

Abstract
Individuals with a ring 15 chromosome [r(15)] and those with Russell-Silver syndrome have short stature, developmental delay, triangular face, and clinodactyly. To assess whether the apparent phenotypic overlap of these conditions reflects a common genetic cause, the extent of deletions in chromosome 15q was determined in 5 patients with r(15), 1 patient with del 15q26.1–qter, and 5 patients with Russell- Silver syndrome. All patients with Russell- Silver syndrome were diploid for genetic markers in distal 15q, indicating that Russell-Silver syndrome in these individuals was unlikely to be related to the expression of single alleles at these or linked genetic loci. At least 3 distinct sites of chromosome breakage close to the telomere were found in the r(15) and del 15q25.1–qter patients, with 1 r(15) patient having both a terminal and an interstitial deletion. Although the patient with del 15q25.1–qter exhibited the largest deletion and the most profound growth retardation, the degree of growth impairment among the r(15) patients was not correlated with the size of the deleted interval. Rather, the parental origin of the ring chromosome in several patients was associated with phenotypes that are also seen in patients with either Prader-Willi (PWS) or Angelman (AS) syndromes, conditions that result from uniparental expression of genes on chromosome 15. In fact, unequal representation of chromosome 15 alleles in 1 patient with r(15) suggests the possibility that a mosaic karyotype composed of the constitutional cell line and cell line(s) possibly deficient in the ring chromosome might be present. The PWS-like or AS-like phenotypes could be explained by postzygotic loss of the ring chromosome, leading to uniparental inheritance of the intact chromosome in some tissues of r(15) patients.
Keywords: ring 15 chromosome syndrome, Russell-Silver syndrome, chromosome 15, insulin-like growth factor receptor, gene dosage, growth retardation, uniparental inheritance, genomic imprinting, Prader-Willi syndrome, Angelman syndrome
INTRODUCTION
Ring 15 chromosome syndrome [r(15)] is characterized by growth retardation, microcephaly, triangular face, hypertelorism, brachydactyly, variable mental retardation, and speech delay [Butler et al., 1988; Roback et al., 1991]. These patients exhibit de novo hemizygous deletions of the distal subbands of 15q (q26.3, q26.2, and/or q26.1) [Wilson et al., 1985; Butler et al., 1988]. Growth hormone levels have been normal in r(15) patients [Wilson et al., 1985; Butler et al., 1988], but several patients with r(15) syndrome have had a deletion of one copy of the insulin-like growth factor receptor gene (IGF1R) [Francke et al., 1988]. Many of the manifestations of r(15) syndrome were reported previously in a patient with a terminal deletion of the long arm of chromosome 15(q26.1 →qter), in whom there was also loss of a copy of IGF1R [Roback et al., 1991]. To address the possibility that hemizygous deletions of IGF1R could be related to growth retardation in these patients, we defined the extent of terminal deletions of chromosome 15 and examined the relationship between the size of the hemizygous interval and growth in these patients.
Similar clinical findings have also been noted in a patient with Russell-Silver syndrome and an r(15) karyotype [Wilson et al., 1985]. Patients with Russell-Silver and r(15) syndrome may exhibit growth deficiency, triangular face, digital anomalies, and café au lait spots. Microcephaly, developmental/speech delay, minor facial anomalies, limb anomalies, and cardiac defects are more striking in r(15) patients, whereas hemihypertrophy and abnormal genitalia are thought to be more characteristic of Russell-Silver syndrome. No consistent Mendelian or chromosomal basis for Russell-Silver syndrome has been established, although IGF1R has been suggested as a candidate gene [Donlon, 1992]. The present study investigates the loss of a single IGF1R gene copy and other distal 15qter genetic loci as possible explanations for the overlapping changes of Russell-Silver and r(15) syndromes. Analysis of markers from the 15q25→qter region in 5 patients with Russell-Silver syndrome and 6 patients with either deletion 15q or r(15) karyotypes indicates that these conditions probably have distinct genetic causes.
MATERIALS AND METHODS
Patients
Five patients (4 males and 1 female) were studied, ranging in age from 9 months to 14 years with the characteristic cytogenetic and clinical findings of ring 15 chromosome syndrome, including growth retardation (Table I), microcephaly, triangular face, and variable mental retardation [Butler et al., 1988]. Growth retardation was measured in terms of standard deviations (SD) below the age-related, mean height for the general population. Five patients (3 females and 2 males) ranging in age from 1–11 years were ascertained with a diagnosis of Russell-Silver syndrome. The subjects with Russell-Silver syndrome displayed growth retardation of prenatal onset, small triangular face, normocephaly, clinodactyly, asymmetric growth, and normal karyotypes.
TABLE I.
Correlation of Growth Parameters with Estimates of IGF1R Copy Number*
| Subject | Height (SD below mean for age) | IGF1R copy number (by analysis of dosage) | FES proto-oncogene number of alleles(PCR amplification) |
|---|---|---|---|
| Ring chromosome 15: | |||
| 14-year-old male (A) | −3 | 1.4a | 2 |
| 10-year-old male (B) | −4 | 1.0 | 2 |
| 8-year-old male (C) | −4 | 1.2 | 2 |
| 3-year-old female (D) | −4 | 0.9 | 2 |
| 9-month-old male (E) | −5 | Not done | 2 |
| Russell-Silver syndrome: | |||
| 3-year-old male (F) | −3 | 2.3 | 2 |
| 4-year-old female (G) | −2 | 2.1 | 2 |
| 9-year-old female (H) | 0 | 2.1 | 2 |
| 11-year-old female (I) | −4 | 2.3 | 2 |
| 1-year-old female (J) | −2 | Not available | 2 |
| One-year-old female with 15q26.1–qter deletion (K) | −7 | 0.7 | 1c |
| Control individuals: | |||
| Prader-Willi syndrome: | |||
| 9-year-old male with 15q11–q13 deletion (L) | −2 | 0.7 for D15S10b | 2 |
| 4-year-old male with normal chromosomes (M) | −1 | 1.7 for D15S10b | 2 |
| 28-year-old normal male (N) | 0 | 2.0 | 2 |
IGF1R gene dosage was determined by comparing band intensities in r(15), Russell-Silver, and del 15q patients with individuals who were diploid at this locus (patients L–N). The number of copies of IGF1R was calculated according to Tantravahi et al. [1989] and Roback et al. [1991], and was based on both densitometry of Southern blots and on radioactivity standards. A proximal 15q control probe, D15S10, was used to calibrate copy number by comparison of band intensities from a control subject who was diploid at this locus (patient N) and patient L, who was hemizygous. Copy number of IGF1R in r(15), del 15q, and Russell-Silver patients was determined from the ratio of the IGF1R and D15S10 band intensities, based on the average of two or more independent experiments. IGFR1 gene dosage was not estimated for patient E.
Copy number does not fall within deletion range by dosage analysis; PCR amplification of IGF1R polymorphism shows heterozygosity at this locus [Meloni et al., 1992].
Copy number was calculated for D15S10 using IGF1R as the control probe for Prader-Willi syndrome patients.
Patient was uninformative at FES proto-oncogene locus due to homozygous inheritance of an allele common to both parents. Her mother was homozygous and her father was heterozygous for this locus.
The height of patient A with r(15) syndrome was −4 SD at age 11 years, −3.5 SD at age 12 years, and −3 SD at age 14 years. Patient B exhibited several manifestations of Prader-Willi syndrome (PWS), including high- arched palate, narrow bifrontal diameter, hypogenitalism, small hands and feet, developmental delay (IQ = 52), poor suck reflex, and feeding difficulties, in addition to the r(15) syndrome phenotype. He was initially diagnosed with Russell-Silver syndrome, but because of delayed onset of hyperphagia and marked obesity at age 10 years, the diagnosis of PWS was entertained. His growth was −4 SD at age 10 years, −5 SD at age 16 years, and −4.5 SD at age 18 years. Patient C exhibited short stature, developmental delay, absent speech, and a triangular face. He has been described elsewhere [Butler et al., 1988]. His height was −4 SD at birth and at age 2, 8, and 10 years, and −5 SD at age 6 years. Patient D exhibited a triangular face with microcephaly, micrognathia, short stature, clinodactyly, small hands and feet, hyperpigmentation, speech impairment, and developmental delay. Length of this patient was −2 SD at birth, and height at age 3 years was −4 SD. Patient E is an infant with feeding difficulties, delayed development, downslanting palpebral fissures, downturned corners of the mouth, and intermittent strabismus. His length was −2.5 SD at age 1 month and − 5 SD at age 9 months.
The patient with del 15q26.1→qter (K) was described by Roback et al. [1991]. A cell line derived from patient A (GM10173) was obtained from the NIGMS Mutant Cell Line Repository (Camden, NJ); a detailed history was unavailable on this individual. Complete genetic analyses of patients D, F, and G were not possible due to limited quantities of genomic DNA from these individuals. Cell lines for all patients and the parents of patients B, C, and K were established by transforming peripheral lymphocytes with Epstein-Barr virus [Neitzel, 1986].
Cytogenetics
Chromosome studies on lymphocytes were carried out using standard G-banding techniques. Fluorescence in situ hybridization studies were performed on patient C with r(15) syndrome with an all-human telomere probe using conditions recommended by the manufacturer (Oncor, Gaithersburg, MD).
Southern Hybridization
Quantitative Southern hybridization of genomic DNA derived from leukocytes or lymphoblastoid cell lines was carried out according to previously described methods [Mbikay et al., 1988; Roback et al., 1991; Butler et al., 1993]. Filters were simultaneously probed with a 0.7-kilobase (kb) EcoRI fragment of the cDNA for human IGF1R (15q26.1→qter) [Ullrich et al., 1986; Roback et al., 1991] and pTD3-21 (D15S10), which maps to proximal 15q [Nicholls et al., 1989a; Tantravahi et al., 1989], pTD3-21 was used as an internal control for densitometric quantitation of diploid copy number. Patients with terminal deletions of chromosome 15 were also analyzed by hybridization of genomic DNA to probes derived from the polymorphic genetic markers D15S3 [Brissenden et al., 1986] and D15S86 [Armour et al., 1990].
Amplification of Short Tandem Repeat Polymorphisms
For each patient, the number of copies of distal 15q sequences was also determined using a series of short tandem repeat polymorphisms (STRP) that have been ordered on the chromosome 15 genetic index map [Malcolm and Donlon, 1994; Beckmann et al., 1993]. IGF1R was not present on this map and has been localized in the present study. Polymerase chain reaction (PCR) amplification and polyacrylamide gel electrophoresis of 32(P) end-labeled amplification products were analyzed according to modifications of the method described by Weber and May [1989]. Oligonucleotide primer sequences for PCR were obtained from the National Institute of Health/Department of Energy Genome Database (Johns Hopkins University) and from the report of the Second International Chromosome 15 Workshop [Malcolm and Donlon, 1994].
RESULTS AND DISCUSSION
The copy numbers of the IGF1R and FES genes and the genotypes of polymorphic markers from the proximal (D15S541 and D15S10) and distal long arm of chromosome 15, including D15S95, D15S111, FES, IP15M9, D15S100, D15S130, D15S230, D15S107, D15S120, D15S87, D15S86, D15S3, and D15S642, were determined in 5 patients with r(15) chromosomes (patients A, B, C, D, E), 5 patients with Russell-Silver syndrome (F, G. H, I, J), and 1 patient with del 15q26.1→qter (patient K).
The patients with Russell-Silver syndrome had two copies of the IGF1R gene, as judged by either quantitative Southern hybridization (patients F, G, H, I, J, Table I) or heterozygosity at this and other 15q loci (patients H, I, J, Table II). These observations are in contrast with a report of a single patient with manifestations of Russell-Silver syndrome, who carried a deletion of one copy of this gene [Tamura et al., 1993]. Russell-Silver syndrome may be genetically heterogeneous. Maternal uniparental disomy of chromosome 7 has been reported in 4 individuals with Russell-Silver syndrome [Kotzot et al., 1995], a genotype that was previously identified in a patient with cystic fibrosis and primordial growth retardation [Spence et al., 1989].
TABLE II.
15q Genotypes of Selected Patients Diagnosed With Russell-Silver Syndrome*
| cen-//-15q12-//-
|
15q25
|
15q26
|
D15S107 | -qter-//-tel
|
||||
|---|---|---|---|---|---|---|---|---|
| Patient | D15S11 | -D15S111- | [FES, | IP15M9] | -D15S100- | -D15S87 | IGF1R | |
| H | H | H | H | H | ni | nd | H | Ia |
| I | H | H | H | ni | H | nd | ni | Ia |
| J | H | H | H | H | nd | nd | H | H,b Ia |
H, heterozygous (parents not available); I, intact (parental origin indeterminate); ni, not informative; nd, no data available. Locus order is derived from Beckmann et al. [1993], Malcolm and Donlon [1994], and the Genome Database.
Copy number based on calibrated gene dosage.
STRP polymorphic locus.
Several distinct sites of chromosome breakage that resulted in terminal deletions in distal 15q were inferred among the patients with r(15) and del 15q26.1–qter. With the exception of patient L (a control individual with Prader-Willi syndrome), each individual was diploid at D15S10 (Table I) and other 15q11–q13 genetic loci, including D15S541 (Table III, and other data not shown); this indicates that the ring chromosome deletions do not extend into the proximal q arm.
TABLE III.
Definition of Deletions in r(15) and del 15q26.1→qter Syndrome Patients*
| Locus | Patient
|
||||
|---|---|---|---|---|---|
| A | B | C | E | K | |
| 15q11 | |||||
| D15S541- | H | M/P | H | H | ni |
| 15q25- | |||||
| D15S95- | nd | nd | nd | nd | M/P |
| D15S111- | H | H | ni | ni | M/P |
| [FES, | Ha, Ib | M/Pa, Ib | Ha, Ib | Ha | nia, Ib |
| IP15M9]- | H | M/P | M/P | nd | ni |
| D15S100- | H | ni | M/P | M/P | PD |
| D15S130- | ni | nd | M/P | ni | nd |
| D15S230- | H | M/P | ni | M/P | PD |
| D15S107- | nd | Pd | ni | M/P | PD |
| D15S120 | H | M/P | H | M/P | PD |
| D15S87- | H | PD | ni | ni | PD |
| D15S86- | nd | nd | MD | nd | nd |
| D15S3- | H | ni | MD | nd | ni |
| D15S642 | ni | ni | ni | PD | PD |
| -qter | |||||
| IGF1Rc | Ha | Db | Db | nia | PDab |
M/P, biparental inheritance; H, heterozygous (either parent not available, or inheritance of common parental allele by proband); PD, paternally-derived deletion; MD, maternally-derived deletion; D, deleted (parental origin indeterminate); I, intact (parental origin indeterminate); ni, not informative; nd, no data available. Locus order is derived from Beckmann et al. [1993], and the Genome Database.
STRP polymorphic locus.
Copy number based on calibrated gene dosage.
IGF1R has been localized to 15q26.1, but has not been assigned on the genetic linkage map.
Patient B exhibited paternally-derived 15q deletions at D15S107 (Fig. 1A) and D15S87 (Table III). D15S120, a marker which lies between these two loci, was inherited biparentally in this patient, indicating the presence of both interstitial and terminal deletions of chromosome 15 (Table III). Concerted loss of both chromosomal domains could have resulted from an aborted, terminal paracentric inversion involving D15S107 and adjacent sequences. Failure to complete and resolve the distal exchange in the chromosomal intermediate could have resulted in the loss of both the interstitial segment (containing D15S107) and the telomeric domain (containing D15S87). The ring chromosome could then have been formed by ligation of the newly-formed terminus to the p arm or centromere.
Fig. 1.

Representative STRP polymorphism results for r(15) and del 15q26.1→qter patients. Alleles are independently designated in each family according to size. Loss of paternal alleles in the ring chromosome is evident at locus D15S107 in patient B (A) and at D15S642 in patient E (D). B: Paternally-derived deletion of D15S100 in patient K. C: Maternally-derived deletion at D15S86 in patient C.
Patient C carried a maternally-derived ring chromosome which was deleted at D15S107, D15S3, IGF1R, and D15S86 (Fig. 1C; Table III). Since patient C is heterozygous for D15S120, IGF1R is presumably distal to this locus, consistent with the known linkage of this gene to D15S3 [Poduslo et al., 1991]. In addition, fluorescence in situ hybridization with an all-human telomere probe failed to detect a telomere in the ring chromosome of patient C (results not shown).
The ring chromosome in patient E was intact at D15S107 and deleted at D15S642, indicating that he harbored a smaller deletion than patient B (Fig. 1D). Patient A was heterozygous at all informative 15q loci examined (including IGF1R; Table I), and is presumed to carry the smallest deletion of the patients with r(15) chromosomes. The genotype of IGF1R in this patient suggests that his ring chromosome shows mitotic instability (Fig. 2; discussed below). Patient K was hemizygous at D15S100 (Fig. 1B), D15S230, D15S107, D15S87, D15S642 (Table III), and IGF1R, thus exhibiting a larger 15q deletion than in any of the patients with r(15) chromosomes. With the exception of patient B, the deduced order of genetic markers based on the locations of the deletion breakpoints in these patients agrees with the chromosome 15 linkage map, which did not include IGF1R [Beckmann et al., 1993].
Fig. 2.
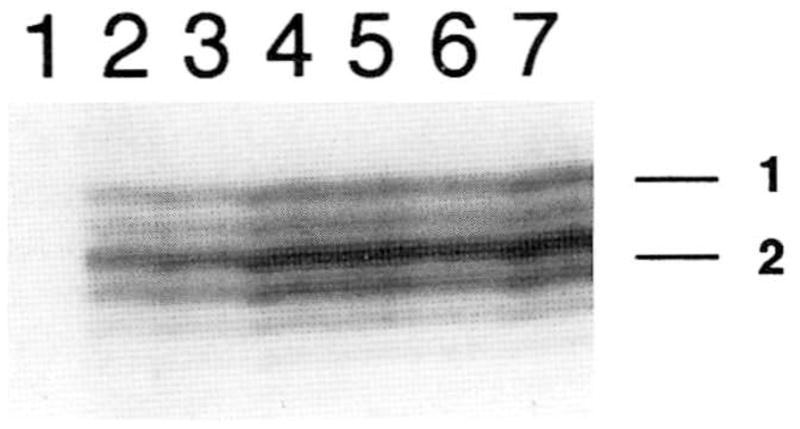
Unequal representation of intact and ring chromosome 15 alleles for patient A. Results indicate significantly increased PCR amplification of allele 2 compared to allele 1 at D15S111 for a series of DNA template concentrations. Similar results were also observed at IGF1R and D15S100 in this patient. Serial dilutions of genomic DNA are as follows: lane 1, 0 ng; lane 2, 10 ng; lane 3, 15 ng; lane 4, 20 ng; lane 5, 25 ng; lane 6, 30 ng; lane 7, 35 ng. As a control, both alleles at heterozygous loci on a distinct chromosome were amplified with equal efficiency (D21S1434 and D21S1437; not shown).
The size of the deleted interval, the IGF1R copy number, and the degree of growth retardation in each of the del 15q and r(15) patients were compared to determine whether their growth impairment was related to the number of copies of IGF1R that were present, as previously suggested [Francke et al., 1988]. Four of 5 r(15) patients (B, C, D, E) and the patient with del 15q26.1→qter (patient K) were hemizygous at IGF1R (Table I). Although patients B and C exhibited similar degrees of growth retardation (−4 to −5 SD), the ring chromosome in patient B carried a larger deletion. Comparable growth retardation was seen in patients E and C, despite the fact that patient E harbors a larger ring chromosome (i.e., a smaller deletion). Since all loci studied in patient A were heterozygous (including IGF1R), the presumed deletion on the ring chromosome of this individual was smaller than those present in patients B and C, yet the degree of growth retardation seen in r(15) patients A, B, and C is also similar (Table I). Thus, growth retardation in this syndrome is not always associated with an IGF-1 receptor defect. Low levels of IGF-1 have been found in another patient with r(15), whereas normal or elevated amounts of this ligand might have been expected if receptor expression had been reduced [Nuutinen et al., 1995]. Furthermore, transgenic mice carrying a single functional copy of IGF1R grow at a normal rate, possibly because the remaining allele is upregulated [Liu et al., 1993; Baker et al., 1993]. With the exception of patient K (who had the largest deletion and more profound growth retardation (−7 SD), organ dysfunction, and developmental delay than any of the r(15) patients), the size of the distal 15q deletion would appear to be uncorrelated with the degree of growth retardation in these patients with terminal chromosome 15 deletions.
The growth retardation and some unusual manifestations described in these patients may be due, in part, to missegregation and loss of the ring chromosome during early fetal development. The loss of the ring chromosome probably occurs by mitotic anaphase lag and produces a mosaic genotype [Kosztolányi, 1987]. The resultant monosomy 15 cell line may not always be viable, and uniparental disomy of this chromosome may arise by a second somatic nondisjunction event [Petersen et al., 1992]. Either monosomy or disomy of the intact chromosome would be expected to document unequal representation of maternally- and paternally-derived alleles in the mosaic cell line(s). In fact, unequal signals corresponding to each allele were consistently detected at different 15q genetic loci in patient A, including D15S111 (Fig. 2), D15S100, and IGF1R (results not shown). By contrast, heterozygous chromosome 21 genetic loci showed equivalent amplification of each allele in this patient.
Based on the constellation of phenotypes seen in these individuals and the parental origins of their ring chromosomes, we propose that mitotic instability of the ring chromosome in patients B, C, and E may have led to partial uniparental inheritance of chromosome 15. Clinical findings typically seen in PWS [Nicholls et al., 1989b; Butler and Meaney, 1987; Butler, 1994] or Angelman syndrome (AS) [Knoll et al., 1989] are inconsistently noted in patients with r(15) syndrome. Of those manifestations which distinguish between PWS and AS, absent/delayed speech (seen in AS) occurs in 39%, cryptorchidism in 30%, small hands (seen in PWS) in 23%, and micrognathia (not seen in AS) in 29% of r(15) patients [Butler et al., 1988]. Patients B and E carried paternally-derived ring chromosomes and presented with clinical findings that are often noted in patients with PWS and infrequently seen in AS. By contrast, patient C had a maternally-derived ring chromosome and absent speech (by contrast, all of the other patients in this study with r(15) syndrome had developed speech). Postzygotic missegregation of the ring chromosome may have, respectively, resulted in maternal uniparental inheritance of chromosome 15 in some tissues of patients B and E, and partial, paternal uniparental inheritance in patient C.
Apart from patient A, a mosaic genotype consistent with mitotic loss of the ring chromosome was not detected in leukocyte genomic DNA of the other patients with r(15) syndrome. This would not preclude the presence of a cell line with monosomy or uniparental disomy 15 in these individuals. Monosomy or disomy chromosome 15 may have been significantly more prevalent in other tissues, or may have been present during development. A mosaic cell line consisting of only 29% cells with a 15q11–q13 deletion in a patient with PWS [Mowrey-Rushton et al., 1994] would appear to suggest that a low threshold of uniparental inheritance in the appropriate tissue and stage may be sufficient for expression of imprinted phenotypes [Nicholls, 1993].
Despite the apparent relationship between the parental origin of the ring chromosome and phenotypes, our results do not exclude the possibility that the variable clinical presentation may be associated with the loss of 15q sequences close to the telomere or the genetic background of each patient. Hemizygous expression of genes close to the telomere of chromosome 15 (distal to D15S3 or D15S642) that are important for growth could explain the absence of a correlation between the size of the 15q deletion and growth impairment in r(15) syndrome. Recessive alleles unmasked by the deletion could also contribute to this phenotype. As an example, recessive inheritance of Bloom syndrome causes prenatal and postnatal growth retardation, and the corresponding gene is linked to distal 15q [Woodage et al., 1994]. However, none of the patients in this study exhibited karyotypic or other phenotypic abnormalities of this disorder, nor is it likely that recessive alleles at this or other 15q loci would be inherited consistently by patients with r(15).
Correlation of specific phenotypes with the parental origin of the ring may provide evidence of uniparental gene expression on other chromosomes. Our observations of syndromic phenotypes with known genomic imprinting effects in patients with ring 15 syndrome may stimulate efforts to understand the variable clinical presentation of other ring chromosome syndromes.
Acknowledgments
The authors thank the families of our patients for their interest in and encouragement of these studies. This work was supported by grants from the National Institutes of Health (P30-HD15052 to M.G.B., R55-HD29098-01 to P.K.R.), from the March of Dimes Birth Defects Foundation (to P.K.R.), and from the David S. and Amy S. Goldberg Memorial Fund for Pediatric Research (to P.K.R.).
Footnotes
This paper was presented at the 43rd annual meeting of the American Society of Human Genetics, October 5–9, 1993, and at the Second International Chromosome 15 Workshop, Oxford, UK, April 2–4, 1994.
References
- Armour JAL, Povey S, Jeremiah S, Jeffreys AJ. Systemic cloning of human minisatellites from ordered array charomid libraries. Genomics. 1990;8:501–512. doi: 10.1016/0888-7543(90)90037-u. [DOI] [PubMed] [Google Scholar]
- Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- Beckmann JS, Tomfohrde J, Barnes RI, Williams M, Broux O, Richard I, Weissenbach J, Bowcock AM. A linkage map of human chromosome 15 with an average resolution of 2 cM and containing 55 polymorphic microsatellites. Hum Mol Genet. 1993;2:2019–2030. doi: 10.1093/hmg/2.12.2019. [DOI] [PubMed] [Google Scholar]
- Brissenden JE, Page DC, deMartinville B, Trousdale J, Botstein D, Francke U. Regional assignments of three polymorphic DNA segments on human chromosome 15. Genet Epidemiol. 1986;3:231–239. doi: 10.1002/gepi.1370030404. [DOI] [PubMed] [Google Scholar]
- Butler MG. Prader-Willi and Angelman syndromes: Examples of genetic imprinting in man. In: Seth PK, Seth S, editors. Human Genetics. New Delhi: Omega Scientific Publishers; 1994. pp. 185–200. [Google Scholar]
- Butler MG, Meaney FJ. Standards for selected anthropometric measurements in Prader-Willi syndrome. Pediatrics. 1987;88:853–860. [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Fogo AB, Fuchs DA, Collins FS, Dev VG, Phillips JA. Brief clinical report and review: Two patients with ring chromosome 15 syndrome. Am J Med Genet. 1988;29:149–154. doi: 10.1002/ajmg.1320290119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler MG, Dahir GA, Schwartz HS. Molecular analysis of transforming growth factor beta in giant cell tumor of bone. Cancer Genet Cytogenet. 1993;66:108–112. doi: 10.1016/0165-4608(93)90237-g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlon TA. Report of the first international workshop on human chromosome 15 mapping. Cytogenet Cell Genet. 1992;61:162–166. [PubMed] [Google Scholar]
- Francke U, Darras BT, Foellmer BE. Loss of IGF-1 receptor gene in patients with ring chromosome 15 is related to Russell-Silver like phenotype. Proceedings of the 9th Annual David W. Smith Workshop on Malformations and Morphogenesis; Oakland, California. August 1988.1988. [Google Scholar]
- Knoll JHM, Nicholls RD, Magenis RE, Graham JM, Lalande M, Latt S. Angelman and Prader-Willi syndromes share a common chromosome 15 deletion, but differ in parental origin of the deletion. Am J Med Genet. 1989;32:285–290. doi: 10.1002/ajmg.1320320235. [DOI] [PubMed] [Google Scholar]
- Kosztolányi G. Does “ring syndrome” exist? Analysis of 207 case reports on patients with a ring autosome. Hum Genet. 1987;75:174–179. doi: 10.1007/BF00591082. [DOI] [PubMed] [Google Scholar]
- Kotzot D, Schmitt S, Bernasconi F, Robinson W, Lurie I, Llyina H, Méhes K, Hamel B, Otten B, Hergersberg M, Werder E, Schoenle E, Schinzel A. Uniparental disomy 7 in Silver-Russell syndrome and primordial growth retardation. Hum Mol Genet. 1995;4:583–587. doi: 10.1093/hmg/4.4.583. [DOI] [PubMed] [Google Scholar]
- Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r) Cell. 1993;75:59–72. [PubMed] [Google Scholar]
- Malcolm S, Donlon TA. Report of the second international workshop on human chromosome 15 mapping. Cytogenet Cell Genet. 1994;67:2–14. doi: 10.1159/000133717. [DOI] [PubMed] [Google Scholar]
- Mbikay M, Linard CG, Sirois F, Lazure C, Seidah NG, Chrétien M. Tissue-specific expression of the prostatic secretory protein PSP94 in cyanomolgus monkey (Macaca fascicularis) Cell Mol Biol. 1988;34:387–398. [PubMed] [Google Scholar]
- Meloni R, Fougerousse F, Roudaut C, Beckmann JS. Trinucleotide repeat polymorphism at the human insulin-like growth factor I receptor gene (IGF1R) Nucleic Acids Res. 1992;20:1427. doi: 10.1093/nar/20.6.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowery-Rushton PA, Hanchett JM, Surti W. Identification of Prader-Willi syndrome mosaicism by fluorescence in situ hybridization. Am J Hum Genet. 55:113. 194. [Google Scholar]
- Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet. 1986;73:320–326. doi: 10.1007/BF00279094. [DOI] [PubMed] [Google Scholar]
- Nicholls RD. Genomic imprinting and uniparental disomy in Angelman and Prader-Willi syndromes: A review. Am J Med Genet. 1993;46:16–25. doi: 10.1002/ajmg.1320460106. [DOI] [PubMed] [Google Scholar]
- Nicholls RD, Knoll JH, Glatt K, Hersh JH, Brewster TD, Graham JM, Wurster-Hill D, Wharton R, Latt SA. Restriction fragment length polymorphisms within proximal 15q and their use in molecular cytogenetics and the Prader-Willi syndrome. Am J Med Genet. 1989a;33:66–77. doi: 10.1002/ajmg.1320330109. [DOI] [PubMed] [Google Scholar]
- Nicholls RD, Knoll JHM, Butler MG, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in non-deletion Prader-Willi syndrome. Nature. 1989b;342:281–285. doi: 10.1038/342281a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuutinen M, Kouvalainen K, Knip M. Good growth response to growth hormone treatment in the ring chromosome 15 syndrome. J Med Genet. 1995;32:486–487. doi: 10.1136/jmg.32.6.486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen MB, Bartsch O, Adelsberger PA, Mikkelsen M, Schwinger E, Antonarakis SE. Uniparental isodisomy due to duplication of chromosome 21 occurring in somatic cells monosomic for chromosome 21. Genomics. 1992;13:269–274. doi: 10.1016/0888-7543(92)90242-k. [DOI] [PubMed] [Google Scholar]
- Poduslo SE, Dean M, Kolch U, O’Brien S. Detecting high-resolution polymorphisms in human coding loci by combining PCR and single-strand conformation polymorphism (SSCP) analysis. Am J Hum Genet. 1991;49:106–111. [PMC free article] [PubMed] [Google Scholar]
- Roback EW, Barakat AY, Dev VG, Mbikay M, Chrétien M, Butler MG. An infant with deletion of the distal long arm of chromosome 15 (q26.1→qter) and loss of insulin-like growth factor I receptor gene. Am J Med Genet. 1991;38:74–79. doi: 10.1002/ajmg.1320380117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence JE, Percinccante RG, Greig GM, Huntington FW, Ledbetter DH, Hejtmancik JF, Pollack MS, Obrien WE, Beaudet AL. Uniparental disomy as a mechanism for human genetic disease. Am J Hum Genet. 1989;42:217–226. [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Tohma T, Ohta T. Ring chromosome 15 involving deletion of the insulin-like growth factor receptor gene in a patient with features of Silver-Russell syndrome. Clin Dysmorphol. 2:106–113. 193. [PubMed] [Google Scholar]
- Tantravahi U, Nicholls RD, Stroh H, Ringer S, Neve RL, Kaplan L, Wharton R, Wurster-Hill D, Graham JM, Cantu ES, Frias JL, Kousseff B, Latt SA. Quantitative calibration and use of DNA probes for investigating chromosome abnormalities in Prader-Willi syndrome. Am J Med Genet. 1989;33:78–87. doi: 10.1002/ajmg.1320330110. [DOI] [PubMed] [Google Scholar]
- Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Hentzell W, Le Bon T, Kathuria S, Chen E, Jacobs S, Francke U, Ramanchandran J, Fugita-Hamagushi Y. Insulin-like growth factor receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986;5:2501–2512. doi: 10.1002/j.1460-2075.1986.tb04528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JL, May PE. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet. 1989;44:388–396. [PMC free article] [PubMed] [Google Scholar]
- Wilson GN, Sauder SE, Bush M, Beitins IZ. Phenotypic delineation of ring chromosome 15 and Russell-Silver syndromes. J Med Genet. 1985;22:233–236. doi: 10.1136/jmg.22.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodage T, Prasad M, Dixon JW, Selby RE, Romain DR, Columbano-Green LM, Graham D, Rogan PK, Seip JR, Smith A, Trent RJ. Bloom syndrome and maternal uniparental disomy for chromosome 15. Am J Hum Genet. 1994;55:74–80. [PMC free article] [PubMed] [Google Scholar]