

Abstract
The possible implication of transcription factor EB (TFEB) as a therapeutic target in Parkinson’s disease has gained momentum since it was discovered that TFEB controls lysosomal biogenesis and autophagy and that its activation might counteract lysosomal impairment and protein aggregation. However, the majority of putative direct targets of TFEB described to date is linked to a range of biological processes that are not related to the lysosomal-autophagic system. Here, we assessed the effect of overexpressing TFEB with an adeno-associated viral vector in mouse substantia nigra dopaminergic neurons. We demonstrate that TFEB overexpression drives a previously unknown bona fide neurotrophic effect, giving rise to cell growth, higher tyrosine hydroxylase levels, and increased dopamine release in the striatum. TFEB overexpression induces the activation of the mitogen-activated protein kinase 1/3 (MAPK1/3) and AKT pro-survival pathways, phosphorylation of mTORC1 effectors 4E-binding protein 1 (4E-BP1) and S6 kinase B1 (S6K1), and increased protein synthesis. We show that TFEB overexpression prevents dopaminergic cell loss and counteracts atrophy and the associated protein synthesis decline in the MPTP mouse model of Parkinson’s disease. Our results suggest that increasing TFEB activity might prevent neuronal death and restore neuronal function in Parkinson’s disease and other neurodegenerative diseases through different mechanisms.
Keywords: TFEB, neurotrophic, Parkinson's disease, neuroprotection, neurorescue, dopamine, MPTP, neuronal atrophy, prosurvival pathways
Graphical Abstract
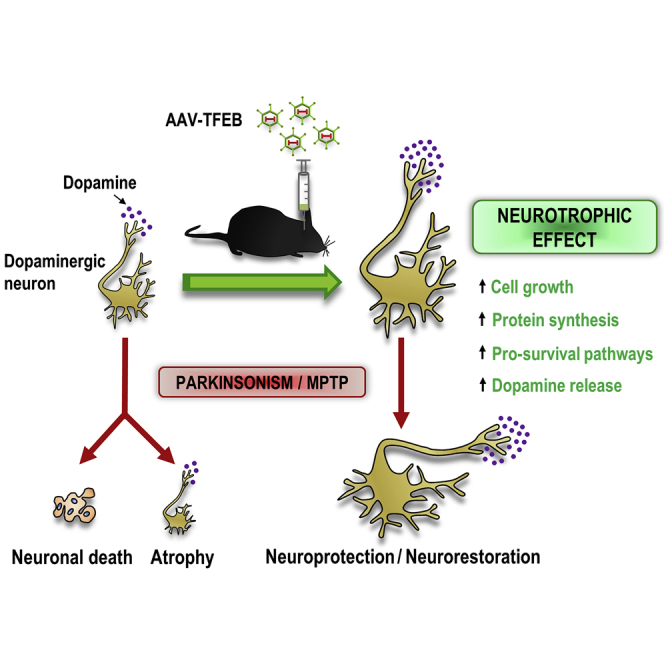
Torra et al. demonstrate that transcription factor EB (TFEB) overexpression drives a bona fide neurotrophic effect in mice dopaminergic neurons that counteracts neuronal death, atrophy, and dysfunction in a Parkinson’s disease model. They show that TFEB overexpression effect goes beyond autophagy and involves the activation of different pro-survival pathways.
Introduction
Parkinson’s disease is a multifactorial neurodegenerative disease that has been linked to several pathogenic neuron-autonomous events like mitochondrial dysfunction, oxidative stress, impairment of the degradation pathways, protein aggregation, and programmed cell death. We and others have demonstrated that autophagy impairment due to lysosomal depletion contributes to Parkinson’s disease neurodegeneration and Lewy body formation.1, 2, 3 Given that transcription factor EB (TFEB) promotes lysosomal biogenesis and autophagy4, 5, 6 by binding to the coordinated lysosomal expression and regulation (CLEAR) element, we proposed TFEB activation as a strategy to reverse Parkinson’s disease lysosomal/autophagy dysfunction. In this regard, we reported that TFEB overexpression in a neuroblastoma cell line counteracts lysosomal depletion and cell death mediated by the parkinsonian toxin MPP+.1 Subsequently, TFEB overexpression was proven to be neuroprotective in the Parkinson’s disease animal model based on the overexpression of human alpha-synuclein.7 Even though an effect on the lysosomal/autophagy system was demonstrated in both cases, only 64 out of 471 putative direct targets of TFEB described by Palmieri and collaborators5 are linked to this system. Hundreds of the other genes controlled by TFEB are linked to different biological processes like mitochondrial metabolism, regulation of gene expression, cellular response to stress, and cell cycle or translation, among others. The effect of overexpressing or activating TFEB on these other processes has never been characterized and may account for its neuroprotective effect. In fact, the microphthalmia (MiT) family of transcription factors, which includes TFEB, is a physiologic regulator of proliferation, differentiation, and survival in several non-nervous tissues.8 Although TFEB is the least studied member, it has been linked to renal carcinoma9 and pancreatic ductal adenocarcinoma,10 suggesting that it also has a role in the proliferation and survival of dividing cells. The TFEB putative direct targets mitogen-activated protein kinases (MAPK) 1 and 3,5 also known as extracellular signal-regulated kinase (ERK) 2 and 1, may explain this proliferative effect because they are known to transmit signals from many extracellular stimuli to regulate these processes.11 The MAPK1/3 pathways also play relevant roles in neuronal differentiation, plasticity, and survival.12
In light of the TFEB putative direct targets described beyond those related to lysosomal/autophagy function, we assessed here, at the histological, functional, and molecular levels, the effect of overexpressing TFEB in mouse substantia nigra dopaminergic neurons using an adeno-associated viral vector (AAV-TFEB). We demonstrate that TFEB overexpression drives a pleiotropic neurotrophic effect that mimics the effect mediated by glial cell line-derived neurotrophic factor (GDNF) or neurturin on dopaminergic neurons13 and prevents atrophy and dopaminergic neuronal loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease.
Results
Overexpressed TFEB Translocates to the Nucleus and Increases Autophagy-Lysosomal Markers in Substantia Nigra Dopaminergic Neurons
AAV-TFEB injection in mice ventral midbrain yielded a 17-fold increase of TFEB expression after 5 weeks of administration (Figure 1A). TFEB overexpression in dopaminergic neurons induced its translocation to the nucleus, as shown by a strong nuclear immunofluorescence staining (Figure 1B). TFEB translocates to the nucleus to trigger the transcription of its target genes only when is activated; therefore, its nuclear localization confirms that TFEB is activated.14 It is well established that TFEB binds to the promoter regions of numerous autophagy-lysosomal genes to induce autophagosome and lysosome biogenesis and autophagosome-lysosome fusion.6 To confirm that TFEB overexpression was able to boost the autophagy-lysosomal degradation pathway machinery in mice dopaminergic neurons, we measured by western blot the levels of two lysosomal markers, LAMP1 and cathepsin D (CTSD), and the levels of the autophagic vacuole marker LC3-II, expressed as the ratio LC3-II/LC3-I. As expected, protein levels of both lysosomal markers were clearly raised, particularly in the case of cathepsin D, which was increased 5-fold (Figures 1C and 1D). The increase of lysosomal markers confirms previously published results, in which TFEB was overexpressed in vivo in neurons and lysosomal proteins or mRNA levels were raised.7, 15 LC3-II/LC3-I ratio was also raised (226%) (Figure 1E), corroborating that TFEB overexpression is also inducing autophagosome formation in dopaminergic neurons in vivo.
Figure 1.

Overexpressed TFEB Translocates to the Nucleus and Increases Autophagy-Lysosomal Markers
(A) Tfeb cDNA levels measured by qPCR in ventral midbrain homogenates of mice after vehicle (n = 6) or AAV-TFEB nigral injection (n = 5); Mann-Whitney test. *p < 0.05 compared to control. (B) Immunofluorescence for tyrosine hydroxylase (red), Flag (green), and nucleus (blue) in substantia nigra sections showing 3xFlag-TFEB translocation to the nucleus after AAV-TFEB nigral injection. Scale bar, 25 μm. (C–E) Representative western blots and protein levels in ventral midbrain homogenates from mice overexpressing TFEB (n = 6) compared to vehicle-injected mice (n = 5–6) of (C) LAMP1, (D) CTSD, and (E) LC3-I and -II. Mann-Whitney test, *p < 0.05 compared to control. In all panels, samples were collected 5 weeks after AAV-TFEB or vehicle injections. All data are represented as mean ± SEM.
TFEB Overexpression Drives a Neurotrophic Effect that Involves Cell Growth and Higher Amounts of Releasable Dopamine
After performing tyrosine hydroxylase immunohistochemistry, we observed a patent enlargement of the AAV-TFEB-injected substantia nigra pars compacta (SNpc), accompanied by an increase in dendritic arborization (Figure 2A). We determined that this enlargement represents a 30% increase in the total volume of the SNpc when compared to the contralateral or vehicle-injected SNpc (Figure 2B). This increase of the SNpc volume was due to a 40% increase in the average cell body area of the dopaminergic neurons (Figure 2C), which was also apparent on visual inspection of cells under the light microscope (Figure 2A). No increase in neuronal cell size was observed when an empty AAV (AAV-EV) was injected in the SNpc (Figure S1A). Stereological cell counts of the total number of dopaminergic neurons in the TFEB-overexpressing SNpc were no different than the number estimated in the contralateral or vehicle-injected SNpc (Figure 2D), ruling out not only a deleterious effect of overexpressing TFEB but also an effect on neurogenesis.
Figure 2.

TFEB Neurotrophic and Functional Effects in Substantia Nigra Dopaminergic Neurons
(A) Representative photomicrograph of a tyrosine-hydroxylase-immunostained substantia nigra section of a mouse injected with AAV-TFEB into the right substantia nigra. Scale bar, 25 μm. (B) Stereological volumes of both SNpc of unilateral vehicle-injected (n = 7) and AAV-TFEB-injected mice (n = 8); two-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control injected side. #p < 0.05 compared to AAV-TFEB non-injected side. (C) Average cell area of SNpc dopaminergic neurons of vehicle-injected (n = 7) and AAV-TFEB-injected mice (n = 8); two-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control injected side. #p < 0.05 compared to AAV-TFEB non-injected side. (D) Stereological cell counts of dopaminergic neurons in substantia nigra pars compacta of mice injected with vehicle (n = 7) or AAV-TFEB (n = 9); two-way ANOVA, post hoc Tukey’s. (E) Intraneuronal optical densitometry of tyrosine hydroxylase immunoreactivity; two-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control injected side. #p < 0.05 compared to AAV-TFEB non-injected side. (F) Content of dopamine and its metabolites DOPAC and HVA and DOPAC/DA and HVA/DA ratios in striatal homogenates measured by HPLC of vehicle-injected (n = 6) and AAV-TFEB-injected mice (n = 6); Mann-Whitney test, *p < 0.05 compared to control. (G) Striatal DA relative amounts in AAV-TFEB and vehicle-injected mice (n = 4–6 per group) measured by microdialysis in the ipsilateral striatum following local amphetamine administration by reverse dialysis; ANOVA for repeated measures, post hoc Tukey’s. *p < 0.05 compared to control. In all panels, samples were collected 5 weeks after AAV-TFEB or vehicle injections. All data are represented as mean ± SEM.
We demonstrated that TFEB overexpression triggers a trophic effect that resembles the one elicited by neurotrophic factors. To determine whether TFEB overexpression was mimicking neurotrophic factors like GDNF or neurturin-increasing phenotypic markers and enhancing neuronal function,16, 17, 18 we assessed by high-pressure liquid chromatography (HPLC) the relative amount of intraneuronal tyrosine hydroxylase and the content of dopamine (DA) and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) in the striatum. We determined the optical density of tyrosine hydroxylase-positive neurons and found that it was 15% higher in TFEB-overexpressing neurons (Figure 2E). Regarding HPLC analysis, TFEB-overexpressing mice exhibited a decrease in both dopamine (20%) and DOPAC (18%) content, but an increase in postsynaptic metabolite HVA content (36%) (Figure 2F). The DOPAC/DA ratio, which represents dopamine metabolism, did not change, whereas the HVA/DA ratio, which represents dopamine release, was almost doubled in the striatum of TFEB-overexpressing mice (Figure 2F). Interestingly, the same pattern is achieved when GDNF16 or BDNF19 are chronically delivered, with only increases in HVA levels and the HVA/DA ratio having been reported. To further confirm that dopaminergic neurons were functional and that the releasable pool of dopamine was increased in AAV-TFEB mice, we carried out in vivo microdialysis studies to measure intrastriatal D-amphetamine-evoked dopamine overflow. D-amphetamine elevates dopamine extracellular levels by three major mechanisms. First, it is a substrate for the dopamine transporter (DAT) that competitively inhibits dopamine uptake; second, it facilitates the movement of dopamine out of vesicles; and third, it mediates DAT-mediated reverse transport of dopamine into the synaptic cleft.20 D-amphetamine-evoked increase of extracellular dopamine levels in the striatum of TFEB-overexpressing mice doubled the increase evoked in control mice (Figure 2G), demonstrating that TFEB expression increases the available pool of dopamine.
All together, this demonstrates that TFEB overexpression in dopaminergic neurons triggers a neurotrophic effect that is accompanied by an increase in the releasable amount of dopamine.
Protein Synthesis Inducers EIF4E and S6K1 Are Both Activated in TFEB-Overexpressing Neurons
Cell growth involves increasing cellular biomass, which is achieved by enhanced protein synthesis that is controlled by eukaryotic initiation factor 4E (EIF4E) or ribosomal protein S6 kinase B1 (S6K1).21, 22 EIF4E is a component of the complex EIF4F and binds the 5′ cap in translation initiation. Binding of EIF4F to the cap is hindered by EIF4E binding protein (4E-BP), which, when hypophosphorylated, sequesters EIF4E and prevents translation initiation. Once 4E-BP is phosphorylated, it removes this inhibition, allowing the formation of EIF4F and the subsequent initiation of translation.23 Because EIF4E has been identified as a TFEB direct target, we expected the involvement of this initiation factor in TFEB-induced cell growth.5 To test this hypothesis, we carried out the necessary western blot analysis with ventral midbrain protein homogenates. As anticipated, we found an increase in EIF4E levels (43%) in AAV-TFEB-injected mice (Figure 3A). We next determined the ratio of Ser65-phosphorylated to total 4E-BP as an indicator of free and active EIF4E. This ratio was much higher (58%) in TFEB-overexpressing SNpc, showing a translation initiation enhancement (Figure 3B). Regarding S6K1, once it is phosphorylated, mRNA translation is promoted by the phosphorylation of or binding to multiple proteins, including ribosomal protein S6 (RPS6).24 A prominent increase in activated/phosphorylated S6K1 (176%) and the phosphorylated/total ratio (100%) was detected in ventral midbrain homogenates of TFEB-overexpressing mice (Figure 3C). This was accompanied by an increase in the intensity of phosphorylated/activated RPS6 immunohistochemistry in the SNpc (Figure 3D). These results demonstrate that the 4E-BP/EIF4E and S6K1 pathways collaborate to increase the protein synthesis necessary for the TFEB-induced neurotrophic effect. We confirm that TFEB overexpression promotes protein synthesis in neurons, as we have also recently demonstrated with other cell types.25
Figure 3.

Activation of EIF4E/4E-BP1 and S6K1 Protein Synthesis Pathways
(A–C) Representative western blots and protein levels in ventral midbrain homogenates from mice overexpressing TFEB (n = 6) compared to control mice (n = 5 to 6) of (A) EIF4E, (B) phosphorylated and non-phosphorylated 4E-BP1, p4E-BP1S65, expressed as a ratio, and (C) endogenous ribosomal protein S6K1, its activated form (pS6K1T389), and the ratio. Mann-Whitney test, *p < 0.05 compared to control. (D) Representative photomicrograph of a substantia nigra section immunostained for the phosphorylated/activated RPS6, pRPS6S235/236, of a mouse injected with AAV-TFEB into the right substantia nigra. Scale bar, 50 μm. In all panels, samples were collected 5 weeks after AAV-TFEB or vehicle injections. All data are represented as mean ± SEM.
TFEB Overexpression Activates MAPK1/3 and AKT Pro-survival Pathways
Our initial hypothesis was that the MAPK1/3 pathway might be activated after TFEB overexpression because MAPK 1 and 3 have been identified as TFEB-direct targets.5 It has been demonstrated that the MAPK1/3 pathway is involved in several physiological functions of neurons, including cell growth, survival, and regulation of response to various growth factors.12, 17, 26, 27, 28 Its activation could therefore explain the neurotrophic effect we had found. To test this hypothesis, we determined the total levels and the phosphorylated/activated form of MAPK1/3. Not only were the total MAPK1/3 levels upregulated (43%), we also found a 3-fold increase in the activated form (Figure 4A). To further confirm MAPK1/3 pathway activation, we measured the levels of phosphorylated ribosomal protein S6 kinase α−1 (P90RSK), which is known to be a ubiquitous and versatile mediator of MAPK1/3 signal transduction.29 We again found a 3-fold increase of activated P90RSK in TFEB-overexpressing SNpc. Phosphorylated protein/total protein ratio was also increased (213%) (Figure 4B).
Figure 4.

Activation of MAPK1/3 and AKT Pro-survival Pathways
(A–E) Representative western blots and protein levels in ventral midbrain homogenates from mice overexpressing TFEB (n = 6) compared to control mice (n = 5 to 6) of (A) MAPK 1 and 3, their activated forms (pMAPK1/3Thr202/Tyr204), and the ratio; (B) 90-kDa ribosomal protein S6 kinase (P90RSK) and its phosphorylated/activated form (pP90RSKT359/S363) and the ratio; (C) total AKT and phosphorylated/activated AKT (pAKTS473) and the ratio; (D) total NDRG1, its phosphorylated/activated form (pNDRG1T346), and the ratio; and (E) TBK1, its phosphorylated/activated form (pTBK1S172), and the ratio. Mann-Whitney test, *p < 0.05 compared to control. In all panels, samples were collected 5 weeks after AAV-TFEB or vehicle injections. All data are represented as mean ± SEM.
Next, to determine whether the PI3K/AKT pathway, which is the other relevant signaling pathway that mediates neurotrophic activity,30, 31, 32 was also involved in the TFEB-mediated effects, we measured the relative amount of AKT as well as phosphorylated/activated AKT levels. Total amounts of AKT were downregulated by half, but phosphorylated AKT at S473 was dramatically increased (233%) in TFEB-overexpressing neurons as well as the phosphorylated/total protein ratio (603%) (Figure 4C). To determine whether mTORC2, known to phosphorylate AKT at S473,33 was responsible for the strong activation of AKT, we measured the total and phosphorylated/activated form of N-Myc downstream regulated 1 protein (NDRG1), which is a proxy indicator of mTORC2 activity.34 Total NDRG1 was downregulated (47%) and activated NDRG1 was decreased even further (72%) (Figure 4D), suggesting that the mTORC2 pathway was not involved. We subsequently looked for a putative TFEB direct target that could be responsible for this activation/phosphorylation.5 Among the possible options, TANK-binding kinase 1 (TBK1) is known to phosphorylate/activate AKT independently of PDPK1 and mTORC2.35 We therefore assessed both TBK1 total and phosphorylated/activated levels and found a 3-fold increase of both (Figure 4E), demonstrating that TBK1 is not only overexpressed but activated, probably by autophosphorylation.36 TBK1 only binds to AKT when activated and non-phosphorylated TBK1 is not interfering the activation of AKT;35 therefore, TBK1 is a strong candidate for AKT phosphorylation. Moreover, we are also demonstrating that despite phosphorylation of mTORC1 effectors, the mTORC2 pathway is not activated.
Our results demonstrate that MAPK1/3 and AKT pro-survival pathways are activated after TFEB overexpression. Further studies should be conducted to demonstrate the contribution of each pathway to the elicited neurotrophic effect.
TFEB Overexpression Counteracts Atrophy and Preserves Neuronal Integrity and Function in the MPTP Model
TFEB overexpression has been used as a strategy to eliminate protein accumulation in a model of synuclenopathy7 and in a model of tauopathy,15 resulting in a neuroprotective effect. Here, we wanted to assess the neuroprotective effect of overexpressing TFEB in a Parkinson’s disease model, in which neurodegeneration is not induced by the accumulation of a protein. For this purpose, we used the MPTP mouse model, in which neuronal cell death is triggered by the inhibition of mitochondrial complex I.37 Stereological dopaminergic cell counts showed that MPTP intoxication induced a 30% loss of dopaminergic neurons in the SNpc, which was completely prevented on the side in which TFEB was overexpressed (Figure 5A). Vehicle stereotaxic injection had no effect on the MPTP-induced neuronal death. To determine whether this total preservation of dopaminergic cell bodies was accompanied by a preservation of the dopaminergic terminals in the striatum, we measured the optical density of tyrosine hydroxylase staining in this region. MPTP intoxication induced a 75% reduction of the optical density in the striatum, and again, we found a complete preservation of the fibers in the TFEB-injected side (Figure 5B). The non-injected side appears partially preserved. Anatomic and functional studies have demonstrated the existence of contralateral nigrostriatal projections,38, 39 which may explain this result.
Figure 5.

Neuroprotective Effect of TFEB Overexpression in the MPTP Mouse Model of Parkinson’s Disease
(A) Top: representative photomicrographs of tyrosine-hydroxylase-immunostained substantia nigra of MPTP-intoxicated mice previously injected with AAV-TFEB (n = 14) or vehicle (n = 10), and control mice treated with saline and previously injected with vehicle (n = 7). Scale bar, 200 μm. Bottom: stereological cell counts of dopaminergic neurons in substantia nigra pars compacta of all groups of animals; two-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control non-injected side. #p < 0.05 compared to control injected side. $p < 0.05 compared to MPTP+sham injected side. ˆp < 0.05 compared to MPTP+AAV-TFEB non-injected side. (B) Left: representative photomicrographs of tyrosine-hydroxylase-immunostained striata of MPTP-intoxicated mice previously injected with AAV-TFEB (n = 14) or vehicle (n = 10) and control mice treated with saline and previously injected with vehicle (n = 7). Scale bar, 500 μm. Right: optical densitometry of striatal TH-positive dopaminergic fibers of all groups of animals; two-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control non-injected side. #p < 0.05 compared to control injected side. +p < 0.05 compared to MPTP+sham non-injected side. $p < 0.05 compared to MPTP+sham injected side. ˆp < 0.05 compared to MPTP+AAV-TFEB non-injected side. (C) Average cell area of dopaminergic neurons of all groups of animals (n = 6–10 mice per group); two-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control non-injected side. #p < 0.05 compared to control injected side. $p < 0.05 compared to MPTP+sham injected side. ˆp < 0.05 compared to MPTP+AAV-TFEB non-injected side. (D) Cumulative rotations induced by systemic administration of amphetamine during 10-min intervals 14 days after the last administration of MPTP. The net contralateral rotations were obtained as follows: total left − total right 360° turns. The test was performed with MPTP-intoxicated mice previously injected with AAV-TFEB (n = 9) or vehicle (n = 13) and control mice treated with saline and previously stereotaxically injected with vehicle (n = 10); two-way ANOVA for repeated-measures, post hoc Tukey’s. *p < 0.05 compared to MPTP+sham. #p < 0.05 compared to control. (E) Striatal DA amount measured by microdialysis in the ipsilateral striatum following local amphetamine administration by reverse dialysis in MPTP-intoxicated mice (21 days after last MPTP injection) previously injected with AAV-TFEB (n = 5) or vehicle (n = 5) and control mice treated with saline and previously injected with vehicle (n = 7); two-way ANOVA for repeated-measures, post hoc Tukey’s. *p < 0.05 compared to control. #p < 0.05 compared to MPTP+sham. All data are represented as mean ± SEM.
Morphological assessment of SNpc dopaminergic neurons in human postmortem samples has demonstrated that cell body shrinkage is linked to programmed cell death in Parkinson’s disease patients.40 To determine whether this cellular atrophy was also occurring in the MPTP mouse model and could be prevented, as we had hypothesized by TFEB overexpression, we measured the cell body size of SNpc dopaminergic neurons in MPTP-intoxicated mice before cell death occurred (1 day post-MPTP). The average cell area of the MPTP-intoxicated mice decreased 20% compared to control mice, with this neuronal atrophy prevented by TFEB overexpression (Figure 5C, left graph). To assess the evolution of these atrophic neurons, we next determined the cell body size of neurons 21 days after the last MPTP administration, when cell counts were performed and cell death was stabilized. We found that the surviving dopaminergic neurons were still atrophic. However, TFEB-overexpressing neurons reached the size that we had previously determined for the TFEB trophic effect, going from 25% to 37% of control neurons (Figure 5C, right graph), suggesting that TFEB overexpression can also reverse neuronal atrophy. To rule out any possible effect of the vector itself, we carried out an experiment with an AAV empty vector. Mice were stereotaxically injected with the empty vector and intoxicated with MPTP following exactly the same protocol that we followed for the AAV-TFEB mice. Empty vector injection did not prevent MPTP-induced cell loss (Figures S1B and S1C) or atrophy (Figure S1A).
To determine whether TFEB expression was not only preserving the integrity of nigral dopaminergic neurons but also their function after MPTP intoxication, we carried out a rotational behavioral test. Because TFEB is only overexpressed in the right substantia nigra, there should be an imbalance between both sides in the MPTP-intoxicated mice if neuronal function is preserved in the AAV-TFEB-injected side. This imbalance between the two dopaminergic systems triggers a measurable rotational behavior when a dopaminomimetic drug is systemically injected. When a drug that stimulates the release of dopamine, such as amphetamine, is administered, the rodent exhibits a contralateral rotation due to the fact that the non-lesioned side is able to release more dopamine than the lesioned side.37 AAV-TFEB MPTP intoxicated mice exhibited a sustained contralateral rotation, unlike sham MPTP intoxicated mice after D-amphetamine systemic administration (Figure 5D). This result demonstrates that TFEB expression is preserving neuronal function in the Parkinson’s disease MPTP mouse model. To further confirm that this rotational behavior was accompanied by an increase of the available pool of dopamine after MPTP intoxication, we also measured the intrastriatal D-amphetamine-evoked dopamine release in microdialysis samples of these animals. TFEB-overexpressing neurons were indeed able to retain their capacity to increase the dopamine pool in the model (Figure 5E).
With these results, we demonstrate that TFEB-induced neurotrophic/neuroprotective effect counteracts atrophy and preserves neuronal integrity and function in the MPTP Parkinson’s disease model.
The Mitochondrial-Lysosomal Deleterious Axis Is Counteracted by TFEB Overexpression in the MPTP Model
There is strong evidence showing that caspase-9 phosphorylation prevents its cleavage and the subsequent activation of the intrinsic apoptotic pathway, which is the cell death pathway involved in the MPTP Parkinson’s disease model.37, 41 Caspase-9 can be phosphorylated at multiple sites. A major inhibitory phosphorylation site in caspase-9 is Thr125. There are three known possible protein kinases that can phosphorylate caspase-9 at Thr125: MAPK1/3, which is activated by extracellular growth/survival signals, CDK1-cyclin B1 in mitosis, and DYRK1A, which regulates apoptosis during development.42 Taking into account that MAPK1/3 is activated in TFEB-overexpressing mice, we assessed phosphorylated caspase-9 levels in ventral midbrain homogenates of mice before cell death occurred. MPTP intoxication reduced T125-phosphorylated caspase-9 (pCASP9) levels by half compared to control mice, whereas TFEB overexpression in MPTP mice not only prevented the MPTP-induced diminution but also showed much higher levels than control mice (Figure 6A). These findings demonstrate that the neuroprotective effect elicited by TFEB overexpression also involves anti-apoptotic mechanisms that target downstream events, like blocking caspase-9, the main instigator of mitochondrial-mediated cell death.
Figure 6.

Mitochondrial-Lysosomal Cell Death Axis Is Blocked by TFEB Overexpression in the MPTP Model of Parkinson’s Disease
(A–C) Representative western blots and protein levels of ventral midbrain homogenates in MPTP-intoxicated mice previously injected with AAV-TFEB (n = 7 to 8) or vehicle (n = 7) and control mice treated with saline and previously injected with vehicle (n = 6) of (A) pCASP9T125, (B) LAMP1, and (C) LC3-I and -II; one-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control. #p < 0.05 compared to MPTP+sham. (D) Confocal images showing immunofluorescence for tyrosine hydroxylase (red), LAMP1 (green), and nuclear staining (blue). Scale bar, 25 μm. (E) mRNA levels in ventral midbrain extracts in MPTP-intoxicated mice previously injected with AAV-TFEB (n = 6 to 7) or vehicle (n = 6) and control mice treated with saline and previously injected with vehicle (n = 6) of mitochondrial genes Ppargc1a and Tfam and lysosomal genes Lamp1 and Ctsd; one-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to MPTP+sham. #p < 0.05 compared to control. All samples were collected 1 day after the last administration of MPTP prior to neuronal cell death taking place. All data are represented as mean ± SEM.
We have previously reported that lysosomal depletion and a subsequent lysosomal-autophagy defect that involves accumulation of autophagic vacuoles precedes cell death in the MPTP model.1, 2 To determine whether TFEB overexpression could also prevent these events, as we have shown in vitro,1 we assessed by western blot protein levels of the lysosomal marker LAMP1 and the autophagic vacuole marker LC3-II. As expected, MPTP intoxication induced a decrease of LAMP1 and an increase in the LC3-II/LC3-I ratio, thus showing defective autophagic degradation. TFEB overexpression was able to prevent the changes seen in these two markers, demonstrating that TFEB counteracts both lysosomal depletion and autophagic vacuole accumulation (Figures 6B and 6C). To confirm that these events were occurring in the dopaminergic neurons, we performed double immunofluorescence experiments for tyrosine hydroxylase and LAMP1. MPTP intoxicated mice indeed showed a clear decrease in LAMP1 immunofluorescence inside the SNpc dopaminergic neurons, which was prevented in TFEB-overexpressing mice (Figure 6D).
We hypothesized that MPTP intoxication would impair mitochondrial and lysosomal biogenesis besides inducing mitochondrial and lysosomal damage, and that TFEB overexpression would be able to counteract these effects. To test this hypothesis, we measured mRNA levels of Ppargc1a and Tfam, which control mitochondrial biogenesis, and mRNA levels of lysosomal proteins Lamp1 and Ctsd. Levels of all four mRNAs were diminished in MPTP-intoxicated mice, although only the decrease in Tfam was statistically significant. However, the levels of all four mRNAs were significantly increased in TFEB-overexpressing mice compared to sham mice after MPTP intoxication, indicating that TFEB overexpression prevents the diminution of all of them (Figure 6E).
TFEB Overexpression Prevents the Decline of Protein Synthesis that Is Observed in the MPTP Model
It is widely accepted that neuronal dysfunction that involves atrophied morphology and loss of tyrosine hydroxylase expression precedes actual neuronal death in Parkinson’s disease. In the MPTP mouse model, we saw that cell soma shrinkage preceded neuronal loss. However, some neurons that underwent atrophy were able to overcome MPTP’s deleterious effects and survive. Some experimental data suggest that neuronal atrophy and programmed cell death can be dissociated in experimental Parkinson’s disease,43 and the same seems to occur in Parkinson’s disease.44 Targeting programmed cell death alone as a strategy to halt the progression of the disease would therefore not prevent neuronal dysfunction and associated symptoms. Consequently, elucidating the mechanisms of neuronal dysfunction and atrophy is essential to develop a therapeutic approach that blocks neurodegeneration in all senses of the word. As previously discussed, cell size is mainly controlled by the 4E-BP/EIF4E and S6K1 pathways. We hypothesized that MPTP-induced neuronal dysfunctional atrophy was mediated by the inhibition of at least one of these two pathways that control protein biosynthesis and that TFEB might be preventing this inhibition. To test this hypothesis, we first assessed total and Ser65-phosphorylated 4E-binding protein 1 (4E-BP1) levels and found that MPTP triggered a dramatic reduction of this phosphorylated form, showing no band in the western blot. This reduction was not prevented by TFEB overexpression at this time point (Figure 7A). When 4E-BP1 is phosphorylated on the sites located in the carboxy terminal to the EIF4E-binding site, including Ser65 and Thr70, it releases from EIF4E. These phosphorylations cannot occur if 4E-BP1 is not previously phosphorylated on Thr37 and Thr46.45 To determine whether these phosphorylations were also altered by MPTP intoxication and modulated by TFEB overexpression, we measured the Thr37/Thr46 phosphorylated-4E-BP1/non-Thr37/Thr46 phosphorylated-4E-BP1 ratio. In this case, we found that MPTP intoxication substantially reduced this ratio and that this reduction was prevented by TFEB overexpression (Figure 7B). Then, to assess the effect on EIF4E, we first measured Eif4e mRNA levels and found that MPTP intoxication reduced the amount by half, whereas TFEB overexpression completely prevented this diminution (Figure 7C). EIF4E phosphorylation, although not required for translation initiation, has been shown to enhance the translation of specific proteins in neurons,46 whereas its dephosphorylation has been linked to apoptosis.47 To determine whether EIF4E activity was also inhibited at a post-translational level, we measured phosphorylated EIF4E levels in the different treatment groups. MPTP intoxication reduced phosphorylated EIF4E levels by half compared to control mice, whereas TFEB overexpression prevented this diminution (Figure 7D). The fact that EIF4E phosphorylation is known to be mediated by MNK1,48 the downstream kinase of the MAPK1/3 signaling pathway, explains this prevention. In line with this, it has been demonstrated that the BDNF-induced upregulation of protein synthesis is triggered by the MNK1-mediated phosphorylation of EIF4E.49 To determine whether the S6K1 pathway was also affected in the MPTP mouse model of Parkinson’s disease, we measured both S6K1 total and phosphorylated/activated protein levels. We found that MPTP intoxication did not induce any statistically significant variation of these forms, but TFEB overexpression in MPTP-intoxicated mice increased both of them; in particular, it doubled the activated form and increased the activated/total protein ratio (Figure 7E). These results demonstrate that neuronal atrophy is mediated by inhibition of the 4E-BP/EIF4E pathway in the MPTP mouse model and that TFEB counteracts atrophy by the activation of both the EIF4E and S6K1 pathways.
Figure 7.

TFEB Overexpression Prevents the Decline of Protein Synthesis that Occurs in the MPTP Model of Parkinson’s Disease
(A, B, D, and E) Representative western blot and protein levels of ventral midbrain homogenates in MPTP-intoxicated mice previously injected with AAV-TFEB (n = 7 to 8) or vehicle (n = 7) and control mice treated with saline and previously injected with vehicle (n = 5 to 6) of (A) Ser65-phosphorylated and total 4E-BP1, expressed as a ratio; (B) Thr-37/Thr-46 phosphorylated-4E-BP1/non-Thr-37/Thr-46 phosphorylated-4E-BP1 ratio; (D) phosphorylated eukaryotic translation initiation factor 4E, pEIF4ES209; and (E) endogenous ribosomal protein S6K1, its activated form (pS6K1T389), and the ratio; one-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control. #p < 0.05 compared to MPTP+sham group. (C) Eif4e mRNA levels of ventral midbrain homogenates in MPTP-intoxicated mice previously injected with AAV-TFEB (n = 7) or vehicle (n = 6) and control mice treated with saline and previously injected with vehicle (n = 6); one-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control. #p < 0.05 compared to MPTP+sham group. All samples were collected 1 day after the last administration of MPTP prior to neuronal cell death taking place. All data are represented as mean ± SEM.
Knocking Down the Master Transcriptional Repressor of Autophagy ZKSCAN3 Does Not Prevent MPTP-Induced Neurodegeneration
TFEB-sustained overexpression triggers a pleiotropic neurotrophic effect that involves a myriad of direct and indirect targets of this transcription factor. To elucidate which are the most relevant events or pathways and how they work synergistically to build up this neurotrophic/neuroprotective effect is not an easy task. So far, as we discussed above, it is believed that the TFEB neuroprotective effect is mainly due to its effect on the autophagy-lysosomal pathway. Our results demonstrate that TFEB is indeed boosting this degradation system but also activating pro-survival pathways that could explain the neuroprotective effect by themselves. Therefore, we believe that it is very relevant for the neurodegenerative diseases field to determine the actual neuroprotective extent of inducing autophagy and lysosomal biogenesis. ZKSCAN3 is a master transcriptional repressor of autophagy and lysosome biogenesis/function and is regulated in an opposite manner than TFEB by starvation.50 As previously demonstrated, knocking down ZKSCAN3 activates autophagy and increases lysosomal biogenesis. Therefore, we decided to knock down ZKSCAN3 in mice dopaminergic neurons with an adeno-associated vector containing the sequence coding a short double-stranded hairpin RNA (shRNA) directed against rodent ZKSCAN3, and test its neuroprotective effect in the same MPTP Parkinson’s disease model that we tested the TFEB neuroprotective effect. To demonstrate that we were really knocking down ZKSCAN3, we performed an immunofluorescence assay that showed a strong nuclear staining in the control group that was dramatically reduced in the shZKSACN3 group. The reduction of the intensity elicited was more than 60% (Figure 8A). Then, we assessed the levels of the lysosomal marker LAMP1 and the mTORC1 activity marker Ser235/Ser236 phosphorylated RPS6 by means of measuring substantia nigra intensity in midbrain sections after performing immunohistochemistry. We found a 30% increase in LAMP1 intensity (Figure 8B) and a 40% decrease of phospho-RPS6 intensity (Figure 8C) in shZKSCAN3 ventral midbrains. These results are compatible with an increase of lysosomal biogenesis and activation of autophagy that have been described in vitro with several cell lines, including a neuronal cell line, after knocking down ZKSCAN3.50
Figure 8.

Knocking Down the Master Transcriptional Repressor of Autophagy ZKSCAN3 Does Not Prevent MPTP-Induced Atrophy or Neuronal Death
(A) Left: representative images showing immunofluorescence for tyrosine hydroxylase (green), the transcriptional repressor of autophagy ZKSCAN3 (red), and nuclear staining (blue) in substantia nigra sections in mice after vehicle or AAV-shZKSCAN3 nigral injection. Scale bar, 50 μm. Right: quantification of intracellular ZKSCAN3 immunolabeling intensity of both groups of animals (n = 5 to 6 per group); Mann-Whitney test, *p < 0.05 compared to control. (B) Left: representative photomicrographs of LAMP1-immunostained substantia nigra in mice after vehicle or AAV-shZKSCAN3 nigral injection. Scale bar, 200 μm. Right: quantification of intracellular LAMP-1 immunolabeling intensity of both groups of animals (n = 4 per group); Mann-Whitney test, *p < 0.05 compared to control. (C) Left: representative photomicrographs of S235/236-phosphorylated RPS6-immunostained substantia nigra in mice after vehicle or AAV-shZKSCAN3 nigral injection. Scale bar, 150 μm. Right: quantification of intracellular S235/236-phosphorylated RPS6-immunolabeling intensity of both groups of animals (n = 4 per group); Mann-Whitney test, *p < 0.05 compared to control. (D) Stereological cell counts of dopaminergic neurons in substantia nigra pars compacta in MPTP-intoxicated mice previously injected with AAV-shZKSCAN3 (n = 5) or vehicle (n = 5) and mice treated with saline and previously injected with vehicle (n = 6) or AAV-shZKSCAN3 (n = 5); two-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control non-injected side. #p < 0.05 compared to control injected side. $p < 0.05 compared to saline AAV-shZKSCAN3 non-injected side. ˆp < 0.05 compared to saline AAV-shZKSCAN3 injected side. (E) Optical densitometry of striatal TH-positive dopaminergic fibers in MPTP-intoxicated mice previously injected with AAV-shZKSCAN3 (n = 8) or vehicle (n = 7) and mice treated with saline and previously injected with vehicle (n = 7) or AAV-shZKSCAN3 (n = 7); two-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control non-injected side. #p < 0.05 compared to control injected side. $p < 0.05 compared to saline AAV-shZKSCAN3 non-injected side. ˆp < 0.05 compared to saline AAV-shZKSCAN3 injected side. (F) Average cell area of dopaminergic neurons in MPTP-intoxicated mice previously injected with AAV-shZKSCAN3 (n = 5) or vehicle (n = 5) and mice treated with saline and previously injected with vehicle (n = 6) or AAV-shZKSCAN3 (n = 6); two-way ANOVA, post hoc Tukey’s. *p < 0.05 compared to control non-injected side. #p < 0.05 compared to control injected side. $p < 0.05 compared to saline AAV-shZKSCAN3 non-injected side. ˆp < 0.05 compared to saline AAV-shZKSCAN3 injected side. (G) Left: representative photomicrographs of Thr202/Tyr204-phosphorylated MAPK1/3-immunostained substantia nigra in mice after vehicle or AAV-shZKSCAN3 nigral injection. Scale bar, 125 μm. Right: quantification of intracellular Thr202/Tyr204-phosphorylated MAPK1/3-immunolabeling intensity of both groups of animals (n = 4 per group); Mann-Whitney test. (H) Left: representative photomicrographs of S473-phosphorylated AKT-immunostained substantia nigra in mice after vehicle or AAV-shZKSCAN3 nigral injection. Scale bar, 125 μm. Right: quantification of intracellular S473-phosphorylated AKT-immunolabeling intensity of both groups of animals (n = 4 per group); Mann-Whitney test. In all panels, quantifications were performed 21 days after the last MPTP or saline administration. All data are represented as mean ± SEM.
Then, we assessed neuronal cell death and atrophy in the MPTP model in control and AAV-shZKSCAN3 mice. Knocking down ZKSCAN3 did not prevent MPTP-induced neuronal loss in the SNpc (Figure 8D), fiber loss in the striatum (Figure 8E), or neuronal atrophy (Figure 8F), suggesting that boosting the autophagy-lysosomal pathway does not suffice to counteract neurodegeneration in this Parkinson’s disease model. However, we cannot rule out that this mechanism is contributing somehow to neuroprotection. To confirm that lysosomal biogenesis was not accompanied by the activation of the MAPK1/3 and AKT pathways, we measured the intensities after phospho-MAPK1/3 (Figure 8G) and phospho-AKT (Figure 8H) immunohistochemistries, and no difference was detected between control and AAV-shZKSCAN3 mice.
Discussion
None of the clinical trials carried out to date to halt the progression of Parkinson’s disease have been fully satisfactory. One possible explanation for this systematic failure is that the majority of the strategies tested, with the exception of the neurotrophic factors, target only one of the pathogenic mechanisms involved in the development of the disease. It is precisely because of the holistic effect of the neurotrophic factors that neurotrophic factor-based therapy stands as the most promising disease-modifying therapeutic approach for Parkinson’s disease. Extensive evidence from experimental animal models of neurological diseases suggests that neurotrophic factors are both neuroprotective and neurorestorative.51, 52 Several early phase clinical trials with GDNF and neurturin have shown the potential of neurotrophic factors; however, no neurotrophic factor-based protein or gene therapy has yet demonstrated sufficient efficacy in humans.51 This bewildering poor success could be explained by two facts: impaired axonal transport52 and downregulation of GDNF/neurturin receptor RET.53 Once GDNF or neurturin binds to the presynaptic striatal RET receptors, delivering signaling molecules to the soma relies on retrograde axonal transport.54 Consequently, the axonal impairment that occurs in Parkinson’s disease may interfere with the activation of pro-survival pathways. RET downregulation in nigral dopaminergic neurons observed in Parkinson’s disease patients also compromises the therapeutic effect of the GDNF family of ligands, and has encouraged scientists in the field to seek RET-independent neurotrophic factors. An alternative to delivering neurotrophic factors to overcome these hurdles is to directly activate the intracellular signaling pathways responsible for their effect.55 It is known that RET stimulation activates the RAS/MAPK and PI3K/AKT signaling pathways,28, 30 both of which lead to the activation of several transcription factors and increase the expression of genes involved in neurite outgrowth, neuronal survival, synaptic plasticity, and the expression of enzymes involved in dopamine biosynthesis.52, 56 Here, we demonstrate that TFEB overexpression in nigral dopminergic neurons mediates a bona fide neurotrophic effect that involves cell growth, neurite outgrowth, increased tyrosine hydroxylase levels, and higher amounts of dopamine to be released in the striatum, thus mimicking RET-mediated effects.13, 16, 17, 18, 52, 56 We also demonstrate that TFEB overexpression activates pro-survival MAPK1/3 and AKT pathways. Even though it seems plausible that these pathways contribute to the neurotrophic effect, further experiments should be carried out to prove a causative effect.
mTORC1 and MAPK1 are the main protein kinases known to phosphorylate/inactivate TFEB.14 However, as we show, TFEB translocates to the nucleus after being overexpressed, confirming its activation. This seeming paradox can be explained by the fact that when TFEB is overexpressed, it saturates the ability of mTORC1 and MAPK1 to phosphorylate it. Thus, some of the overexpressed TFEB escapes mTORC1 and MAPK1 phosphorylation and this is the reason why TFEB is found in the nucleus in normally fed neurons.57
We found that TFEB overexpression protected dopaminergic neurons of the SNpc in the MPTP mouse model of Parkinson’s disease, both at the cell body level as well as striatal dopaminergic terminals. We also demonstrate that TFEB overexpression is able to counteract the deleterious events that, like lysosomal depletion, mitochondrial dysfunction, and caspase-9 activation, are particularly relevant to MPTP neurotoxicity and Parkinson’s disease. Our results suggest that the TFEB neuroprotective effect could be due to several mechanisms that go beyond autophagy. In this regard, we show that knocking down the master transcriptional repressor of autophagy, ZKSCAN3, does not protect dopaminergic neurons in the MPTP mouse model of Parkinson disease, suggesting that boosting the autophagy-lysosomal system does not suffice per se to neuroprotect in this model. However, further studies should be conducted to dissect all the relevant neuroprotective mechanisms triggered by TFEB overexpression, including autophagy. In fact, TFEB overexpression has been proven to exert a neuroprotective effect in other models,7, 15 suggesting that the neuroprotective effect of TFEB is independent of the pathogenic mechanisms underlying neurodegeneration, as in the case of neurotrophic factors.
In Parkinson’s disease, neuronal dysfunction and atrophy accompanied by a loss of phenotype that involves tyrosine hydroxylase downregulation precede actual neuronal death.58 These neurons are still viable and are possible targets for neurorestorative therapies. Our results suggest that TFEB overexpression is capable of restoring the activity and phenotype of dopaminergic neurons. We demonstrate that TFEB mediates an increase of tyrosine hydroxylase expression and boosts dopamine release in the striatum, and probably also triggers the expression of other proteins relevant to the dopaminergic phenotype that were not assessed here. Moreover, we demonstrate that TFEB overexpression counteracts the diminution of protein synthesis and mitochondrial/lysosomal biogenesis associated with neuronal atrophy that occurs in neurodegeneration.
Overall, our results suggest that TFEB gene therapy or TFEB pharmacological activation are alternative candidates to neurotrophic-factor-based therapies for Parkinson’s disease and other neurodegenerative diseases. Further research should be conducted to elucidate the safest and most efficient ways to translate these results into clinical practice.
Materials and Methods
Animals
8- to 11-week-old male C57BL/6Ncrl mice were used for all the experiments. The acquisition, care, housing, use, and disposition of laboratory animals in this research procedure have been in compliance with applicable Catalan and Spanish laws and regulations, institutional policies, and international conventions to which the European Union is a party. The study was approved by the Ethical Committee of Animal Experimentation of the Vall d’Hebron Research Institute.
Stereotaxic Delivery of Adeno-associated Viral Vectors
Viral vectors were stereotaxically injected into the region immediately above mice right SNpc (−2.9 mm anteroposterior, 1.3 mm lateral, and −4.5 mm dorsoventral from Bregma). Control mice received vehicle.
AAV serotype 2/9 containing the murine Tfeb cDNA fused to 3 Flag epitopes under control of the cytomegalovirus (CMV) promoter (concentration: 3 × 1012 genome copies [gc]/mL) was produced at TIGEM AAV Vector Core Facility (Italy), as previously described,6 and was used for the overexpression of TFEB in dopaminergic neurons of the substantia nigra pars compacta (SNpc). An empty vector with noncoding stuffer DNA was also used.
An AAV containing the sequence coding a double-stranded shRNA directed against rodent ZKSCAN3 has been used to knock down ZKSCAN3. To generate AAV vectors expressing shZKSCAN3, oligonucleotides containing 19 targeting nucleotides were synthesized. shZKSCAN3: 5′-AGCTTCCGGTCAGACTTGGAACTCTTTTCAAGAGAAAGAGTTCCAAGTCTGACCTTTTTGGAAG-3′. The shRNA was cloned downstream of the H1 RNA polymerase III promoter, and the construct was cloned into a self-complementary pAAV2 backbone to generate pAAV2/9-shZKSCAN3. Recombinant AAV2/9 was produced by polyethylenimine (PEI)-mediated triple transfection of low passage HEK293T/17 cells (ATCC, Molsheim, France; cat number CRL-11268). The AAV expression plasmids (pAAV2-H1-shZKSCAN3-pA) were co-transfected with the adeno helper pAd Delta F6 plasmid (Penn Vector Core, Philadelphia, USA; cat # PL-F-PVADF6) and AAV Rep Cap pAAV2/9 plasmid (Penn Vector Core, Philadelphia, USA; cat # PL-T-PV008). AAV vectors were purified as previously described.59 Cells are harvested 72 hr post-transfection, resuspended in lysis buffer (150 mM NaCl and 50 mM Tris-HCl, pH 8.5), and lysed by 3 freeze-thaw cycles (37°C/−80°C). The cell lysate is treated with 150 U/mL Benzonase (Sigma, St Louis, USA) for 1 hr at 37°C, and the crude lysate is clarified by centrifugation. Vectors are purified by iodixanol step gradient centrifugation and concentrated and buffer exchanged into Lactated Ringer’s solution (Baxter, Deerfield, USA) using the vivaspin20 100-kDa cut off concentrator (Sartorius Stedim, Goettingen, Germany). Titrations were performed at the platform study of the transcriptome (Neurocenter Magendie, INSERM U862, Bordeaux, France). The genome-containing particle (gcp) titer was determined by real-time qPCR using the Light Cycler 480 SYBR green master mix (Roche, Meylan, France; cat # 04887352001), with primers specific for the AAV2 inverted terminal repeats (ITRs) (fwd 5′-GGAACCCCTAGTGATGGAGTT-3′; rev 5′-CGGCCTCAGTGAGCGA-3′)60 on a Light Cycler 480 instrument. Purity assessment of vector stocks was estimated by loading 10 μL of vector stock on 10% SDS acrylamide gels, and total proteins were visualized using the Krypton Infrared Protein Stain according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, USA). Finally, the titer of the AAV batch used for the in vivo application in this study was 2.58 × 1013 gc/mL for AAV2/9-shZKSCAN3.
MPTP Intoxication
An intraperitoneal (i.p.) injection of MPTP-HCl (30 mg/kg/day of free base; Sigma-Aldrich) was administrated for 5 consecutive days 4 weeks after the stereotaxic administration of AAV-TFEB, vehicle, or AAV-EV. Control mice received saline injections instead of MPTP. Mice were euthanized 1 or 21 days after the last MPTP injection.
Quantitative Morphology
Mice were deeply anesthetized and perfused through the ascending aorta with physiological saline (0.9% NaCl), followed by 4% ice-cold paraformaldehyde. The brains were removed, post-fixed, and crioprotected with 30% sucrose. Midbrain sections (thickness of 30 μm) were immunostained against tyrosine hydroxylase (TH) to assess different parameters of the SNpc and their dopaminergic neurons using a computerized stereology system (StereoInvestigator, MBF Bioscience). The researcher was blind to the experimental condition of the animal being studied.
SNpc Volume
The volume of the SNpc was estimated on the basis of SNpc area obtained from contour measurements of every fourth section of the SNpc.
Neuronal Cell Body Area
The mean area of the dopaminergic neurons was measured with a stereological nucleator probe. Neurons randomly selected from four sections, including the SNpc, were measured for each animal.
Intraneuronal Tyrosine Hydroxylase Optical Density
The intracellular optical density of transmitted light microscopy images of TH-positive neurons from four sections was analyzed using ImageJ. Transversely cut neurons were selected for the analysis.
Nigral Dopaminergic Neuron Counts
The total number of tyrosine-hydroxylase-positive neurons was estimated by stereology using the optical fractionator method in the different groups of mice. Every fourth section of the whole SNpc was counted. In the MPTP experiments, the total number of neurons was estimated at 21 days after the last MPTP or saline injection.
Optical Density of TH-Positive Fibers in the Striatum
Striatal TH-positive fiber density was measured by optical densitometry at four coronal levels from each animal using SigmaScan Pro 5.0 software (Systat Software, USA). The measured values were corrected for non-specific background staining by subtracting values obtained from the corpus callosum.
In Vivo Microdialysis and HPLC Measurements
Striatal homogenate levels of dopamine and metabolites, DOPAC and HVA, were measured by HPLC with electrochemical detection (Waters model 2465; +0.7 V), as previously described.61 Mice were killed and their brains were quickly removed and placed over a cold plate. Caudate and putamen were carefully dissected out, weight, frozen on dry ice, and kept at −80°C until assayed. The tissue was homogenized in 200 μL of buffer containing 0.4M perchloric acid containing 0.1% sodium metabisulphite, 0.01% EDTA, 0.1% cysteine, and centrifuged at 12,000 × g for 30 min. Aliquots of supernatants were then filtered through 0.45-μm filters (Millex, Barcelona, Spain) and analyzed by HPLC as described. The mobile phase consisted of 0.1 M KH2PO4, 1 mM octyl sodium sulfate, 0.1 mM EDTA (pH 2.65), and 18% methanol. DA and their metabolites were separated on a Mediterranean Sea (18, 3 μm, 10 cm × 6.4 mm) (Teknokroma, ref TR010042, Barcelona, Spain).
To assess local effects of D-amphetamine sulfate on striatal DA release in microdialysis experiments, it was dissolved in artificial cerebrospinal fluid (aCSF: 125 mM NaCl, 2.5 mM KCl, 1.26 mM CaCl2, and 1.18 mM MgCl2) and administered by reverse dialysis at the stated concentration (uncorrected for membrane recovery).
Extracellular DA concentration was measured by in vivo microdialysis, as previously described,62 at 5 weeks after AAV-TFEB or vehicle injections. In the MPTP experiments, microdialysis was performed at 21 days after the last MPTP or saline injection. Briefly, one concentric dialysis probe (Cuprophan membrane; 6,000 Da molecular weight cutoff; 1.5 mm long) was implanted in the right striatum (coordinates in mm: AP, 0.5; ML, −1.7; DV, −4.5) of pentobarbital-anaesthetized mice (40 mg/kg i.p.). Microdialysis experiments were performed 24–48 hr after surgery in freely moving mice. The aCSF was pumped (WPI model, SP220i) at 1.5 μL/min, and 20-min samples were collected. Following an initial 100-min stabilization period, five or six baseline samples were collected (20 min each) before local drug application by reverse dialysis and then successive dialysate samples were collected. The concentration of DA in dialysate samples was determined by HPLC with electrochemical detection (Hewlett Packard 1049, Palo Alto, CA, USA). Striatal dialysates were collected on microvials containing 5 μL of 10 mM perchloric acid and were rapidly injected into the HPLC. DA was detected at 5–7.5 min, with a limit of detection of 3 fmol/sample using an oxidation potential of +0.75 V.
Western Blot Analysis
Ventral midbrains were dissected and stored at −80°C. Tissues were homogenized in 1 M Tris HCl (pH 7.5), 5 M NaCl, 0.5 M EDTA, 1% SDS, Nonidet P-40, protease inhibitors (Complete Mini; Roche Diagnostics), and protease/phosphatase inhibitors (Cell Signaling Technology). Proteins were separated by SDS-PAGE and transferred to nitrocellulose membranes. The following antibodies were used: LAMP1 (1:750; GeneTex), Cathepsin D (1:1,000; Sta. Cruz), and LC3 (1:750; Novus). The following antibodies from Cell Signaling Technology were also used: EIF4E (1:1,000), phospho-4E-BP1S65 (1:1,000), 4E-BP1 (1:1,000), S6K1 (1:1,000), phospho-S6K1T389 (1:1,000), MAPK1/3 (1:1,000), phospho-MAPK1/3Thr202/Tyr204 (1:1,000), phospho-P90RSKT359/S363 (1:1,000), P90RSK (1:500), AKT (1:1,000), phospho-AKTS473 (1:1,000), NDRG1 (1:500), phospho-NDRG1T346 (1:1,000), TBK1 (1:500), phospho-Caspase9T125 (1:1,000), phospho-4E-BP1T37/46 (1:1,000), non-phospho-4E-BP1 (1:1,000), and phospho-EIF4ES209 (1:1,000). Other antibodies were also used: phospho-TBK1S172 (1:1,000; Millipore). Membranes were reprobed for different antibodies. Antibodies against GAPDH (1:3,000; Chemicon) and Tubulin (1:5,000; Sigma-Aldrich) were used as loading controls.
Immunofluorescence/Immunohistochemistry and Optical Density Quantification
Immunohistochemistry and immunofluorescence were performed on free-floating 30-μm sections, including the substantia nigra or the striatum.
For immunofluorescence, the following antibodies were used: TH (1:1,000; Calbiochem), FLAG (1:1,000; Sigma-Aldrich), TH (1:1,000; Chemicon), LAMP1 (1:1,000; Abcam), and ZKSCAN3 (1:50; Proteintech). Secondary antibodies Alexa Fluor (Life Technologies) 594 (anti-rabbit, goat, 1:1,000), Alexa Fluor 488 (anti-mouse, goat, 1:1,000), Alexa Fluor 594 (anti-mouse, goat, 1:1,000), and Alexa Fluor 488 (anti-rabbit, goat, 1:1,000). Nuclei were stained with Hoechst (1:2,000; Life Technologies). Fluorescence was analyzed using Olympus FV1000 confocal microscopy.
For immunohistochemistry, antibodies against TH (1:2,000 for substantia nigra [SN] and 1:5,000 for striatum; Calbiochem), phospho-RPS6S235/236 (1:300; Cell Signaling Technology), LAMP1 (1:250; GeneTex), phospho-MAPK1/3Thr202/Tyr204 (1:200; Cell Signaling Technology), and phospho-AKTS473 (1:200; Cell Signaling Technology) were used. 3,3′-Diaminobenzidine was used as a chromogen.
The intraneuronal intensity of fluorescence or transmitted-light microscopy images of SNpc sections after ZKSCAN3, LAMP1, phospho-RPS6S235/236, phospho-MAPK1/3Thr202/Tyr204, or phospho-AKTS473 immunohistochemistry was analyzed using ImageJ. Neurons transversely cut were selected for the analysis. All quantifications were performed by an investigator blinded to the experimental groups.
RNA Extraction and Gene Expression Analysis by RT-qPCR
Total RNA was extracted from injected ventral midbrains using RNeasy Lipid Tissue Mini Kit (QIAGEN). Total RNA from mouse tissue was isolated using QIAzol lysis reagent (QIAGEN) according to the supplier’s recommendations. RNA concentration was determined using the NanoDrop assay (Thermo Scientific), and RNA integrity was assessed by running the samples on an Agilent RNA 6000 Nano chip on an Agilent 2100 BioAnalyzer (Agilent Technologies). 1 μg of RNA was used for reverse transcription with oligo(dT)12–18 primers (Invitrogen) and SuperScriptII reverse transcriptase (Invitrogen). Real-time qPCR was performed with Taqman Gene Expression Master Mix (Applied Biosystems) and Taqman gene expression assays (Applied Biosystems) for Tfeb (Mm00448968_m1), Ppargc1a (Mm01208835_m1), Tfam (Mm00447485_m1), Lamp1 (Mm00495262_m1), Ctsd (Mm00515586_m1), and Eif4e (Mm00725633_s1) mouse genes. Fold change was calculated with the ΔΔCt-method and normalized to Gapdh and Rpl19 gene expression.
Amphetamine-Induced Rotation Test
Mice were placed in a cylinder in a closed room to avoid any environmental disturbance and allowed to habituate for 1 hr. Then, mice were injected intraperitoneally with D-amphetamine (5 mg/kg; Tocris) and recorded for 60 min. Each subject was scored for full body rotations in 10-min intervals. The net contralateral rotations were obtained as follows: total left − total right 360° turns.
Statistical Analysis
All values are expressed as the mean ± SEM. Statistical comparisons were performed with GraphPad Prism software (v6, GraphPad Software, USA) using the appropriate statistical tests, as indicated in each figure legend. Differences among means were analyzed by using the Mann-Whitney test and 1- or 2-way ANOVA. When ANOVA showed significant differences, pairwise comparisons between means were subjected to Tukey post hoc test. In all analyses, the null hypothesis was rejected at the 0.05 level.
Author Contributions
Conceptualization, M.V., J.B.; Methodology, A.T., A.P., T.C., B.R-G, E.R-B., J.B.; Formal Analysis, A.T., A.P., T.C., B.R-G., E.R-B., A. Bortolozzi; Writing – Original Draft, J.B.; Writing – Review and Editing, A.T., M.V., J.B.; Resources, A. Ballabio, A. Bortolozzi; Visualization, A.T.; Supervision, J.B.; Funding Acquisition, M.V., J.B.
Conflicts of Interest
None of the authors have a conflict of interest.
Acknowledgments
This work was supported by Fondo de Investigación Sanitaria-Instituto de Salud Carlos III grants and CIBERNED (PI13/01897 to M.V. and CP11/00229 and PI15/01937 to J.B.) or CIBERSAM (PI13/01390 and SAF2016-75797-R to A. Bortolozzi) co-financed by ERDF (European Regional Development Fund) “A way to build Europe” and an AGAUR-GRC grant (2014-SGR-1609 to M.V.). A.T. is a recipient of a predoctoral fellowship of Fondo de Investigación Sanitaria-Instituto de Salud Carlos III (PFIS, FI14/00613). AAV2/9-shZKSCAN3 viral particles used in this study were a kind gift of Dr. Benjamin Dehay. We thank Iria Carballo-Carbajal for her technical support.
Footnotes
Supplemental Information includes one figure and can be found with this article online at https://doi.org/10.1016/j.ymthe.2018.02.022.
Contributor Information
Miquel Vila, Email: miquel.vila@vhir.org.
Jordi Bové, Email: jordi.bove@vhir.org.
Supplemental Information
References
- 1.Dehay B., Bové J., Rodríguez-Muela N., Perier C., Recasens A., Boya P., Vila M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bové J., Martínez-Vicente M., Dehay B., Perier C., Recasens A., Bombrun A., Antonsson B., Vila M. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy. 2014;10:889–900. doi: 10.4161/auto.28286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu Y., Dodiya H., Aebischer P., Olanow C.W., Kordower J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009;35:385–398. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 4.Sardiello M., Palmieri M., di Ronza A., Medina D.L., Valenza M., Gennarino V.A., Di Malta C., Donaudy F., Embrione V., Polishchuk R.S. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 5.Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M., Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011;20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 6.Settembre C., Di Malta C., Polito V.A., Garcia A.M., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Decressac M., Mattsson B., Weikop P., Lundblad M., Jakobsson J., Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl. Acad. Sci. USA. 2013;110:E1817–E1826. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haq R., Fisher D.E. Biology and clinical relevance of the micropthalmia family of transcription factors in human cancer. J. Clin. Oncol. 2011;29:3474–3482. doi: 10.1200/JCO.2010.32.6223. [DOI] [PubMed] [Google Scholar]
- 9.Kauffman E.C., Ricketts C.J., Rais-Bahrami S., Yang Y., Merino M.J., Bottaro D., Srinivasan R., Linehan W.M. Molecular genetics and cellular characteristics of TFE3 and TFEB fusion kidney cancers. Nat. Rev. Urol. 2014;11:465–475. doi: 10.1038/nrurol.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perera R.M., Stoykova S., Nicolay B.N., Ross K.N., Fitamant J., Boukhali M., Lengrand J., Deshpande V., Selig M.K., Ferrone C.R. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature. 2015;524:361–365. doi: 10.1038/nature14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meloche S., Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26:3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 12.Hetman M., Gozdz A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. Eur. J. Biochem. 2004;271:2050–2055. doi: 10.1111/j.1432-1033.2004.04133.x. [DOI] [PubMed] [Google Scholar]
- 13.Kramer E.R., Liss B. GDNF-Ret signaling in midbrain dopaminergic neurons and its implication for Parkinson disease. FEBS Lett. 2015;589(24 Pt A):3760–3772. doi: 10.1016/j.febslet.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 14.Napolitano G., Ballabio A. TFEB at a glance. J. Cell Sci. 2016;129:2475–2481. doi: 10.1242/jcs.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polito V.A., Li H., Martini-Stoica H., Wang B., Yang L., Xu Y., Swartzlander D.B., Palmieri M., di Ronza A., Lee V.M. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 2014;6:1142–1160. doi: 10.15252/emmm.201303671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grondin R., Zhang Z., Yi A., Cass W.A., Maswood N., Andersen A.H., Elsberry D.D., Klein M.C., Gerhardt G.A., Gash D.M. Chronic, controlled GDNF infusion promotes structural and functional recovery in advanced parkinsonian monkeys. Brain. 2002;125:2191–2201. doi: 10.1093/brain/awf234. [DOI] [PubMed] [Google Scholar]
- 17.Herzog C.D., Dass B., Holden J.E., Stansell J., 3rd, Gasmi M., Tuszynski M.H., Bartus R.T., Kordower J.H. Striatal delivery of CERE-120, an AAV2 vector encoding human neurturin, enhances activity of the dopaminergic nigrostriatal system in aged monkeys. Mov. Disord. 2007;22:1124–1132. doi: 10.1002/mds.21503. [DOI] [PubMed] [Google Scholar]
- 18.Hyman C., Juhasz M., Jackson C., Wright P., Ip N.Y., Lindsay R.M. Overlapping and distinct actions of the neurotrophins BDNF, NT-3, and NT-4/5 on cultured dopaminergic and GABAergic neurons of the ventral mesencephalon. J. Neurosci. 1994;14:335–347. doi: 10.1523/JNEUROSCI.14-01-00335.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altar C.A., Boylan C.B., Jackson C., Hershenson S., Miller J., Wiegand S.J., Lindsay R.M., Hyman C. Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc. Natl. Acad. Sci. USA. 1992;89:11347–11351. doi: 10.1073/pnas.89.23.11347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fleckenstein A.E., Volz T.J., Riddle E.L., Gibb J.W., Hanson G.R. New insights into the mechanism of action of amphetamines. Annu. Rev. Pharmacol. Toxicol. 2007;47:681–698. doi: 10.1146/annurev.pharmtox.47.120505.105140. [DOI] [PubMed] [Google Scholar]
- 21.Fingar D.C., Salama S., Tsou C., Harlow E., Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schipany K., Rosner M., Ionce L., Hengstschläger M., Kovacic B. eIF3 controls cell size independently of S6K1-activity. Oncotarget. 2015;6:24361–24375. doi: 10.18632/oncotarget.4458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gingras A.C., Raught B., Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999;68:913–963. doi: 10.1146/annurev.biochem.68.1.913. [DOI] [PubMed] [Google Scholar]
- 24.Ruvinsky I., Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci. 2006;31:342–348. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 25.Di Malta C., Siciliano D., Calcagni A., Monfregola J., Punzi S., Pastore N., Eastes A.N., Davis O., De Cegli R., Zampelli A. Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science. 2017;356:1188–1192. doi: 10.1126/science.aag2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomes E., Papa L., Hao T., Rockwell P. The VEGFR2 and PKA pathways converge at MEK/ERK1/2 to promote survival in serum deprived neuronal cells. Mol. Cell. Biochem. 2007;305:179–190. doi: 10.1007/s11010-007-9542-2. [DOI] [PubMed] [Google Scholar]
- 27.Nicole O., Ali C., Docagne F., Plawinski L., MacKenzie E.T., Vivien D., Buisson A. Neuroprotection mediated by glial cell line-derived neurotrophic factor: involvement of a reduction of NMDA-induced calcium influx by the mitogen-activated protein kinase pathway. J. Neurosci. 2001;21:3024–3033. doi: 10.1523/JNEUROSCI.21-09-03024.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melillo R.M., Santoro M., Ong S.H., Billaud M., Fusco A., Hadari Y.R., Schlessinger J., Lax I. Docking protein FRS2 links the protein tyrosine kinase RET and its oncogenic forms with the mitogen-activated protein kinase signaling cascade. Mol. Cell. Biol. 2001;21:4177–4187. doi: 10.1128/MCB.21.13.4177-4187.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frödin M., Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol. Cell. Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 30.Besset V., Scott R.P., Ibáñez C.F. Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J. Biol. Chem. 2000;275:39159–39166. doi: 10.1074/jbc.M006908200. [DOI] [PubMed] [Google Scholar]
- 31.Hsuan S.L., Klintworth H.M., Xia Z. Basic fibroblast growth factor protects against rotenone-induced dopaminergic cell death through activation of extracellular signal-regulated kinases 1/2 and phosphatidylinositol-3 kinase pathways. J. Neurosci. 2006;26:4481–4491. doi: 10.1523/JNEUROSCI.4922-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinet C., Pellerin L. Brain-derived neurotrophic factor enhances the expression of the monocarboxylate transporter 2 through translational activation in mouse cultured cortical neurons. J. Cereb. Blood Flow Metab. 2010;30:286–298. doi: 10.1038/jcbfm.2009.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarbassov D.D., Guertin D.A., Ali S.M., Sabatini D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 34.Weiler M., Blaes J., Pusch S., Sahm F., Czabanka M., Luger S., Bunse L., Solecki G., Eichwald V., Jugold M. mTOR target NDRG1 confers MGMT-dependent resistance to alkylating chemotherapy. Proc. Natl. Acad. Sci. USA. 2014;111:409–414. doi: 10.1073/pnas.1314469111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ou Y.H., Torres M., Ram R., Formstecher E., Roland C., Cheng T., Brekken R., Wurz R., Tasker A., Polverino T. TBK1 directly engages Akt/PKB survival signaling to support oncogenic transformation. Mol. Cell. 2011;41:458–470. doi: 10.1016/j.molcel.2011.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shu C., Sankaran B., Chaton C.T., Herr A.B., Mishra A., Peng J., Li P. Structural insights into the functions of TBK1 in innate antimicrobial immunity. Sctructure. 2014;21:1137–1148. doi: 10.1016/j.str.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bové J., Perier C. Neurotoxin-based models of Parkinson’s disease. Neuroscience. 2012;211:51–76. doi: 10.1016/j.neuroscience.2011.10.057. [DOI] [PubMed] [Google Scholar]
- 38.Jaeger C.B., Joh T.H., Reis D.J. The effect of forebrain lesions in the neonatal rat: survival of midbrain dopaminergic neurons and the crossed nigrostriatal projection. J. Comp. Neurol. 1983;218:74–90. doi: 10.1002/cne.902180105. [DOI] [PubMed] [Google Scholar]
- 39.Fox M.E., Mikhailova M.A., Bass C.E., Takmakov P., Gainetdinov R.R., Budygin E.A., Wightman R.M. Cross-hemispheric dopamine projections have functional significance. Proc. Natl. Acad. Sci. USA. 2016;113:6985–6990. doi: 10.1073/pnas.1603629113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anglade P., Vyas S., Javoy-Agid F., Herrero M.T., Michel P.P., Marquez J., Mouatt-Prigent A., Ruberg M., Hirsch E.C., Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 41.Perier C., Bové J., Vila M. Mitochondria and programmed cell death in Parkinson’s disease: apoptosis and beyond. Antioxid. Redox Signal. 2012;16:883–895. doi: 10.1089/ars.2011.4074. [DOI] [PubMed] [Google Scholar]
- 42.Allan L.A., Clarke P.R. Apoptosis and autophagy: regulation of caspase-9 by phosphorylation. FEBS J. 2009;276:6063–6073. doi: 10.1111/j.1742-4658.2009.07330.x. [DOI] [PubMed] [Google Scholar]
- 43.Kim T.W., Moon Y., Kim K., Lee J.E., Koh H.C., Rhyu I.J., Kim H., Sun W. Dissociation of progressive dopaminergic neuronal death and behavioral impairments by Bax deletion in a mouse model of Parkinson’s diseases. PLoS One. 2011;6:e25346. doi: 10.1371/journal.pone.0025346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng H.C., Ulane C.M., Burke R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010;67:715–725. doi: 10.1002/ana.21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gingras A.C., Gygi S.P., Raught B., Polakiewicz R.D., Abraham R.T., Hoekstra M.F., Aebersold R., Sonenberg N. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 1999;13:1422–1437. doi: 10.1101/gad.13.11.1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao R., Gkogkas C.G., de Zavalia N., Blum I.D., Yanagiya A., Tsukumo Y., Xu H., Lee C., Storch K.F., Liu A.C. Light-regulated translational control of circadian behavior by eIF4E phosphorylation. Nat. Neurosci. 2015;18:855–862. doi: 10.1038/nn.4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bushell M., Poncet D., Marissen W.E., Flotow H., Lloyd R.E., Clemens M.J., Morley S.J. Cleavage of polypeptide chain initiation factor eIF4GI during apoptosis in lymphoma cells: characterisation of an internal fragment generated by caspase-3-mediated cleavage. Cell Death Differ. 2000;7:628–636. doi: 10.1038/sj.cdd.4400699. [DOI] [PubMed] [Google Scholar]
- 48.Waskiewicz A.J., Johnson J.C., Penn B., Mahalingam M., Kimball S.R., Cooper J.A. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol. Cell. Biol. 1999;19:1871–1880. doi: 10.1128/mcb.19.3.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Genheden M., Kenney J.W., Johnston H.E., Manousopoulou A., Garbis S.D., Proud C.G. BDNF stimulation of protein synthesis in cortical neurons requires the MAP kinase-interacting kinase MNK1. J. Neurosci. 2015;35:972–984. doi: 10.1523/JNEUROSCI.2641-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chauhan S., Goodwin J.G., Chauhan S., Manyam G., Wang J., Kamat A.M., Boyd D.D. ZKSCAN3 is a master transcriptional repressor of autophagy. Mol. Cell. 2013;50:16–28. doi: 10.1016/j.molcel.2013.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Domanskyi A., Saarma M., Airavaara M. Prospects of neurotrophic factors for Parkinson’s disease: comparison of protein and gene therapy. Hum. Gene Ther. 2015;26:550–559. doi: 10.1089/hum.2015.065. [DOI] [PubMed] [Google Scholar]
- 52.Olanow C.W., Bartus R.T., Volpicelli-Daley L.A., Kordower J.H. Trophic factors for Parkinson’s disease: to live or let die. Mov. Disord. 2015;30:1715–1724. doi: 10.1002/mds.26426. [DOI] [PubMed] [Google Scholar]
- 53.Decressac M., Kadkhodaei B., Mattsson B., Laguna A., Perlmann T., Björklund A. α-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci. Transl. Med. 2012;4:163ra156. doi: 10.1126/scitranslmed.3004676. [DOI] [PubMed] [Google Scholar]
- 54.Tomac A., Widenfalk J., Lin L.F., Kohno T., Ebendal T., Hoffer B.J., Olson L. Retrograde axonal transport of glial cell line-derived neurotrophic factor in the adult nigrostriatal system suggests a trophic role in the adult. Proc. Natl. Acad. Sci. USA. 1995;92:8274–8278. doi: 10.1073/pnas.92.18.8274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ries V., Henchcliffe C., Kareva T., Rzhetskaya M., Bland R., During M.J., Kholodilov N., Burke R.E. Oncoprotein Akt/PKB induces trophic effects in murine models of Parkinson’s disease. Proc. Natl. Acad. Sci. USA. 2006;103:18757–18762. doi: 10.1073/pnas.0606401103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lei Z., Jiang Y., Li T., Zhu J., Zeng S. Signaling of glial cell line-derived neurotrophic factor and its receptor GFRα1 induce Nurr1 and Pitx3 to promote survival of grafted midbrain-derived neural stem cells in a rat model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2011;70:736–747. doi: 10.1097/NEN.0b013e31822830e5. [DOI] [PubMed] [Google Scholar]
- 57.Medina D.L., Fraldi A., Bouche V., Annunziata F., Mansueto G., Spampanato C., Puri C., Pignata A., Martina J.A., Sardiello M. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell. 2011;21:421–430. doi: 10.1016/j.devcel.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kordower J.H., Olanow C.W., Dodiya H.B., Chu Y., Beach T.G., Adler C.H., Halliday G.M., Bartus R.T. Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain. 2013;136:2419–2431. doi: 10.1093/brain/awt192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zolotukhin S., Byrne B.J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R.J., Muzyczka N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999;6:973–985. doi: 10.1038/sj.gt.3300938. [DOI] [PubMed] [Google Scholar]
- 60.Aurnhammer C., Haase M., Muether N., Hausl M., Rauschhuber C., Huber I., Nitschko H., Busch U., Sing A., Ehrhardt A. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods. 2012;23:18–28. doi: 10.1089/hgtb.2011.034. [DOI] [PubMed] [Google Scholar]
- 61.Bortolozzi A., Artigas F. Control of 5-hydroxytryptamine release in the dorsal raphe nucleus by the noradrenergic system in rat brain. Role of alpha-adrenoceptors. Neuropsychopharmacology. 2003;28:421–434. doi: 10.1038/sj.npp.1300061. [DOI] [PubMed] [Google Scholar]
- 62.Díaz-Mataix L., Scorza M.C., Bortolozzi A., Toth M., Celada P., Artigas F. Involvement of 5-HT1A receptors in prefrontal cortex in the modulation of dopaminergic activity: role in atypical antipsychotic action. J. Neurosci. 2005;25:10831–10843. doi: 10.1523/JNEUROSCI.2999-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.