantovani A. (2012) Immunology in the clinic review series; focus on cancer: Tumour-associated macrophages: Undisputed stars of the inflammatory tumour microenvironment. Clin. Exp. Immunol. 167, 195–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Palsson-Mcdermott E. M., et al. (2015) Pyruvate kinase M2 regulates hif-1α activity and il-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 21, 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tarassishin L., Suh H.-S., and Lee S. C. (2011) Interferon regulatory factor 3 plays an anti-inflammatory role in microglia by activating the PI3K/Akt pathway. J. Neuroinflammation 8, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nance E., et al. (2014) Brain-penetrating nanoparticles improve paclitaxel efficacy in malignant glioma following local administration. ACS Nano 8, 10655–10664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liu Y., et al. (2014) Paclitaxel loaded liposomes decorated with a multifunctional tandem peptide for glioma targeting. Biomaterials 35, 4835–4847 [DOI] [PubMed] [Google Scholar]
- 60. Hu Q., et al. (2013) Glioma therapy using tumor homing and penetrating peptide-functionalized PEG-PLA nanoparticles loaded with paclitaxel. Biomaterials 34, 5640–5650 [DOI] [PubMed] [Google Scholar]
- 61. Chirio D., et al. (2014) Positive-charged solid lipid nanoparticles as paclitaxel drug delivery system in glioblastoma treatment. Eur. J. Pharm. Biopharm. 88, 746–758 [DOI] [PubMed] [Google Scholar]
- 62. Jahraus A., et al. (2001) ATP-dependent membrane assembly of F-actin facilitates membrane fusion. Mol. Biol. Cell 12, 155–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guerin I. (2000) Disruption of the actin filament network affects delivery of endocytic contents marker to phagosomes with early endosome characteristics: The case of phagosomes with pathogenic mycobacteria. Eur. J. Cell Biol. 79, 735–749 [DOI] [PubMed] [Google Scholar]
- 64. Zhu Q., Han X., Peng J., Qin H., and Wang Y. (2012) The role of CXC chemokines and their receptors in the progression and treatment of tumors. J. Mol. Histol. 43, 699–713 [DOI] [PubMed] [Google Scholar]
- 65. Keeley E. C., Mehrad B., and Strieter R. M. (2010) CXC chemokines in cancer angiogenesis and metastases. Adv. Cancer Res. 106, 91–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang B., et al. (2009) A key role for early growth response-1 and nuclear factor-kappaB in mediating and maintaining GRO/CXCR2 proliferative signaling in esophageal cancer. Mol. Cancer Res. 7, 755–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Plaza-Menacho I., et al. (2006) Ras/ERK1/2-mediated STAT3 Ser727 Phosphorylation by Familial Medullary Thyroid Carcinoma-associated RET Mutants Induces Full Activation of STAT3 and Is Required for c-fos Promoter Activation, Cell Mitogenicity, and Transformation. J. Biol. Chem. 282, 6415–6424 [DOI] [PubMed] [Google Scholar]
- 68. Kees T., et al. (2012) Microglia isolated from patients with glioma gain antitumor activities on poly (I:C) stimulation. Neuro. Oncol. 14, 64–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mora R., et al. (2009) TNF-α- and TRAIL-resistant glioma cells undergo autophagy-dependent cell death induced by activated microglia. Glia 57, 561–581 [DOI] [PubMed] [Google Scholar]
- 70. Vizcaíno J. A., et al. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Vizcaíno J. A., et al. (2013) The Proteomics Identifications (PRIDE) database and associated tools: Status in 2013. Nucleic Acids Res. 41, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets and the Perseus result files used for analysis were deposited at the ProteomeXchange Consortium (70) (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (71) with the dataset identifier PXD004444 for cellular extracts.