

Abstract
Fungal infections are spreading widely worldwide, and the types of treatment are limited due to the lack of diverse therapeutic agents and their associated side effects and toxicity. The discovery of new antifungal classes is vital and critical. We discovered the antifungal activity of abscisic acid through a rational drug design methodology that included the building of homology models for fungal chorismate mutases and a pharmacophore model derived from a transition state inhibitor. Ligand-based virtual screening resulted in some hits that were filtered using molecular docking and molecular dynamic simulations studies. Both in silico methods and in vitro antifungal assays were used as tools to select and validate the abscisic acid repurposing. Abscisic acid inhibition assays confirmed the inhibitory effect of abscisic acid on chorismate mutase through the inhibition of phenylpyruvate production. The repositioning of abscisic acid, the well-known and naturally occurring plant growth regulator, as a potential antifungal agent because of its suggested action as an inhibitor to several fungal chorismate mutases was the main result of this work.
Introduction
The shikimate pathway provides one of the main channels for the biosynthesis of aromatic compounds, primarily the aromatic amino acids phenylalanine, tyrosine, and tryptophan, in plants and microorganisms (Fig. 1). The pathway converts phosphoenolpyruvate and erythrose 4-phosphate to chorismate through seven enzymatically catalysed steps. Chorismate serves as a precursor for the synthesis of a variety of aromatic compounds, such as p-aminobenzoic, 2,3-dihydroxybenzoic, prephenic and anthranilic acids (Fig. 1)1–3.
Figure 1.

Shikimic acid biosynthetic pathway as a precursor for essential amino acids in plants, fungi and bacteria.
This pathway was originally discovered in plants. However, it is now established that several essential amino acids are biosynthesized in many organisms such as bacteria4,5, Mycobacterium tuberculosis6, fungi, and parasites7 via such a pathway. The absence of this pathway in animals, which obtain these essential amino acids through their diet, makes the enzymes of this pathway ideal targets for the development of new antimicrobial agents and nonhazardous herbicides8. For example, glyphosate, in addition to its wide use as a commercial herbicide, is an effective anti-microbial and anti-parasitic drug that inhibits 5-enolpyruvyl shikimate-3-phosphate (EPSP) synthase7,9.
Chorismate is a central metabolite in the shikimate pathway and a branch point for at least five different metabolic pathways in microorganisms (Fig. 1)4. The aromatic amino acids L-phenylalanine and L-tyrosine are formed from chorismate via prephenate, which undergoes either decarboxylation/dehydration or decarboxylation/dehydrogenation, followed by transamination to form the corresponding amino acids (Fig. 1). Chorismate mutase/prephenate dehydrogenase is a bifunctional enzyme that occurs as a homodimer with a molecular weight of approximately 78,000 in Escherichia coli, and it is involved in phenylalanine and tyrosine biosynthesis in bacteria and fungi4,10,11. To summarize, the absence of this type of enzyme in mammals, including humans, makes it a promising target for the development of new anti-mycobacterial, antifungal and antibacterial drugs12,13.
Molecular modelling tools have recently been discovered and developed for use in the design, modification and discovery of new chemical entities. These tools have been used to predict the biological activities of unknown compounds compared to reference drugs. Recent advances in computer-aided drug design have enabled scientists to interpret the mode of action of many enzyme inhibitors. The use of such tools in the process of drug repurposing may be of great interest, providing a powerful way to find new medical uses either for old drugs or for those with nonmedical use in new dosage forms. Drug discovery tools help to identify new targets of known drugs in new diseases14. Rational drug design is defined as the development of small molecules with desired properties for targets, biomolecules (proteins or nucleic acids), whose functional roles in cellular processes and 3D structural information are known15. It is one of the commonly used tools for drug discovery, and it has been used widely in pharmaceutical industries.
The main aim of this work was to discover a novel chorismate mutase inhibitor as a potential antifungal agent using ligand-based virtual screening, homology modelling, molecular docking and molecular dynamic simulations as a rational approach. The results of the in silico studies were supported by in vitro antifungal and enzyme inhibition assay investigations.
Results
The design of inhibitors based on structural information derived from chorismate mutase enzymes, which are present in microorganisms (archaebacteria, eubacteria, and yeast), fungi, and plants but not in animals and humans, provides the potential for the discovery of new selective antifungal agents. Unfortunately, the crystal structures of the chorismate mutases for most fungal strains, such as Candida albicans, Candida parapsilosis, Aspergillus niger, Trychophyton rubrum and Trychophyton mentagrophytes, are not available. In addition, the absence of any data related to either FDA-approved drugs or novel lead compounds that target the chorismate mutase enzyme makes the process more difficult to find at least one potential compound for further development.
Establishment of homology models for chorismate mutase in different fungal strains
To search for chorismate mutase inhibitors that could be used as potential antifungal drugs, the first step was to build the homology model for chorismate mutases of C. albicans, C. parapsilosis, A. niger, T. rubrum and T. mentagrophytes strains, which represent different types of fungal species. The FASTA sequences for the previous fungal strains were downloaded from the UniPort protein data bank. However, the sequence for T. mentagrophytes could not be retrieved. A sequence similarity search was performed using MOE 2014.09 software to determine the best template with the highest identity to be used for building the homology models, and the crystal structure of Saccharomyces cerevisiae was acquired as the most suitable template (Table 1).
Table 1.
Percentage of identity of different fungal chorismate mutase protein in respect to S. cerevisiae chorismate mutase.
| Fungal Strain | UniProt Code | Percentage of identity | Number of residues |
|---|---|---|---|
| C. albicans | Q59TS4 | 62.45% | 268 |
| C. prapsilosis | G8BD21 | 58.36% | 267 |
| T. rubrum | F2SCL7 | 36.778% | 325 |
| A. niger | A2R3Z4 | 44.00% | 266 |
| S. cerevisiae | P32178 | — | 256 |
These results were obtained using a sequence similarity search using MOE 2014.09 software.
Homology models for C. albicans, C. parapsilosis, A. niger and T. rubrum were constructed using MOE 2014.09 software. The resulting models were validated by computing the root-mean-square deviation (RMSD) from the template and analysis of the Ramachandran plot results for each model (Supplementary Data).
Determination of the active substrate-binding site
To determine the active substrate-binding site, protein-sequence alignment for different chorismate mutase enzymes of the previous fungi strains was performed. The results (Fig. 2) identified conservative regions in all sequences (155-SRRIHFGKFVAE-166) that should be required for the enzyme activity and represent the active site of substrate binding.
Figure 2.

Protein sequence alignment for chorismate mustase proteins of different fungi strains, showing different identical sites. This sequence alignment was done using Clustal omega software (http://www.ebi.ac.uk/Tools/msa/clustalo/). The fungal stains under investigation are C. albicans (Q59T54), C. prapsilosis (G88D21), T. rubrum (F2SCL7), A. niger (A2R3Z4), S. cerevisiae (P32178).
Design of a pharmacophore model for fungal chorismate mutase inhibitors
Previous studies sought an explanation of the mechanism of chorismate mutase enzyme inhibition. In these studies, they found that endo-oxabicyclic and aza-bicyclic transition state inhibitors were the most effective simulators for the enzyme. In addition, these inhibitors provide good insights into the requirements for an ideal inhibitor. However, these endo-oxabicyclic and aza-bicyclic inhibitors were not further developed, possibly due to the difficulty of chemical synthesis. Our aim was to derive the pharmacophoric features of these inhibitors (ligand-based approach) by building a pharmacophore model to be used in a virtual screening process. With the aim of designing a ligand-based pharmacophore model for the discovery of novel fungal chorismate mutase inhibitors, the crystal structure of yeast chorismate mutase in complex with an endo-oxabicyclic transition-state analogue inhibitor16 was studied. The complex provided very important insights into the inhibitor’s binding mode to the enzyme. The same endo-oxabicyclic transition-state analogue inhibitor was also reported in complex with different chorismate mutases for both Bacillus subtilis17 and E. coli18, indicating that this inhibitor has all the pharmacophores required for the inhibition of such enzymes (Fig. 3A). In addition, other transition-state analogue inhibitors were discovered for yeast chorismate mutase (Fig. 3B). However, they share similar structural features as endo-oxabicyclic.
Figure 3.

(A) The possible binding mode of endo-oxabicyclic inhibitor inside the S. cerevisiae chorismate mutase. (B) Chemical structures of different transition state analogue inhibitors.
The analysis of the endo-oxabicyclic and aza-bicyclic inhibitors mode of binding revealed that the two anionic and/or acceptor pharmacophoric features of the two carboxylate groups were essential in supporting the salt bridge formation with Arg157, Arg16 and Lys168. The hydroxyl group acts as an acceptor with its oxygen atom to form a strong hydrogen bond with Asn194 and as a hydrogen donor to form another hydrogen bond with Glu198 (Fig. 3A). These data were retrieved from the inactive form of the yeast chorismate mutase bound to the Tyr amino acid allosteric inhibitor. Neither the oxygen atom nor the nitrogen atom in the bicyclic system of inhibitors was involved in any binding. In addition, the six-membered cyclohexene ring was the most common in the inhibition of the chorismate mutases of B. subtilis, E. coli and S. cerevisiae19. The endo-oxabicyclic inhibitor has 0.007 µM inhibitory activity on chorismate mutase. Currently, there are no inhibitors for fungal chorismate mutases. Therefore, data from the yeast inhibitors were used to build a pharmacophore model for the virtual screening process. The established model comprised four pharmacophoric features, as shown in Fig. 4.
Figure 4.
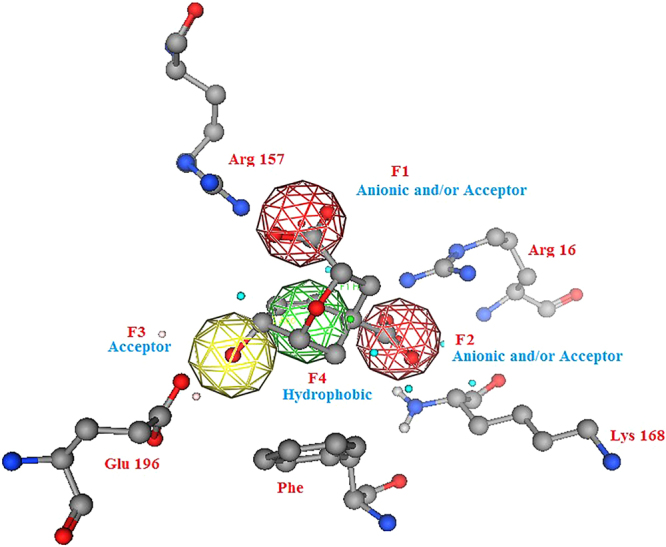
A Pharmacophore model for fungal chorismate mutase inhibitors. The model was designed after studying the crystal structure of yeast chorismate mutase in complex with endo-oxabicyclic transition-state analogue inhibitor16. The established model composed of four pharmacophoric features.
The pharmacophore query model was used to screen of a number of databases, such as the MOE 2014.09 and ZINC databases. The screening involved 160,000 lead-like compounds, and a conformational import was conducted to derive all possible conformations of the compounds screened. The selection in the final output was filtered according to Lipinski’s rule of five for drug-like properties. As a result, 25 compounds were selected as top-ranked hits with an RMSD < 1.00 and retention of all pharmacophore features. According to the RMSD values resulting from the virtual screening process, hits 1, 2, 4, 5, 7, 9, 10, 12, 13, 16, and 20 showed the lowest RMSD values (Supplementary Material: Fig. SI).
Docking of potential fungal chorismate mutase inhibitors
Molecular docking of the top-ranked hits with low RMSD values was performed inside the transition-state binding site of the four chorismate mutase homology models of C. albicans, C. parapsilosis, A. niger and T. rubrum. The RMSD values of the hits and docking results are summarized in Table 2. According to the docking results, hits with one C=C bond in the cyclohexene ring showed higher docking scores than those with two C=C bonds. In addition, hits with pentanoic acid side chains such as compound (13) and methylpent-2-enoic acid such as compounds (9) and (10) showed higher free binding energies and low clash scores than propionic acid side chains. Compound (13) showed the highest docking sore (−38.91 kcal/mol) and a relatively low (4.15) clash score. Unfortunately, compound (13) is not available commercially. The second two compounds in ranking were (9) and (10), with free binding energy scores of (−31.65 kcal/mol) and (−31.45 kcal/mol), respectively. They showed exactly the same binding mode that was similar to the transition-state analogue inhibitor. Similarly, neither compound is available commercially because they both have two chiral centres that may result in four isomers. The presence of several isomers could affect the biological and pharmacological activities of these compounds and may result in severe side effects due to the toxicity of one of the isomers.
Table 2.
Docking results of the top-ranked 11 compounds resulted from database screening by the established pharmacophore model for fungal chorismate mutase inhibitors.
| Compounds | RMSD (Ǻ) | C. albicans | C. parapsilosis | A. niger | T. rubrum | ||||
|---|---|---|---|---|---|---|---|---|---|
| ΔG (Kcal/mol) | Clash score | ΔG (Kcal/mol) | Clash score | ΔG (Kcal/mol) | Clash score | ΔG (Kcal/mol) | Clash score | ||
| Compound 1 | 0.95 | −27.18 | 5.75 | −28.13 | 5.30 | −26.93 | 4.62 | −24.91 | 5.52 |
| Compound 2 | 0.86 | −31.46 | 6.11 | −30.45 | 4.56 | −28.17 | 3.36 | −26.99 | 3.44 |
| Compound 4 | 0.82 | −29.19 | 5.85 | −19.11 | 4.82 | −29.15 | 3.65 | −26.35 | 3.49 |
| Compound 5 | 0.98 | −21.35 | 7.24 | −19.89 | 5.60 | −27.56 | 4.84 | −23.68 | 3.30 |
| Compound 7 | 0.96 | −28.73 | 6.95 | −20.05 | 4.90 | −26.95 | 5.28 | −27.00 | 4.08 |
| Compound 9 | 0.72 | −31.65 | 4.28 | −30.85 | 4.18 | −30.89 | 3.25 | −28.45 | 3.85 |
| Compound 10 | 0.75 | −31.45 | 4.30 | −31.11 | 4.56 | −31.02 | 3.33 | −28.70 | 3.75 |
| Compound 12 | 0.84 | −19.85 | 8.40 | −20.85 | 4.25 | −19.85 | 4.51 | −23.27 | 4.73 |
| Compound 13 | 0.65 | −38.91 | 4.15 | −35.14 | 4.45 | −31.55 | 3.46 | −29.08 | 3.81 |
| Compound16 | 1.01 | −16.85 | 5.21 | −17.65 | 4.45 | −15.65 | 4.25 | −25.68 | 5.60 |
| Compound 20 | 1.00 | −14.97 | 9.80 | −17.19 | 6.08 | −19.86 | 4.51 | −18.20 | 4.85 |
| Endo-oxabicyclic transition-state analogue | −40.52 | 2.59 | −38.31 | 2.54 | −31.53 | 3.92 | −33.16 | 4.87 | |
Docking was done by Leadit 2.1.8. The free energy of binding is represented by ΔG (Kcal/mol).
A structure-similarity search based on compounds (9) and (10) using both SciFinder and PubChem midlines was performed to obtain a highly similar compound that could be easily synthesized and/or purchased. Both searches resulted in one common compound with 98% similarity: abscisic acid.
Stereoisomers of abscisic acid
ABA has a 5-(1-hydroxy-2,6,6-trimethyl-4-oxo-2-cyclohexene-1-yl)-3-methyl-pentadienoic acid scaffold, which may be in (S) or (R) stereo-configuration, and both isomers are commercially available (Supplementary Material: Fig. SII). To verify the activity of abscisic acid as a fungal chorismate mutase inhibitor and to identify which of its two isomers have the best activity, a molecular docking study was performed for both abscisic acid isomers (Fig. 5) against the four homology models and compared to the docking of endo-oxabicyclic transition-state analogue inhibitors.
Figure 5.
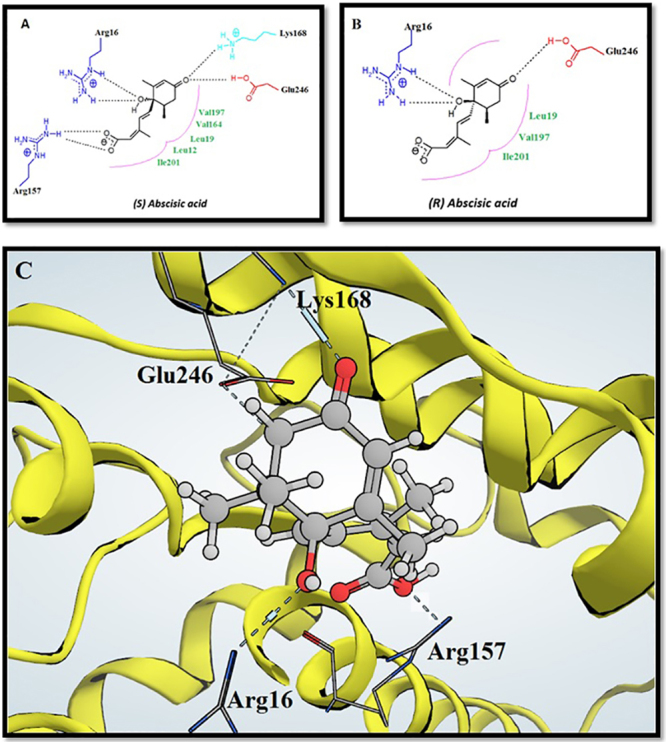
(A) Best binding mode of (S) Abscisic acid in C. albicans chorismate mutase substrate binding site. (B) Best binding mode of (R) Abscisic acid in C. albicans chorismate mutase substrate binding site. (C) The 3D binding mode of (S) Abscisic acid in C. albicans chorismate mutase.
Table 3 shows the docking results that indicate that the (S) configuration was better than the (R) configuration in terms of the binding mode, free energy binding score and lowest clash, which resembles those of the endo-oxabicyclic transition state-analogue inhibitor. Figure 6 shows one of the generated conformations for the abscisic acid (S) isomer, which was matched with the previously established pharmacophore model for fungal chorismate mutase inhibitors.
Table 3.
Docking results of Abscisic acid isomers using Leadit 2.1.8. The free energy of binding is represented by ΔG (Kcal/mol).
| ΔG Free binding energy kcal/mol | Lipophilic contribution score | Clash score | Ligand entropy conformation score | |
|---|---|---|---|---|
| C. albicans chorismate mutase | ||||
| (S) configuration | −32.22 | −7.89 | 4.24 | 2.80 |
| (R) configuration | −28.84 | −8.09 | 6.11 | 2.80 |
| C. parapsilosis chorismate mutase | ||||
| (S) configuration | −32.11 | −7.89 | 4.20 | 2.80 |
| (R) configuration | −29.89 | −8.09 | 5.95 | 2.80 |
| A.niger chorismate mutase | ||||
| (S) configuration | −32.25 | −7.89 | 4.22 | 2.80 |
| (R) configuration | −28.54 | −8.09 | 6.14 | 2.80 |
| T. rubrum chorismate mutase | ||||
| (S) configuration | −35.45 | −7.89 | 4.22 | 2.80 |
| (R) configuration | −27.80 | −8.09 | 5.60 | 2.80 |
Figure 6.
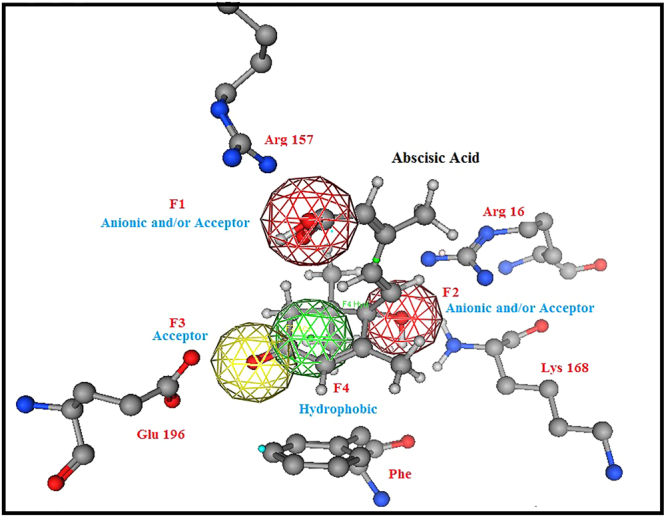
One of the generated conformations for ABA (S) isomer, matched with the established pharmacophore model for fungal chorismate mutase inhibitors.
The best configuration of the (S) configuration was fixed in each homology model of C. albicans, C. parapsilosis, A. niger and T. rubrum after the docking process. Then, the four chorismate mutase-ligand complexes were subjected to a molecular dynamic simulation to test the stability of (+) (S) abscisic acid (ABA) binding inside the model constructed.
Molecular dynamics
A molecular dynamic simulation study has been conducted for the four ligand-protein complexes resulted from the docking to obtain more insights into the validity of the docking results achieved and to select phases using the GROMOS force field20. The Cα RMSD was calculated and plotted for all of the systems starting from the end of the equilibration phase (Fig. 7I). Through the simulations period, no significant fluctuations were observed in the backbone of the proteins, implying that the binding of ABA at the active sites of the proteins is not only stable and strong but also does not disturb the protein backbone stability.
Figure 7.

(I) Time dependence of root mean square deviations (RMSDs) of the backbone of protein complexes against the initial structures during 1,000 ps molecular dynamics (MD) simulation.: (A) ABA- C. albicans complex, (B) ABA- C. parapsilosis complex, (C) ABA- A. niger complex, (D) ABA- T. rubrum complex. (II) Time dependence of root mean square deviations (RMSDs) of the drug candidates against the initial structures during 1,000 ps molecular dynamics (MD) simulation.: (E) ABA- C. albicans complex, (F) ABA- C. parapsilosis complex, (G) ABA- A. niger complex, (H) ABA- T. rubrum complex.
The ligand positional RMSD of each model was generated and analysed to ensure the binding stability of the ABA in the active site of proteins (Fig. 7II). Both the C. albicans and C. parapsilosis complexes showed stable and strong binding, while the A. niger and T. rubrum complexes showed more and continuous fluctuations.
The MD analysis of the proteins and the selected drug candidate’s complex stability were monitored during the trajectory period to determine the stability of hydrogen bonds with the binding site of protein. Hydrogen bond profiles were calculated using the g_h bond utility of GROMACS (Supplementary Material: Fig. SIII). This analysis revealed that the C. albicans-abscisic acid complex comprises 5–6 (highest) average H bonds during the simulation period, while the C. parapsilosis-abscisic acid, A. niger-abscisic acid and T. rubrum-abscisic acid complexes showed poor H bond interactions with 2–3 H bonds on average during the trajectory period.
Two different approaches were carried out in order to estimate the binding association between drug candidates and proteins. First, the crude interaction energy was estimated based on the short-range energy of the system. Next, to determine the binding affinity between the protein and drug complexes, the MM-GBSA energy of the complexes was scored21. Re-scoring of complexes using g_mmpbsa shows that the selected drug compounds have a different interaction energy of −181.913 kJ/mol, −60.563 kJ/mol, −81.687 kJ/mol, −56.899 kcal/mol in the C. albicans-ABA, C. parapsilosis-ABA, A. niger-ABA, and T. rubrum-ABA complexes, respectively. The C. albicans-ABA complex displayed a higher binding free energy.
According to the molecular dynamic results, it was strongly predicted that the ABA had a high stability towards blocking the C. albicans chorismate mutase active site. It was also predicted that ABA had very good binding to A. niger, C. parapsilosis and T. rubrum.
In vitro antifungal screening for abscisic acid
The previously collected results from virtual screening, molecular docking, and molecular dynamics simulation suggested that ABA had antifungal activity against C. albicans, C. parapsilosis, A. niger, and T. rubrum through the inhibition of chorismate mutase enzyme. Therefore, the in vitro antifungal activity of ABA against these strains was determined to support the previous in silico finding. The in vitro antifungal results (Table 4) indicated good antifungal activity of ABA against all the fungal strains, especially C. parapsilosis and T. rubrum, in comparison with amphotericin B as a positive control.
Table 4.
The in vitro antifungal results for ABA against four strains of fungi; C. albicans (RCMB 05036), C. parapsilosis (RCMB 05064), A. niger (RCMB 02568), and T. rubrum (RCMB 010162).
| Fungal strain | MIC (µg/mL) | |
|---|---|---|
| (S+)-Abscisic acid | Amphotericin B | |
| C. albicans | 125 | 1.95 |
| C. parapsilosis | 62.5 | 0.98 |
| A. niger | 125 | 1.95 |
| T.rubrum | 62.5 | 7.81 |
The results was expressed in MIC (µg/mL) in comparison with Amphotericin B as a positive control.
Chorismate mutase assay
Wild-type yeast, Saccharomyces cerevisiae, contains a single copy of the chorismate mutase gene, which allows the yeast to convert chorismic acid into prephenic acid (Fig. 1). The latter can be converted into an equivalent concentration of phenylpyruvate through the addition of acid. Thus, the fluctuation in phenylpyruvate or chorismic acid concentrations could be used to indicate yeast chorismate mutase activity. In this assay, yeast chorismate mutase catalysed the conversion of externally added chorismic acid to prephenic acid when favourable conditions were applied. The prephenic acid was converted, non-enzymatically, to phenylpyruvate through the addition of 0.2 N HCl. Phenylpyruvate absorbs UV radiation more effectively in an alkaline medium. Therefore, excess NaOH was added to the reaction medium. HPLC was used to track the amount of chorismic acid utilized as a substrate or/and the amount of phenylpyruvate generated as a product. The established HPLC system enabled the assay of nearly all compounds under investigation: chorismic acid, phenylpyruvate, tyrosine and ABA. However, the tyrosine absorbance and peak resolution were not optimal (Supplementary Materials: Fig. SIV). The system could efficiently separate all of the required metabolites from other yeast metabolites and thus quantify them (Supplementary Materials: Fig. SV).
Time of reaction
The time needed to reach the maximum product concentration was investigated in the yeast-chorismate-mutase assay system. Chorismic acid was added to the wild-type yeast culture to a final concentration of 1 mM and left at 30 °C. The enzymatic reaction was terminated by the addition of 0.2 N HCl, which simultaneously converted the prephenic acid produced to phenylpyruvate. The acid was added after 5, 10, 20, 45, 60, 90, 120 and 240 minutes. The medium was rendered alkaline using excess NaOH, and the concentration of chorismic acid and phenylpyruvate were identified using HPLC (Supplementary Materials: Fig. SVI). The ideal reaction time to obtain the maximal concentration of the product was 60 minutes (Supplementary Materials: Fig. SVIB). After 240 minutes, there were other products that could be separated by the HPLC system (Supplementary Materials: Fig. SVIA). However, we were not able to identify these products, even by using hyphenated mass spectrometry and library searches. The newly compounds seem to be byproducts of phenylpyruvate, since the amount of phenylpyruvate produced declined.
Abscisic acid inhibition assay
To test the inhibitory effect of ABA on chorismate mutase, the established yeast-chorismate-mutase assay system was used. Keeping the concentration of the chorismic acid initially added to 1 mM, various concentrations of ABA were added (500, 250, 100, 50 and 10 µM) to the yeast system. The concentrations of chorismic acid and phenylpyruvate were assessed using the previously established HPLC system, and the results are shown in Fig. 8. The results suggested an inhibitory activity of ABA on chorismate mutase through the inhibition of phenylpyruvate production. The results also indicated that the best concentration of ABA to be used as an inhibitor to yeast chorismate mutase in this system is from 200 to 300 µM.
Figure 8.
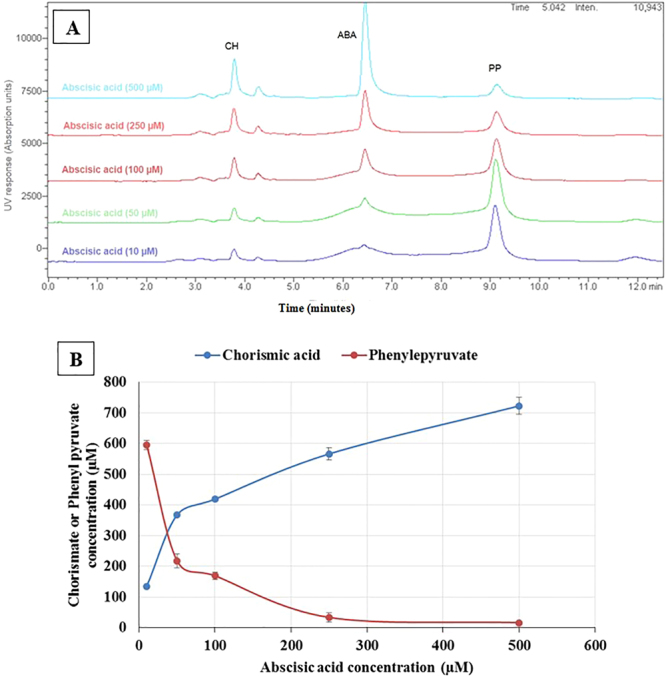
Concentrations of Chorismic acid (CH) and phenyl pyruvate (PP) in response to the addition of Abscisic acid (ABA) in the yeast chorismate mutase assay system. (A) HPLC Chromatograms of ABA addition to the yeast-chorismate-mutase-assay system in different concentrations. (B) Scatter analysis of chorismic acid and phenylpyruvate concentrations in response to the increase of ABA in the yeast-chorismate-mutase-assay system. The establishment of the yeast-chorismate-mutase-assay system is described in methodology section. Final concentration of CH added was 1000 µM.
Tyrosine inhibition assay
Tyrosine is a known allosteric inhibitor of chorismate mutase. Tyrosine was tested as a chorismate mutase inhibitor in an experiment similar to that of ABA. Various concentrations of tyrosine (500, 250, 100, 50 and 10 µM) were added to the yeast chorismate mutase system. The concentrations of chorismic acid and phenylpyruvate were evaluated (Supplementary Materials: Fig. SVII). The results indicated the inhibitory effect of tyrosine on chorismate mutase through the decline in the phenylpyruvate produced. The optimal concentration for tyrosine to work in this system was 100 µM. This tyrosine experiment can be considered as a validation for the established system of the chorismate mutase assay. Upon comparing the inhibitory activity of tyrosine to that of ABA, it was concluded that tyrosine is 2 to 3 times more potent than ABA.
Discussion
Fungal infections are highly correlated with increasing rates of mortality, especially among immunocompromised patients22. Current treatment choices are limited and hampered by a narrow spectrum, poor bioavailability, toxicity, drug-interactions and by compounds that have fungistatic rather than fungicidal activity23,24. Additionally, current medications are ineffective against some fungal infections, such as aspergillosis, and the emergence of multiple drug resistance is also a serious concern25. Therefore, the importance of the development of new effective antifungal drugs is imperative.
In this study, the rational drug design approach resulted in repurposing a well-known plant growth-regulating agent, ABA, as a potential fungal chorismate mutase inhibitor, since it might elicit an antifungal effect. Using the previous approach, homology models for chorismate mutase in different fungal strains, C. albicans, C. parapsilosis, A. niger, and T. rubrum, were established depending on the crystal structure of the same enzyme in S. cerevisiae as a template. The active site for substrate binding was determined through the alignment of chorismate mutase protein sequences of previous fungal strains. The crystal structure of yeast chorismate mutase in complex with endo-oxabicyclic transition-state analogue inhibitor16 was used to build a pharmacophore model for fungal chorismate mutase inhibitors. The pharmacophore model was used to screen more than 160,000 compounds for their potential anti-chorismate mutase activities, and the top 11 compounds were docked inside the transition-state binding site of the four chorismate mutase homology models of previous fungal strains. Structure similarity searches for the best compounds in previous docking studies led to ABA as a potential fungal chorismate mutase inhibitor. Another molecular docking study was performed for ABA (S) and (R) isomers against the four homology models and compared to the docking of endo-oxabicyclic transition-state analogue inhibitors. The results indicated that the (S) isomer is more active and fit to the pharmacophore model. The molecular docking results were supported by the molecular dynamics and in vitro antifungal studies. The molecular dynamics studies favoured the interaction between ABA and C. albicans homology model of chorismate mutase and gave it the lowest interaction energy. These results not only imply a stable drug-protein interaction but also suggest a long duration of action as well. This property can affect its therapeutic concentration and its dose regimen. The in vitro antifungal activity of ABA against the previous fungi strains indicated activity in the microgram range of concentrations that indicates good antifungal activity considering that ABA is not a toxic compound. The established yeast chorismate mutase assay system in this study depended on the release of yeast chorismate mutase after cell disruption, using sonication and in the presence of protease inhibitors. The assay could efficiently separate all the required metabolites. The ideal reaction time needed to reach the product concentration was 60 minutes. The inhibitory concentration of ABA against yeast chorismate mutase ranged from 200 to 300 µM which is a confirmation of the inhibition. Tyrosine, which is a well-known chorismate mutase inhibitor, was compared to ABA and found to be 2 to 3 times more potent than ABA.
ABA is a well-known, naturally occurring sesquiterpene phytohormone that plays fundamental and important roles in plants. ABA regulates plant growth and development, seed and bud dormancy, senescence and abscission, and it exhibits a vital role in plant responses to stresses26–29. ABA is produced through the isoprenoid biosynthetic pathway via the degradation of C40 β-carotene derivatives such as zeaxanthin and neoxanthin. The splitting of such products produces xanthoxin, which is the precursor of ABA30,31. Although ABA is a major signalling molecule in plants, it was identified in many other organisms such as algae32, cyanobacteria33 and fungi34. ABA was identified in a wide range of lower animals, such as sponges35, as well as in higher mammals36, including various human tissues and cells30,37. Recently, some of the medicinal activities of ABA were identified38, including anti-inflammatory39, haemopoietic growth factor40, antituberculosis41, antiatherosclerosis42 and anticancer activities30. ABA has structural similarities to thiazolidinedione, and consequently it increases the expression of PPAR-γ43,44. It is also considered to be an endogenous stimulator of insulin release from human pancreatic islets45. These two characteristics contribute to its anti-diabetic activity30. ABA exhibits antifungal activity in plants46 through the promotion of callose (a β-1,3-glucan polymer) deposition47. However, this study suggests another mechanism of ABA as an antifungal through the direct inhibition of the fungal chorismate mutase. ABA is a well-studied molecule that has been shown to be found naturally in the human body30,37 and in edible plants. It is a safe and nontoxic with no evidence for carcinogenicity or teratogenicity48, which makes it a good target for the pharmaceutical industry. ABA possesses an LD50 of more than 5,000 mg/kg in rats48, reflecting a wide therapeutic index of the molecule. The compound has only two isomers, which facilitate its synthesis and purification, and it is commercially available.
Conclusion
ABA was repositioned as a possible anti-chorismate mutase agent with an antifungal activity as a result of a rational drug discovery. This could introduce a new class of antifungal agents, which can contribute to the treatment of life-threatening fungal infections and avoid the severe side effects and toxicity attributed to most currently known antifungal agents. Once again, the rational drug design approach proved to be one of the successful approaches in drug discovery, and it may help the pharmaceutical industry develop and discover more drugs. The effect of the compound on chorismate mutase was confirmed by a chorismate mutase assay in which ABA showed (200 to 300 µM) inhibition, and these results were validated by comparing it to tyrosine which is known to be an allosteric inhibitor of chorismate mutase.
Methods
Materials
(+)-Abscisic acid (5 mg, ≥98% (HPLC), m.p. 160 °C) was purchased from Sigma-Aldrich. The compound was tested using its melting point, 1H and 13C NMR spectroscopy, mass spectrometry and HPLC for structure and purity authentication.
Sequence similarity search
The pdb search module provided in MOE 2014.09 was used for searching for the best template. Gap start was used as default (−12), Gap extent (−2), E-value cutoff (10) E-value accept (1e-012), Z iterations (100) and Z-cutoff (6).
Molecular alignment
The molecular alignment was conducted using a Clustal Omega tool provided in the UniProt server (http://www.ebi.ac.uk/Tools/msa/clustalo/). The alignment was conducted between the chorismate mutase FASTA sequence of Candida albicans (UniProt ID: Q59TS4), strain (SC5314/ATCC MYA-2876), Candida parapsilosis (UniProt ID: G8BD21) strain (CDC 317/ATCC MYA-4646), Aspergillus niger (UniProt ID: A2R3Z4) strain (CBS 513.88/FGSC A1513), Trichophyton rubrum (UniProt ID: F2SCL7) strain (ATCC MYA-4607/CBS 118892) and Saccharomyces cerevisiae (UniProt ID: P32178).
Homology modelling
The crystal structure of S. cerevisiae chorismate mutase (PDB entry code 4CSM) was used as a template with sequence identity 62.82%. S. cerevisiae chorismate mutase was used as template chain A, and C. albicans, C. parapsilosis, A. niger, and T. rubrum chorismate mutase sequences were used as queries. AMBER99 was used as a force field, and all of the settings were kept as the default in which strain cutoff 1.5 and distance cutoff 1.2 were used.
Molecular Docking Studies
The molecular docking studies were conducted using MOE 2014.09 and Leadit 2.1.2 software. Molecular Docking Studies with MOE 2014.09.
The Molecular Operating Environment (MOE) 2014.09 package license was purchased from Chemical Computing Group Inc., Sherbooke St, Montreal, QC, Canada49, and the Leadit 2.1.2 software license was purchased from BioSolveIT GmbH, Germany50. All compounds were built and saved as moe. Rigid receptor was used as a docking protocol. Both receptor-solvent were kept as a “receptor”. Triangle matcher was used as a placement method. Two rescoring were computed; rescoring 1 was selected as London dG. Rescoring 2 was selected as affinity. Force field was used as a refinement.
Molecular Docking Studies with Leadit 2.1.2
All of the compounds were built and saved as Mol2. The crystal structure of S. cerevisiae chorismate mutase in complex with endo-oxabicyclic transition state analogue inhibitor was downloaded from a protein databank (pdb code = 4CSM). The protein was loaded into Leadit 2.1.2, and the receptor components were chosen by the selection of chain A as a main chain, which is in complex with the endo-oxabicyclic transition state analogue inhibitor. The binding site was defined by choosing the inhibitor as a reference ligand to which all coordinates were computed. Amino acids within a radius of 6.5 Å were selected in the binding site. All chemical ambiguities of residues were left as default. Ligand binding was driven by enthalpy (classic Triangle matching). All default settings were restored for scoring. Intra-ligand clashes were computed by using clash factor = 0.6. Maximum number of solutions per iteration = 200. Maximum of solution per fragmentation = 200. The base placement method was used as a docking strategy.
Molecular dynamics simulations for protein ligand complexes
Molecular dynamics (MD) simulations were performed using Gromacs 4.5.520. Drug candidates were evaluated within the protein binding sites. The topology file for the selected small molecules were generated using the automated topology builder server PRODRG251 in the framework of GROMOS 53A6 force field52. The protein-ligand complexes were then solvated with TIP3P explicit water molecules and placed in the centre of an octahedral box of size 24324 3 24 Å3. A minimum 1.0 Å distance was maintained between the protein and the edge of the simulation box, and the periodic boundary condition was used, so that protein can fully immerse with water and rotate freely. Then, the Particle Mesh Ewald (PME) method53 was used for the electrostatic energy calculation. It permits the use of the Ewald summation at a computational cost comparable to that of a simple truncation method of 10 Å or less, and the linear constraint solver (LINCS)54. An algorithm was used to determine covalent bond constraints. Before minimization, the system was neutralized by the addition of Na+ ions. The steepest descent approach (1000 ps) was used for each protein-ligand complex to minimize energy. Further NVT was performed for 20 ps to equilibrate the system with protein and ligand for constant volume, pressure (1 atm) and temperature (300 K). The final MD run was set to 1000 ps for each protein-ligand complex, and trajectories were saved for further analysis using Xmgrace and VMD software55. Interaction energy and Gibbs free energy were calculated using Gromacs and g_mmpbsa software, respectively21. The interaction energy for the proteins and drug complexes was calculated by estimating the short range Lennard-Jones and short range Coulomb energies using the g_energy analysis tool of Gromacs software. It estimates the stability of proteins and drug candidate complexes. The flowchart that describes all computational steps can be found in (Supplementary Materials: Fig. SVIII).
In vitro antifungal activity
The antifungal assay was conducted as described by Clancy and Nguyen56 in comparison with amphotericin B. The minimum inhibitory concentrations were determined at the Regional Center for Mycology and Biotechnology, Al-Azhar University, Cairo, Egypt. The antifungal activity was tested against five fungal strains: Candida albicans (RCMB 05036), Candida parapsilosis (RCMB 05064), Aspergillus niger (RCMB 02568), Trichophyton rubrum (RCMB 010162), and Trichophyton mentagrophytes (RCMB 010173). An MTT micro dilution assay was used to evaluate the antifungal activity through the determination of the MIC using the serial dilution concentrations of 0.49 µg/mL, 0.98 µg/mL, 1.95 µg/mL, 3.9 µg/mL, 7.81 µg/mL, 15.63 µg/mL, 31.25 µg/mL, 62.5 µg/mL, and 125 µg/mL. The antifungal potency of the drug was calculated as MIC (µg/ml) in comparison with amphotericin B as a positive control.
Assay of anti-chorismate mutase activity
Yeast extract (Y1625), glucose (G8270), adenine hemisulphate (A3159), Tris-HCl (T5941), benzamidine HCL (434760), EDTA (E9884), PMSF (78830), chorismic acid (C1259), tyrosine (T3754), phenylpyruvate (P8001) and abscisic acid (A4906) were obtained from Sigma-Aldrich.
Yeast cells
Wild type yeast, Saccharomyces cerevisiae, was obtained from the Date and Palm Center of Excellence (DPCE), University of King Faisal, Al-Ahsaa, Kingdom of Saudi Arabia. The yeast strain had been previously identified by experts in the Center, and its purity was ensured through sequential streaking and single colony selection.
Growth of yeast cells
A single colony of yeast cells was grown in liquid YPAD medium (2% bactopeptone, 1% yeast extract, 2% glucose and 0.04% adenine sulphate). Cultures were grown at 28 °C with shaking at 200 RPM for 48 hours.
Production of yeast extracts
Yeast was extracted as described previously with some modifications57,58. Briefly, yeast cells were harvested by centrifugation at 4000 × g for 15 minutes and washed with washing buffer (Tris-HCl (50 mM, pH 8.0), benzamidine (1 mM), EDTA (1 mM) and PMSF (1 mM, freshly prepared)) twice. The yeast cells were suspended in this buffer and diluted until the optical density reached an absorbance of 2.0 at 600 nm. Sonication was used to disrupt the yeast cells as described previously59,60. Sonication was conducted using a Wise clean sonicator (model WUC-D06H) in 30 °C, 20 kHz (50% of full power). The sonication cycle consisted of 5 minutes of sonication and a 2-minute break, and for 80% yeast cell disruption, 10 cycles were required. Cell disruption was confirmed visually by counting the cells under the microscope in three different fields and comparing with the original cell count before and after sonication. Yeast cell extract was prepared fresh for every experiment.
Chorismate mutase assays
The chorismate mutase was assayed as described previously with some modifications.57,58 For each experiment, 150 µL of previously made yeast extract was mixed with 100 µL Chorismic acid (10 mM). For experiments using inhibitors, tyrosine (10 mM) or abscisic acid (10 mM), with various volumes according to the experiment, were added to the reaction mixture. The whole components were vortexed and incubated at 30 °C at 150 RPM for various times. The conversion of chorismic acid to prephenic acid was detected through the conversion of the acid produced to the more stable and UV detectable phenylpyruvate by the addition of 250 µL 0.2 N HCl. The reaction was left at room temperature for 10 minutes, and then 500 µL 1 N NaOH was added. The reaction mixture was brought to 1 mL by the addition of Tris-HCl (pH 8) if needed. The reaction mixture was centrifuged at 12,000 RPM for 10 minutes, and the supernatant was subjected to HPLC analysis.
HPLC analysis
HPLC analysis, including chorismic acid, phenylpyruvate, tyrosine and abscisic acid separation and quantification, was performed using a Shimadzu Prominence HPLC system equipped with a CBM-20A controller, LC-20A solvent unit, SIL-20A auto-sampler, CTO-20A column oven and SPD-20A UV-VIS detector. The compounds were separated using a Luna-C18 (L1, Phenomenex, 150 mm × 4.6 mm × 5 μm). Mobile phases were a combination of 0.1% trifluoroacetic acid in water: acetonitrile: methanol (50:40:10). The flow rate was adjusted to 0.5 mL/minute, and the UV detector was adjusted to 320 nm. Calibration curves for chorismic acid, phenylpyruvate, tyrosine and abscisic acid were performed using standards.
Identification of Phenylpyruvate
To identify phenylpyurate, HPLC analysis was performed using an Agilent LC-MS platform (1200 series) comprising a pump system, photodiode array detector and a Quadrupole mass spectrometer (model 6120, detecting between 150 and 600 m/z units, with electrospray positive ionization (ESI+)). The same chromatographic conditions described above were applied. The phenylpyruvate produced by the yeast cultures was identified through the recognition of the new peak produced after the addition of chorismic acid to the cultures. The mass chromatogram of the new peak was compared to mass spectral data in Wiley Registry of Mass Spectral Data 10th Edition (April 2013) and NIST 11 Mass Spectral Library (NIST11/2011/EPA/NIH). Spiking experiments with authentic phenylpyruvate were also used to confirm the identity of the newly produced peak. The flowchart that describes the chorismate mutase in vitro assay can be found in (Supplementary Materials: Fig. SVIX).
Electronic supplementary material
Acknowledgements
The authors, MAK and MEM are Grateful to the college of Clinical Pharmacy, King Faisal University for the financial and facilities support. The college has no involvement in the study design, the collection, analysis, interpretation of data, writing of manuscript and in the decision to submit the manuscript for publication.
Author Contributions
M.A.K. and M.E.M. designed, performed the study. M.A.K., designed the pharmacophore model, built the homology models and performed the docking studies. M.E.M., performed the Chorismate mutase assay, Abscisic acid inhibition assay, Tyrosine inhibition assay. A.M., contributed by performing, analyzing and writing the molecular dynamics part. All the authors contributed in writing the initial draft of the manuscript. All the authors read and approved the final draft of the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-26998-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Herrmann KM, Weaver LM. THE SHIKIMATE PATHWAY. Annual review of plant physiology and plant molecular biology. 1999;50:473–503. doi: 10.1146/annurev.arplant.50.1.473. [DOI] [PubMed] [Google Scholar]
- 2.Tzin V, Galili G. The Biosynthetic Pathways for Shikimate and Aromatic Amino Acids in Arabidopsis thaliana. The Arabidopsis book/American Society of Plant Biologists. 2010;8:e0132. doi: 10.1199/tab.0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mir, R., Jallu, S. & Singh, T. P. The shikimate pathway: Review of amino acid sequence, function and three-dimensional structures of the enzymes. Critical reviews in microbiology, 10.3109/1040841x.2013.813901 (2013). [DOI] [PubMed]
- 4.Dosselaere F, Vanderleyden J. A metabolic node in action: chorismate-utilizing enzymes in microorganisms. Critical reviews in microbiology. 2001;27:75–131. doi: 10.1080/20014091096710. [DOI] [PubMed] [Google Scholar]
- 5.Tzin V, Malitsky S, Aharoni A, Galili G. Expression of a bacterial bi-functional chorismate mutase/prephenate dehydratase modulates primary and secondary metabolism associated with aromatic amino acids in Arabidopsis. The Plant journal: for cell and molecular biology. 2009;60:156–167. doi: 10.1111/j.1365-313X.2009.03945.x. [DOI] [PubMed] [Google Scholar]
- 6.Dias MV, et al. Structure of chorismate synthase from Mycobacterium tuberculosis. Journal of structural biology. 2006;154:130–143. doi: 10.1016/j.jsb.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Roberts CW, et al. The shikimate pathway and its branches in apicomplexan parasites. The Journal of infectious diseases. 2002;185(Suppl 1):S25–36. doi: 10.1086/338004. [DOI] [PubMed] [Google Scholar]
- 8.Kancharla PK, Doddi VR, Kokatla H, Vankar YD. A concise route to (−)-shikimic acid and (−)-5-epi-shikimic acid, and their enantiomers via Barbier reaction and ring-closing metathesis. Tetrahedron Letters. 2009;50:6951–6954. doi: 10.1016/j.tetlet.2009.09.111. [DOI] [Google Scholar]
- 9.Schonbrunn E, et al. Interaction of the herbicide glyphosate with its target enzyme 5-enolpyruvylshikimate 3-phosphate synthase in atomic detail. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:1376–1380. doi: 10.1073/pnas.98.4.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hudson GS, Wong V, Davidson BE. Chorismate mutase/prephenate dehydrogenase from Escherichia coli K12: purification, characterization, and identification of a reactive cysteine. Biochemistry. 1984;23:6240–6249. doi: 10.1021/bi00320a054. [DOI] [PubMed] [Google Scholar]
- 11.Sasso S, Ramakrishnan C, Gamper M, Hilvert D, Kast P. Characterization of the secreted chorismate mutase from the pathogen Mycobacterium tuberculosis. FEBS Journal. 2005;272:375–389. doi: 10.1111/j.1742-4658.2004.04478.x. [DOI] [PubMed] [Google Scholar]
- 12.Schneider CZ, Parish T, Basso LA, Santos DS. The two chorismate mutases from both Mycobacterium tuberculosis and Mycobacterium smegmatis: biochemical analysis and limited regulation of promoter activity by aromatic amino acids. Journal of bacteriology. 2008;190:122–134. doi: 10.1128/JB.01332-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arcuri HA, Palma MS. Understanding the structure, activity and inhibition of chorismate synthase from Mycobacterium tuberculosis. Current medicinal chemistry. 2011;18:1311–1317. doi: 10.2174/092986711795029528. [DOI] [PubMed] [Google Scholar]
- 14.Gad, S. C. Drug Discovery Handbook. 1st edn, (Wiley-interscience, John Wiley & Sons. Inc., 2005).
- 15.Mandal S, Moudgil MN, Mandal SK. Rational drug design. European Journal of Pharmacology. 2009;625:90–100. doi: 10.1016/j.ejphar.2009.06.065. [DOI] [PubMed] [Google Scholar]
- 16.Strater N, Schnappauf G, Braus G, Lipscomb WN. Mechanisms of catalysis and allosteric regulation of yeast chorismate mutase from crystal structures. Structure (London, England: 1993) 1997;5:1437–1452. doi: 10.1016/S0969-2126(97)00294-3. [DOI] [PubMed] [Google Scholar]
- 17.Chook YM, Ke H, Lipscomb WN. Crystal structures of the monofunctional chorismate mutase from Bacillus subtilis and its complex with a transition state analog. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:8600–8603. doi: 10.1073/pnas.90.18.8600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burschowsky D, et al. Electrostatic transition state stabilization rather than reactant destabilization provides the chemical basis for efficient chorismate mutase catalysis. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:17516–17521. doi: 10.1073/pnas.1408512111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hediger MED. synthesis, and evaluation of aza inhibitors of chorismate mutase. Bioorganic & medicinal chemistry. 2004;12:4995–5010. doi: 10.1016/j.bmc.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 20.Pronk S, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics (Oxford, England) 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumari R, Kumar R, Lynn A. g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. Journal of chemical information and modeling. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 22.Calderone R, et al. Antifungal drug discovery: the process and outcomes. Future microbiology. 2014;9:791–805. doi: 10.2217/fmb.14.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butts A, Krysan DJ. Antifungal drug discovery: something old and something new. PLoS pathogens. 2012;8:e1002870. doi: 10.1371/journal.ppat.1002870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mejia R. Evolution-based drug discovery: antifungal also disrupts blood vessel formation. PLoS biology. 2012;10:e1001380. doi: 10.1371/journal.pbio.1001380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gallo-Ebert C, et al. Novel antifungal drug discovery based on targeting pathways regulating the fungus-conserved Upc2 transcription factor. Antimicrobial agents and chemotherapy. 2014;58:258–266. doi: 10.1128/AAC.01677-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao FY, Yoshioka K, Desveaux D. The roles of ABA in plant-pathogen interactions. Journal of plant research. 2011;124:489–499. doi: 10.1007/s10265-011-0409-y. [DOI] [PubMed] [Google Scholar]
- 27.Nakashima K, Yamaguchi-Shinozaki K. ABA signaling in stress-response and seed development. Plant cell reports. 2013;32:959–970. doi: 10.1007/s00299-013-1418-1. [DOI] [PubMed] [Google Scholar]
- 28.Nonogaki H. Seed dormancy and germination-emerging mechanisms and new hypotheses. Frontiers in plant science. 2014;5:233. doi: 10.3389/fpls.2014.00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregersen PL, Culetic A, Boschian L, Krupinska K. Plant senescence and crop productivity. Plant molecular biology. 2013;82:603–622. doi: 10.1007/s11103-013-0013-8. [DOI] [PubMed] [Google Scholar]
- 30.Li HH, et al. Occurrence, function and potential medicinal applications of the phytohormone abscisic acid in animals and humans. Biochemical pharmacology. 2011;82:701–712. doi: 10.1016/j.bcp.2011.06.042. [DOI] [PubMed] [Google Scholar]
- 31.Milborrow BV. The pathway of biosynthesis of abscisic acid in vascular plants: a review of the present state of knowledge of ABA biosynthesis. Journal of Experimental Botany. 2001;52:1145–1164. doi: 10.1093/jexbot/52.359.1145. [DOI] [PubMed] [Google Scholar]
- 32.Lu Y, et al. Antagonistic roles of abscisic acid and cytokinin during response to nitrogen depletion in oleaginous microalga Nannochloropsis oceanica expand the evolutionary breadth of phytohormone function. The Plant journal: for cell and molecular biology. 2014;80:52–68. doi: 10.1111/tpj.12615. [DOI] [PubMed] [Google Scholar]
- 33.Marsalek B, Simek M. Abscisic acid and its synthetic analog in relation to growth and nitrogenase activity of Azotobacter chroococcum and Nostoc muscorum. Folia microbiologica. 1992;37:159–160. doi: 10.1007/BF02836623. [DOI] [PubMed] [Google Scholar]
- 34.Gong T, et al. Isolation of genes related to abscisic acid production in Botrytis cinerea TB-3-H8 by cDNA-AFLP. Journal of basic microbiology. 2014;54:204–214. doi: 10.1002/jobm.201200311. [DOI] [PubMed] [Google Scholar]
- 35.Zocchi E, et al. The temperature-signaling cascade in sponges involves a heat-gated cation channel, abscisic acid, and cyclic ADP-ribose. Proceedings of the National Academy of Sciences. 2001;98:14859–14864. doi: 10.1073/pnas.261448698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Le Page-Degivry MT, Bidard JN, Rouvier E, Bulard C, Lazdunski M. Presence of abscisic acid, a phytohormone, in the mammalian brain. Proceedings of the National Academy of Sciences. 1986;83:1155–1158. doi: 10.1073/pnas.83.4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bellotti M, et al. Synthesis, structural characterization and effect on human granulocyte intracellular cAMP levels of abscisic acid analogs. Bioorganic & medicinal chemistry. 2015;23:22–32. doi: 10.1016/j.bmc.2014.11.035. [DOI] [PubMed] [Google Scholar]
- 38.Bassaganya-Riera J, et al. Mechanisms of action and medicinal applications of abscisic Acid. Current medicinal chemistry. 2010;17:467–478. doi: 10.2174/092986710790226110. [DOI] [PubMed] [Google Scholar]
- 39.Bassaganya-Riera J, et al. Abscisic acid regulates inflammation via ligand-binding domain-independent activation of peroxisome proliferator-activated receptor gamma. The Journal of biological chemistry. 2011;286:2504–2516. doi: 10.1074/jbc.M110.160077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scarfi S, et al. The plant hormone abscisic acid stimulates the proliferation of human hemopoietic progenitors through the second messenger cyclic ADP-ribose. Stem cells (Dayton, Ohio) 2009;27:2469–2477. doi: 10.1002/stem.173. [DOI] [PubMed] [Google Scholar]
- 41.Clark TN, Ellsworth K, Li H, Johnson JA, Gray CA. Isolation of the plant hormone (+)-abscisic acid as an antimycobacterial constituent of the medicinal plant endophyte Nigrospora sp. Natural product communications. 2013;8:1673–1674. [PubMed] [Google Scholar]
- 42.Magnone M, et al. Abscisic acid released by human monocytes activates monocytes and vascular smooth muscle cell responses involved in atherogenesis. The Journal of biological chemistry. 2009;284:17808–17818. doi: 10.1074/jbc.M809546200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu P, Bevan DR, Lewis SN, Hontecillas R, Bassaganya-Riera J. Molecular modeling of lanthionine synthetase component C-like protein 2: a potential target for the discovery of novel type 2 diabetes prophylactics and therapeutics. Journal of molecular modeling. 2011;17:543–553. doi: 10.1007/s00894-010-0748-y. [DOI] [PubMed] [Google Scholar]
- 44.Guri AJ, Hontecillas R, Bassaganya-Riera J. Abscisic acid synergizes with rosiglitazone to improve glucose tolerance and down-modulate macrophage accumulation in adipose tissue: Possible action of the cAMP/PKA/PPAR γ axis. Clinical Nutrition. 2010;29:646–653. doi: 10.1016/j.clnu.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bruzzone S, et al. Abscisic acid is an endogenous stimulator of insulin release from human pancreatic islets with cyclic ADP ribose as second messenger. The Journal of biological chemistry. 2008;283:32188–32197. doi: 10.1074/jbc.M802603200. [DOI] [PubMed] [Google Scholar]
- 46.Trusov Y, et al. Heterotrimeric G proteins-mediated resistance to necrotrophic pathogens includes mechanisms independent of salicylic acid-, jasmonic acid/ethylene- and abscisic acid-mediated defense signaling. The Plant journal: for cell and molecular biology. 2009;58:69–81. doi: 10.1111/j.1365-313X.2008.03755.x. [DOI] [PubMed] [Google Scholar]
- 47.Oide S, et al. A novel role of PR2 in abscisic acid (ABA) mediated, pathogen-induced callose deposition in Arabidopsis thaliana. The New phytologist. 2013;200:1187–1199. doi: 10.1111/nph.12436. [DOI] [PubMed] [Google Scholar]
- 48.Authority EFS. Conclusion on the peer review of the pesticide risk assessment of the active substance S-abscisic acid. EFSA Journal. 2013;11:3341–3419. doi: 10.2903/j.efsa.2013.3341. [DOI] [Google Scholar]
- 49.Molecular Operating Environment (moe) v. 2013.08 (Chemical Computing Group Inc., 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2014).
- 50.Rarey M, Kramer B, Lengauer T, Klebe G. A fast flexible docking method using an incremental construction algorithm. Journal of molecular biology. 1996;261:470–489. doi: 10.1006/jmbi.1996.0477. [DOI] [PubMed] [Google Scholar]
- 51.Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta crystallographica. Section D, Biological crystallography. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 52.Oostenbrink C, Villa A, Mark AE, van Gunsteren WF. A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. Journal of computational chemistry. 2004;25:1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 53.Wang H, Dommert F, Holm C. Optimizing working parameters of the smooth particle mesh Ewald algorithm in terms of accuracy and efficiency. The Journal of Chemical Physics. 2010;133:034117. doi: 10.1063/1.3446812. [DOI] [PubMed] [Google Scholar]
- 54.Amiri S, Sansom MS, Biggin PC. Molecular dynamics studies of AChBP with nicotine and carbamylcholine: the role of water in the binding pocket. Protein engineering, design & selection: PEDS. 2007;20:353–359. doi: 10.1093/protein/gzm029. [DOI] [PubMed] [Google Scholar]
- 55.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of molecular graphics. 1996;14(33-38):27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 56.Clancy CJ, Nguyen MH. Comparison of a photometric method with standardized methods of antifungal susceptibility testing of yeasts. Journal of Clinical Microbiology. 1997;35:2878–2882. doi: 10.1128/jcm.35.11.2878-2882.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schnappauf G, Lipscomb WN, Braus GH. Separation of inhibition and activation of the allosteric yeast chorismate mutase. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:2868–2873. doi: 10.1073/pnas.95.6.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mobley EM, Kunkel BN, Keith B. Identification, characterization and comparative analysis of a novel chorismate mutase gene in Arabidopsis thaliana. Gene. 1999;240:115–123. doi: 10.1016/S0378-1119(99)00423-0. [DOI] [PubMed] [Google Scholar]
- 59.Bzducha-Wrobel A, et al. Evaluation of the efficiency of different disruption methods on yeast cell wall preparation for beta-glucan isolation. Molecules (Basel, Switzerland) 2014;19:20941–20961. doi: 10.3390/molecules191220941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu D, Zeng X-A, Sun D-W, Han Z. Disruption and protein release by ultrasonication of yeast cells. Innovative Food Science & Emerging Technologies. 2013;18:132–137. doi: 10.1016/j.ifset.2013.02.006. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.