

Abstract
FoxO transcription factors serve as the central regulator of cellular homeostasis and are tumor suppressors in human cancers. Recent studies have revealed that, besides their classic functions in promoting cell death and inducing cell cycle arrest, FoxOs also regulate cancer metabolism, an emerging hallmark of cancer. In this review, we summarize the regulatory mechanisms employed to control FoxO activities in the context of cancer biology, and discuss FoxO function in metabolism reprogramming in cancer and interaction with other key cancer metabolism pathways. A deeper understanding of FoxOs in cancer metabolism may reveal novel therapeutic opportunities in cancer treatment.
Keywords: FoxO, cancer metabolism, tumor suppression, mTOR, Myc
Introduction
In order to maintain cellular homeostasis, metazoans have evolved an intricate signaling network which senses and adapts to the changes in the extracellular or intracellular environment. Successful adaptation and long-term survival under altered physiological conditions are dependent on the precise regulation of gene expression, which is mainly controlled by the specific recruitment of transcription factors (TFs) to the target gene promoters or enhancer regions. Therefore, TFs serve as the ultimate effector molecules and even define the fate of a cell [1]. The human genome encodes around 2,000 TFs, which regulate a diverse array of cellular processes ranging from cell division to cell death [2]. Dysregulation of TFs results in various pathological conditions including cancer. Indeed, TFs have often been found to be mutated, deleted, or amplified in many cancers, and therefore have been considered as attractive therapeutic targets for cancer treatment [3].
The forkhead box O (FoxO) family of TFs are the central regulator to the metazoan physiology and have diverse cellular functions including cell cycle, cell growth, apoptosis, autophagy, stress resistance, protection from aggregate toxicity, DNA repair, tumor suppression, and metabolism [4–8]. They have also been implicated in the regulation of organ development, stem cell maintenance, and cell differentiation, suggesting their crucial roles in development [9–11]. FoxOs belong to the superfamily of TFs known as forkhead box TFs and are characterized by the presence of an evolutionarily conserved winged-helix DNA binding motif and the forkhead domain [8]. The expression of FoxO target genes is regulated by selective recruitment of FoxOs to the consensus DNA sequence TTGTTAC and their interactions with other TFs [8]. FoxOs are evolutionarily conserved and have a single orthologue in invertebrates, such as DAF-16 in Caenorhabditis elegans and dFOXO in Drosophila melanogaster. In contrast, mammalian FoxOs consist of at least four members: FoxO1, FoxO3, FoxO4, and FoxO6. The expression of specific FoxO members in mammals varies among different tissues and is regulated in a spatiotemporal manner during various developmental stages [12, 13]. In addition, FoxO TFs sense the changes in the extracellular or intracellular environment and their activities are also regulated by different types of signaling stimuli, including growth factors that activate the phosphatidyl-inositol-3-kinases (PI3K)-AKT (also known as PKB) pathway and different stress signaling, such as oxidative stress [6]. Tight regulation of FoxO transcriptional activity by complex signaling networks ensures that specific gene expression switch coordinates with environmental cues. FoxOs have been implicated in various diseases, including cancer. FoxOs generally exert tumor suppression functions by promoting cell cycle arrest, apoptosis, stress resistance, and DNA repair in cancer cells, and are inactivated in various human cancers [4, 14]. In addition, FoxOs act as a central regulator of cellular metabolism and longevity [5, 15], thereby placing FoxOs at the crossroad of cancer and metabolism.
In this review, we first present a detailed discussion on the intricate regulatory mechanisms employed to fine-tune the transcriptional activities of FoxOs in cancer, followed by a discussion of the biological roles of FoxOs in tumor suppression. We then focus on the new insights in FoxO regulation of cancer metabolism. Finally, we discuss the cross talks between FoxOs and other important pathways/cellular processes involved in cancer metabolism. It is important to note that FoxO regulation of metabolism also plays pivotal roles in insulin resistance, diabetes, and obesity [7, 16, 17]. However, this topic will be beyond the scope of this review focusing on metabolic function of FoxOs in cancer.
Molecular mechanisms of FoxO regulation
FoxO TFs are regulated by the coordinated actions of multiple signaling pathways at several mechanistic levels, including regulation of FoxO transcriptional activity, cellular localization, protein stability, and mRNA levels (Fig. 1). These multiple modes of regulation ensure the condition-specific activation or inhibition of FoxOs. Notably, many of the FoxO regulators also play instrumental roles in cancer biology, and the regulatory modes to control FoxOs are often dysregulated in cancer.
Figure 1. FoxO regulation by growth factor and stress signaling.
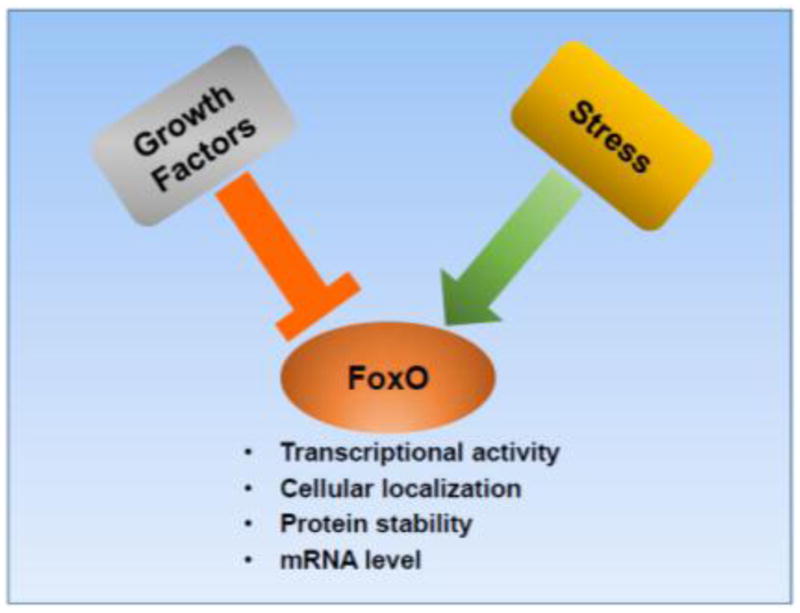
Growth factors inhibit FoxOs whereas stress signaling generally activates FoxOs. FoxOs are modulated at several mechanistic levels as indicated.
Regulation by growth factor-PI3K-AKT signaling
Classically, FoxO activity is regulated via an evolutionarily conserved pathway that involves the negative regulation of FoxOs by growth factor-PI3K-AKT pathway. Under normal physiological conditions, binding of insulin or other growth factors to receptor tyrosine kinases (RTKs) results in RTK activation through autophosphorylation. Activated RTK recruits PI3K via phosphotyrosine sites in the cytoplasmic tail of RTKs or adaptor proteins. This recruitment leads to activation of PI3K, which in turn catalyzes the production of lipid second messenger phosphatidylinositol-3,4,5-triphosphate (PIP3) from its substrate phosphatidylinositol-4,5-bisphosphate (PIP2) [18, 19]. Generation of PIP3 marks the initiation of a signaling cascade that regulates various aspects of cellular physiology including cell cycle, cell survival, metabolism, apoptosis, and DNA repair. Specifically, PIP3 functions as a docking site for pleckstrin homology (PH) domain-containing proteins including AKT and phosphoinositide-dependent kinase-1 (PDK1) [20]. Recruitment of PDK1 and AKT to the plasma membrane facilitates partial activation of AKT via phosphorylation at Thr308 by PDK1. In addition, phosphorylation at Ser473 on AKT by mammalian target of rapamycin complex 2 (mTORC2, also known as mechanistic target of rapamycin complex 2) leads to full activation of AKT [21]. AKT activation can be reversed by the action of phosphatase and tensin homolog (PTEN), which functions as a lipid phosphatase to convert PIP3 back to PIP2 [22].
Key mechanisms of FoxO regulation were first identified in 1999. A series of classical experiments demonstrated that AKT phosphorylates FoxOs at three residue (Thr24, Ser256, Ser391 for FoxO1, Thr32, Ser253, Ser315 for FoxO3, and Thr28, Ser193, Ser258 for FoxO4), and this phosphorylation event negatively regulates FoxO nuclear localization, thereby preventing FoxO association with TF binding sites on DNA and inhibiting its transcriptional activity [23–27]. One current model proposes that AKT-mediated FoxO phosphorylation prevents FoxO association with DNA by facilitating their binding to nuclear 14-3-3 protein, resulting in enhanced export from nucleus and diminished re-entry into nucleus possibly by masking the nuclear localization signal (NLS) in FoxOs [28]. Aside from AKT, FoxOs is also inactivated by phosphorylation via AKT-related serum and glucocorticoid-inducible kinase (SGK), which, similar to AKT, can be activated by PDK1-mediated phosphorylation [29]. Although both AKT and SGK share similar FoxO phosphorylation sites, they show preferential activity at selective residues [29]. In contrast to other FoxO members discussed above, the PI3K-AKT pathway fails to prevent FoxO6 nuclear shuttling due to a lack of carboxyl-terminal AKT-dependent phosphorylation sites in FoxO6. However, phosphorylation at the other two residues in FoxO6 disrupts its binding affinity towards DNA, leading to its inactivation [30, 31].
Regulation by AKT-independent phosphorylation
In contrast to growth factor-PI3K-AKT signaling which induces the nuclear exclusion of FoxOs, cellular oxidative stress generally promotes nuclear localization of FoxOs, which then mediates antioxidant response by regulating the expression of key antioxidant genes such as catalase, superoxide dismutases (SODs), and sestrins [5]. Nuclear translocation of FoxOs following oxidative stress is mediated by an evolutionarily conserved signaling that involves c-Jun N-terminal kinase (JNK) [32, 33]. Specifically, following oxidative stress, JNK phosphorylates FoxO4 at Thr447 and Thr451, resulting in its nuclear translocation even in presence of the PI3K-AKT signaling [34]. JNK can also indirectly regulate FoxO activity by dissociating FoxO from 14-3-3 via phosphorylating 14-3-3 at Ser184 [35]. Nuclear translocation of FoxOs and expression of pro-apoptotic genes following oxidative stress can also be mediated by mammalian sterile 20-like kinase-1 (MST1). MST1 phosphorylates FoxO3 at Ser207, causing disruption of FoxO3 interaction with 14-3-3 [36]. Interestingly, the upstream signaling resulting in MST1 activation might also involve its phosphorylation at Ser82 by JNK [37].
FoxOs also serve as a key component of the nutrient sensing circuit and have been implicated in extending lifespan along with dietary restriction. AMP-activated protein kinase (AMPK) is a central regulator of energy homeostasis. High cellular AMP to ATP ratio following energy deprivation results in AMPK activation [38, 39]. Activated AMPK then phosphorylates a variety of substrates promoting cellular catabolism and simultaneously inhibiting cellular anabolism [38]. AMPK activation also alters the expression of many genes to adapt to energy stress partly through FoxOs. Specifically, AMPK phosphorylates FoxO3 at six different residues (Thr179, Ser399, Ser413, Ser439, Ser555, and Ser588), causing upregulation of genes involved in antioxidant response and energy utilization pathways [40]. In contrast to the kinases described above that often affect nuclear-cytoplasmic shuttling of FoxOs, AMPK-mediated FoxO3 phosphorylation does not affect FoxO3 cellular localization, but may regulate FoxO3 binding on the target genes under energy stress [40]. Notably, AMPK-mediated FoxO phosphorylation also plays a role in FoxO regulation of organismal longevity following dietary restriction in C. elegans, highlighting that this is an evolutionarily conserved mechanism [41]. Regulation of hepatic gluconeogenesis following prolonged starvation was also found to be mediated by AMPK-FoxO signaling. Specifically, TGF-β/Smad3 signaling inhibits AMPK phosphorylation and promotes FoxO1 activation, resulting in upregulation of gluconeogenic genes [42].
Several other kinases, including cyclin-dependent kinase (CDK), Ataxia telangiectasia mutated (ATM), ATM and Rad3-related (ATR), dual-specificity tyrosine-phosphorylated and regulated kinase 1A (DYRK1A), IκB kinase (IKK), and MAPKs, can also target FoxOs in response to various signaling cues. Different CDKs have been shown to phosphorylate FoxOs. CDK2 inhibits FoxO activity via phosphorylation at Ser249 residue, resulting in its nuclear exclusion. However, this inactivation is reversed in response to DNA damage with inhibition of CDK2 activity [43]. In neuronal cells FoxO1 phosphorylation by CDK1 abolishes FoxO1 interaction with 14-3-3 proteins, resulting in FoxO1 nuclear translocation and gene expression, whereas in prostate cancer cells CDK1-mediated phosphorylation of FoxO1 abrogated its transcriptional activity [44, 45], thus it seems that CDK1 regulation of FoxO1 is cell type specific. In addition, recently it has been shown that CDK6 phosphorylates and stabilizes FoxO3, resulting in unresponsiveness of epithelial ovarian cancer cells to platinum therapy [46]. FoxO activity is also regulated by kinases of MAPK family such as p38. p38 phosphorylates FoxO3 in response to DNA damage or energy stress [47, 48]. p38 physically interacts with FoxO3 and phosphorylates FoxO3 at Ser7 after treatment with doxorubicin, preventing FoxO3 cytoplasmic localization [47]. FoxO3 can also be phosphorylated by other kinases involved in MAPK signaling, including MAPK-activated protein kinase 5 (MK5) [49]. ATM and ATR are the key components involved in DNA damage response, in which DNA damage activates ATM and ATR to initiates downstream signaling cascades responsible for DNA repair. FoxOs have been identified as a substrate of ATM/ATR [50]; however, the physiological consequence of this interaction is not clear. Interestingly, it has been shown that FoxOs also promote ATM phosphorylation and regulate ATM expression [51–53], suggesting a reciprocal regulation between FoxOs and ATM.
Another cell survival factor IKK has also been shown to interact with and phosphorylate FoxO3 [54]. Phosphorylation of FoxO3 by IKK marks FoxO3 for ubiquitin-dependent proteasomal degradation, thus promoting cell proliferation. Apart from targeting FoxO3 for degradation, IKK can also regulate FoxO3 cytoplasmic retention in acute myeloid leukemia (AML) [55]. Similarly, DYRK1A can phosphorylate FoxO1 and FoxO3 thereby inhibiting their nuclear translocation and target gene expression [56]. Finally, intracellular protein homeostasis requires complex equilibrium between protein synthesis, folding, and degradation, and defective protein homeostasis compromises cell survival. For example, disturbance in the equilibrium of protein synthesis and folding causes endoplasmic reticulum (ER) stress and triggers the ER-associated degradation (ERAD) pathway [57]. ER stress enhances FoxO activities partly through protein kinase RNA-like endoplasmic reticulum kinase (PERK) [58]. In contrast to AKT-mediated FoxO phosphorylation, PERK-mediated FoxO phosphorylation promotes FoxO activities.
In brief summary, FoxOs can be phosphorylated and regulated by a diverse array of kinases that sense specific environmental cues, such as growth factors or different types of stress (Fig. 2). In general, kinases activated by growth factors that promote tumor development (such as AKT) inhibit FoxO function, whereas stress-activated kinases that often (but not always) exert tumor suppressive functions (such as JNK, AMPK, and PERK) promote FoxO function. FoxO phosphorylation controls FoxO function through three major mechanisms, namely regulating FoxO nuclear localization, transcriptional activity, and protein stability. Below we will further discuss the regulation of FoxO stability by ubiquitin-proteasome degradation.
Figure 2. FoxO regulation by post-translational modifications.

Multiple pathways converge to regulate FoxO activity. Growth factor signaling inhibits FoxO activity by promoting AKT activation. AKT-mediated inactivation of FoxOs is antagonized by stress-induced FoxO activation through various stress-induced kinases. FoxO activity is also modulated by a plethora of other kinases in response to particular stimuli such as DNA damage. Other post-translational modifications such as ubiquitylation, acetylation and methylation also regulate FoxO activity.
Regulation by ubiquitin-proteasome degradation
Ubiquitin-proteasome degradation system regulates the half-lives of proteins under normal and stress conditions [59]. FoxO protein level is also subjected to regulation via ubiquitin-proteasome degradation system (Fig. 2). Classical regulation of FoxOs by AKT can also promote FoxO degradation via skp1/cul1/F-box protein (SCF) ubiquitin ligase complex. Skp2, the F-box protein in the SCF complex, interacts with and degrades FoxO1 upon FoxO1 phosphorylation by AKT; therefore AKT signaling maximizes FoxO inhibition by degrading FoxO apart from promoting its nuclear exclusion [60–62]. Another upstream FoxO kinase ERK can also regulate FoxO degradation via murine double minute 2 (MDM2), an E3 ubiquitin ligase [63]. MDM2 interacts with and ubiquitinates FoxO3 following ERK-mediated phosphorylation of FoxO3 at Ser 294, Ser 344 and Ser 425 [63]. MDM2 can also sense FoxO phosphorylation by AKT to promote FoxO degradation [64]. C-terminus of Hsc70-interacting protein (CHIP) is a dual-function co-chaperone and ubiquitin ligase. The ubiquitin ligase activity of CHIP is responsible for FoxO1 degradation in response to TNF-α in smooth muscle cells. Interaction of CHIP with FoxO1 is mediated by FoxO1 phosphorylation at Ser256 [65]. Finally, insulin signaling can also promote FoxO degradation via another E3 ubiquitin ligase, COP1. Insulin upregulates the expression of COP1, which in turn interacts with and polyubiquitinates FoxO1, resulting in its degradation. However, the role of insulin-induced FoxO phosphorylation in COP1-FoxO1 interaction remains less clear [66]. In summary, FoxO stability can be regulated by ubiquitin-proteasome degradation system via various ubiquitin ligases that interact with FoxOs and promote FoxO degradtion, and the interactions between ubiquitin ligases and FoxOs are often modulated by FoxO phosphorylation by upstream kinases in response to particular stimuli.
Regulation by other post-translational modifications
In addition to phosphorylation and ubiquitination, upstream regulators of FoxO can modify FoxO activity by other post-translational modifications including acetylation, methylation, glycosylation, and hydroxylation (Fig. 2). Acetylation of cellular proteins is an important post-translational modification covering 80–90% of the total cellular proteome [67]. FoxOs interact with different cellular acetyltransferases and deacetylases, and are subjected to acetylation. Acetylation of FoxOs affects FoxO function in a dual manner, by altering FoxO binding on DNA as well as FoxO nuclear exclusion [68, 69]. Unlike other post-translational modifications, FoxO acetylation can result in different cellular outputs. For example, p300/CBP can interact with and acetylate FoxO1, resulting in enhanced FoxO1 transcriptional activity [70]. However, FoxO4 acetylation by CBP inhibits its transcriptional activity [71]. Acetylation is a reversible process that can be erased by deacetylases. At least three members of the sirtuin family (namely SIRT1, SIRT2, and SIRT3) have been shown to deacetylate FoxOs. SIRT1 was the first sirtuin that had been identified as FoxO deacetylase [71] [72]. FoxO3 is acetylated in response to oxidative stress resulting in its binding to SIRT1. SIRT1 then deacetylates FoxO3 to induce cell cycle arrest but prevent cell death. This dual regulation of FoxO to differentially regulate cell cycle and cell death by SIRT1 can be attributed to its altered DNA binding ability [68]. Similar to SIRT1, SIRT2 and SIRT3 can also deacetylate FoxOs, causing cell type-specific regulation of gene expression [73–75]. Finally, a recent study showed that HDAC3 can be recruited to FoxO3 by Geminin. HDAC3 then deacetylates FoxO3 and inhibits FoxO3 transcriptional activity [76].
FoxOs were also shown to be methylated by different methyltransferases. Protein arginine methyltransferase 1 (PRMT1) can transfer methyl group from S-adenosylmethionine (SAM) to conserved residues Arg248 and Arg250 on FoxO1. These methylated arginines are present in the conserved phosphorylation motif for AKT, therefore attenuating AKT-mediated FoxO phosphorylation and inhibition [77]. Oxidative and other cellular stresses that facilitate FoxO nuclear translocation result in increased FoxO methylation to prevent FoxO phosphorylation by AKT [77]. In oppose to FoxO methylation at arginine residues, FoxO3 methylation by SET9 inhibits its DNA binding activity and transactivation [78, 79]. In addition, FoxOs were reported to be glycosylated, in which FoxO1 O-glycosylation promotes its transcriptional activity without affecting its nuclear-cytoplasmic localization, although the exact mechanism underlying O-glycosylation regulation of FoxO transcriptional activity is unclear [80, 81]. In addition to these modifications, FoxOs can also be regulated by prolyl hydroxylation. Specifically, proline residues in FoxO3 (Pro426 and Pro437) were shown to be hydroxylated by EglN prolyl hydroxylases 2 (EglN2). This prolyl hydroxylation of FoxO3 abrogates its interaction with USP9x deubiquitinase, resulting in the destabilization of FoxO3 [82].
Regulation by transcriptional and posttranscriptional mechanisms
Although many studies have focused on the regulation of FoxO activities as a consequence of different post-translational modifications, mRNA levels of FoxOs can also be altered depending on different environmental cues or developmental stages. Recent investigations have identified different transcription factors that regulate FoxO transcription, including E2F-1, p53, hypoxia-inducible factor-1α (HIF-1α), and FoxO itself. E2F-1, a member of E2F family of transcription factors was identified to upregulate the expression of FoxO1 and FoxO3 [83]. E2F-1 binding to the FoxO promoter is cell type- and tissue-specific and upregulates FoxO expression following ischemia/reperfusion injury to promote cell death [84]. Tumor suppressor p53 and its homolog p73 can also occupy FoxO promoter region and upregulate its expression. p53 was shown to be activated in response to DNA damage and specifically activate FoxO3 transcription [85, 86]. FoxO promoter also contains HIF-1α binding sites. It has been show that HIF-1α drives FoxO transcription in response to hypoxic stress to inhibit HIF-1α-induced apoptosis as a negative feedback loop [87]. Surprisingly, FoxOs can also transcriptionally activate other members of the FoxO family as demonstrated by that FoxO3 can upregulate the expression of FoxO1 and FoxO4 [88]. All three FoxOs were downregulated upon growth factor treatment, possibly through the inhibition of FoxO3 by growth factor-induced FoxO3 phosphorylation [88]. However, this forward activation loop can be context dependent as FoxO3 inhibits FoxO1 transcription in prostate cancer cells [89]. Interestingly, MYST family histone acetyltransferase complex (MYS-1/TRR-1 complex) also promotes DAF-16 expression by histone acetylation at its promoter region [90].
mRNA stabilization by different RNA binding proteins has emerged as another regulatory mechanism to control FoxO mRNA levels. HuR, an RNA binding protein, has been shown to stabilize many target mRNAs including FoxO1. 3′ untranslated region (3′UTR) of FoxO1 mRNA is enriched with HuR binding sites, which in turn recruit HuR to stabilize FoxO1 mRNA [91]. In contrast, another RNA binding protein Quaking (QKI) represses FoxO1 post-transcriptionally through binding to FoxO1 3′UTR [92]. Other mechanisms governing FoxO at the post-transcriptional levels include FoxO regulation by non-coding RNAs including micro RNA (miRNAs) and long non-coding RNA (LncRNAs). Various miRNAs including miR-27a, miR-96, miR-182, miR-9, miR-27, miR-96, miR-153, miR-182, miR-183, miR-186, miR-139, miR-486, and miR-155 can target expression of different FoxO members [6]. Another class of non-coding RNA lncRNAs can also affect FoxO expression levels. For example, linc00598 was found to interact with FoxO1, regulate the transcriptional activity of FoxO1, and in turns regulate the expression of cell cycle regulator cyclin D2 [93]. Another lncRNA lncFoxO1 has been shown to function as a breast cancer suppressor by increasing the expression of FoxO1 [94]. Finally, lncRNA H19, an important regulator of myogenesis, was demonstrated to negatively regulate Sirt1/FoxO to promote myoblast differentiation [95].
FoxOs in cancers
The relevance of FoxOs to human cancers was initially realized when FoxOs were identified as fusion products that resulted from chromosomal translocation in different cancers, including FoxO1 in alveolar rhabdomyosarcomas, and FoxO3 and FoxO4 in AML [96–98]. Since then, FoxOs have been implicated in the pathogenesis of various cancers. FoxO generally function as tumor suppressors in cancers, and the tumor suppressive roles of FoxOs are supported by three major lines of evidence. First, FoxOs are found to be deleted or inactivated in many human cancers. For example, both FoxO1 and FoxO3 are deleted in 15% to 20% prostate cancer, indicating their tumor suppressive function in prostate cancer [99]. FoxOs are also extensively down-regulated or inactivated in human cancers by numerous regulatory mechanisms as discussed in the previous session. It should be noted that FoxOs are rarely mutated in human cancers. The potential reason for this will be further discussed below. Second, studies from genetically engineered mouse models (GEMMs) have revealed that deletion of FoxOs in vivo leads to tumor prone phenotypes in different contexts [100, 101]. Importantly, these studies revealed that FoxOs are redundant tumor suppressors in vivo, such that deletion of all six alleles of FoxO1, 3, and 4 is required to engender dramatic tumor phenotypes in vivo [100]. This might provide a possible explanation to the lack of FoxO mutations in human cancers: given the redundant tumor suppressive function of FoxOs in cancer, it will be very difficult to simultaneously select loss-of-function mutations in all three FoxO members during tumor development. In contrast, FoxOs are usually inactivated through various post-translational mechanisms in cancers, as the regulatory mechanisms to control FoxO activities are often similar among different FoxO members. For example, hyperactivation of the PI3K-AKT signaling in cancers results in the inactivation of FoxO1, 3, and 4 simultaneously. Finally, many cell line studies have revealed that FoxOs function to limit various hallmarks of cancer. In this session, we will briefly discuss the roles of FoxOs in limiting several traditional hallmarks of cancer, including inhibiting cell proliferation, inducing apoptosis and senescence, and limiting angiogenesis and invasion. In later sessions, we will further discuss the roles of FoxOs in restraining tumor metabolism.
FoxO TFs can directly regulate the expression of components involved in both intrinsic and extrinsic apoptosis pathways to promote cell death, and this pro-cell death role is partly responsible for the tumor suppressive functions of FoxOs. FoxO can activate the extrinsic arm of apoptosis by promoting the expression of Fas ligand (FasL), TNF-related apoptosis-inducing ligand (TRAIL), and TNF receptor-associated death domain (TRADD) [102]. Expression of TRAIL can be attenuated by the transcriptional repressor NFIL3 (nuclear factor interleukin 3-regulated), which prevents FoxOs from binding the TRAIL promoter [103]. FoxO activation can also induce the intrinsic apoptosis by regulating the expression of Bcl-2 family members including Bim, BNIP3 and Puma [48, 104, 105]. FoxO regulation of various components in apoptosis pathways play critical roles in chemotherapy response. For example, FoxO-mediated TRADD expression sensitizes lung cancer cells to chemotherapeutic drugs [106]. In breast cancer cells, FoxO regulation of Bim expression modulates cancer cell sensitivity to Paclitaxel [107].
The ability of FoxO TFs to restrict tumor growth is also attributed to their roles in cell cycle arrest. Upregulation or activation of FoxOs can result in cell cycle arrest at G1-S or G2-M transition. Cyclins and CDKs complexes are the crucial regulators of the cell cycle progression, and their activities are monitored by the CDK inhibitors. FoxO TFs can drive the expression of CDK inhibitors including p27kip1, p21Cip1, p15, p19, and p130 to prevent G1-S transition [102]. Recently, it has been shown that FoxO TFs also regulate the expression of C-terminal domain small phosphatase 2 (CTDSP2), which in turn promotes the expression of several cell cycle regulators including p21 Cip1in a phosphatase dependent manner[108]. In addition, FoxO TFs can stimulate cell cycle arrest by suppressing the expression of cyclin D1 and D2 [109, 110]. Finally, FoxO-mediated cell cycle arrest at G2-M transition involves the up-regulation of the growth arrest and DNA damage-inducible protein 45 (GADD45)[111].
Induction of cellular senescence is another mechanism through which FoxOs suppress tumor growth. It has been shown that melanoma cells carrying BRAFV600E mutation undergo cellular senescence partly through FoxO4-mediated upregulation of p21cip1 [112]. FoxO3 has been reported to initiate cellular senescence by inducing the expression of vitamin D3 up-regulated protein 1 (VDUP1) in fibroblast cells [113]. However, other reports also revealed a senescence-antagonistic role of FoxOs. For example, FoxO inhibition via constitutive activation of AKT triggers cellular senescence in human endothelial cells [114], whereas overexpression of FoxO3 suppresses cellular senescence by increasing the expression of its downstream targets SOD and catalase as well as inhibiting ROS generation [115]. Thus, it seems that FoxOs regulate cellular senescence in a context-dependent manner.
Sustained angiogenesis is a crucial requirement for the physically constrained tumor cells to acquire oxygen and nutrients during tumor invasion and metastasis. It has been demonstrated that overexpression of FoxO1 and FoxO3 inhibits angiogenesis by restricting endothelial cell migration and tube formation, whereas their knockdown reverses these effects [116]. In addition, cytoplasmic restriction and inhibition of FoxO1 in endothelial cells alters metabolism, resulting in enhanced proliferation in a Myc-dependent manner [117]. Recent studies have also revealed important roles of FoxOs in regulating cancer cell metastasis. In hepatocellular carcinomas, the expression of FoxO1 is inversely correlated with the intracellular levels of the key players of epithelial-to-mesenchymal transition (EMT), a crucial process involved in cancer metastasis [118]. In line with this, in clear-cell renal cell carcinoma (ccRCC), downregulation of FoxO3 results in SNAIL1-dependent induction of EMT and metastasis [119]. Crosstalk between ERK and PI3K signaling via MEK1/2 and AKT interaction also alters FoxO1 activation and metastasis [120]. In breast cancer, HDAC3-mediated FoxO3 deacetylation blocks FoxOs transcriptional activity, resulting in downregulation of FoxO3 target Dicer and promotion of breast cancer metastasis [76].
While most studies support a tumor suppressive function of FoxOs in cancer, other studies have also revealed a tumor promoting function of FoxOs in certain contexts. First, FoxOs play important roles in maintaining cancer stem cells in several cancers. Specifically, it has been shown that FoxOs regulate the maintenance of leukemia-initiating cells (LICs) in CML and AML [121]. LICs are the leukemic stem cells that are implicated in the reoccurrence or drug resistance in CML and AML. It was reported that LICs are enriched with decreased AKT activation and increased nuclear localization of FoxO3, and FoxO3 deficiency significantly inhibits the ability of LICs to generate CML [122]. Another study revealed that proto-oncogene BCL6 serves as an important downstream effector of FoxOs in LIC maintenance in CML, and BCL6 regulates LIC maintenance by repressing Arf and p53 in CML cells [123]. Similarly, FoxOs were found to be active in AML patients and FoxO deletion or AKT activation inhibited leukemic cell growth and significantly decreased LIC function in vivo [124]. Apart from their role in maintaining cancer stem cells, FoxO TFs can also exhibit pro-tumorigenic effects by mediating drug resistance in various cancers. For example, FoxO1 can confer drug resistance towards adriamycin and doxorubicin by inducing the expression of multidrug resistance 1 (MDR1) protein [125]. Feedback regulation of PI3K activation by FoxO3 confers the doxorubicin resistance and is mediated by the upregulating the expression PI3K catalytic subunit p110alpha [126]. Notably, cancer cells adapt to drugs inhibiting the PI3K-AKT pathway by FoxO-mediated reactivation of PI3K-AKT [127–129]. For example, FoxOs promote, rather than inhibit, renal tumor growth in response to PI3K or AKT inhibitor treatment by upregulating Rictor and promoting AKT activation [39]. In addition, FoxO3 can also promote tumor invasion by upregulating the expression of matrix metalloproteinases (MMP-9 & MMP13), key components of the proteolytic machinery responsible for extracellular matrix remodeling and tumor invasion [130]. Finally, FoxOs can also promote the expression of Isocitrate dehydrogenase 1 (IDH1) and its mutant form, a key metabolic enzyme involved in the conversion of isocitrate to α-KG and NADPH [131]. Mutation in the IDH1 gene has been linked with the generation of oncometabolite 2-hydroxyglutarate that promotes tumor development. Thus, in this particular context, FoxOs promote cancer cell proliferation by upregulating oncometabolite.
In summary, FoxOs generally function as tumor suppressors due to their roles in promoting apoptosis and inhibiting cell cycle progression, angiogenesis, and metastasis. However, under certain contexts FoxOs might also promote tumor development by maintaining cancer stem cells, promoting drug resistance, or reactivating the PI3K-AKT pathway through negative feedback regulation (Fig. 3).
Figure 3. Role of FoxO TFs in cancer biology.

(A) FoxOs mainly function as tumor suppressors, and inhibit tumor development through repressing various hallmarks of cancer. (B) In certain contexts, FoxOs can also promote tumor growth by maintaining cancer stem cells, inducing drug resistance, and reactivating the PI3K-AKT pathway.
Roles of FoxOs in cancer metabolism
Metabolic rewiring in cancer cells has been recognized as a new hallmark of cancer in recent years [132]. In order to support their increased biosynthetic and bioenergetic needs, to maintain the redox balance, and to survive under metabolic stress conditions resulting from tumor growth and poor vasculature within tumor mass, cancer cells extensively remodel their metabolic networks, partly through reprogramming gene transcription [133]. For example, many cancer cells upregulate the expression of glucose transporters, most notably GLUT1, to facilitate glucose uptake and energy generation via anaerobic glycolysis. Recent studies have highlighted the important roles of FoxO TFs in the regulation of cancer cell adaptation under metabolic stress conditions. In this session, we will focus on the roles of FoxOs in glucose, amino acid, lipid and ROS metabolism in the context of cancer biology.
Glucose is a key nutrient required to fulfill the major energy demand as well as to support biosynthetic pathways in cancer cells [134]. It has been well documented that glucose metabolism is significantly reprogrammed in most cancer cells. While normal non-proliferating cells mainly utilize oxidative phosphorylation to generate ATP under aerobic conditions and only extensively utilize glycolysis for energy generation under nonaerobic conditions, cancer cells switch to use glycolysis for ATP generation even under aerobic conditions [135]. This observation, now best known as the Warburg effect, was originally noticed by Otto Warburg almost a century ago, and since then, has been confirmed by numerous studies [135]. The current model proposes that cancer cells utilize the Warburg effect to direct a significant proportion of glucose to other biosynthetic pathways, including pentose phosphate, hexosamine, and amino acid anabolism pathways, which are essential to meet the increased biosynthetic needs and to support cell growth and survival in cancer cells. Although glycolysis is a much less efficient way for ATP production than oxidative phosphorylation, cancer cells in general are not limited with ATP generation due to increased glucose uptake [136]. It is well documented that several major oncogenes promote the Warburg effect, among which Myc probably is the most notable example. Myc is a master transcription factor of glucose metabolism and upregulates the transcription of essentially all metabolic enzymes involved in glycolysis pathway [137]. In contrast, various studies have revealed that FoxOs can inhibit glycolysis and suppress the Warburg effect partly through FoxO-mediated antagonism with Myc (Fig. 4) (the detailed molecular mechanisms by which FoxO antagonize with Myc will be discussed in the next session). For example, in renal cancer cells FoxOs inhibit the expression of glycolysis genes and glucose uptake and lactate production partly via inhibiting Myc [138]. Mechanistically, in response to energy stress, FoxOs upregulate the expression of a lncRNA called FoxO-induced long non-coding RNA 1 (FILNC1), which then suppresses Myc mRNA translation and inhibits Myc-mediated energy metabolism in renal cancer cells. FILNC1 knockdown upregulates the expression of several glycolysis genes and promotes glucose uptake and lactate production via Myc, resulting in enhanced renal cancer cell survival under glucose starvation and markedly increased renal tumor development [138]. In glioblastoma, mTORC2 inhibits FoxOs through FoxO acetylation, resulting in Myc activation and upregulation of the Warburg effect [139]. In line with this, in vivo studies with inducible deletion or overexpression of FoxO1 in mice revealed that FoxO1 represses glycolysis and mitochondrial respiration through inhibiting Myc [117]. Conversely, FoxO1 promotes gluconeogenesis, the metabolism pathway that generates glucose from non-carbohydrate substrates such as lactate and glucogenic amino acids, by upregulating key metabolic enzymes involved in gluconeogenesis including glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK) [140–142]. The roles of FoxO in gluoconeogenesis have been extensively studied in the context of liver metabolism and insulin resistance, although its relevance in cancer biology remains less understood.
Figure 4. FoxOs modulate cancer metabolism via multiple mechanisms.

FoxOs can either promote or inhibit various metabolism processes in cancer cells as indicated.
Amino acids are important for tumor cell proliferation, growth, and survival, as they provide the building blocks for proteins. In addition, some amino acids, such as serine, glycine, glutamine, and aspartate, contribute to nucleotide biosynthesis, which is required for sustaining high proliferative demand of tumor cells [143]. Glutamine is the most abundant amino acid in blood and cell culture medium, and arguably, the most important amino acid in supporting tumor growth. Once uptaken into cells, glutamine is converted into glutamate, which then serves as the precursor for several important metabolism processes critical for tumor growth, including tricarboxylic acid (TCA) cycle, biosynthesis of nucleotide, glutathione, and other nonessential amino acids. Glutamate can be converted back to glutamine by glutamine synthetase [144]. Various enzymes involved in glutamine metabolism have been reported to be significantly altered in various cancers. Transcriptional network analysis of the PI3K-AKT-FoxO pathway revealed that FoxO3 and FoxO4 regulate the expression of glutamine synthetase (Fig. 4). Activation of FoxO TFs results in mTOR inhibition by regulating its lysosomal translocation via glutamine synthetase, therefore promoting cellular autophagy [145]. FoxO1-induced expression of glutamine synthetase has also been shown in vivo from FoxO1 KO mouse model and a transgenic mouse model with FoxO1 overexpression in skeletal muscle [146]. It was shown that FoxO1 overexpression increased whereas FoxO1 deletion decreased the expression level of glutamine synthetase in skeletal muscle. Glutamine synthetase mediates the condensation reaction of glutamate and ammonia to form glutamine. Consistent with this, FoxO1 deletion increased blood ammonia concentration, potentially by decreasing glutamine synthetase expression [146]. Amino acids also in turn regulate FoxO activities. For example, extracellular amino acids inactivate FoxOs via activation of mTORC2 and AKT in mammalian cells [147]. In addition, it has been shown that in Drosophila melanogaster amino acid starvation increases FoxO activities and FoxO deficiency results in reduced survival of both larvae and adult files under amino acid starvation conditions [148].
Lipids are an essential component of mammalian cells and play a crucial role in the maintenance of cellular physiology by compartmentalizing cellular components, serving energy source and functioning as secondary messengers [149]. Many cancer cells reprogram lipid metabolism partly by promoting fatty acid synthesis from acetyl-CoA, a key metabolite generated from the TCA cycle intermediate citrate [133, 150]. Sterol regulatory element-binding proteins (SREBPs) are a family of transcription factors that function as the master regulator of lipid metabolism by regulating the expression of genes involved in fatty acid and cholesterol biosynthesis [151]. Recently it has been shown that FoxO1 can negatively regulate the expression of SREBP1c by occupying the SREBP1c promoter and disrupting the assembly of the transcriptional initiation complex on the SREBP-1c promoter [152]. In addition, FoxO1 can promote lipolysis by upregulating the expression on key lipolytic enzyme adipose triacylglycerol lipase (ATGL) (Fig. 4) [153]. However, the relevance of FoxO regulation of lipogenesis and lipolysis to cancer biology still remains largely unknown.
Under normal physiological conditions, intracellular reactive oxygen species (ROS) levels are maintained by an intricate ROS detoxification system, which involves both non-enzymatic (glutathione, vitamins, and flavonoids) and enzymatic (catalase, superoxide dismutase, and glutathione peroxidase) mechanisms [154]. Cancer cells often exhibit aberrant ROS levels due to their altered metabolic activities, and ROS can either promote or inhibit tumor development in a context-dependent manner [155, 156]. As discussed above, oxidative stress induces FoxO nuclear translocation and activates FoxO-mediated transcription through various stress-activated kinases such as JNK. In response to oxidative stress, FoxOs in turn induce the expression of classic target genes involved in ROS detoxification, such as manganese superoxide dismutase (MnSOD), catalase, and sestrins [5], by which FoxOs confer cell resistance to oxidative resistance (Fig. 4). It has also been shown that FoxO3 downregulates the expression of nuclear-encoded genes with mitochondrial function by inhibiting c-Myc, thereby decreasing intracellular ROS levels [157]. FoxO regulation of transcriptional targets involved in ROS detoxification requires FoxO coordination with other transcription factors that often also play important roles in tumor biology. For example, FoxO1 interacts with Yes-associated protein (YAP), a downstream effector of the Hippo pathway that regulates organ size and tumor suppression, and this interaction is critical for FoxO upregulation of MnSOD and catalase expression during oxidative stress response [158]. While there are extensive studies revealing the important roles of FoxO-mediated ROS detoxification in promoting longevity [6], the exact roles of FoxO in oxidative stress response in cancer biology remain less clear.
FoxO crosstalk with other major cancer metabolism pathways
Altered cellular metabolism now is widely recognized as a hallmark of cancer [132]. However, except a few notable examples (such as IDH1 mutations in gliomas and AML [159]), metabolic enzymes are generally not mutated in human cancers. Instead, deregulated cellular metabolism in cancer is mainly driven by cancer signaling pathways that reprogram metabolism networks in cancer cells, prominent among which are the PI3K-AKT pathway, the LKB1-AMPK pathway, mTOR, and several transcription factors such as Myc [135]. In the preceding sessions, we have already discussed the biochemical mechanisms by which FoxOs are regulated by some of the major cancer metabolism pathways, including the PI3K-AKT and AMPK pathways. In this session, we will focus on discussing how FoxOs interact with and control other cancer metabolic pathways, including mTOR and Myc, as well as autophagy.
FoxO and mTOR
mTOR is the master regulator of metabolic homeostasis [160]. Extracellular and intracellular nutrients, such as amino acids and glucose, can be sensed by mTOR, which in turn modulates various downstream targets to regulate cellular metabolism and growth [160]. mTOR exists as two distinct multiprotein complexes based on their composition, namely mTOR complex 1 (mTORC1) and mTORC2. mTORC1 consists of mTOR, Raptor, mLST8, PRAS40 and Deptor, whereas mTORC2 is composed of mTOR, Rictor, mSIN1, Protor1, mLST8 and Deptor [160]. mTORC1 promotes cell growth and proliferation by phosphorylating downstream targets including eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and p70 ribosomal S6 kinase 1 (S6K1). mTORC2, on the other hand, regulates cell survival and cytoskeleton reorganization through phosphorylating AKT and other substrates [160]. While how mTORC2 is controlled by upstream signaling still remains less understood, the mechanisms by which mTORC1 is regulated by various upstream stimuli have been extensively characterized. Briefly, the small G protein Rheb is a direct upstream activator of mTORC1. Tumor suppressors tuberous sclerosis complex 1 (TSC1) and TSC2 form a complex, which functions as a GTPase activating protein (GAP) toward Rheb and inhibits Rheb-mediated mTORC1 activation. The TSC1-TSC2 complex serves as a critical node to coordinate multiple upstream signaling inputs including growth factor, energy and oxygen levels with mTORC1 activation [161]. Amino acids, on the other hand, are sensed by mTORC1 largely through TSC-independent mechanisms that involve another small G protein Rag and Rag-associated protein complexes located on lysosome [162].
Various studies have reported that FoxOs negatively regulate mTORC1. It has been shown that energy stress-mediated mTORC1 inactivation is partially alleviated in FoxO deficient cells. Mechanistically, in response to energy stress, FoxOs upregulate the expression of BNIP3, which in turn interacts with Rheb and inhibits Rheb-mediated mTORC1 activation [48]. FoxO also inhibits Rheb-mTORC1 signaling axis through transcriptional upregulation of TSC1, an upstream negative regulator of Rheb [163]. As discussed above, Sestrin 3 is a transcriptional target of FoxOs [164]. Besides its function in ROS detoxification, Sestrin3 also negatively regulates mTORC1 through its interaction with AMPK and the GATOR complex that mediates amino acid sensing by mTORC1 [165–167]. Thus, FoxOs can inhibit mTORC1 through multiple parallel mechanisms (Fig. 5A). mTORC1 can also reciprocally regulate FoxOs through feedback loop regulation (Fig. 5A), in which mTORC1 hyperactivation downregulates upstream PI3K-AKT signaling and subsequently activates FoxOs through S6K-mediated inhibition of PI3K-AKT activators such as IRS-1 [168]. Consistent with this, benign polycystic kidneys with mTORC1 hyperactivation from TSC1 KO mice exhibit robust FoxO activation with predominant FoxO nuclear localization, but renal carcinomas developed from TSC1 deficient mice show markedly reduced FoxO staining, indicating that mTORC hyperactivation-induced FoxO activation through feedback loop restrains renal tumor development in TSC1 KO mice, and there is selection pressure to inactivate FoxOs to allow renal tumor development in this mouse model [101]. In supportive of model, dual deletion of both TSC1 and FoxOs dramatically promotes renal tumor development in vivo [101]. Another study showed that the anti-tumor effect of the mTOR inhibitor rapamycin can be diminished due to rapamycin-induced FoxO activation through feedback loop, and FoxO inhibition sensitizes cancer cells to rapamycin treatment [169]. Thus, mTORC1 hyperactivation-mediated FoxO activation through feedback loop regulation plays important roles in both tumor development and treatment.
Figure 5. FoxO crosstalk with other cancer metabolism pathways.

(A) FoxOs negatively regulate mTORC1 by inducing the expression of BNIP3, Sestrins, and TSC1, which inhibit mTORC1. Reciprocally, mTORC1 can promote FoxO activation by inhibiting the PI3K-AKT signaling via negative feedback loop. While mTORC2 inhibit FoxOs through AKT, FoxOs can also promote mTORC2 activation through upregulating Rictor expression. (B) The reciprocal antagonism between Myc and FoxOs. FoxOs inhibit Myc through multiple mechanisms as indicated. (C) FoxOs promote autophagy through both transcriptional dependent and independent mechanisms.
Finally, FoxOs can promote mTORC2 activation and mTORC2-mediated AKT phosphorylation through upregulating the expression of Rictor, a component of mTORC2 [164]. Correspondingly, it has been shown that FoxO deficiency sensitizes cancer cells to PI3K or AKT inhibitor treatment by attenuating PI3K/AKT inhibition-induced Rictor expression and AKT Ser473 phosphorylation [129]. In summary, current data support an intriguing model of reciprocal regulation between FoxOs and mTORC1/2: FoxOs inhibit mTORC1, while mTORC1 activates FoxOs through feedback regulation. On the other hand, mTORC2 inhibits FoxOs through AKT, while FoxOs activate mTORC2 through feedback regulation of Rictor (Fig. 5A).
FoxO and Myc
Myc proto-oncogene is a master transcription factor that controls cell proliferation, growth, and metabolism. For example, it has been well established that Myc promotes glycolysis and glutaminolysis by upregulating the expression of many metabolic enzymes involved in glucose and glutamine metabolism [137]. Increased Myc expression, through either gene amplification or overexpression, has been reported in as many as 70% of all cancers, and aberrant expression of Myc promotes tumor development in many human cancers [170]. Accordingly, mammalian cells have developed an intricate regulatory mechanisms to fine-tune Myc expression and activity [171], one of which is the reciprocal antagonism between Myc and FoxOs (Fig. 5B).
Myc can antagonize FoxO-mediated transcription of certain target genes by occupying FoxO promoters [172]. Multiple studies have also documented that FoxOs reciprocally antagonize Myc function through diverse mechanisms. First, it has been shown that FoxO TFs antagonize Myc function by occupying the promoters of and inhibiting expression of certain Myc targeted genes [173] or through upregulation of the MAX-interacting protein 1 (Mxi1), a member of Mad/Mxd family of transcriptional repressors that mediates antagonistic actions on Myc [174]. In addition, FoxO can suppress Myc function via upregulating the expression of microRNAs that inhibit Myc mRNA stability or translation, such as mir145 and mir34b/c [49, 101]. A recent study also showed that FoxOs inhibit Myc through a lncRNA-involved mechanism [138]. Specifically, in response to energy stress, FoxOs upregulate the expression of lncRNA FILNC1, which then interact with AUF1 protein in an energy stress-dependent manner. AUF1 is a RNA-binding protein that binds to 3′ untranslated region (UTR) of Myc mRNA and promotes Myc translation. FILNC1 functions as a molecular decoy to sequester AUF1 from binding to Myc mRNA, thus inhibiting Myc translation [138]. Finally, FoxO3 activation promotes polyubiquitination-dependent degradation of Myc in certain context [157]. In summary, FoxO antagonize Myc function at multiple levels, including negative regulating Myc mRNA stability, mRNA translation, protein stability, and transcriptional activity (Fig. 5B). It is possible that these different mechanisms are employed in tissue-, cancer type- or stimuli-dependent manners to ensure tight regulation of Myc level and activity.
FoxO and Autophagy
Autophagy is a complex cellular process which involves regulated degradation of damaged proteins and organelles in lysosomes and recycling of intracellular nutrients to support metabolism and cell survival under diverse stress conditions [175]. Although mammalian cells recycle damaged organelles and proteins via basal autophagy, many cancer cells display upregulation of autophagic flux to support their growth [176–178]. Multiple studies support a double-edged sword role of autophagy in cancer such that autophagy inhibits tumor initiation but promotes tumor maintenance in advanced cancer [178]. Several multiprotein complexes act in a sequential manner and form a signaling cascade to regulate autophagy [179]. Autophagy is also intimately linked to several major cancer metabolism pathways. For example, AMPK pathway promotes whereas mTORC1 inhibits autophagy [179]. Different studies have shown that FoxOs generally promote autophagy by regulating the expression of various components involved in autophagy. For example, FoxOs can also induce the expression of LC3 and BNIP3 to promote cellular autophagy [180]. A recent study showed that induction of autophagy by FoxO3 also involves upregulation of decidual protein induced by progesterone (C10ORF10/DEPP) in neuroblastoma cells [181]. As discussed above, FoxO3 and FoxO4 can promote autophagy by upregulating the expression of glutamine synthetase, which then promotes mTOR dissociation from the lysosomal membrane [145]. FoxOs can also regulate autophagy through transcription-independent mechanisms. It has been shown that cytosolic FoxO1 can induce autophagic flux by interacting with Atg7, a crucial regulator of autophagy. FoxO1 interaction with Atg7 and FoxO1-mediated autophagy induction results in increased apoptosis, which is associated with FoxO1’s tumor suppressive function [182]. Together, FoxOs promote autophagy through both transcriptional–dependent and –independent mechanisms (Fig. 5C). Given the complex roles of autophagy in cancer, the exact roles of FoxO regulation of autophagy in tumor biology remain less clear.
Conclusion and perspectives
Emerging evidence supports the critical roles of FoxO TFs in the regulation of cancer metabolism. FoxOs are regulated by a diverse array of signaling networks that sense nutrient status and mediate stress response in the cells, and these intricate regulatory modes to control FoxO activity are significantly altered in human cancers. FoxOs repress tumor metabolism by inhibiting glycolysis and other metabolism processes for biosynthesis as well as by modulating other key cancer metabolism pathways. Several important questions remain concerning the roles of FoxOs in cancer metabolism. In vivo functions of FoxOs revealed by GEMMs have been extensively characterized in many other biological contexts and diseases, such as stem cell biology, insulin resistance, and tissue homeostasis. However, most current data on FoxO function in cancer metabolism have been derived from cell line studies, and the relevant GEMM studies still remain limited. Future studies should be directed to develop appropriate GEMMs to further study FoxO function in cancer metabolism in vivo. Another important focus in FoxO research would be to develop therapeutic strategies to target FoxO function in cancer metabolism for cancer treatment. As FoxOs generally function as tumor suppressors that are often lost or inactivated in human cancers, one needs to restore FoxO-mediated tumor suppression function in cancer therapy, which is technically challenging (in contrast, it is more feasible to design drugs to block oncogene function that are hyperactivated in cancers). Despite these challenges, there have been recent progresses to target FoxOs in disease treatment. In one remarkable example, a recent study showed that disruption of FoxO4 interaction with p53 by a FoxO4 peptide can selectively induce apoptosis in senescent cells, mitigate chemotoxicity, and restore tissue fitness in several aging mouse models [183]. We envision the future studies will employ similar approaches to target FoxOs in cancer therapies.
Acknowledgments
We apologize to the many collogues whose related work cannot be cited due to space limitations. We are grateful for funding support from the Andrew Sabin Family Fellow Award and Institutional Research Grant from the University of Texas MD Anderson Cancer Center, National Institutes of Health (CA181196 and CA190370), Anna Fuller Fund, and Ellison Medical Foundation (AG-NS-0973-13). B. G. is an Ellison Medical Foundation New Scholar and an Andrew Sabin Family Fellow.
Footnotes
Conflict of interest
The authors have no conflict of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Iwafuchi-Doi M, Zaret KS. Cell fate control by pioneer transcription factors. Development. 2016;143(11):1833–7. doi: 10.1242/dev.133900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee TI, Young RA. Transcriptional regulation and its misregulation in disease. Cell. 2013;152(6):1237–51. doi: 10.1016/j.cell.2013.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhagwat AS, Vakoc CR. Targeting Transcription Factors in Cancer. Trends Cancer. 2015;1(1):53–65. doi: 10.1016/j.trecan.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Gan B, Liu D, Paik JH. FoxO family members in cancer. Cancer Biol Ther. 2011;12(4):253–9. doi: 10.4161/cbt.12.4.15954. [DOI] [PubMed] [Google Scholar]
- 5.van der Horst A, Burgering BM. Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol. 2007;8(6):440–50. doi: 10.1038/nrm2190. [DOI] [PubMed] [Google Scholar]
- 6.Eijkelenboom A, Burgering BM. FOXOs: signalling integrators for homeostasis maintenance. Nat Rev Mol Cell Biol. 2013;14(2):83–97. doi: 10.1038/nrm3507. [DOI] [PubMed] [Google Scholar]
- 7.Maiese K. FoxO Transcription Factors and Regenerative Pathways in Diabetes Mellitus. Curr Neurovasc Res. 2015;12(4):404–13. doi: 10.2174/1567202612666150807112524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27(16):2276–88. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- 9.Bartell SM, Kim HN, Ambrogini E, Han L, Iyer S, Serra Ucer S, Rabinovitch P, Jilka RL, Weinstein RS, Zhao H, O’Brien CA, Manolagas SC, Almeida M. FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation. Nature communications. 2014;5:3773. doi: 10.1038/ncomms4773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, Stone EL. FOXO transcription factors throughout T cell biology. Nat Rev Immunol. 2012;12(9):649–61. doi: 10.1038/nri3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tothova Z, Kollipara R, Huntly BJ, Lee BH, Castrillon DH, Cullen DE, McDowell EP, Lazo-Kallanian S, Williams IR, Sears C, Armstrong SA, Passegue E, DePinho RA, Gilliland DG. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Hoekman MF, Jacobs FM, Smidt MP, Burbach JP. Spatial and temporal expression of FoxO transcription factors in the developing and adult murine brain. Gene Expr Patterns. 2006;6(2):134–40. doi: 10.1016/j.modgep.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Furuyama T, Yamashita H, Kitayama K, Higami Y, Shimokawa I, Mori N. Effects of aging and caloric restriction on the gene expression of Foxo1, 3, and 4 (FKHR, FKHRL1, and AFX) in the rat skeletal muscles. Microsc Res Tech. 2002;59(4):331–4. doi: 10.1002/jemt.10213. [DOI] [PubMed] [Google Scholar]
- 14.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24(50):7410–25. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 15.Accili D, Arden KC. FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell. 2004;117(4):421–6. doi: 10.1016/s0092-8674(04)00452-0. [DOI] [PubMed] [Google Scholar]
- 16.Pajvani UB, Accili D. The new biology of diabetes. Diabetologia. 2015;58(11):2459–68. doi: 10.1007/s00125-015-3722-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol. 2014;220(2):T1–T23. doi: 10.1530/JOE-13-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanhaesebroeck B, Stephens L, Hawkins P. PI3K signalling: the path to discovery and understanding. Nat Rev Mol Cell Biol. 2012;13(3):195–203. doi: 10.1038/nrm3290. [DOI] [PubMed] [Google Scholar]
- 19.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell. 2017;170(4):605–635. doi: 10.1016/j.cell.2017.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4(9):a011189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 22.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–8. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 23.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96(6):857–68. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 24.Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96(13):7421–6. doi: 10.1073/pnas.96.13.7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature. 1999;398(6728):630–4. doi: 10.1038/19328. [DOI] [PubMed] [Google Scholar]
- 26.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274(24):17179–83. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 27.Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274(24):16741–6. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- 28.Tzivion G, Dobson M, Ramakrishnan G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim Biophys Acta. 2011;1813(11):1938–45. doi: 10.1016/j.bbamcr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 29.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21(3):952–65. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JP, Hoekman MF, Smidt MP. FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. J Biol Chem. 2003;278(38):35959–67. doi: 10.1074/jbc.M302804200. [DOI] [PubMed] [Google Scholar]
- 31.van der Heide LP, Jacobs FM, Burbach JP, Hoekman MF, Smidt MP. FoxO6 transcriptional activity is regulated by Thr26 and Ser184, independent of nucleo-cytoplasmic shuttling. Biochem J. 2005;391(Pt 3):623–9. doi: 10.1042/BJ20050525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang MC, Bohmann D, Jasper H. JNK extends life span and limits growth by antagonizing cellular and organism-wide responses to insulin signaling. Cell. 2005;121(1):115–25. doi: 10.1016/j.cell.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 33.Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, Tissenbaum HA. JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc Natl Acad Sci U S A. 2005;102(12):4494–9. doi: 10.1073/pnas.0500749102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, Burgering BM. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23(24):4802–12. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J Cell Biol. 2005;170(2):295–304. doi: 10.1083/jcb.200409117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villen J, Becker EB, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125(5):987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 37.Bi W, Xiao L, Jia Y, Wu J, Xie Q, Ren J, Ji G, Yuan Z. c-Jun N-terminal kinase enhances MST1-mediated pro-apoptotic signaling through phosphorylation at serine 82. J Biol Chem. 2010;285(9):6259–64. doi: 10.1074/jbc.M109.038570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Xiao ZD, Han L, Zhang J, Lee SW, Wang W, Lee H, Zhuang L, Chen J, Lin HK, Wang J, Liang H, Gan B. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat Cell Biol. 2016;18(4):431–42. doi: 10.1038/ncb3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greer EL, Oskoui PR, Banko MR, Maniar JM, Gygi MP, Gygi SP, Brunet A. The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem. 2007;282(41):30107–19. doi: 10.1074/jbc.M705325200. [DOI] [PubMed] [Google Scholar]
- 41.Greer EL, Dowlatshahi D, Banko MR, Villen J, Hoang K, Blanchard D, Gygi SP, Brunet A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr Biol. 2007;17(19):1646–56. doi: 10.1016/j.cub.2007.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yadav H, Devalaraja S, Chung ST, Rane SG. TGF-beta1/Smad3 Pathway Targets PP2A-AMPK-FoxO1 Signaling to Regulate Hepatic Gluconeogenesis. J Biol Chem. 2017;292(8):3420–3432. doi: 10.1074/jbc.M116.764910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314(5797):294–7. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- 44.Liu P, Kao TP, Huang H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene. 2008;27(34):4733–44. doi: 10.1038/onc.2008.104. [DOI] [PubMed] [Google Scholar]
- 45.Yuan Z, Becker EB, Merlo P, Yamada T, DiBacco S, Konishi Y, Schaefer EM, Bonni A. Activation of FOXO1 by Cdk1 in cycling cells and postmitotic neurons. Science. 2008;319(5870):1665–8. doi: 10.1126/science.1152337. [DOI] [PubMed] [Google Scholar]
- 46.Dall’Acqua A, Sonego M, Pellizzari I, Pellarin I, Canzonieri V, D’Andrea S, Benevol S, Sorio R, Giorda G, Califano D, Bagnoli M, Militello L, Mezzanzanica D, Chiappetta G, Armenia J, Belletti B, Schiappacassi M, Baldassarre G. CDK6 protects epithelial ovarian cancer from platinum-induced death via FOXO3 regulation. EMBO Mol Med. 2017;9(10):1415–1433. doi: 10.15252/emmm.201607012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ho KK, McGuire VA, Koo CY, Muir KW, de Olano N, Maifoshie E, Kelly DJ, McGovern UB, Monteiro LJ, Gomes AR, Nebreda AR, Campbell DG, Arthur JS, Lam EW. Phosphorylation of FOXO3a on Ser-7 by p38 promotes its nuclear localization in response to doxorubicin. J Biol Chem. 2012;287(2):1545–55. doi: 10.1074/jbc.M111.284224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin A, Yao J, Zhuang L, Wang D, Han J, Lam EW, Network TR, Gan B. The FoxO-BNIP3 axis exerts a unique regulation of mTORC1 and cell survival under energy stress. Oncogene. 2014;33(24):3183–94. doi: 10.1038/onc.2013.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kress TR, Cannell IG, Brenkman AB, Samans B, Gaestel M, Roepman P, Burgering BM, Bushell M, Rosenwald A, Eilers M. The MK5/PRAK kinase and Myc form a negative feedback loop that is disrupted during colorectal tumorigenesis. Mol Cell. 2011;41(4):445–57. doi: 10.1016/j.molcel.2011.01.023. [DOI] [PubMed] [Google Scholar]
- 50.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 51.Tsai WB, Chung YM, Takahashi Y, Xu Z, Hu MC. Functional interaction between FOXO3a and ATM regulates DNA damage response. Nat Cell Biol. 2008;10(4):460–7. doi: 10.1038/ncb1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yalcin S, Zhang X, Luciano JP, Mungamuri SK, Marinkovic D, Vercherat C, Sarkar A, Grisotto M, Taneja R, Ghaffari S. Foxo3 is essential for the regulation of ataxia telangiectasia mutated and oxidative stress-mediated homeostasis of hematopoietic stem cells. J Biol Chem. 2008;283(37):25692–705. doi: 10.1074/jbc.M800517200. [DOI] [PubMed] [Google Scholar]
- 53.Chung YM, Park SH, Tsai WB, Wang SY, Ikeda MA, Berek JS, Chen DJ, Hu MC. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nature communications. 2012;3:1000. doi: 10.1038/ncomms2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004;117(2):225–37. doi: 10.1016/s0092-8674(04)00302-2. [DOI] [PubMed] [Google Scholar]
- 55.Chapuis N, Park S, Leotoing L, Tamburini J, Verdier F, Bardet V, Green AS, Willems L, Agou F, Ifrah N, Dreyfus F, Bismuth G, Baud V, Lacombe C, Mayeux P, Bouscary D. IkappaB kinase overcomes PI3K/Akt and ERK/MAPK to control FOXO3a activity in acute myeloid leukemia. Blood. 2010;116(20):4240–50. doi: 10.1182/blood-2009-12-260711. [DOI] [PubMed] [Google Scholar]
- 56.Woods YL, Rena G, Morrice N, Barthel A, Becker W, Guo S, Unterman TG, Cohen P. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem J. 2001;355(Pt 3):597–607. doi: 10.1042/bj3550597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vembar SS, Brodsky JL. One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol. 2008;9(12):944–57. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang W, Hietakangas V, Wee S, Lim SC, Gunaratne J, Cohen SM. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013;27(4):441–9. doi: 10.1101/gad.201731.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9(9):679–90. doi: 10.1038/nrm2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003;278(14):12361–6. doi: 10.1074/jbc.M213069200. [DOI] [PubMed] [Google Scholar]
- 61.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003;100(20):11285–90. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang H, Regan KM, Wang F, Wang D, Smith DI, van Deursen JM, Tindall DJ. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A. 2005;102(5):1649–54. doi: 10.1073/pnas.0406789102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC, Huang H, Kuo HP, Lee DF, Li LY, Lien HC, Cheng X, Chang KJ, Hsiao CD, Tsai FJ, Tsai CH, Sahin AA, Muller WJ, Mills GB, Yu D, Hortobagyi GN, Hung MC. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008;10(2):138–48. doi: 10.1038/ncb1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fu W, Ma Q, Chen L, Li P, Zhang M, Ramamoorthy S, Nawaz Z, Shimojima T, Wang H, Yang Y, Shen Z, Zhang Y, Zhang X, Nicosia SV, Zhang Y, Pledger JW, Chen J, Bai W. MDM2 acts downstream of p53 as an E3 ligase to promote FOXO ubiquitination and degradation. J Biol Chem. 2009;284(21):13987–4000. doi: 10.1074/jbc.M901758200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li F, Xie P, Fan Y, Zhang H, Zheng L, Gu D, Patterson C, Li H. C terminus of Hsc70-interacting protein promotes smooth muscle cell proliferation and survival through ubiquitin-mediated degradation of FoxO1. J Biol Chem. 2009;284(30):20090–8. doi: 10.1074/jbc.M109.017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kato S, Ding J, Pisck E, Jhala US, Du K. COP1 functions as a FoxO1 ubiquitin E3 ligase to regulate FoxO1-mediated gene expression. J Biol Chem. 2008;283(51):35464–73. doi: 10.1074/jbc.M801011200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Drazic A, Myklebust LM, Ree R, Arnesen T. The world of protein acetylation. Biochim Biophys Acta. 2016;1864(10):1372–401. doi: 10.1016/j.bbapap.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 68.Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J Biol Chem. 2010;285(35):27396–401. doi: 10.1074/jbc.M110.140228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci U S A. 2005;102(32):11278–83. doi: 10.1073/pnas.0502738102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perrot V, Rechler MM. The coactivator p300 directly acetylates the forkhead transcription factor Foxo1 and stimulates Foxo1-induced transcription. Mol Endocrinol. 2005;19(9):2283–98. doi: 10.1210/me.2004-0292. [DOI] [PubMed] [Google Scholar]
- 71.van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2(SIRT1) J Biol Chem. 2004;279(28):28873–9. doi: 10.1074/jbc.M401138200. [DOI] [PubMed] [Google Scholar]
- 72.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303(5666):2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 73.Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119(9):2758–71. doi: 10.1172/JCI39162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang F, Nguyen M, Qin FX, Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging cell. 2007;6(4):505–14. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 75.Tseng AH, Shieh SS, Wang DL. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic Biol Med. 2013;63:222–34. doi: 10.1016/j.freeradbiomed.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 76.Zhang L, Cai M, Gong Z, Zhang B, Li Y, Guan L, Hou X, Li Q, Liu G, Xue Z, Yang MH, Ye J, Chin YE, You H. Geminin facilitates FoxO3 deacetylation to promote breast cancer cell metastasis. J Clin Invest. 2017;127(6):2159–2175. doi: 10.1172/JCI90077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, Mukai H, Kasuya Y, Fukamizu A. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell. 2008;32(2):221–31. doi: 10.1016/j.molcel.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 78.Calnan DR, Webb AE, White JL, Stowe TR, Goswami T, Shi X, Espejo A, Bedford MT, Gozani O, Gygi SP, Brunet A. Methylation by Set9 modulates FoxO3 stability and transcriptional activity. Aging. 2012;4(7):462–79. doi: 10.18632/aging.100471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xie Q, Hao Y, Tao L, Peng S, Rao C, Chen H, You H, Dong MQ, Yuan Z. Lysine methylation of FOXO3 regulates oxidative stress-induced neuronal cell death. EMBO Rep. 2012;13(4):371–7. doi: 10.1038/embor.2012.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kuo M, Zilberfarb V, Gangneux N, Christeff N, Issad T. O-glycosylation of FoxO1 increases its transcriptional activity towards the glucose 6-phosphatase gene. FEBS letters. 2008;582(5):829–34. doi: 10.1016/j.febslet.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 81.Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, Puigserver P, Hart GW. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem. 2008;283(24):16283–92. doi: 10.1074/jbc.M802240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zheng X, Zhai B, Koivunen P, Shin SJ, Lu G, Liu J, Geisen C, Chakraborty AA, Moslehi JJ, Smalley DM, Wei X, Chen X, Chen Z, Beres JM, Zhang J, Tsao JL, Brenner MC, Zhang Y, Fan C, DePinho RA, Paik J, Gygi SP, Kaelin WG, Jr, Zhang Q. Prolyl hydroxylation by EglN2 destabilizes FOXO3a by blocking its interaction with the USP9x deubiquitinase. Genes Dev. 2014;28(13):1429–44. doi: 10.1101/gad.242131.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nowak K, Killmer K, Gessner C, Lutz W. E2F-1 regulates expression of FOXO1 and FOXO3a. Biochim Biophys Acta. 2007;1769(4):244–52. doi: 10.1016/j.bbaexp.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 84.Angelis E, Zhao P, Zhang R, Goldhaber JI, Maclellan WR. The role of E2F-1 and downstream target genes in mediating ischemia/reperfusion injury in vivo. J Mol Cell Cardiol. 2011;51(6):919–26. doi: 10.1016/j.yjmcc.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kurinna S, Stratton SA, Tsai WW, Akdemir KC, Gu W, Singh P, Goode T, Darlington GJ, Barton MC. Direct activation of forkhead box O3 by tumor suppressors p53 and p73 is disrupted during liver regeneration in mice. Hepatology. 2010;52(3):1023–32. doi: 10.1002/hep.23746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Renault VM, Thekkat PU, Hoang KL, White JL, Brady CA, Kenzelmann Broz D, Venturelli OS, Johnson TM, Oskoui PR, Xuan Z, Santo EE, Zhang MQ, Vogel H, Attardi LD, Brunet A. The pro-longevity gene FoxO3 is a direct target of the p53 tumor suppressor. Oncogene. 2011;30(29):3207–21. doi: 10.1038/onc.2011.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bakker WJ, Harris IS, Mak TW. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol Cell. 2007;28(6):941–53. doi: 10.1016/j.molcel.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 88.Essaghir A, Dif N, Marbehant CY, Coffer PJ, Demoulin JB. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J Biol Chem. 2009;284(16):10334–42. doi: 10.1074/jbc.M808848200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhu WL, Tong H, Teh JT, Wang M. Forkhead box protein O3 transcription factor negatively regulates autophagy in human cancer cells by inhibiting forkhead box protein O1 expression and cytosolic accumulation. PLoS One. 2014;9(12):e115087. doi: 10.1371/journal.pone.0115087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ikeda T, Uno M, Honjoh S, Nishida E. The MYST family histone acetyltransferase complex regulates stress resistance and longevity through transcriptional control of DAF-16/FOXO transcription factors. EMBO Rep. 2017 doi: 10.15252/embr.201743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li Y, Yu J, Du D, Fu S, Chen Y, Yu F, Gao P. Involvement of post-transcriptional regulation of FOXO1 by HuR in 5-FU-induced apoptosis in breast cancer cells. Oncol Lett. 2013;6(1):156–160. doi: 10.3892/ol.2013.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yu F, Jin L, Yang G, Ji L, Wang F, Lu Z. Post-transcriptional repression of FOXO1 by QKI results in low levels of FOXO1 expression in breast cancer cells. Oncol Rep. 2014;31(3):1459–65. doi: 10.3892/or.2013.2957. [DOI] [PubMed] [Google Scholar]
- 93.Jeong OS, Chae YC, Jung H, Park SC, Cho SJ, Kook H, Seo S. Long noncoding RNA linc00598 regulates CCND2 transcription and modulates the G1 checkpoint. Sci Rep. 2016;6:32172. doi: 10.1038/srep32172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xi J, Feng J, Li Q, Li X, Zeng S. The long non-coding RNA lncFOXO1 suppresses growth of human breast cancer cells through association with BAP1. Int J Oncol. 2017;50(5):1663–1670. doi: 10.3892/ijo.2017.3933. [DOI] [PubMed] [Google Scholar]
- 95.Xu X, Ji S, Li W, Yi B, Li H, Zhang H, Ma W. LncRNA H19 promotes the differentiation of bovine skeletal muscle satellite cells by suppressing Sirt1/FoxO1. Cell Mol Biol Lett. 2017;22:10. doi: 10.1186/s11658-017-0040-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG, Rauscher FJ., 3rd The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol. 1995;15(3):1522–35. doi: 10.1128/mcb.15.3.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Borkhardt A, Repp R, Haas OA, Leis T, Harbott J, Kreuder J, Hammermann J, Henn T, Lampert F. Cloning and characterization of AFX, the gene that fuses to MLL in acute leukemias with a t(X;11)(q13;q23) Oncogene. 1997;14(2):195–202. doi: 10.1038/sj.onc.1200814. [DOI] [PubMed] [Google Scholar]
- 98.Hillion J, Le Coniat M, Jonveaux P, Berger R, Bernard OA. AF6q21, a novel partner of the MLL gene in t(6;11)(q21;q23), defines a forkhead transcriptional factor subfamily. Blood. 1997;90(9):3714–9. [PubMed] [Google Scholar]
- 99.N. Cancer Genome Atlas Research. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128(2):309–23. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gan B, Lim C, Chu G, Hua S, Ding Z, Collins M, Hu J, Jiang S, Fletcher-Sananikone E, Zhuang L, Chang M, Zheng H, Wang YA, Kwiatkowski DJ, Kaelin WG, Jr, Signoretti S, DePinho RA. FoxOs enforce a progression checkpoint to constrain mTORC1-activated renal tumorigenesis. Cancer Cell. 2010;18(5):472–84. doi: 10.1016/j.ccr.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.van der Vos KE, Coffer PJ. The extending network of FOXO transcriptional target genes. Antioxid Redox Signal. 2011;14(4):579–92. doi: 10.1089/ars.2010.3419. [DOI] [PubMed] [Google Scholar]
- 103.Keniry M, Pires MM, Mense S, Lefebvre C, Gan B, Justiano K, Lau YK, Hopkins B, Hodakoski C, Koujak S, Toole J, Fenton F, Calahan A, Califano A, DePinho RA, Maurer M, Parsons R. Survival factor NFIL3 restricts FOXO-induced gene expression in cancer. Genes Dev. 2013;27(8):916–27. doi: 10.1101/gad.214049.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203(7):1657–63. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10(19):1201–4. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 106.Rokudai S, Fujita N, Kitahara O, Nakamura Y, Tsuruo T. Involvement of FKHR-dependent TRADD expression in chemotherapeutic drug-induced apoptosis. Mol Cell Biol. 2002;22(24):8695–708. doi: 10.1128/MCB.22.24.8695-8708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sunters A, Fernandez de Mattos S, Stahl M, Brosens JJ, Zoumpoulidou G, Saunders CA, Coffer PJ, Medema RH, Coombes RC, Lam EW. FoxO3a transcriptional regulation of Bim controls apoptosis in paclitaxel-treated breast cancer cell lines. J Biol Chem. 2003;278(50):49795–805. doi: 10.1074/jbc.M309523200. [DOI] [PubMed] [Google Scholar]
- 108.Kloet DE, Polderman PE, Eijkelenboom A, Smits LM, van Triest MH, van den Berg MC, Groot Koerkamp MJ, van Leenen D, Lijnzaad P, Holstege FC, Burgering BM. FOXO target gene CTDSP2 regulates cell cycle progression through Ras and p21(Cip1/Waf1) Biochem J. 2015;469(2):289–98. doi: 10.1042/BJ20140831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR. A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell. 2002;2(1):81–91. doi: 10.1016/s1535-6108(02)00086-7. [DOI] [PubMed] [Google Scholar]
- 110.Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol. 2002;22(22):7842–52. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem. 2002;277(30):26729–32. doi: 10.1074/jbc.C200256200. [DOI] [PubMed] [Google Scholar]
- 112.de Keizer PL, Packer LM, Szypowska AA, Riedl-Polderman PE, van den Broek NJ, de Bruin A, Dansen TB, Marais R, Brenkman AB, Burgering BM. Activation of forkhead box O transcription factors by oncogenic BRAF promotes p21cip1-dependent senescence. Cancer Res. 2010;70(21):8526–36. doi: 10.1158/0008-5472.CAN-10-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhuo de X, Niu XH, Chen YC, Xin DQ, Guo YL, Mao ZB. Vitamin D3 up-regulated protein 1(VDUP1) is regulated by FOXO3A and miR-17-5p at the transcriptional and post-transcriptional levels, respectively, in senescent fibroblasts. J Biol Chem. 2010;285(41):31491–501. doi: 10.1074/jbc.M109.068387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Miyauchi H, Minamino T, Tateno K, Kunieda T, Toko H, Komuro I. Akt negatively regulates the in vitro lifespan of human endothelial cells via a p53/p21-dependent pathway. EMBO J. 2004;23(1):212–20. doi: 10.1038/sj.emboj.7600045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Qi XF, Chen ZY, Xia JB, Zheng L, Zhao H, Pi LQ, Park KS, Kim SK, Lee KJ, Cai DQ. FoxO3a suppresses the senescence of cardiac microvascular endothelial cells by regulating the ROS-mediated cell cycle. J Mol Cell Cardiol. 2015;81:114–26. doi: 10.1016/j.yjmcc.2015.01.022. [DOI] [PubMed] [Google Scholar]
- 116.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J Clin Invest. 2005;115(9):2382–92. doi: 10.1172/JCI23126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wilhelm K, Happel K, Eelen G, Schoors S, Oellerich MF, Lim R, Zimmermann B, Aspalter IM, Franco CA, Boettger T, Braun T, Fruttiger M, Rajewsky K, Keller C, Bruning JC, Gerhardt H, Carmeliet P, Potente M. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016;529(7585):216–20. doi: 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dong T, Zhang Y, Chen Y, Liu P, An T, Zhang J, Yang H, Zhu W, Yang X. FOXO1 inhibits the invasion and metastasis of hepatocellular carcinoma by reversing ZEB2-induced epithelial-mesenchymal transition. Oncotarget. 2017;8(1):1703–1713. doi: 10.18632/oncotarget.13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ni D, Ma X, Li HZ, Gao Y, Li XT, Zhang Y, Ai Q, Zhang P, Song EL, Huang QB, Fan Y, Zhang X. Downregulation of FOXO3a promotes tumor metastasis and is associated with metastasis-free survival of patients with clear cell renal cell carcinoma. Clin Cancer Res. 2014;20(7):1779–90. doi: 10.1158/1078-0432.CCR-13-1687. [DOI] [PubMed] [Google Scholar]
- 120.Procaccia S, Ordan M, Cohen I, Bendetz-Nezer S, Seger R. Direct binding of MEK1 and MEK2 to AKT induces Foxo1 phosphorylation, cellular migration and metastasis. Sci Rep. 2017;7:43078. doi: 10.1038/srep43078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang X, Rielland M, Yalcin S, Ghaffari S. Regulation and function of FoxO transcription factors in normal and cancer stem cells: what have we learned? Curr Drug Targets. 2011;12(9):1267–83. doi: 10.2174/138945011796150325. [DOI] [PubMed] [Google Scholar]
- 122.Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, Nakao S, Motoyama N, Hirao A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676–80. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- 123.Hurtz C, Hatzi K, Cerchietti L, Braig M, Park E, Kim YM, Herzog S, Ramezani-Rad P, Jumaa H, Muller MC, Hofmann WK, Hochhaus A, Ye BH, Agarwal A, Druker BJ, Shah NP, Melnick AM, Muschen M. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J Exp Med. 2011;208(11):2163–74. doi: 10.1084/jem.20110304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, Ferraro F, Mercier F, Singh H, Brumme KM, Acharya SS, Scholl C, Tothova Z, Attar EC, Frohling S, DePinho RA, Armstrong SA, Gilliland DG, Scadden DT. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146(5):697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Han CY, Cho KB, Choi HS, Han HK, Kang KW. Role of FoxO1 activation in MDR1 expression in adriamycin-resistant breast cancer cells. Carcinogenesis. 2008;29(9):1837–44. doi: 10.1093/carcin/bgn092. [DOI] [PubMed] [Google Scholar]
- 126.Hui RC, Gomes AR, Constantinidou D, Costa JR, Karadedou CT, Fernandez de Mattos S, Wymann MP, Brosens JJ, Schulze A, Lam EW. The forkhead transcription factor FOXO3a increases phosphoinositide-3 kinase/Akt activity in drug-resistant leukemic cells through induction of PIK3CA expression. Mol Cell Biol. 2008;28(19):5886–98. doi: 10.1128/MCB.01265-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19(1):58–71. doi: 10.1016/j.ccr.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, Brugge JS. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell. 2012;21(2):227–39. doi: 10.1016/j.ccr.2011.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lin A, Piao HL, Zhuang L, Sarbassov DD, Ma L, Gan B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacological inhibition of the PI3K-AKT pathway. Cancer Res. 2014 doi: 10.1158/0008-5472.CAN-13-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Storz P, Doppler H, Copland JA, Simpson KJ, Toker A. FOXO3a promotes tumor cell invasion through the induction of matrix metalloproteinases. Mol Cell Biol. 2009;29(18):4906–17. doi: 10.1128/MCB.00077-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Charitou P, Rodriguez-Colman M, Gerrits J, van Triest M, Groot Koerkamp M, Hornsveld M, Holstege F, Verhoeven-Duif NM, Burgering BM. FOXOs support the metabolic requirements of normal and tumor cells by promoting IDH1 expression. EMBO Rep. 2015;16(4):456–66. doi: 10.15252/embr.201439096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 133.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23(1):27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hay N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat Rev Cancer. 2016;16(10):635–49. doi: 10.1038/nrc.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell. 2017;168(4):657–669. doi: 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Stine ZE, Walton ZE, Altman BJ, Hsieh AL, Dang CV. MYC, Metabolism, and Cancer. Cancer Discov. 2015;5(10):1024–39. doi: 10.1158/2159-8290.CD-15-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xiao ZD, Han L, Lee H, Zhuang L, Zhang Y, Baddour J, Nagrath D, Wood CG, Gu J, Wu X, Liang H, Gan B. Energy stress-induced lncRNA FILNC1 represses c-Myc-mediated energy metabolism and inhibits renal tumor development. Nature communications. 2017;8(1):783. doi: 10.1038/s41467-017-00902-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, Iwanami A, Liu F, Villa GR, Gu Y, Campos C, Zhu S, Yang H, Yong WH, Cloughesy TF, Mellinghoff IK, Cavenee WK, Shaw RJ, Mischel PS. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. 2013;18(5):726–39. doi: 10.1016/j.cmet.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007;6(3):208–16. doi: 10.1016/j.cmet.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 141.Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27(16):2320–36. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- 142.Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, Matika R, Xiao X, Franks R, Heidenreich KA, Sajan MP, Farese RV, Stolz DB, Tso P, Koo SH, Montminy M, Unterman TG. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem. 2006;281(15):10105–17. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 143.Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16(10):650–62. doi: 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- 144.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–34. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.van der Vos KE, Coffer PJ. Glutamine metabolism links growth factor signaling to the regulation of autophagy. Autophagy. 2012;8(12):1862–4. doi: 10.4161/auto.22152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kamei Y, Hattori M, Hatazawa Y, Kasahara T, Kanou M, Kanai S, Yuan X, Suganami T, Lamers WH, Kitamura T, Ogawa Y. FOXO1 activates glutamine synthetase gene in mouse skeletal muscles through a region downstream of 3′-UTR: possible contribution to ammonia detoxification. American journal of physiology. Endocrinology and metabolism. 2014;307(6):E485–93. doi: 10.1152/ajpendo.00177.2014. [DOI] [PubMed] [Google Scholar]
- 147.Tato I, Bartrons R, Ventura F, Rosa JL. Amino acids activate mammalian target of rapamycin complex 2 (mTORC2) via PI3K/Akt signaling. J Biol Chem. 2011;286(8):6128–42. doi: 10.1074/jbc.M110.166991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kramer JM, Slade JD, Staveley BE. foxo is required for resistance to amino acid starvation in Drosophila. Genome. 2008;51(8):668–72. doi: 10.1139/G08-047. [DOI] [PubMed] [Google Scholar]
- 149.Sunshine H, Iruela-Arispe ML. Membrane lipids and cell signaling. Curr Opin Lipidol. 2017;28(5):408–413. doi: 10.1097/MOL.0000000000000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279(15):2610–23. doi: 10.1111/j.1742-4658.2012.08644.x. [DOI] [PubMed] [Google Scholar]
- 151.Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nature reviews. Endocrinology. 2017 doi: 10.1038/nrendo.2017.91. [DOI] [PubMed] [Google Scholar]
- 152.Deng X, Zhang W, OSI, Williams JB, Dong Q, Park EA, Raghow R, Unterman TG, Elam MB. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. J Biol Chem. 2012;287(24):20132–43. doi: 10.1074/jbc.M112.347211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chakrabarti P, Kandror KV. FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J Biol Chem. 2009;284(20):13296–300. doi: 10.1074/jbc.C800241200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Panieri E, Santoro MM. ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis. 2016;7(6):e2253. doi: 10.1038/cddis.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15(6):411–21. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 156.Storz P. Reactive oxygen species in tumor progression. Front Biosci. 2005;10:1881–96. doi: 10.2741/1667. [DOI] [PubMed] [Google Scholar]
- 157.Ferber EC, Peck B, Delpuech O, Bell GP, East P, Schulze A. FOXO3a regulates reactive oxygen metabolism by inhibiting mitochondrial gene expression. Cell Death Differ. 2012;19(6):968–79. doi: 10.1038/cdd.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shao D, Zhai P, Del Re DP, Sciarretta S, Yabuta N, Nojima H, Lim DS, Pan D, Sadoshima J. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nature communications. 2014;5:3315. doi: 10.1038/ncomms4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102(13):932–41. doi: 10.1093/jnci/djq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;169(2):361–371. doi: 10.1016/j.cell.2017.03.035. [DOI] [PubMed] [Google Scholar]
- 161.Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol. 2013;15(6):555–64. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wolfson RL, Sabatini DM. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab. 2017;26(2):301–309. doi: 10.1016/j.cmet.2017.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Khatri S, Yepiskoposyan H, Gallo CA, Tandon P, Plas DR. FOXO3a regulates glycolysis via transcriptional control of tumor suppressor TSC1. J Biol Chem. 2010;285(21):15960–5. doi: 10.1074/jbc.M110.121871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen CC, Jeon SM, Bhaskar PT, Nogueira V, Sundararajan D, Tonic I, Park Y, Hay N. FoxOs inhibit mTORC1 and activate Akt by inducing the expression of Sestrin3 and Rictor. Dev Cell. 2010;18(4):592–604. doi: 10.1016/j.devcel.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Parmigiani A, Nourbakhsh A, Ding B, Wang W, Kim YC, Akopiants K, Guan KL, Karin M, Budanov AV. Sestrins inhibit mTORC1 kinase activation through the GATOR complex. Cell Rep. 2014;9(4):1281–91. doi: 10.1016/j.celrep.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chantranupong L, Wolfson RL, Orozco JM, Saxton RA, Scaria SM, Bar-Peled L, Spooner E, Isasa M, Gygi SP, Sabatini DM. The Sestrins interact with GATOR2 to negatively regulate the amino-acid-sensing pathway upstream of mTORC1. Cell Rep. 2014;9(1):1–8. doi: 10.1016/j.celrep.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134(3):451–60. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167(3):399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Abdelnour-Berchtold E, Cerantola Y, Roulin D, Dormond-Meuwly A, Demartines N, Dormond O. Rapamycin-mediated FOXO1 inactivation reduces the anticancer efficacy of rapamycin. Anticancer research. 2010;30(3):799–804. [PubMed] [Google Scholar]
- 170.Dang CV. MYC on the path to cancer. Cell. 2012;149(1):22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med. 2014;4(1):a014357. doi: 10.1101/cshperspect.a014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Amente S, Zhang J, Lavadera ML, Lania L, Avvedimento EV, Majello B. Myc and PI3K/AKT signaling cooperatively repress FOXO3a-dependent PUMA and GADD45a gene expression. Nucleic Acids Res. 2011;39(22):9498–507. doi: 10.1093/nar/gkr638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bouchard C, Marquardt J, Bras A, Medema RH, Eilers M. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J. 2004;23(14):2830–40. doi: 10.1038/sj.emboj.7600279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Delpuech O, Griffiths B, East P, Essafi A, Lam EW, Burgering B, Downward J, Schulze A. Induction of Mxi1-SR alpha by FOXO3a contributes to repression of Myc-dependent gene expression. Mol Cell Biol. 2007;27(13):4917–30. doi: 10.1128/MCB.01789-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 176.Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, Mautner J, Tonon G, Haigis M, Shirihai OS, Doglioni C, Bardeesy N, Kimmelman AC. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–29. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD, White E. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–70. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30(17):1913–30. doi: 10.1101/gad.287524.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–93. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6(6):458–71. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 181.Salcher S, Hermann M, Kiechl-Kohlendorfer U, Ausserlechner MJ, Obexer P. C10ORF10/DEPP-mediated ROS accumulation is a critical modulator of FOXO3-induced autophagy. Mol Cancer. 2017;16(1):95. doi: 10.1186/s12943-017-0661-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol. 2010;12(7):665–75. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 183.Baar MP, Brandt RM, Putavet DA, Klein JD, Derks KW, Bourgeois BR, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, van IWF, Houtsmuller AB, Pothof J, de Bruin RW, Madl T, Hoeijmakers JH, Campisi J, de Keizer PL. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell. 2017;169(1):132–147 e16. doi: 10.1016/j.cell.2017.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]