

Abstract
A hallmark of positive-sense RNA viruses is the formation of membranous shelters for safe replication in the cytoplasm. Once considered invisible to the immune system, these viral shelters are now found to be antagonized through the cooperation of autophagy proteins and anti-microbial GTPases. This coordinated effort of autophagy proteins guiding GTPases functions against not only the shelters of viruses but also cytoplasmic vacuoles containing bacteria or protozoa, suggesting a broad immune-defense mechanism against disparate vacuolar pathogens. Fundamental questions regarding this process remain: how the host recognizes these membranous structures as a target, how the autophagy proteins bring the GTPases to the shelters, and how the recruited GTPases disrupt these shelters. In this review we discuss these questions, the answers to which will significantly advance our understanding of the response to vacuole-like structures of pathogens, thereby paving the way for the development of broadly effective anti-microbial strategies for public health.
Keywords: replication compartment, positive-sense RNA virus, LC3 conjugation, interferon-inducible GTPases, autophagy, pathogen-containing vacuoles, vacuolar pathogen
1. Introduction
The success of an intracellular pathogen depends upon its ability to subvert host defenses. In many cases this is achieved simply by localizing in cellular compartments inaccessible to host defense machinery.[1] The replication compartment or complex (RC), characteristic of positive-sense RNA [(+)RNA] viruses, exemplifies this concept.[2] About one-third of all known viruses possess (+)RNA genomes,[3] allowing for immediate production of proteins upon successful entry. Viral proteins reorganize host membranes from various organelles, such as the ER and Golgi, rapidly forming the RC.[2] The RC sequesters the viral genome and proteins in a subcellular enclave, facilitating replication while simultaneously shielding viral components from innate recognition.[4], [5] Accordingly, formation of the RC is critical in the life cycle of (+)RNA viruses. While much is known about the molecular mechanisms that regulate the formation of viral RCs,[5], [6], [7], [8] whether the host antagonizes RCs has been less clear.
Using murine norovirus (MNV) as a model (+)RNA virus, we identified a cell autonomous defense mechanism mounted against RCs. This mechanism was dependent upon elements of two evolutionarily conserved defense systems: autophagy and interferon.[9], [10] We found that viral RCs were marked with microtubule-associated-protein-1-light-chain-3 (LC3, a.k.a. ATG8) homologs, the classic marker of autophagosomes. Upon interferon signaling, several interferon (IFN)-inducible GTPases were induced and localized to the LC3-marked RCs. Recruitment of these dynamin-like GTPases resulted in disruption of viral RCs and inhibition of viral replication. A similar process was previously associated with cellular control of bacteria, protozoa, and fungi, in which IFN-inducible GTPases demarcate, and induce the destruction of, pathogen-containing vacuoles.[11] The addition of MNV to the growing list of targeted pathogens suggests a potential pan-antimicrobial strategy in response to intracellular infection.
A major unanswered question of the LC3-mediated targeting of IFN-inducible GTPases is what property or properties of the RC enable its recognition, despite being composed of host membranes. Several questions remain regarding the precise molecular events that constitute this process. Unlike canonical autophagy, this process is independent of lysosomal degradation. Understanding the factors that lead to this noncanonical outcome–i.e. the recruitment of IFN-inducible GTPases to the LC3-marked structure rather than lysosomal degradation–will be a major focus of future studies. Moreover, the IFN-inducible GTPases central to this process have been studied predominantly in the context of bacterial and protozoan infection. Whether these dynamin-like proteins inhibit viral replication through previously observed mechanisms or through a novel function will be an important question to answer.[11]
Herein, we present the context of these questions and current thinking in the field, and provide our up-to-date hypotheses and speculations on the mechanisms of intracellular pathogen targeting.
2. Viral replication compartments: an unexplored target of antiviral activity
Viruses with a (+)RNA genome comprise a large group of plant and animal viruses,[12] including many medically important viruses, such as Zika virus, West Nile virus, and norovirus.[3] In order to replicate their genome, (+)RNA viruses must synthesize and utilize a complementary negative-sense RNA template through a double-strand RNA (dsRNA) intermediate.[13] Host recognition of the dsRNA intermediate by innate immune sensors results in activation of the antiviral defense system, particularly through the production of interferons and subsequent induction of a multitude of antiviral genes. These genes and gene products have been shown to inhibit nearly all steps of the (+)RNA viral life cycle, except one: the formation of the RC. Often visualized as vacuole-like membrane compartments (Fig. 1), the formation and maintenance of the RC is an essential part of the (+)RNA virus life cycle by increasing replication efficiency and avoiding innate recognition.[2], [3], [14], [15], [16] Despite its importance, it was largely unknown whether an antiviral response against the RC exists.[17], [18] Given the structural conservation of RCs across (+)RNA viruses, the RC represents an attractive target for a general antiviral strategy.
Figure 1. Schematic of replication complexes of (+) RNA viruses.
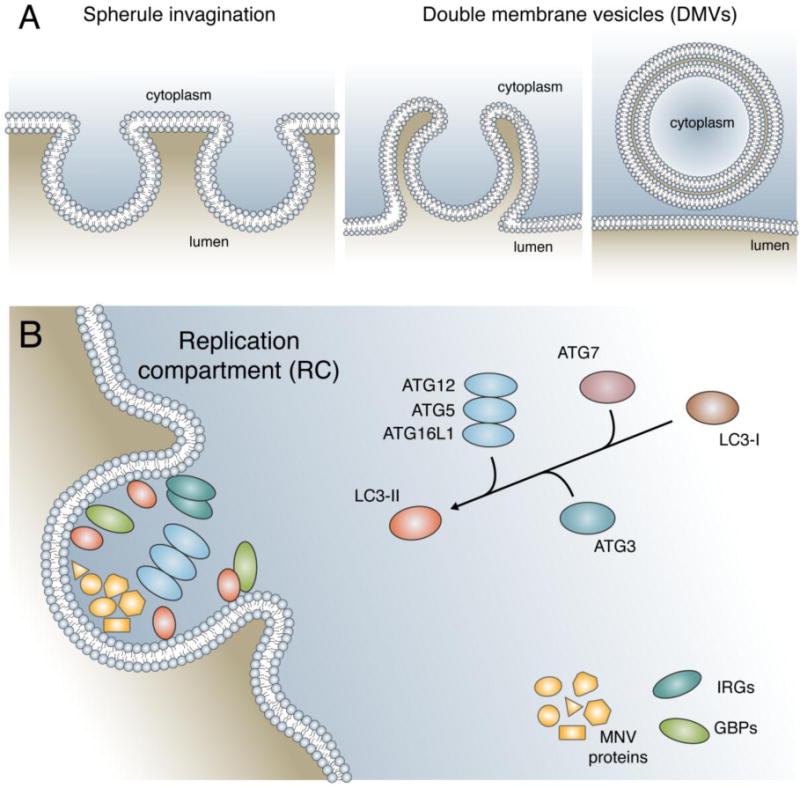
A) Through the reorganization of host membranes, the (+) RNA virus forms a membranous compartment known as the replication complex (RC). The two major forms of RCs are spherule invaginations and double membrane vesicles (DMVs). Spherule invaginations (left) are formed by the small invagination of a host organelle away from the cytosol. DMVs (middle and right) are formed by the extrusion of host membrane followed by invagination and subsequent vesicular budding from the point of extrusion. B) The RC formed by MNV is targeted by the LC3 conjugation system of autophagy. The ubiquitin-like conjugation system composed of ATG7, ATG3, and ATG12–ATG5-ATG16L1 convert LC3-I to the lipidated form, LC3-II. Marking of the MNV RC with LC3-II leads to the recruitment of IFN-inducible GTPases (IRGs and GBPs) which are critical in the subsequent block in viral replication.
Upon infection, (+)RNA viruses immediately express viral proteins,[19] which can interact directly with membranes and a variety of host proteins responsible for lipid manipulation to form the RC.[2], [19] Different viruses utilize unique sets of cellular factors and preferred membrane sources, but in all cases the membranes of RCs are solely derived from cellular organelles, or even the plasma membrane.[5] The RC creates an environment where viral components are in a relatively small place, ensuring maximum likelihood of interaction between viral proteins, viral RNA, and co-opted cellular components, facilitating viral replication.[2], [3], [19] Importantly, RCs act as a shelter to avoid innate immune detection.[15], [16] Not only are cytosolic dsRNA sensors excluded from RCs,[4] but also RNAs within RCs are protected from nuclease treatment, suggesting that the RCs of (+)RNA viruses sequester dsRNA intermediates from escaping the RC, further thwarting immune recognition.[15], [16]
To date, two types of RC structures have been identified: spherule invaginations and double membrane vesicles (DMVs) (Fig. 1A).[2], [19] The spherule form of RCs is generated through invaginations into the negative curvature of a membrane into the lumen of an organelle, away from the cytoplasm. These structures maintain a small neck that connects the RC interior to the cytoplasm, believed to selectively restrict traffic in and out of the structure.[2], [20] In contrast, DMVs are formed from the near closure of a single membrane vesicle after exvagination accompanied by invaginations (Fig. 1A). An open connection to the cytoplasm is only observed in a minority of DMVs undergoing active replication. A connection should be present for necessary molecular exchange between the cytoplasm and DMV RCs, but how this works for the majority of DMVs is unclear. Of note, hepatitis C virus (HCV) recruits nuclear pore complex machinery to their DMV RCs, suggesting a potential mechanism for selective RC entry and exit.[4], [21] Due to the structural similarity between autophagosomes and viral RCs, many groups speculated that autophagy supports (+)RNA virus RC formation and therefore replication of some viruses.[22], [23]
3. Control of MNV replication depends on autophagy proteins
Originally described by Christian de Duve as “bulk segregation and digestion of portions of the cytoplasm”, autophagy is an essential cellular homeostasis process that removes obsolete proteins, organelles, and other macromolecules.[24], [25], [26] By definition, macroautophagy (henceforth, simply ‘autophagy’) is an evolutionarily conserved cellular pathway that sequesters cytosolic materials in a double-membrane-bound autophagosome and delivers them to the lysosome for degradation.[27] Autophagy has often been considered an antimicrobial pathway through the capture and degradation of cytosolic invaders, a process known as xenophagy.[28] In addition to xenophagy of viral particles, autophagy can contribute to antiviral immunity through delivery of viral genomes to endosomal immune sensors and presentation of viral antigens for the adaptive immune response.[22], [29], [30] To combat this, several viruses have evolved mechanisms to block autophagy, or even co-opt the machinery for their own benefit. For many viruses, autophagosomes can act as a source of membrane for RC formation, though in many cases autophagy is not essential for viral replication.[22], [23] Unexpectedly, while studying a potential relationship between autophagy and murine norovirus (MNV), we found a novel antiviral role of autophagy proteins against viral RCs.
The formation of a globular autophagosome requires the cooperation of many proteins that execute three main steps: initiation, nucleation and elongation. The elongation step of autophagosome formation requires a ubiquitin-like system known as the LC3 conjugation system, which functions to conjugate LC3 and its homologs to the lipid phosphatidylethanolamine (PE) in membranes (Fig 1B). For this ubiquitin-like conjugation, ATG7, ATG3, and the ATG12–ATG5-ATG16L1 complex (henceforth, ATG5 complex) function as the E1-like activating enzyme, E2-like conjugating enzyme, and E3-like ligase, respectively.[27] Using the MNV model, we found that the ATG5 complex is required for interferon-gamma (IFNG) mediated control of MNV replication, via inhibiting the formation and/or maintenance of the MNV RC, in cells and mice.[9] Proteins involved in the initiation and nucleation of the autophagosome, as well as lysosomal degradation, were dispensable for this antiviral response. Additional experiments detailing the IFNG signaling requirements favored a model in which a group of IFNG-regulated effector proteins would cooperate with the ATG5 complex, but the specific proteins were unknown.[31]
4. Cooperation of autophagy and IFN-inducible GTPases restricts Toxoplasma gondii
An antimicrobial process dependent on ATG5 but not on canonical autophagy had previously been identified for the protozoan parasite, Toxoplasma gondii.[32] This IFNG-mediated response results in the disruption of the membranous niche of T. gondii, the parasitophorous vacuole (PV) (Fig. 2). Disruption of the PV membrane (PVM) occurs as a result of the localization of several interferon-inducible GTPases such as interferon inducible GTPase 1 (IIGP1, a.k.a. Immunity-related GTPase family membrane a6 [IRGA6]). In the absence of ATG5, IRGA6 forms aggregates in the cytosol, rather than being targeted to the PVM of T. gondii,[33] leading to uncontrolled parasite replication.[32], [34], [35]
Figure 2. Role of LC3 conjugation in the intracellular response to Toxoplasma gondii.
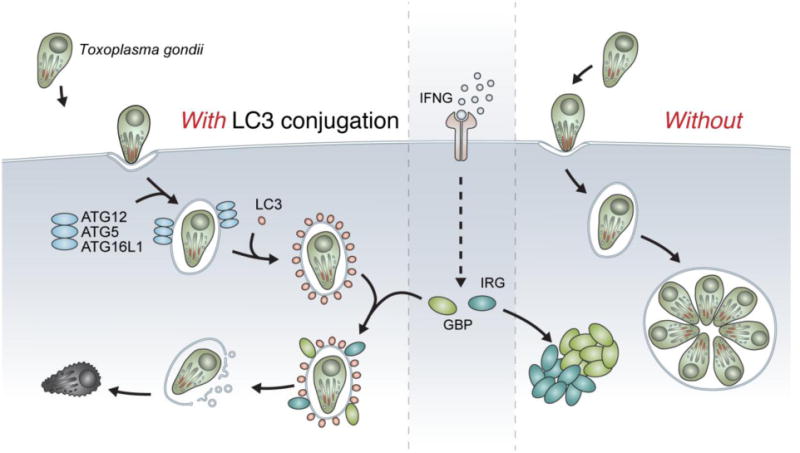
(Left) Upon invasion of the cell, T. gondii resides within its replicative niche, the parasitophorous vacuole (PV). Shortly after infection, the ATG12–ATG5-ATG16L1 complex of autophagy localizes to the vacuole, leading to LC3 deposition at the PV. In the presence of IFNγ, IRGs and GBPs are expressed and recruited to the PV. These dynamin-like GTPases vesiculate the PV, leading to pathogen exposure to the cytosol where it is ultimately killed. (Right) In the absence of LC3 conjugation, upon infection by T. gondii the PV is not decorated by LC3. Without LC3 conjugation, IFN-inducible GTPases are expressed but form aggregates in the cytosol. Consequently, T. gondii is not controlled and the parasite replicates freely in the PV.
Only the LC3 conjugation system of autophagy (ATG7, ATG3, and the ATG5 complex) is required for targeting of the IFN-inducible GTPases to the T. gondii PV and the IFNG-mediated control of T. gondii infection in cells and mice.[36] The upstream role of ATG5 and the fact that LC3 localizes on the PVM of T. gondii even in the absence of IFNG treatment pointed to a role for LC3 in the recruitment of IFN-inducible GTPases.[36] In fact, all known LC3 homologs, LC3 and gamma-aminobutyric acid receptor-associated protein (GABARAP) subfamilies, can be detected on the PVM of T. gondii.[37], [38], [39]
Furthermore, the ATG5 complex is both necessary and sufficient to recruit the IFN-inducible GTPases to a target membrane. By relocating the E3-like ligase ATG5 complex to the mitochondrial outer membrane or the plasma membrane, the GTPases are recruited to those membranes rather than the PVM of T. gondii.[39] Collectively, though the mechanistic details remain poorly understood (further discussed below), these data clearly show that the ATG5 complex can determine the target membrane of IFN-inducible GTPases. Thus, we termed this process Targeting by AutophaGy proteins (TAG).[39]
5. IFN-inducible GTPases attack and destroy pathogen containing vacuoles
The IFN-inducible GTPase superfamily is composed of four subfamilies: immunity related GTPases (IRGs), guanylate binding proteins (GBPs), myxovirus resistance proteins (Mx), and the very large IFN-inducible GTPases (VLIGs).[11] Only IRGs and GBPs were known to attack the vacuole-like shelters containing bacteria, protozoa, or fungi.[40], [41], [42] Initial observations of IRG knockout mice established the critical role of IRGs in the control of intracellular pathogens.[43], [44] Not all IRGs are created equally however. Knockouts of different IRGs led to non-redundant susceptibilities to various pathogens, suggesting unique roles for members of the IRG family. Not long after, the GBP family was discovered to also be involved in the control of intracellular bacteria and protozoa.[45], [46]
Together, these families of proteins accumulate on the PV and disrupt the membrane structure as demonstrated by immunoelectron microscopy.[32], [33], [46] Due to their structural similarity to dynamin, it was hypothesized that these proteins function equivalently, remodeling the membrane via mechanical forces exerted through GTP hydrolysis, though this has not been formally demonstrated biochemically.[47] The targeted membranes are subsequently vesiculated to the point of vacuole rupture, leading to the death of the now-exposed pathogens (Fig. 2).[40], [48], [49] While some studies have established links between antiviral activity and the IRGs and GBPs,[50], [51], [52] in many cases the underlying mechanisms are unknown.
6. Autophagy proteins bring the IFN-inducible GTPases to viral shelters
Although T. gondii and MNV are two disparate pathogens, i.e., a eukaryotic parasite and a small RNA virus, the parallel dependence of IFNG and ATG5 suggested a possible common effector mechanism. Thus, we investigated the potential role of IFN-inducible GTPases in the antiviral function of the ATG5 complex against the MNV RC. Indeed, the ATG5 complex is detected on the MNV RC in the absence of IFNG signaling,[9] suggesting that the complex might recruit the IFN-inducible GTPases to the RC.
LC3 and the IFN-inducible GTPases are detected on the MNV RC in wild type but not in Atg5-deficient cells,[10] consistent with an Atg5-dependent phenotype. Using models of a non-functional IRG system and a non-functional GBP system,[46], [53] we also confirmed that these GTPases are indeed required to control MNV replication in cells and mice.[10] Together, these data decisively established that IRGs and GBPs are the IFN-inducible effectors brought to the MNV RC by the ATG5 complex to disrupt the RC formation and replication of MNV (Fig. 1).
Characterization of the Atg5-dependent IFN-inducible GTPase targeting process has mainly been confined to studies in mice, which possess a different armament of IFN-inducible GTPases than humans. Though the autophagy proteins and GBPs are well conserved in both mouse and human,[47], [54] the IRG system is substantially contracted in the human system.[55] Compared to the mouse genome, which contains over 20 IRG genes, humans possess only two orthologs: IRGM and IRGC. Both lack the IFN stimulated response element, and therefore are not IFN inducible.[56] Despite this major difference, we found that the system was functionally intact in human cells. Both LC3 and GBPs not only localize to the RC of MNV but also are required for IFNG to control MNV replication in human cells.[10]
LC3 localization is not unique to the RC of MNV as the RCs of several other (+)RNA viruses have been reported to exhibit colocalization.[57], [58], [59], [60], [61], [62], [63] In some cases, LC3 on the RC is indicative of the viruses co-opting the autophagy pathway/machinery for their own benefit.[64], [65] However, LC3 on the RC, like a double-edged sword, may aid viral replication, while simultaneously recruiting the destructive GTPases to the RCs upon their induction. Indeed, analogous to MNV, both LC3 and the IFN-inducible GTPases are targeted to the RC of another (+)RNA virus, encephalomyocarditis virus (EMCV).[10] We hypothesize that other (+)RNA viruses are targeted by LC3 and IFN-inducible GTPases based on the shared effect of the TAG across even disparate pathogens (viruses as well as bacteria and protozoa). Identification of other susceptible (+)RNA viruses will solidify TAG as a major antiviral mechanism.
Two major obstacles may complicate straightforward identification of additional sensitive (+)RNA viruses: alternative antiviral effects of IFNG and potential immune evasion of viruses. IFNG can exert antiviral activity through multiple mechanisms and TAG is only one arm of this immune defense. We have shown previously that IFNG inhibits the replication of other (+)RNA viruses such as murine hepatitis virus and West Nile virus in the absence of Atg5.[9] Thus, dissecting the antiviral role of TAG in such cases is confounded by the potential of other antiviral mechanisms initiated by IFNG. Alternatively, it is possible that (+)RNA viruses have evolved mechanisms to actively evade TAG. Indeed, there is precedent for this in the field of IFN-inducible GTPases. Many pathogens including virulent strains of T. gondii possess counter measures for disabling the IFN-inducible GTPases.[66], [67] Thus, it is reasonable to speculate that certain (+)RNA viruses are capable of evading the LC3-mediated targeting of IFN-inducible GTPases entirely.
7. How do the autophagy proteins recognize viral RCs as a target?
Work demonstrating the crucial role of the TAG process in controlling intracellular pathogens has raised a critical question:[39] how does TAG recognize a target membrane (Fig. 3)? It is now appreciated that in addition to non-self recognition, the immune system may perceive changes in self–e.g. missing- or altered-self–that alert the host of infection.[68] Thus, there might be a defined molecular signature of the RC that enables the host immune system to detect the structure. In the context of the multi-pathogen targeting ability of TAG, another important question is whether a universal or pathogen-specific mechanism determines the target.
Figure 3. Three models for the innate recognition of replication complexes.
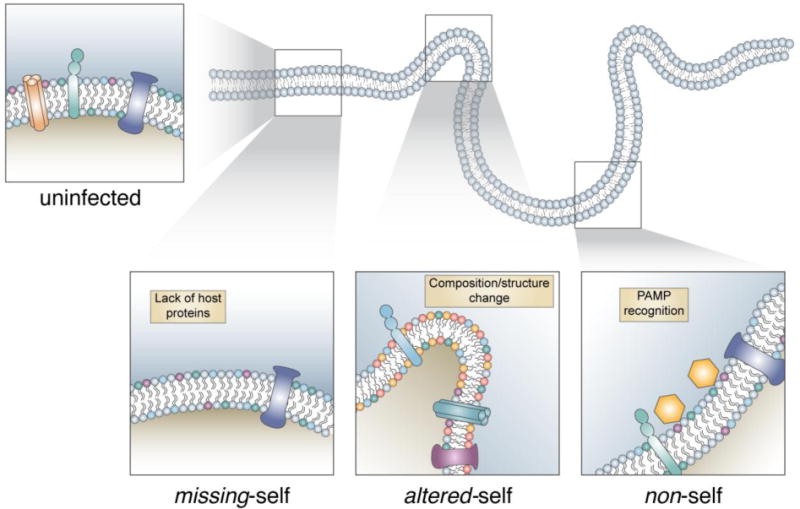
Despite host origin, the replication complex can be recognized and targeted by the autophagy proteins. Models of recognition include recognition of missing-self, altered-self, or non-self. (Bottom left) The missing-self model proposes that the RC lacks host proteins normally found on the membrane. This array of host proteins would typically prevent the activation of this response, therefore the RC lacking these proteins would be susceptible to targeting. (Bottom center) The altered-self model proposes that the RC differs significantly from homeostatic conditions, either by composition changes in the lipid/protein makeup or by distinct structural changes such as curvature. (Bottom right) The non-self model proposes that a microbial PAMP at the membrane is responsible for the targeting of autophagy proteins. As it is unlikely one PAMP is shared between the various pathogens known to be targeted by this pathway, the non-self model suggests different pathogen specific PAMPs would all converge on the activation of targeting by autophagy proteins.
Initial studies established that T. gondii, through an active invasion process, stripped the resultant PV of most host proteins.[69] Thus, it was hypothesized that the PV might be a distinct compartment that could be sensed as a target due to “missing-self” (Fig. 3).[68] The discovery of IRG targeting to the vacuoles of the bacterium Chlamydia trachomatis and the fungus Encephalitozoon cuniculi further supported this hypothesis.[42], [70] Devoid of the normal array of host proteins, these pathogen-containing vacuoles could be recognized by the lack of proteins, thereby allowing targeting by IRGs/GBPs.[68] A subset of IRGs, known as GMS IRGs (so named for their GXS motif in their GTPase domain),[71], [72], [73] are known to decorate most membrane structures within a cell and to function as guanine dissociation inhibitors that keep the rest of the IRGs, known as GKS IRGs, in their inactive GDP-bound forms.[74] Thus, the absence of GMS IRGs on PVs would lead to a de-regulated state, thereby allowing the effector GKS IRGs and GBPs to accumulate on the membrane.[70] However, this model was challenged when the localization of IRGs and GBPs to the vacuoles containing T. gondii or C. trachomatis was found to be dependent upon the LC3 conjugation system of autophagy.[39], [36], [75], [76] Furthermore, the demonstration that the same targeting mechanism functions against viral RCs,[10] composed of a mixture of proteins and lipids from the ER and the Golgi,[77], [78] also argued against this model.
In contrast to “missing self”, an alternate mode of target membrane recognition is the sensing of “altered-self” (Fig. 3). In order to form the vacuole-like replication niches, pathogens may need to alter host membranes, thereby creating an altered membrane structure, which may be no longer recognizable as “self”. An altered composition of proteins, lipids, or structure of the membrane due to pathogen proteins or by recruited host proteins could all contribute to the emergence of a unique compartment, not native to the host in homeostatic conditions. The ATG5 complex is reported to bind lipids of unknown specificity through ATG5.[79] Thus, it is tempting to speculate that ATG5 may recognize and bind specific lipid moieties enriched at the membranes of pathogen-containing vacuoles. In line with this, it is noteworthy that T. gondii proteins secreted into the host cytoplasm during invasion can traffic back onto the PVM,[80], [81] suggesting a unique signature of the PVM. Whether these proteins of T. gondii can also traffic to the replication vacuoles of other intracellular pathogens, like the RC of MNV, is an appealing possibility.
The last mode of target recognition is sensing of “non-self” (Fig. 3). To form the vacuole-like shelters, pathogen proteins need to actively modify the host membrane structure and embed themselves in the membranes. These pathogen-derived proteins, or something equivalent, may be recognized as a pathogen-associated signal by the ATG5 complex or an upstream sensor that can recruit the ATG5 complex. This model would suggest a distinct way of sensing different classes of pathogens, which may converge on the LC3 conjugation system to recruit the downstream anti-microbial effectors like IRGs and GBPs. Given the broad range of microbial target membranes, we speculate that many different upstream sensing pathways (with possible redundancies) are utilized to identify a target for the TAG-mediated recruitment of effectors. Once LC3 homologs have marked a target membrane, however, the same membranolytic pathway may be initiated, thereby destroying the structure.
8. How do autophagy proteins recruit the GTPases to viral RCs?
The location of the ATG5 complex is sufficient to determine the destination of the IRGs and GBPs.[36], [39] How the ATG5 complex brings the IFN-inducible GTPases to the pathogen-filled, LC3 homolog-marked compartments is a major outstanding question. Presumably, all the LC3 homologs conjugated to membranes by the ATG5 complex may recruit the IFN-inducible GTPases. In support of this, the LC3 homologs have been shown to be necessary for the recruitment of the GTPases to the target membrane.[10], [39] However, only in the situation where all homologs are knocked-out/down is the GTPase recruitment affected, indicating a redundant function of the LC3 and GABARAP subfamilies. This contrasts with previous studies in canonical autophagy establishing distinct functions for each subfamily despite high structural conservation. The sufficiency of the LC3 homologs in the recruitment of the IFN-inducible GTPases, independent of the ATG5 complex, has yet to be examined. However, a recent report implicates an essential role of GABARAPL2, but not the LC3 subfamily, in the proper localization and function of the IFN-inducible GTPases through its association with ADP-ribosylation factor 1 (ARF1).[82] Hence, the role of the LC3 homologs in directing the GTPases is still uncertain.
The simplest working mechanism, as briefly mentioned above, is that one or all of the LC3 homologs conjugated by the ATG5 complex onto the target membrane recruit(s) the GTPases (Fig. 4). LC3 homologs may recruit effector IRGs first and then GBPs through ubiquitin and p62, as previously proposed.[11], [83] Based on the recruitment of GBPs to the MNV RC in the absence of the functional IRGs in the human system,[10] it is also possible that GBPs are recruited by the LC3 homolog(s) independently of the IRGs. In line with this, certain IRGs and GBPs have putative LC3-interacting region (LIR) motifs,[84] suggestive of a direct protein-protein interaction with the LC3 homologs. Despite this, no direct interaction between LC3 homologs and GTPases has been shown experimentally. Alternatively, the LC3 homologs may associate with another mediator (e.g. ubiquitin, ARF1, etc.) on the target membrane to indirectly recruit the GTPases. A major caveat of this working hypothesis is that, if LC3s on a membrane are sufficient for the recruitment of the GTPases, the LC3-labeled autophagosome would be just as susceptible to targeting, and disruption, by the GTPases. In fact, IFNs enhance, rather than inhibit, autophagy,[85], [86] suggesting that autophagosomes are not disrupted by the IFN-inducible GTPases. It is possible that the presence of autophagy proteins and/or GMS IRGs remaining on the autophagosome, the membrane curvature, or the double-membraned nature of autophagosomes prevents autophagosomes from being targeted, or even if targeted, disrupted by the GTPases.[75], [87], [88]
Figure 4. Potential mechanisms of IFN-inducible GTPase targeting to the LC3-marked membrane.
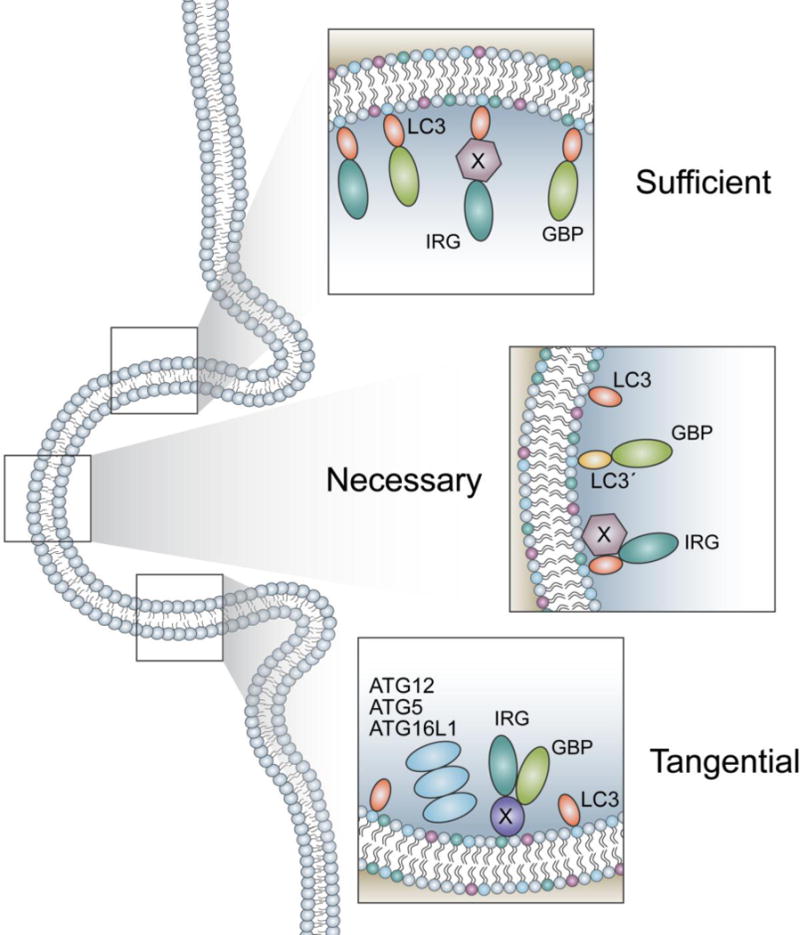
(Top) LC3 may be sufficient to recruit the GTPases to a given membrane. Whether LC3 alone, or another mediator is required between LC3 and the GTPases is unknown. (Center) LC3 may be necessary but not sufficient to recruit the GTPases. A second protein may be required to facilitate the recruitment of GTPases, or LC3 may need to be modified by a post translational modification to bind IRGs or GBPs. (Bottom) LC3 may be tangential to the recruitment of IRGs and GBPs and have no role. The localization of the ATG12–ATG5-ATG16L1 complex may recruit a protein to the membrane that is responsible for the IRG and GBP binding. As a consequence of the complex localizing to the membrane LC3 also becomes conjugated at the site.
Given these facts, an alternative yet complementary working mechanism is that LC3 homologs are not sufficient and additional factors are needed for the recruitment (Fig. 4). Potential necessary factors that may include other proteins brought to the target membranes by the ATG5 complex or post-translational modifications of the LC3 homolog(s) on the membrane. Simply, LC3 homologs on the membrane of the autophagosome and pathogen-containing vacuoles might differ functionally through their association with other proteins or specific post-translational modifications. In this regard, it is noteworthy that re-localizing the ATG5 complex to the mitochondrial membrane requires T. gondii infection for efficient recruitment of the GTPases to that membrane, whereas re-localizing the complex to the plasma membrane does not.[39] Given that LC3s can undergo post-translational modifications and no GMS IRG is associated with the plasma membrane, a “triple-check” model of IFN, LC3, and infection has been hypothesized.[75], [89] The model proposes LC3 homologs are conjugated to a given membrane, T. gondii infection removes inhibitory GMS IRGs from the LC3-marked membranes, and IFN modifies LC3s to function as a guanine nucleotide exchange factor for the local activation of the GTPases. Nevertheless, it is still unverified whether such a modification happens specifically to LC3 homologs on the membrane of pathogen-containing vacuoles.
In contrast to the above scenarios, it is also entirely possible that the LC3 homologs are not involved in GBP recruitment to the target membrane at all (Fig. 4). Rather, the additional factors brought by the ATG5 complex recruit the GTPases, and the localization of LC3 homologs on the membrane of pathogen-containing vacuole is merely a consequence of the ATG5 complex localization. Based on the necessity of LC3 homologs, this model suggests the LC3 homologs would be required somewhere else to maintain a functional status of the GTPases (e.g. through activation of ARF1).[82] This possibility stems from the concern that the frequency of the LC3 homologs on the target membrane is significantly lower than that of the GTPases.[36], [39], [82] Further studies on the sufficiency of LC3 homologs on the membrane will clarify the role of LC3 homologs in the recruitment of the GTPases and consequently the recruitment mechanism by the ATG5 complex.
While it is tempting to hypothesize one universal mechanism that facilitates GTPase recruitment to these pathogen-containing vacuoles, there may exist multiple mechanisms to recruit GTPases to membranes. In alignment with this idea, the GKS effector IRG, IRGB10, was recently shown to bind directly to the membrane of a bacterium via a putative transmembrane amphipathic helix,[90] suggesting a potential propensity of IRGB10 toward bacterial membranes. This putative transmembrane helix is shared by most other IRGs, indicating this may be a common targeting mechanism of IRGs to bacterial membranes. Similarly, GBPs were also shown to directly attack the membrane of T. gondii itself.[91] In addition, mouse GBP2 can be recruited to the breached vacuole, via a mechanism independent of regulatory IRGs or the LC3 conjugation system.[92] Collectively, these data suggest that GTPases can localize to membranes through various mechanisms and that these mechanisms may function in parallel for the most efficient defense against various pathogen invasions.
9. How do the recruited GTPases disrupt viral RCs?
The representative anti-microbial function of both IRGs and GBPs involves the pinching off of small vesicles from the membranes of pathogen-containing vacuoles.[11] The IRGs and GBPs, phylogenetically related to dynamin, are known for their ability to vesiculate their target membranes. In addition to their dynamin-like function, these GTPases have more recently been appreciated to have a multitude of roles within cell-autonomous defenses. GBPs are also known to bring various anti-microbial effector proteins or to stunt bacterial spread by blocking bacterial association with actin.[45], [93], [94] Furthermore, GBPs can enhance inflammasome formation, which produces inflammatory cytokines in response to pathogen invasion and cellular damages.[47], [95], [49]
Consistent with their localization in the cytoplasm or on the cytoplasmic side of a membrane, IRGs and GBPs colocalize with viral polymerase on the cytoplasmic side of the MNV RC.[10] Although their localization on RCs is positively correlated with disruption of the compartment and consequent inhibition of viral replication, the functional mechanism of such inhibition is unknown. It is also unclear whether the GTPases are recruited onto the membrane during or after RC formation, thereby inhibiting the formation, or disrupting the pre-formed structure, respectively (Fig. 5). Of course, these possibilities are not mutually exclusive and elucidating the mechanism will lead to a better understanding of the potential immune evasion function of the RC and will facilitate the development of broad antiviral strategies against these structures.
Figure 5. Three models for the IRG/GBP mediated inhibition of viral RCs.
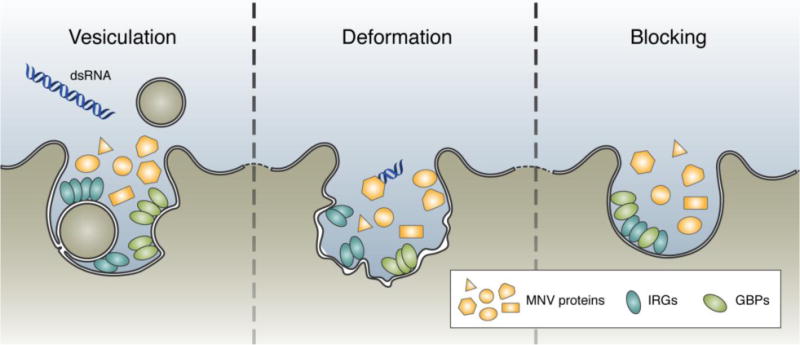
Based on previous characterization of the IRGs and GBPs in the control of pathogens, there are three potential effector mechanisms of these proteins in relation to the RC. (Left) Expanding upon the dynamin-like role assigned to the IRGs and GBPs in the control of T. gondii, these proteins may function to vesiculate the RC, releasing viral proteins and PAMPs into the open cytosol for innate recognition. (Center) Alternatively, the GTPases may function to deform the membrane structure of the RC. Deformation of the RC may inhibit the formation of viral protein complexes important for the efficient replication of the genome, and therefore preventing viral replication. (Right) Finally, the GTPases may function to sterically block the MNV proteins from associating with the membrane and preventing viral replication.
Extrapolating from other pathogens targeted by the GTPases, a straightforward potential mechanism is that the GTPases may disrupt pre-formed RCs through vesiculation (Fig. 5). However, the small size of the viral RC complicates this possibility.[96] The average size of vesicles created during the IRG/GBP attack on the PVM of T. gondii is nearly as large as the entire replication compartment of MNV.[97] Thus, it is unlikely for IRGs/GBPs to disrupt the viral RC through vesiculation.
Instead, a more plausible mechanism is that these proteins may exert direct antiviral effects by compromising the integrity of viral RC membranes (Fig. 5). Since both subfamilies of GTPases can bind to and oligomerize on membranes, they may still be able to distort membranes significantly, deforming the structure and leading to inefficient viral replication and/or structural maintenance. Alternatively, but not exclusively, such membrane association of the GTPases may block the access of the membrane to viral proteins, such that viral proteins cannot form a necessary complex on the membrane to replicate (Fig. 5). Deciphering the functional requirement of the GTPases (e.g. GTPase activity, oligomerization, membrane binding) will be essential for a mechanistic understanding of their antiviral activity against viral RCs.[98], [99]
10. Conclusions and Prospects
The recent discovery of an antiviral strategy against the RCs of a model (+)RNA virus has uncovered new and exciting avenues of study in the field of cell-autonomous immunity.[9], [10], [31], [97] This antiviral response combines two evolutionarily conserved systems, autophagy and IFN, through the work of the LC3 conjugation system and the IFN-inducible GTPases, respectively. Parallel studies using MNV, T. gondii, and C. trachomatis have brought to light core mechanistic details shared between these distinct pathogens, yet much still remains to be understood.
Given the host organelle membrane composition of viral RCs, the existence of an antiviral immune defense mechanism against viral RCs challenges the fundamental dogma of self/non-self differentiation. Moreover, the IFN-inducible GTPases emerge as a powerful group of effector proteins, with broad anti-microbial functions against the vacuole-like structures of pathogens including viruses, bacteria, protozoa, and fungi. Follow-up studies illuminating the specific mechanism of sensing target membranes, recruiting effector GTPases, and breaking viral RCs will be critical in understanding the extensive role of this system in response to pathogens. Of particular interest, proteomic and lipidomic comparisons among the vacuole-like structures of pathogens, like the RC of MNV, the PV of T. gondii, and the inclusion body of C. trachomatis, may reveal the unique and shared nature of handling the pathogen-containing vacuoles by the immune system. Last but not least, many viruses have evolved to directly antagonize the interferon signaling pathway;[100] we expect many (+)RNA viruses have also evolved strategies to counteract this autophagy and IFN-mediated attack of viral RCs. As usual, fundamental biology as well as treatment of infectious diseases will benefit from understanding the co-evolution of the immune defense system of host and the immune evasion strategies of pathogens.
Acknowledgments
This work was supported in part by the Brinson Foundation Junior Investigator Grant, Cancer Research Foundation Young Investigator Award, an Institutional Research Grant (IRG-58-004-53-IRG) from the American Cancer Society, the University of Chicago Digestive Diseases Research Core Center (NIDDK P30DK42086), the University of Chicago Comprehensive Cancer Center Support Grant (P30 CA14599), the National Center for Advancing Translational Sciences of the National Institutes of Health (NIH UL1 TR000430), NIH R01AI127518, and NIH DP2CA225208. H.M.B. was supported in part by NIH T32 AI007090 and S.B.B. was supported in part by NIH T32 GM007183.
Abbreviations
- RC
replication compartment or replication complex
- MNV
murine norovirus
- PV
parasitophorous vacuole
- IRG
immunity-related GTPase
- GBP
guanylate-binding protein
- TAG
Targeting by AutophaGy Proteins
- IFN
interferon
Footnotes
All authors declare no conflict of interest.
References
- 1.Randow F, MacMicking JD, James LC. Science. 2013;340:701. doi: 10.1126/science.1233028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harak C, Lohmann V. Virology. 2015;479–480:418. doi: 10.1016/j.virol.2015.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.den Boon JA, Diaz A, Ahlquist P. Cell Host Microbe. 2010;8:77. doi: 10.1016/j.chom.2010.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neufeldt CJ, Joyce MA, Van Buuren N, Levin A, Kirkegaard K, Gale M, Jr, Tyrrell DL, Wozniak RW. PLoS Pathog. 2016;12:e1005428. doi: 10.1371/journal.ppat.1005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romero-Brey I, Bartenschlager R. Viruses. 2014;6:2826. doi: 10.3390/v6072826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Diaz A, Wang X, Ahlquist P. Proc Natl Acad Sci USA. 2010;107:16291. doi: 10.1073/pnas.1011105107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagy PD. J Virol. 2015;89:5196. doi: 10.1128/JVI.02973-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagy PD, Pogany J. Nat Rev Microbiol. 2012;10:137. doi: 10.1038/nrmicro2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, Shrestha B, Diamond MS, Dani A, Sosnovtsev SV, Green KY, Lopez-Otin C, Xavier RJ, Thackray LB, Virgin HW. Cell Host Microbe. 2012;11:397. doi: 10.1016/j.chom.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biering SB, Choi J, Halstrom RA, Brown HM, Beatty WL, Lee S, McCune BT, Dominici E, Williams LE, Orchard RC, Wilen CB, Yamamoto M, Coers J, Taylor GA, Hwang S. Cell Host Microbe. 2017;22:74. doi: 10.1016/j.chom.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pilla-Moffett D, Barber MF, Taylor GA, Coers J. J Mol Biol. 2016;428:3495. doi: 10.1016/j.jmb.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.den Boon JA, Ahlquist P. Annu Rev Microbiol. 2010;64:241. doi: 10.1146/annurev.micro.112408.134012. [DOI] [PubMed] [Google Scholar]
- 13.Yoneyama M, Onomoto K, Jogi M, Akaboshi T, Fujita T. Curr Opin Immunol. 2015;32:48. doi: 10.1016/j.coi.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 14.Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. Biol Cell. 2005;97:147. doi: 10.1042/BC20040058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Overby AK, Popov VL, Niedrig M, Weber F. J Virol. 2010;84:8470. doi: 10.1128/JVI.00176-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uchida L, Espada-Murao LA, Takamatsu Y, Okamoto K, Hayasaka D, Yu F, Nabeshima T, Buerano CC, Morita K. Sci Rep. 2014;4:7395. doi: 10.1038/srep07395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oudshoorn D, van der Hoeven B, Limpens RWAL, Beugeling C, Snijder EJ, Bárcena M, Kikkert M. mBio. 2016;7:e01991. doi: 10.1128/mBio.01991-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scutigliani EM, Kikkert M. Cytokine Growth Factor Rev. 2017;37:17. doi: 10.1016/j.cytogfr.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shulla A, Randall G. Curr Opin Microbiol. 2016;32:82. doi: 10.1016/j.mib.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quinkert D, Bartenschlager R, Lohmann V. J Virol. 2005;79:13594. doi: 10.1128/JVI.79.21.13594-13605.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neufeldt CJ, Joyce MA, Levin A, Steenbergen RH, Pang D, Shields J, Tyrrell DL, Wozniak RW. PLoS Pathog. 2013;9:e1003744. doi: 10.1371/journal.ppat.1003744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jordan TX, Randall G. Microbes Infect. 2012;14:126. doi: 10.1016/j.micinf.2011.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong X, Levine B. J Innate Immun. 2013;5:480. doi: 10.1159/000346388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Duve C, Wattiaux R. Annu Rev Physiol. 1966;28:435. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 25.Codogno P, Mehrpour M, Proikas-Cezanne T. Nat Rev Mol Cell Biol. 2012;13:7. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 26.Zhi X, Feng W, Rong Y, Liu R. Cell Mol Life Sci. 2017;75:815. doi: 10.1007/s00018-017-2657-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noda NN, Inagaki F. Annu Rev Biophys. 2015;44:101. doi: 10.1146/annurev-biophys-060414-034248. [DOI] [PubMed] [Google Scholar]
- 28.Levine MNB, Virgin HW. Nature. 2011;469:323. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A. Science. 2007;315:1398. doi: 10.1126/science.1136880. [DOI] [PubMed] [Google Scholar]
- 30.Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C. Science. 2005;307:593. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 31.Maloney NS, Thackray LB, Goel G, Hwang S, Duan E, Vachharajani P, Xavier R, Virgin HW. J Virol. 2012;86:12655. doi: 10.1128/JVI.01564-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, Schmidt RE, Mizushima N, Deretic V, Sibley LD, Virgin HW. Cell Host Microbe. 2008;4:458. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, Howard JC. PLoS Pathog. 2005;1:e24. doi: 10.1371/journal.ppat.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao YO, Khaminets A, Hunn JP, Howard JC. PLoS Pathog. 2009;5:e1000288. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Selleck EM, Fentress SJ, Beatty WL, Degrandi D, Pfeffer K, Virgin HWt, Macmicking JD, Sibley LD. PLoS Pathog. 2013;9:e1003320. doi: 10.1371/journal.ppat.1003320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, Fujita N, Saitoh T, Akira S, Yoshimori T, Sibley LD, Hwang S, Virgin HW. Immunity. 2014;40:924. doi: 10.1016/j.immuni.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, Akematsu T, Akira S, Al-Younes HM, Al-Zeer MA, Albert ML, Albin RL, Alegre-Abarrategui J, Aleo MF, Alirezaei M, Almasan A, Almonte-Becerril M, Amano A, Amaravadi R, Amarnath S, Amer AO, Andrieu-Abadie N, Anantharam V, Ann DK, Anoopkumar-Dukie S, Aoki H, Apostolova N, Arancia G, Aris JP, Asanuma K, Asare NYO, Ashida H, Askanas V, Askew DS, Auberger P, Baba M, Backues SK, Baehrecke EH, Bahr BA, Bai X-Y, Bailly Y, Baiocchi R, Baldini G, Balduini W, Ballabio A, Bamber BA, Bampton ETW, Bánhegyi G, Bartholomew CR, Bassham DC, Bast RCJ, Batoko H, Bay B-H, Beau I, Béchet DM, Begley TJ, Behl C, Behrends C, Bekri S, Bellaire B, Bendall LJ, Benetti L, Berliocchi L, Bernardi H, Bernassola F, Besteiro S, Bhatia-Kissova I, Bi X, Biard-Piechaczyk M, Blum JS, Boise LH, Bonaldo P, Boone DL, Bornhauser BC, Bortoluci KR, Bossis I, Bost F, Bourquin J-P, Boya P, Boyer-Guittaut M, Bozhkov PV, Brady NR, Brancolini C, Brech A, Brenman JE, Brennand A, Bresnick EH, Brest P, Bridges D, Bristol ML, Brookes PS, Brown EJ, Brumell JH, Brunetti-Pierri N, Brunk UT, Bulman DE, Bultman SJ, Bultynck G, Burbulla LF, Bursch W, Butchar JP, Buzgariu W, Bydlowski SP, Cadwell K, Cahová M, Cai D, Cai J, Cai Q, Calabretta B, Calvo-Garrido J, Camougrand N, Campanella M, Campos-Salinas J, Candi E, Cao L, Caplan AB, Carding SR, Cardoso SM, Carew JS, Carlin CR, Carmignac V, Carneiro LAM, Carra S, Caruso RA, Casari G, Casas C, Castino R, Cebollero E, Cecconi F, Celli J, Chaachouay H, Chae H-J, Chai C-Y, Chan DC, Chan EY, Chang RC-C, Che C-M, Chen C-C, Chen G-C, Chen G-Q, Chen M, Chen Q, Chen SS-L, Chen W, Chen X, Chen X, Chen X, Chen Y-G, Chen Y, Chen Y, Chen Y-J, Chen Z, Cheng A, Cheng CHK, Cheng Y, Cheong H, Cheong J-H, Cherry S, Chess-Williams R, Cheung ZH, Chevet E, Chiang H-L, Chiarelli R, Chiba T, Chin L-S, Chiou S-H, Chisari FV, Cho CH, Cho D-H, Choi AMK, Choi D, Choi KS, Choi ME, Chouaib S, Choubey D, Choubey V, Chu CT, Chuang T-H, Chueh S-H, Chun T, Chwae Y-J, Chye M-L, Ciarcia R, Ciriolo MR, Clague MJ, Clark RSB, Clarke PGH, Clarke R, Codogno P, Coller HA, Colombo MI, Comincini S, Condello M, Condorelli F, Cookson MR, Coppens GHCI, Corbalan R, Cossart P, Costelli P, Costes S, Coto-Montes A, Couve E, Coxon FP, Cregg JM, Crespo JL, Cronjé MJ, Cuervo AM, Cullen JJ, Czaja MJ, D’Amelio M, Darfeuille-Michaud A, Davids LM, Davies FE, De Felici M, de Groot JF, de Haan CAM, De Martino L, De Milito A, De Tata V, Debnath J, Degterev A, Dehay B, Delbridge LMD, Demarchi F, Deng YZ, Dengjel J, Dent P, Denton D, Deretic V, Desai SD, Devenish RJ, Di Gioacchino M, Di Paolo G, Di Pietro C, Díaz-Araya G, Díaz-Laviada I, Diaz-Meco MT, Diaz-Nido J, Dikic I, Dinesh-Kumar SP, Ding W-X, Distelhorst CW, Diwan A, Djavaheri-Mergny M, Dokudovskaya S, Dong Z, Dorsey FC, Dosenko V, Dowling JJ, Doxsey S, Dreux M, Drew ME, Duan Q, Duchosal MA, Duff K, Dugail I, Durbeej M, Duszenko M, Edelstein CL, Edinger AL, Egea G, Eichinger L, Eissa NT, Ekmekcioglu S, El-Deiry WS, Elazar Z, Elgendy M, Ellerby LM, Eng KE, Engelbrecht A-M, Engelender S, Erenpreisa J, Escalante R, Esclatine A, Eskelinen E-L, Espert L, Espina V, Fan H, Fan J, Fan Q-W, Fan Z, Fang S, Fang Y, Fanto M, Fanzani A, Farkas T, Farré J-C, Faure M, Fechheimer M, Feng CG, Feng J, Feng Q, Feng Y, Fesues L, Feuer R, Figueiredo-Pereira ME, Fimia GM, Fingar DC, Finkbeiner S, Finkel T, Finley KD, Fiorito F, Fisher EA, Fisher PB, Flajolet M, Florez-McClure ML, Florio S, Fon EA, Fornai F, Fortunato F, Fotedar R, Fowler DH, Fox HS, Franco R, Frankel LB, Fransen M, Fuentes JM, Fueyo J, Fujii J, Fujisaki K, Fujita E, Fukuda M, Furukawa RH, Gaestel M, Gailly P, Gajewska M, Galliot B, Galy V, Ganesh S, Ganetzky B, Ganley IG, Gao F-B, Gao GF, Gao J, Garcia L, Garcia-Manero G, Garcia-Marcos M, Garmyn M, Gartel AL, Gatti E, Gautel M, Gawriluk TR, Gegg ME, Geng J, Germain M, Gestwicki JE, Gewirtz DA, Ghavami S, Ghosh P, Giammarioli AM, Giatromanolaki AN, Gibson SB, Gilkerson RW, Ginger ML, Ginsberg HN, Golab J, Goligorsky MS, Golstein P, Gomez-Manzano C, Goncu E, Gongora C, Gonzalez CD, Gonzalez R, González-Estévez C, González-Polo RA, Gonzalez-Rey E, Gorbunov NV, Gorski S, Goruppi S, Gottlieb RA, Gozuacik D, Granato GE, Grant GD, Green KN, Gregorc A, Gros F, Grose C, Grunt TW, Gual P, Guan J-L, Guan K-L, Guichard SM, Gukovskaya AS, Gukovsky I, Gunst J, Gustafsson AB, Halayko AJ, Hale AN, Halonen SK, Hamasaki M, Han F, Han T, Hancock MK, Hansen M, Harada H, Harada M, Hardt SE, Harper JW, Harris AL, Harris J, Harris SD, Hashimoto M, Haspel JA, Hayashi S-i, Hazelhurst LA, He C, He Y-W, Hébert M-J, Heidenreich KA, Helfrich MH, Helgason GV, Henske EP, Herman B, Herman PK, Hetz C, Hilfiker S, Hill JA, Hocking LJ, Hofman P, Hofmann TG, Hoehfeld J, Holyoake TL, Hong M-H, Hood DA, Hotamisligil GS, Houwerzijl EJ, Høyer-Hansen M, Hu B, Hu C-AA, Hu H-M, Hua Y, Huang C, Huang J, Huang S, Huang W-P, Huber TB, Huh W-K, Hung T-H, Hupp TR, Hur GM, Hurley JB, Hussain SNA, Hussey PJ, Hwang JJ, Hwang S, Ichihara A, Ilkhanizadeh S, Inoki K, Into T, Iovane V, Iovanna JL, Ip NY, Isaka Y, Ishida H, Isidoro C, Isobe K-i, Iwasaki A, Izquierdo M, Izumi Y, Jaakkola PM, Jäättelä M, Jackson GR, Jackson WT, Janji B, Jendrach M, Jeon J-H, Jeung E-B, Jiang H, Jiang H, Jiang JX, Jiang M, Jiang Q, Jiménez A, Jiang X, Jin M, Jin S, Joe CO, Johansen T, Johnson DE, Johnson GVW, Jones NL, Joseph B, Joseph SK, Joubert AM, Juhász G, Juillerat-Jeanneret L, Jung CH, Jung Y-K, Kaarniranta K, Kaasik A, Kabuta T, Kadowaki M, Kagedal K, Kamada Y, Kaminskyy VO, Kampinga HH, Kanamori H, Kang C, Kang KB, Kang KI, Kang R, Kang Y-A, Kanki T, Kanneganti T-D, Kanno H, Kanthasamy AG, Kanthasamy A, Karantza V, Kaushal GP, Kaushik S, Kawazoe Y, Ke P-Y, Kehrl JH, Kelekar A, Kerkhoff C, Kessel DH, Khalil H, Kiel JAKW, Kiger AA, Kihara A, Kim DR, Kim D-H, Kim D-H, Kim E-K, Kim H-R, Kim J-S, Kim JH, Kim JC, Kim JK, Kim PK, Kim SW, Kim Y-S, Kim Y, Kimchi A, Kimmelman AC, King JS, Kinsella TJ, Kirkin V, Kirshenbaum LA, Kitamoto K, Kitazato K, Klein L, Klimecki WT, Klucken J, Knecht E, Ko BCB, Koch JC, Koga H, Koh J-Y, Koh YH, Koike M, Komatsu M, Kominami E, Kong HJ, Kong W-J, Korolchuk VI, Kotake Y, Koukourakis MI, Flores JBK, Kovács AL, Kraft C, Krainc D, Kraemer H, Kretz-Remy C, Krichevsky AM, Kroemer G, Krueger R, Krut O, Ktistakis NT, Kuan C-Y, Kucharczyk R, Kumar A, Kumar R, Kumar S, Kundu M, Kung H-J, Kurz T, Kwon HJ, La Spada AR, Lafont F, Lamark T, Landry J, Lane JD, Lapaquette P, Laporte JF, László L, Lavandero S, Lavoie JN, Layfield R, Lazo PA, Le W, Le Cam L, Ledbetter DJ, Lee AJX, Lee B-W, Lee GM, Lee J, Lee J-H, Lee M, Lee M-S, Lee SH, Leeuwenburgh C, Legembre P, Legouis R, Lehmann M, Lei H-Y, Lei Q-Y, Leib DA, Leiro J, Lemasters JJ, Lemoine A, Lesniak MS, Lev D, Levenson VV, Levine B, Levy E, Li F, Li J-L, Li L, Li S, Li W, Li X-J, Li Y-b, Li Y-P, Liang C, Liang Q, Liao Y-F, Liberski PP, Lieberman A, Lim HJ, Lim K-L, Lim K, Lin C-F, Lin F-C, Lin J, Lin JD, Lin K, Lin W-W, Lin W-C, Lin Y-L, Linden R, Lingor P, Lippincott-Schwartz J, Lisanti MP, Liton PB, Liu B, Liu C-F, Liu K, Liu L, Liu QA, Liu W, Liu Y-C, Liu Y, Lockshin RA, Lok C-N, Lonial S, Loos B, Lopez-Berestein G, Lopez-Otin C, Lossi L, Lotze MT, Lőw P, Lu B, Lu B, Lu B, Lu Z, Luciano F, Lukacs NW, Lund AH, Lynch-Day MA, Ma Y, Macian F, MacKeigan JP, Macleod KF, Madeo F, Maiuri L, Maiuri MC, Malagoli D, Malicdan MCV, Malorni W, Man N, Mandelkow E-M, Manon S, Manov I, Mao K, Mao X, Mao Z, Marambaud P, Marazziti D, Marcel YL, Marchbank K, Marchetti P, Marciniak SJ, Marcondes M, Mardi M, Marfe G, Mariño G, Markaki M, Marten MR, Martin SJ, Martinand-Mari C, Martinet W, Martinez-Vicente M, Masini M, Matarrese P, Matsuo S, Matteoni R, Mayer A, Mazure NM, McConkey DJ, McConnell MJ, McDermott C, McDonald C, McInerney GM, McKenna SL, McLaughlin B, McLean PJ, McMaster CR, McQuibban GA, Meijer AJ, Meisler MH, Meléndez A, Melia TJ, Melino G, Mena MA, Menendez JA, Menna-Barreto RFS, Menon MB, Menzies FM, Mercer CA, Merighi A, Merry DE, Meschini S, Meyer CG, Meyer TF, Miao C-Y, Miao J-Y, Michels PAM, Michiels C, Mijaljica D, Milojkovic A, Minucci S, Miracco C, Miranti CK, Mitroulis I, Miyazawa K, Mizushima N, Mograbi B, Mohseni S, Molero X, Mollereau B, Mollinedo F, Momoi T, Monastyrska I, Monick MM, Monteiro MJ, Moore MN, Mora R, Moreau K, Moreira PI, Moriyasu Y, Moscat J, Mostowy S, Mottram JC, Motyl T, Moussa CE-H, Mueller S, Muenger K, Muenz C, Murphy LO, Murphy ME, Musarò A, Mysorekar I, Nagata E, Nagata K, Nahimana A, Nair U, Nakagawa T, Nakahira K, Nakano H, Nakataogawa H, Nanjundan M, Naqvi NI, Narendra DP, Narita M, Navarro M, Nawrocki ST, Nazarko TY, Nemchenko A, Netea MG, Neufeld TP, Ney PA, Nezis IP, Nguyen HP, Nie D, Nishino I, Nislow C, Nixon RA, Noda T, Noegel AA, Nogalska A, Noguchi S, Notterpek L, Novak I, Nozaki T, Nukina N, Nuernberger T, Nyfeler B, Obara K, Oberley TD, Oddo S, Ogawa M, Ohashi T, Okamoto K, Oleinick NL, Oliver FJ, Olsen LJ, Olsson S, Opota O, Osborne TF, Ostrander GK, Otsu K, Ou J-hJ, Ouimet M, Overholtzer M, Ozpolat B, Paganetti P, Pagnini U, Pallet N, Palmer GE, Palumbo C, Pan T, Panaretakis T, Pandey UB, Papackova Z, Papassideri I, Paris I, Park J, Park OK, Parys JB, Parzych KR, Patschan S, Patterson C, Pattingre S, Pawelek JM, Peng J, Perlmutter DH, Perrotta I, Perry G, Pervaiz S, Peter M, Peters GJ, Petersen M, Petrovski G, Phang JM, Piacentini M, Pierre P, Pierrefite-Carle V, Pierron G, Pinkas-Kramarski R, Piras A, Piri N, Platanias LC, Poeggeler S, Poirot M, Poletti A, Poues C, Pozuelo-Rubio M, Prætorius-Ibba M, Prasad A, Prescott M, Priault M, Produit-Zengaffinen N, Progulske-Fox A, Proikas-Cezanne T, Przedborski S, Przyklenk K, Puertollano R, Puyal J, Qian S-B, Qin L, Qin Z-H, Quaggin SE, Raben N, Rabinowich H, Rabkin SW, Rahman I, Rami A, Ramm G, Randall G, Randow F, Rao VA, Rathmell JC, Ravikumar B, Ray SK, Reed BH, Reed JC, Reggiori F, Régnier-Vigouroux A, Reichert AS, Reiners JJJ, Reiter RJ, Ren J, Revuelta JL, Rhodes CJ, Ritis K, Rizzo E, Robbins J, Roberge M, Roca H, Roccheri MC, Rocchi S, Rodemann HP, de Cordoba SR, Rohrer B, Roninson IB, Rosen K, Rost-Roszkowska MM, Rouis M, Rouschop KMA, Rovetta F, Rubin BP, Rubinsztein DC, Ruckdeschel K, Rucker EBI, Rudich A, Rudolf E, Ruiz-Opazo N, Russo R, Rusten TE, Ryan KM, Ryter SW, Sabatini DM, Sadoshima J, Saha T, Saitoh T, Sakagami H, Sakai Y, Salekdeh GH, Salomoni P, Salvaterra PM, Salvesen G, Salvioli R, Sanchez AMJ, Sánchez-Alcázar JA, Sánchez-Prieto R, Sandri M, Sankar U, Sansanwal P, Santambrogio L, Saran S, Sarkar S, Sarwal M, Sasakawa C, Sasnauskiene A, Sass M, Sato K, Sato M, Schapira AHV, Scharl M, Schaetzl HM, Scheper W, Schiaffino S, Schneider C, Schneider ME, Schneider-Stock R, Schoenlein PV, Schorderet DF, Schueller C, Schwartz GK, Scorrano L, Sealy L, Seglen PO, Segura-Aguilar J, Seiliez I, Seleverstov O, Sell C, Seo JB, Separovic D, Setaluri V, Setoguchi T, Settembre C, Shacka JJ, Shanmugam M, Shapiro IM, Shaulian E, Shaw RJ, Shelhamer JH, Shen H-M, Shen W-C, Sheng Z-H, Shi Y, Shibuya K, Shidoji Y, Shieh J-J, Shih C-M, Shimada Y, Shimizu S, Shintani T, Shirihai OS, Shore GC, Sibirny AA, Sidhu SB, Sikorska B, Silva-Zacarin ECM, Simmons A, Simon AK, Simon H-U, Simone C, Simonsen A, Sinclair DA, Singh R, Sinha D, Sinicrope FA, Sirko A, Siu PM, Sivridis E, Skop V, Skulachev VP, Slack RS, Smaili SS, Smith DR, Soengas MS, Soldati T, Song X, Sood AK, Soong TW, Sotgia F, Spector SA, Spies CD, Springer W, Srinivasula SM, Stefanis L, Steffan JS, Stendel R, Stenmark H, Stephanou A, Stern ST, Sternberg C, Stork B, Strålfors P, Subauste CS, Sui X, Sulzer D, Sun J, Sun S-Y, Sun Z-J, Sung JJY, Suzuki K, Suzuki T, Swanson MS, Swanton C, Sweeney ST, Sy L-K, Szabadkai G, Tabas I, Taegtmeyer H, Tafani M, Takács-Vellai K, Takano Y, Takegawa K, Takemura G, Takeshita F, Talbot NJ, Tan KSW, Tanaka K, Tanaka K, Tang D, Tang D, Tanida I, Tannous BA, Tavernarakis N, Taylor GS, Taylor GA, Taylor JP, Terada LS, Terman A, Tettamanti G, Thevissen K, Thompson CB, Thorburn A, Thumm M, Tian F, Tian Y, Tocchini-Valentini G, Tolkovsky AM, Tomino Y, Toenges L, Tooze SA, Tournier C, Tower J, Towns R, Trajkovic V, Travassos LH, Tsai T-F, Tschan MP, Tsubata T, Tsung A, Turk B, Turner LS, Tyagi SC, Uchiyama Y, Ueno T, Umekawa M, Umemiya-Shirafuji R, Unni VK, Vaccaro MI, Valente EM, Van den Berghe G, van der Klei IJ, van Doorn WG, van Dyk LF, van Egmond M, van Grunsven LA, Vandenabeele P, Vandenberghe WP, Vanhorebeek I, Vaquero EC, Velasco G, Vellai T, Vicencio JM, Vierstra RD, Vila M, Vindis C, Viola G, Viscomi MT, Voitsekhovskaja OV, von Haefen C, Votruba M, Wada K, Wade-Martins R, Walker CL, Walsh CM, Walter J, Wan X-B, Wang A, Wang C, Wang D, Wang F, Wang F, Wang G, Wang H, Wang H-G, Wang H-D, Wang J, Wang K, Wang M, Wang RC, Wang X, Wang X, Wang Y-J, Wang Y, Wang Z, Wang ZC, Wang Z, Wansink DG, Ward DM, Watada H, Waters SL, Webster P, Wei L, Weihl CC, Weiss WA, Welford SM, Wen L-P, Whitehouse CA, Whitton JL, Whitworth AJ, Wileman T, Wiley JW, Wilkinson S, Willbold D, Williams RL, Williamson PR, Wouters BG, Wu C, Wu D-C, Wu WKK, Wyttenbach A, Xavier RJ, Xi Z, Xia P, Xiao G, Xie Z, Xie Z, Xu D-z, Xu J, Xu L, Xu X, Yamamoto A, Yamamoto A, Yamashina S, Yamashita M, Yan X, Yanagida M, Yang D-S, Yang E, Yang J-M, Yang SY, Yang W, Yang WY, Yang Z, Yao M-C, Yao T-P, Yeganeh B, Yen W-L, Yin J-j, Yin X-M, Yoo O-J, Yoon G, Yoon S-Y, Yorimitsu T, Yoshikawa Y, Yoshimori T, Yoshimoto K, You HJ, Youle RJ, Younes A, Yu L, Yu L, Yu S-W, Yu WH, Yuan Z-M, Yue Z, Yun C-H, Yuzaki M, Zabirnyk O, Silva-Zacarin E, Zacks D, Zacksenhaus E, Zaffaroni N, Zakeri Z, Zeh HJI, Zeitlin SO, Zhang H, Zhang H-L, Zhang J, Zhang J-P, Zhang L, Zhang L, Zhang M-Y, Zhang XD, Zhao M, Zhao Y-F, Zhao Y, Zhao ZJ, Zheng X, Zhivotovsky B, Zhong Q, Zhou C-Z, Zhu C, Zhu W-G, Zhu X-F, Zhu X, Zhu Y, Zoladek T, Zong W-X, Zorzano A, Zschocke J, Zuckerbraun B. Autophagy. 2012;8:445. [Google Scholar]
- 38.Kraft PMC, Hofmann K. Nat Cell Biol. 2010;12:836. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 39.Park S, Choi J, Biering SB, Dominici E, Williams LE, Hwang S. Autophagy. 2016;12:1153. doi: 10.1080/15548627.2016.1178447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim BH, Shenoy AR, Kumar P, Bradfield CJ, MacMicking JD. Cell Host Microbe. 2012;12:432. doi: 10.1016/j.chom.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Howard JC, Hunn JP, Steinfeldt T. Curr Opin Microbiol. 2011;14:414. doi: 10.1016/j.mib.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 42.Ferreira-da-Silva Mda F, Springer-Frauenhoff HM, Bohne W, Howard JC. PLoS Pathog. 2014;10:e1004449. doi: 10.1371/journal.ppat.1004449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor GA, Collazo CM, Yap GS, Nguyen K, Gregorio TA, Taylor LS, Eagleson B, Secrest L, Southon EA, Reid SW, Tessarollo L, Bray M, McVicar DW, Komschlies KL, Young HA, Biron CA, Sher A, Vande Woude GF. Proc Natl Acad Sci USA. 2000;97:751. doi: 10.1073/pnas.97.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collazo CM, Yap GS, Sempowski GD, Lusby KC, Tessarollo L, Vande Woude GF, Sher A, Taylor GA. J Exp Med. 2001;194:181. doi: 10.1084/jem.194.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD. Science. 2011;332:717. doi: 10.1126/science.1201711. [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, Ohshima J, Sasai M, Kayama H, Okamoto T, Huang DC, Soldati-Favre D, Horie K, Takeda J, Takeda K. Immunity. 2012;37:302. doi: 10.1016/j.immuni.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 47.Kim BH, Chee JD, Bradfield CJ, Park ES, Kumar P, MacMicking JD. Nat Immunol. 2016;17:481. doi: 10.1038/ni.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunn JP, Feng CG, Sher A, Howard JC. Mamm Genome. 2011;22:43. doi: 10.1007/s00335-010-9293-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meunier E, Dick MS, Dreier RF, Schurmann N, Kenzelmann Broz D, Warming S, Roose-Girma M, Bumann D, Kayagaki N, Takeda K, Yamamoto M, Broz P. Nature. 2014;509:366. doi: 10.1038/nature13157. [DOI] [PubMed] [Google Scholar]
- 50.Anderson SL, Carton JM, Lou J, Xing L, Rubin BY. Virology. 1999;256:8. doi: 10.1006/viro.1999.9614. [DOI] [PubMed] [Google Scholar]
- 51.Nordmann A, Wixler L, Boergeling Y, Wixler V, Ludwig S. FASEB J. 2012;26:1290. doi: 10.1096/fj.11-189886. [DOI] [PubMed] [Google Scholar]
- 52.Pan W, Zuo X, Feng T, Shi X, Dai J. Virology J. 2012;9:292. doi: 10.1186/1743-422X-9-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Henry SC, Daniell XG, Burroughs AR, Indaram M, Howell DN, Coers J, Starnbach MN, Hunn JP, Howard JC, Feng CG, Sher A, Taylor GA. J Leukoc Biol. 2009;85:877. doi: 10.1189/jlb.1008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC. Annu Rev Biochem. 2016;85:685. doi: 10.1146/annurev-biochem-060815-014556. [DOI] [PubMed] [Google Scholar]
- 55.Bekpen C, Marques-Bonet T, Alkan C, Antonacci F, Leogrande MB, Ventura M, Kidd JM, Siswara P, Howard JC, Eichler EE. PLoS Genet. 2009;5:e1000403. doi: 10.1371/journal.pgen.1000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bekpen C, Hunn JP, Rohde C, Parvanova I, Guethlein L, Dunn DM, Glowalla E, Leptin M, Howard JC. Genome Biol. 2005;6:R92. doi: 10.1186/gb-2005-6-11-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Donnell TB, Hyde JL, Mintern JD, Mackenzie JM. Virology. 2016;492:130. doi: 10.1016/j.virol.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 58.Prentice E, McAuliffe J, Lu X, Subbarao K, Denison MR. J Virol. 2004;78:9977. doi: 10.1128/JVI.78.18.9977-9986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Panyasrivanit M, Khakpoor A, Wikan N, Smith DR. J Gen Virol. 2009;90:448. doi: 10.1099/vir.0.005355-0. [DOI] [PubMed] [Google Scholar]
- 61.Cottam EM, Maier HJ, Manifava M, Vaux LC, Chandra-Schoenfelder P, Gerner W, Britton P, Ktistakis NT, Wileman T. Autophagy. 2011;7:1335. doi: 10.4161/auto.7.11.16642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, Machida K, Ou JH. J Biol Chem. 2012;287:18036. doi: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang Y, Li Z, Ge X, Guo X, Yang H. Autophagy. 2014;7:613. doi: 10.4161/auto.7.6.15267. [DOI] [PubMed] [Google Scholar]
- 64.Reggiori F, Monastyrska I, Verheije MH, Cali T, Ulasli M, Bianchi S, Bernasconi R, de Haan CA, Molinari M. Cell Host Microbe. 2010;7:500. doi: 10.1016/j.chom.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharma M, Bhattacharyya S, Nain M, Kaur M, Sood V, Gupta V, Khasa R, Abdin MZ, Vrati S, Kalia M. Autophagy. 2014;10:1637. doi: 10.4161/auto.29455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fentress SJ, Behnke MS, Dunay IR, Mashayekhi M, Rommereim LM, Fox BA, Bzik DJ, Taylor GA, Turk BE, Lichti CF, Townsend RR, Qiu W, Hui R, Beatty WL, Sibley LD. Cell Host Microbe. 2010;8:484. doi: 10.1016/j.chom.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haldar AK, Piro AS, Finethy R, Espenschied ST, Brown HE, Giebel AM, Frickel EM, Nelson DE, Coers J. mBio. 2016;7:e01417. doi: 10.1128/mBio.01417-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Coers J. PLoS Pathog. 2013;9:e1003538. doi: 10.1371/journal.ppat.1003538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morisaki JH, Heuser JE, Sibley LD. J Cell Sci. 1995;108:2457. doi: 10.1242/jcs.108.6.2457. [DOI] [PubMed] [Google Scholar]
- 70.Haldar AK, Saka HA, Piro AS, Dunn JD, Henry SC, Taylor GA, Frickel EM, Valdivia RH, Coers J. PLoS Pathog. 2013;9:e1003414. doi: 10.1371/journal.ppat.1003414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prakash B, Praefcke GJ, Renault L, Wittinghofer A, Herrmann C. Nature. 2000;403:567. doi: 10.1038/35000617. [DOI] [PubMed] [Google Scholar]
- 72.Ghosh A, Uthaiah R, Howard J, Herrmann C, Wolf E. Molecular Cell. 2004;15:727. doi: 10.1016/j.molcel.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 73.Gao S, von der Malsburg A, Paeschke S, Behlke J, Haller O, Kochs G, Daumke O. Nature. 2010;465:502. doi: 10.1038/nature08972. [DOI] [PubMed] [Google Scholar]
- 74.Hunn JP, Koenen-Waisman S, Papic N, Schroeder N, Pawlowski N, Lange R, Kaiser F, Zerrahn J, Martens S, Howard JC. EMBO J. 2008;27:2495. doi: 10.1038/emboj.2008.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Choi J, Biering SB, Hwang S. Small GTPases. 2016;8:199. doi: 10.1080/21541248.2016.1213090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Haldar AK, Piro AS, Pilla DM, Yamamoto M, Coers J. PLoS One. 2014;9:e86684. doi: 10.1371/journal.pone.0086684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perry JW, Wobus CE. J Virol. 2010;84:6163. doi: 10.1128/JVI.00331-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hyde JL, Sosnovtsev SV, Green KY, Wobus C, Virgin HW, Mackenzie JM. J Virol. 2009;83:9709. doi: 10.1128/JVI.00600-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Romanov J, Walczak M, Ibiricu I, Schüchner S, Ogris E, Kraft C, Martens S. EMBO J. 2012;31:4304. doi: 10.1038/emboj.2012.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dubremetz JF. Cell Microbiol. 2007;9:841. doi: 10.1111/j.1462-5822.2007.00909.x. [DOI] [PubMed] [Google Scholar]
- 81.Reese ML, Boothroyd JC. Traffic. 2009;10:1458. doi: 10.1111/j.1600-0854.2009.00958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sasai M, Sakaguchi N, Ma JS, Nakamura S, Kawabata T, Bando H, Lee Y, Saitoh T, Akira S, Iwasaki A, Standley DM, Yoshimori T, Yamamoto M. Nat Immunol. 2017;18:899. doi: 10.1038/ni.3767. [DOI] [PubMed] [Google Scholar]
- 83.Haldar AK, Foltz C, Finethy R, Piro AS, Feeley EM, Pilla-Moffett DM, Komatsu M, Frickel EM, Coers J. Proc Natl Acad Sci USA. 2015;112:E5628. doi: 10.1073/pnas.1515966112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Birgisdottir AB, Lamark T, Johansen T. J Cell Sci. 2013;126:3237. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- 85.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Cell. 2004;119:753. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 86.Schmeisser H, Bekisz J, Zoon KC. J Interferon Cytokine Res. 2014;34:71. doi: 10.1089/jir.2013.0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fu MM, Nirschl JJ, Holzbaur ELF. Dev Cell. 2014;29:577. doi: 10.1016/j.devcel.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Maric-Biresev J, Hunn JP, Krut O, Helms JB, Martens S, Howard JC. BMC Biol. 2016;14:33. doi: 10.1186/s12915-016-0255-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xie Y, Kang R, Sun X, Zhong M, Huang J, Klionsky DJ, Tang D. Autophagy. 2015;11:28. doi: 10.4161/15548627.2014.984267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Man SM, Karki R, Sasai M, Place DE, Kesavardhana S, Temirov J, Frase S, Zhu Q, Malireddi RKS, Kuriakose T, Peters JL, Neale G, Brown SA, Yamamoto M, Kanneganti TD. Cell. 2016;167:382. doi: 10.1016/j.cell.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kravets E, Degrandi D, Ma Q, Peulen TO, Klumpers V, Felekyan S, Kuhnemuth R, Weidtkamp-Peters S, Seidel CA, Pfeffer K. Elife. 2016;5:e14246. doi: 10.7554/eLife.11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feeley EM, Pilla-Moffett DM, Zwack EE, Piro AS, Finethy R, Kolb JP, Martinez J, Brodsky IE, Coers J. Proc Natl Acad Sci USA. 2017;114:E1698. doi: 10.1073/pnas.1615771114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li P, Jiang W, Yu Q, Liu W, Zhou P, Li J, Xu J, Xu B, Wang F, Shao F. Nature. 2017;551:378. doi: 10.1038/nature24467. [DOI] [PubMed] [Google Scholar]
- 94.Wandel MP, Pathe C, Werner EI, Ellison CJ, Boyle KB, von der Malsburg A, Rohde J, Randow F. Cell Host Microbe. 2017;22:507. doi: 10.1016/j.chom.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, Coers J. Proc Natl Acad Sci USA. 2014;111:6046. doi: 10.1073/pnas.1321700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Doerflinger SY, Cortese M, Romero-Brey I, Menne Z, Tubiana T, Schenk C, White PA, Bartenschlager R, Bressanelli S, Hansman GS, Lohmann V. PLoS Pathog. 2017;13:e1006705. doi: 10.1371/journal.ppat.1006705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Biering SB, Choi J, Brown HM, Hwang S. Autophagy. 2017;13:2010. doi: 10.1080/15548627.2017.1371396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Praefcke GJ, Kloep S, Benscheid U, Lilie H, Prakash B, Herrmann C. J Mol Biol. 2004;344:257. doi: 10.1016/j.jmb.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 99.Vopel T, Hengstenberg CS, Peulen TO, Ajaj Y, Seidel CA, Herrmann C, Klare JP. Biochemistry. 2014;53:4590. doi: 10.1021/bi500524u. [DOI] [PubMed] [Google Scholar]
- 100.Garcia-Sastre A. Cell Host Microbe. 2017;22:176. doi: 10.1016/j.chom.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]