

Abstract
The Berlin Patient represents the first and only functional HIV cure achieved by hematopoietic stem cell transplant (HSCT). In subsequent efforts to replicate this result, HIV rebounded post-HSCT after withdrawal of antiretroviral therapy. Providing HIV-specific immunity through adoptive T cell therapy may prevent HIV rebound post-HSCT by eliminating newly infected cells before they can seed systemic infection. Adoptive T cell therapy has demonstrated success in boosting Epstein-Barr virus and cytomegalovirus-specific immunity post-HSCT, controlling viral reactivation. However, T cell immunotherapies to boost HIV-specific immunity have been limited by single-epitope specificity and minimal persistence or efficacy in vivo. To improve this strategy, we sought to generate allogeneic HIV-specific T cells from human leukocyte antigen (HLA)-A02+ HIV-negative adult or cord blood donors. We focused on HLA-A02+ donors due to well-characterized epitope restrictions observed in HIV+ populations. We show that multi-antigen HIV-specific T cells can be generated from naive T cells of both cord blood and adults using a reproducible good manufacturing practice (GMP)-grade protocol. This product lysed antigen-pulsed targets and suppressed active HIV in vitro. Interestingly, these cells displayed broad epitope recognition despite lacking recognition of the common HLA-A02-restricted HIV epitope Gag SL9. This first demonstration of functional multi-antigen HIV-specific T cells has implications for improving treatment of HIV through allogeneic HSCT.
Keywords: HIV-specific T cells, adoptive T cell therapy, allogeneic transplant
Graphical Abstract
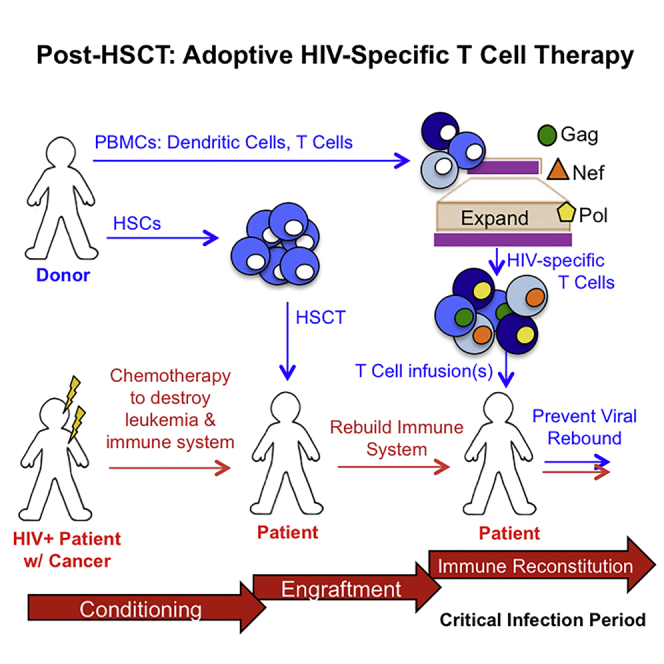
Patel et al. demonstrate the ability to generate HIV-specific T cells from HIV-seronegative adults and cord blood with a good-manufacturing-practice-compliant strategy. These immunotherapies are multi-antigen specific, display cytotoxicity, and suppress HIV in vitro, providing a promising platform for adoptive T cell therapy in a post-transplant setting.
Introduction
Current therapies for HIV are not curative. While antiretroviral therapy (ART) successfully suppresses active infection, it does not eradicate latent HIV reservoirs. HIV cure strategies have focused on replicating the successful allogeneic hematopoietic stem cell transplant (HSCT) strategy used for the Berlin Patient,1, 2 without success due to viral rebound of the patients’ autologous viral reservoirs.3, 4 The delayed viral rebound in the Boston patients suggests large reductions in the HIV reservoir may allow HIV-specific T cells to control viral rebound from the low levels of virus that persist post-transplant.
Several lines of evidence have firmly established that HIV-specific T cell responses play a critical role in controlling HIV replication in infected individuals. First, the emergence of HIV-specific CD8+ T cell responses following infection is temporally associated with a 102- to 103-fold reduction in viremia during acute HIV infection.5, 6, 7 Second, in the rhesus macaque simian immunodeficiency virus (SIV) infection model, the depletion of CD8+ T cells has been shown to lead to a dramatic increase in viral load and rapid progression.8 Third, in infected humans, it has been established that there are strong links between the possession of certain major histocompatibility complex class I (MHC class I) alleles and clinical progression.9, 10 Fourth, CD8+ T cells exert immune pressure on targeted epitopes, driving the emergence of escape mutations, often at a fitness cost to the virus.11, 12 Finally, CD8+ T cells isolated from HIV-infected individuals show a clear ability to eliminate infected cells and suppress viral replication in vitro.13 Thus, there is a strong rationale for enlisting HIV-specific T cells to prevent viral rebound from any residual infected cells that may persist following allogeneic HSCT and achieving a functional cure.
Adoptive T cell therapy post-HSCT has been successful in augmenting anti-viral immunity against chronic infections, such as cytomegalovirus (CMV), Epstein-Barr virus (EBV),14, 15, 16, 17 and associated cancers, emphasizing the critical role T cells play in preventing viral rebound. However, HIV is able to avoid immune pressures more successfully than viral counterparts due to downregulation of MHC class I and CD4 on infected cells, leading to suboptimal anti-HIV CD8+ T cell responses.18 Despite efforts to augment anti-viral immunity against HIV, T cell therapy has shown no efficacy, likely due to infusion of single-epitope-specific clones that are susceptible to immune escape19 or the absence of CD4+ T cells resulting in a lack of persistence of infused cells.20 Furthermore, prevention strategies, such as the HIV vaccine trial RV144,21 have been criticized for the lack of eliciting strong T cell responses needed to achieve sustained anti-HIV immunity.22, 23 Thus, HIV-specific T cell therapies that demonstrate the ability to persist and overcome immune escape through recognition of multiple HIV epitopes will be critical to boosting anti-HIV immunity.
The post-HSCT setting presents a unique opportunity where adoptive HIV T cell therapy could target residual infected cells to prevent rebound from the low levels of virus remaining. Furthermore, these HIV-specific T cells may demonstrate better persistence compared to the previous HIV immunotherapy trials mentioned, which had no conditioning regimen. Based on the successful generation of EBV- and CMV-specific T cells from virus-naive allogeneic donors,24, 25, 26, 27 we sought to generate HIV-specific T cells from HIV-seronegative adults and cord blood naive T cells in a good manufacturing practice (GMP)-compliant manner. Whereas a closely related HIV-negative donor could serve as the source of both the HSCT and the adoptively transferred T cells, we also explored the use of unrelated cord blood donors to generate HIV-specific T cells. There are several benefits associated with the use of cord blood for HSCT, including (1) less restrictive human leukocyte antigen (HLA) matching requirements compared to their adult counterparts, reducing the likelihood of graft-versus-host disease (GvHD);28 (2) rapid availability; (3) flexibility for scheduling transplantation; and (4) lower risk of relapse due to graft-versus-leukemia.28
To develop a widely applicable form of HIV immunotherapy, we focused on HLA-A02+ donors, as this allele has one of the highest frequencies across several ethnic groups and is dominant in HIV+ individuals infected with clade B HIV.29 Many immunodominant HIV A02-restricted cytotoxic T lymphocyte (CTL) epitopes have been identified and well characterized in HIV+ populations.30, 31 Here, we describe a novel approach to generating HIV-specific T cells from HLA-A02+ HIV-naive adults (HNA-T) and cord blood (CB-T), which demonstrate cytolytic capacity, suppress active HIV in vitro, and broadly recognize epitopes from HIV antigens Gag, Nef, and Pol.
Results
HNA-Ts and CB-Ts Are Derived from the Naive T Cell Compartment of HIV-Seronegative Adult Donors and Cord Blood
Based on our previous success generating HIV-specific T cells from HIV seronegative adults (HNA-Ts) (Figure S1),32 we sought to determine whether HNA-Ts were generated from the naive T cell compartment of healthy HIV seronegative adults, as seen in CMV.17 Using magnetic selection, naive T cells were selected for CD3+CD45RA+CCR7+CD62L+ T cells (Figure S2). After the naive selection step, T cells derived from both the naive compartment and non-naive compartment were expanded in parallel using antigen-presenting cells (APCs) pulsed with Gag, Nef, and Pol (GNP) (Figure S1).
Naive T cell-derived HLA-A02+ HNA-T products expanded to clinically relevant numbers (median = 100.5e6 cells; range = 23.8e6–195.3e6 cells; Figure 1A). Interferon-gamma (IFNγ) ELISPOT against GNP pepmix confirmed that HIV-specific T cells were only detected in T cell products derived from the naive T cell compartment, because T cell products derived from non-naive T cells elicited insignificant levels of IFNγ in response to HIV antigens (Figure 1A). The naive T cell-derived HNA-Ts showed HIV specificity against Gag (mean = 158.83 IFNγ SFC/1e5 cells; p < 0.0001), Pol (mean = 122.87 IFNγ SFC/1e5 cells; p = 0.016), and GNP pepmix (mean = 225.75 IFNγ SFC/1e5 cells; p < 0.001) compared to irrelevant controls CTL alone (mean = 3.66 IFNγ SFC/1e5 cells) and actin (mean = 5.37 IFNγ SFC/1e5 cells; two-way ANOVA).
Figure 1.

HNA-Ts and CB-Ts Are Derived from the Naive Compartment, Expand to Clinically Relevant Levels, and Demonstrate HIV Specificity
(A) HNA-T generated from the naive T cell compartment (red) produced HIV-specific T cells compared to the non-naive compartment (blue). HNA-Ts displayed significant IFNγ secretion to GNP stimulation (p < 0.0001) compared to negative controls CTL only and actin. (B) CB-Ts produced significant levels of IFNγ in response to GNP stimulation compared to CTL only (p < 0.0001). (C) HPA-Ts were manufactured from HIV+ HLA-A02 individuals for use in comparative studies later on and demonstrated significant IFNγ secretion to GNP stimulation (p < 0.0001).
To further confirm that HIV-specific T cells can be derived from virus-naive donors, we repeated the approach using cord blood.28 As shown in Figure 1B, CB-Ts were successfully expanded from cord blood (median = 56.8e6 cells; range = 25e6–132e6 cells). IFNγ ELISPOT was performed to evaluate for HIV specificity. Increased IFNγ production was observed in response to HIV Gag (mean = 136.05 IFNγ SFC/1e5 cells; p = 0.023), Nef (mean = 196.86 IFNγ SFC/1e5 cells; p = 0.0028) and Pol (mean = 233.59 IFNγ SFC/1e5 cells; p = 0.0006), as well as the GNP pepmix (mean = 272.73 IFNγ SFC/1e5 cells; p < 0.0001) compared to T cells alone (mean = 4.182 IFNγ SFC/1e5 cells; 2-way ANOVA; Figure 1B).
To evaluate whether the expansion and function of virus-naive donor-derived HIV-specific T cell products differed from products derived from HIV+ adults (HPA-Ts), we also generated HPA-Ts from HIV+ HLA-A02+ donors, based on established and US Food and Drug Administration (FDA)-approved (IND17562 and IND15984) protocols (Figure 1C).33, 34 HPA-T products derived from HIV+ individuals expanded to clinically relevant numbers (median = 95.5e6 cells; range = 65.8e6–171.72e6 cells) similar to naive donor-derived HIV-specific T cells. Similarly, increased IFNγ production was observed in response to HIV GNP pepmix stimulation (mean = 529.6 IFNγ SFC/1e5 cells; p < 0.0001) compared to T cells alone (mean = 5.667 IFNγ SFC/1e5 cells; two-way ANOVA).
HIV-Specific T Cell Products Derived from Virus-Naive Donors Have Higher CD4+ T Cell Frequencies Than Products Derived from HIV+ Donors
Phenotyping analysis of HNA-Ts, CB-Ts, and HPA-Ts revealed differences in the composition of the three product types (Figures 2A–2C). Notably, HIV-specific T cells derived from HIV+ donors (HPA-T products) had low frequencies of CD4+ T cells (mean = 7.65%; range = 0.69%–25.4%; Figure 2C) compared to HNA-T (mean = 38.4%; range = 13.4%–73.6%) and CB-T (mean = 23.35%; range = 3.8%–51.5%) products (Figures 2A and 2B). The frequencies of CD8+ T cells in HPA-T (mean = 44.1%; range = 16.9%–69.6%; n = 7), HNA-T (mean = 26.46%; range = 6.4%–59.8%; n = 6), and CB-T (mean = 49.48%; range = 13.0%–73.0%; n = 6) products were similar between products with higher levels of CD3neg/CD56+ natural killer (NK) cells (mean = 17.29%; range = 3.1%–42.5%) observed in HPA-T products (Figures 2A–2C). The memory populations of all three cohorts were skewed toward an effector memory (EM) phenotype, with a smaller population of T cells detected in all three product types that had a central memory (CM) phenotype (HPA-T-EM mean = 85.71%, range = 61.1%–97.8%; HNA-T-EM mean = 58.13%, range = 41.4%–77.8%; CB-T-EM mean = 69.9%, range = 49.5%–89.6%; Figures 2A–2C).
Figure 2.

Phenotyping of HIV-Specific T Cell Products Reveals Differences in Composition and Minimal Expression of Exhaustion Markers
(A and B) HNA-Ts (A) and CB-Ts (B) display a primarily CD3+ phenotype, comprised of substantial CD4+ and CD8+ subsets. (C) HPA-Ts are comprised of primarily CD8+ T cells and NK/NKT cells, with a negligible CD4+ population. All three cohorts display skewed effector memory responses and low expression of exhaustion markers.
HIV-Specific T Cells from All Sources Show Minimal Expression of Exhaustion Markers
Flow analysis revealed similarly low expression levels of markers associated with exhaustion in HPA-T and HNA-T products (PD-1: HNA-T mean = 10.12% versus HPA-T mean = 10.13%; and TIM-3: HNA-T mean = 7.63% versus HPA-T mean = 9.62%; Figures 2A and 2C). Moreover, CB-T products showed even lower levels of exhaustion marker expression (mean: PD-1 = 6.8%; LAG-3 = 3.45%; TIM-3 = 3.05%; KLRG1 = 0.34%; CD57 = 1.08; Figure 2B).
CB-T, HNA-T, and HPA-T Products Exhibit Similar Cytolytic Ability and Polyfunctionality In Vitro
Because exhaustion marker expression is also associated with cell activation at low levels, we next evaluated the functionality of the HIV-specific T cell products to ensure they all elicited antigen-specific cytolytic activity and were polyfunctional. In a chromium-release cytotoxicity assay, CB-Ts were tested for their ability to lyse autologous lymphoblastoid cell line (LCL) or phytohemagglutinin (PHA) blasts pulsed with GNP pepmix (Figure 3A). CB-Ts (n = 2) were able to lyse LCL pulsed with GNP, with an effector-to-target ratio (E:T) of 40:1 producing a mean specific lysis of 25.28% ± 4.22%. CB-T also displayed lysis against unpulsed LCL (40:1 mean 13.63% ± 1.80%). CB-Ts were manufactured using autologous LCL lines as APCs, which generated products with both HIV and EBV specificity. Similarly, HNA-T products (n = 2) showed a mean lysis of 26.53% ± 4.83% at an E:T ratio of 40:1 against autologous PHA blasts pulsed with GNP, and HPA-T products showed a mean lysis of 34.52% ± 3.75% at 40:1 (n = 3).
Figure 3.

HNA-Ts and CB-Ts Demonstrate Cytotoxicity, Suppression of Active HIV Infection In Vitro, and Secretion of Multiple Cytokines
(A) HNA-Ts and CB-Ts display specific lysis of GNP-pulsed autologous PHAb targets (red) at comparable levels to HPA-T products. CB-Ts demonstrate dual EBV specificity by specific lysis of unpulsed autologous LCLs (green). Error bars represent the SD of triplicate values. (B) HNA-Ts (n = 4) and CB-Ts (n = 4) demonstrate suppression of active HIV infection in vitro at E:T of 40:1 and 20:1 at day 7. Error bars represent the SD of triplicate values. (C) HNA-T, CB-T, and HPA-T products secrete IL-2, IL-8, IFNγ, and TNF-α in response to GNP stimulation, demonstrating product polyfunctionality. Error bars represent the SD from the mean.
To evaluate cytolytic activity against virus-infected cells, CB-Ts were tested in a viral inhibition assay to determine whether these products suppress a laboratory strain of HIV (SF162), in an in vitro model of active HIV infection (Figure 3B). CB-Ts were co-cultured at varying E:Ts with autologous CD8-depleted peripheral blood mononuclear cells (PBMCs) that had been infected with SF162. Supernatants were measured for p24 by ELISA as an indicator of HIV presence on day 7. At E:T ratios of 40:1 and 20:1, CB-Ts were able to significantly suppress HIV through day 7 in vitro (p < 0.0001; two-way ANOVA) compared to CD8-depleted HIV-infected cells alone. This was similar to the levels of HIV suppression we found in HNA-T products, as shown in Figure 3B.
Products from all three cohorts were also tested for product polyfunctionality in response to GNP pepmix stimulation (Figure 3C). T cells were stimulated with either actin (negative control) or GNP pepmix overnight, and cell culture supernatants were tested by multiplex for levels of cytokines interleukin-2 (IL-2), IL-8, IFNγ, and tumor necrosis factor alpha (TNF-α). Actin-stimulated T cell cytokine production levels were negligible (data not shown). The production of similar cytokine levels among the three cohorts in response to GNP stimulation suggests these HIV seronegative, naive-derived T cell products have similar polyfunctional capacity as those products generated from HIV+ individuals.
Epitope Mapping of HPA-T, HNA-T, and CB-T Products Reveals Wide Epitope Breadth in HIV-Seronegative Individuals
We previously showed17 that CMVpp65-specific T cells from cord blood and adult CMV seronegative donors did not recognize the expected typical peptides, such as the HLA-A02-restricted peptide NLVPMVATV. To determine whether this result was unique to CMVpp65 and NLV, we compared the peptide repertoires of HIV-specific T cells from HLA-A02 HNA donors, CB-T donors, and HIV+ donors using HIV gag peptide pools.
Products from all three donor cohorts were epitope mapped using overlapping 15-mer peptides (AIDS Reagent Program). Products were mapped using pool matrices with each pool consisting of 8–10 15-mer peptides. Cross-reactive pools containing the same 15-mer peptide were confirmed by individual 15-mer ELISPOTs, as previously described.32 There were common 15mers mapped among HNA-T (Table 1), CB-T (Table 2), and HPA-T (Table 3) products.
Table 1.
HIV Naive Adult-Derived A02+ HNA-T Epitope Mapping
| Product Name |
HLA Type |
Epitope Mapping |
|||
|---|---|---|---|---|---|
| HNA-T | HLA Class I | HLA Class II | NEF | GAG | POL |
| HNA-T #1 | A02, 68; B07, 35; C04, 07 | DRB1 01, 15; DRB5 01; DQA1 01, 01; DQB1 05, 05; DPB1 04, 04 | WPAVRERIRRTHPAA | – | – |
| RERIRRTHPAAEGVG | |||||
| GFPVRPQVPLRPMTY | |||||
| RPQVPLRPMTYKAAL | |||||
| HNA-T #2 | A02, 03; B07, 50; C06, 07 | DRB1 07, 15; DRB4 01; DRB5 01; DQA1 01, 02; DQB1 02, 06; DPB1 03, 04 | – | IYKRWIILGLNKIVR | – |
| WIILGLNKIVRMYSP | |||||
| HNA-T #3 | A02 known | – | – | HQAISPRTLNAWVKV | – |
| HNA-T #4 | A02, 32; B40, 57; C03, 06 | DRB1 12, 13; DRB3 02, 03; DQA1 01, 05; DQB1 07, 06; DPB1 03, 04 | – | SGGKLDAWEKIRLRP LDAWEKIRLRPGGKK | – |
| IYKRWIILGLNKIVR | |||||
| WIILGLNKIVRMYSP | |||||
| HNA-T #5 | A02, 24; B35, 51; C04, 14 | DRB1 04, 11; DRB3 02; DRB4 01; DQA1 03, 05; DQB1 03, 03; DPB1 01, 14 | – | YRLKHLVWASRELER HLVWASRELERFALN | MGYELHPDKWTVQPI |
| LHPDKWTVQPIVLPE | |||||
| VNDIQKLVGKLNWAS | |||||
| HNA-T #6 | A02, 68; B08, 35; C04, 07 | DRB1 03, 11; DRB3 01, 02; DQA1 05, 05; DQB1 02, 03; DPB1 02, 03 | EVLMWKFDSRLALRH | PVGEIYKRWIILGLN | VGPTPVNIIGRNLLT |
| IYKRWIILGLNKIVR | AYFLLKLAGRWPVKT | ||||
| RDYVDRFFKTLRAEQ | LKLAGRWPVKTIHTD | ||||
| DRFFKTLRAEQATQ | |||||
| HNA-T #7 | A02, 01; B08, 51; C07, 15 | DRB1 04, 11; DRB3 02; DRB4 01; DQA1 03, 05; DQB1 03, 03; DPB1 02, 03 | – | GLNKIVRMYSPVSIL | THLEGKIILVAVHVA |
| GKIILVAVHVASGYI | |||||
| HNA-T #8 | A02, 24; B39, 51; C01, 07 | DRB1 08, 11; DRB3 02; DQA1 04, 05; DQB1 03, 04; DPB1 02, 03 | – | – | MASDFNLPPVVAKEI |
HNA-Ts were mapped with overlapping 15 mers based on HIV-1 consensus sequences of Gag, Nef, and Pol. Known HLA-A02 epitopes (based on LAND) are shown in bold, and confirmed 9-mer epitopes are italic, with unknown HLA restrictions.
Table 2.
HIV Naive Cord-Derived A02+ CB-T Epitope Mapping
| Product Name |
HLA Type |
Epitope Mapping |
|||
|---|---|---|---|---|---|
| CB-T | HLA Class I | HLA Class II | NEF | GAG | POL |
| CB-T #1 | A02, 29; B18, 51; C05, 16 | DRB1 03, 11; DRB3 02, 02; DQA1 05, 05; DQB1 02, 07; DPB1 02, 03 | YPLTFGWCFKLVPV | – | – |
| FKLVPVDPEEVEEAN | |||||
| CB-T #2 | A02, 24; B35, 35; C01, 04 | DRB1 04, 11; DRB3 02; DRB4 01; DQA1 03, 05; DQB1 03, 03; DPB1 04, 17 | EEEVGFPVRPQVPLR | HLVWASRELERFALN | KMIGGIGGFIKVRQY |
| GFPVRPQVPLRPMTY | IVRMYSPVSILDIRQ | VNDIQKLVGKLNWAS | |||
| HTQGYFPDWQNYTPG | RQGPKEPFRDYVDRF | KLPIQKETWEAWWTE | |||
| YFPDWQNYTPGPGIR | class I definitely | WQATWIPEWEFVNTP | |||
| class I definitely | WIPEWEFVNTPPLVK | ||||
| CB-T #3 | A02, 01; B27, 49; C01, 07 | DRB1 01, 01; DQA1 01, 01; DQB1 05, 05; DPB1 04, 04 | – | HQAISPRTLNAWVKV | KMIGGIGGFIKVRQY |
| QKETWEAWWTEYWQA | |||||
| CB-T #4 | A02, 30; B15, 53; C02, 04 | DRB1 01, 11; DRB3 02; DQA1 01, 01; DQB1 05, 06; DPB1 01, 02 | DEEREVLMWKFDSRL | RDYVDRFFKTLRAEQ | AYFLLKLAGRWPVKT |
| DRFFKTLRAEQATQ | LKLAGRWPVKTIHTD | ||||
| CB-T #5 | A02, 68; B35, 51; C04, 08 | DRB1 14, 14; DRB3 01, 01; DQA1 05, 05; DQB1 03, 03; DPB1 04, 04 | – | RDYVDRFFKTLRAEQ | VGPTPVNIIGRNLLT |
| KVYLAWVPAHKGIGG | |||||
| CB-T #6 | A02, 33; B35, 51; C04, 16 | DRB1 13, 15; DRB3 03; DQA1 01, 01; DQB1 06, 06; DPB1 01, 131 | – | – | KMIGGIGGFIKVRQY |
| CB-T #7 | A02, 68; B15, 39; C07, 07 | DRB1 04, 07; DRB4 01, 01; DQA1 02, 03; DQB1 02, 03; DPB1 04, 14 | – | Nef-specific: individual peptides not determined | Pol-specific: individual peptides not determined |
| CB-T #8 | A02, 24; B13, 42; C06, 17 | DRB1 10, 11; DRB3 02; DQA1 01, 01; DQB1 05, 06; DPB1 02, 04 | RPQVPLRPMTYKAAL | HQAISPRTLNAWVKV | YNVLPQGWKGSPAIF |
| PLRPMTYKAALDLSH | NTPPLVKLWYQLEKE | ||||
| LWVYHTQGYFPDWQN | EQVDKLVSAGIRKVL | ||||
| WQNYTPGPGIRYPLT | KLVSAGIRKVLFLDG | ||||
| TPGPGIRYPLTFGW | |||||
CB-Ts were mapped with overlapping 15 mers based on HIV-1 consensus sequences of Gag, Nef, and Pol. Known HLA-A02 epitopes (based on LAND) are shown in bold, and confirmed 9-mer epitopes are italic, with unknown HLA restrictions.
Table 3.
HIV Positive-Derived A02+ HPA-T Epitope Mapping
| Product Name |
HLA Type |
Epitope Mapping |
|||
|---|---|---|---|---|---|
| HPA-T | HLA Class I | HLA Class II | NEF | GAG | POL |
| HPA-T #1 | A02, 03; B07, 40; C03, 07 | DRB1 04, 15; DQB1 03, 06; DRB4 01; DRB5 01; DQA 01, 03 | EEEVGFPVRPQVPLR | RQANFLGKIWPSNKG | PQGWKGSPAIFQSSM |
| GFPVRPQVPLRPMTY | FLGKIWPSNKGRPGN | KGSPAIFQSSMTKIL | |||
| RPQVPLRPMTYKAAL | AIFQSSMTKILEPFR | ||||
| DLSHFLKEKGGLEGL | |||||
| FLKEKGGLEGLIYSK | |||||
| HPA-T #2 | A02, 03; B07, 52; C07, 12 | DRB1 15, 15; DRB5 01, 01; DQA1 01, 01; DQB1 06, 06; DPB1 04, 04 | RPQVPLRPMTYKAAL | VGGPGHKARVLAEAM | KGSPAIFQSSMTKIL |
| TPGPGIRYPLTFGW | SLYNTVATL | AIFQSSMTKILEPFR | |||
| HPA-T #3 | A02, 02; B35, 44; C04, 05 | DRB1 01,04; DRB4 01; DQA1 01, 03; DQB1 05, 03; DPB1 04, 04 | PGIRYPLTFGWCFKL | – | – |
| YPLTFGWCFKLVPV | |||||
| HPA-T #4 | A02, 26; B15, 44; C02, 03 | DRB1 04,11; DRB3 02; DRB4 01; DQA1 03, 05; DQB1 03, 03; DPB1 02, 04 | DLSHFLKEKGGLEGL | ASVLSGGKLDAWEKI | ANRETKLGKAGYVTD |
| FLKEKGGLEGLIYSK | EKIRLRPGGKKKYRL | THLEGKIILVAVHVA | |||
| FSALSEGATPQDLNT | GKIILVAVHVASGYI | ||||
| WIILGLNKIVRMYSP | |||||
| GLNKIVRMYSPVSIL | |||||
| HPA-T #5 | A02, 11; B15, 15; C08, 08 | unknown | RPQVPLRPMTYKAAL | GLNKIVRMYSPVSIL | KKKSVTVLDVGDAYF |
| DRFFKTLRAEQATQ | VTVLDVGDAYFSVPL | ||||
| HPA-T #6 | A02, 32; B15, 40; C02, 03; | DRB1 01, 14; DQB1 05, 05; DRB3 02; DQA 01 | – | LDAWEKIRLRPGGKK | QKQITKIQNFRVYYR |
| EKIRLRPGGKKKYRL | TKIQNFRVYYRDSRD | ||||
| YRLKHLVWASRELER | DDCVASRQDED | ||||
| WIILGLNKIVRMYSP | |||||
| GLNKIVRMYSPVSIL | |||||
| VSILDIRQGPKEPFR | |||||
HPA-Ts were mapped with overlapping 15 mers based on HIV-1 consensus sequences of Gag, Nef, and Pol. HPA-T no. 2 (OM9) is the de-identified HIV+ donor from which SL9-specific T cells are derived from in Figures 4C and 4D. Known HLA-A02 epitopes (based on LAND) are shown in bold with unknown HLA restrictions.
Based on the Los Alamos National Laboratory (LANL) database of known HIV epitopes35 and lists of HLA-associated selection on the HIV proteome,36 the majority of HNA-T and CB-T products recognized known epitopes in HIV+ populations. However, the commonly recognized A02-restricted epitope SLYNTVATL (SL9) was not recognized by any HIV-negative naive-derived products using this screening approach.
HLA A2-Restricted HIV-Epitope-Specific T Cells Derived from Virus-Naive Donors Do Not Recognize the Typical HLA-A02 Gag Epitope SL9 but Have High Functional Avidity
Knowing that SL9-specific T cells were not detected in 21/21 (10 HIV-negative adults and 11 cord donors) HLA-A02+ virus-naive donor-derived HIV-specific T cell products, and given that an estimated 75% of HIV-1-infected HLA-A02+ individuals recognize the Gag-SL9 epitope,37 we next asked whether we could force the expansion of SL9-specific T cells from these naive donors. For this, we stimulated HLA-A02+ HIV-seronegative donor-derived PBMCs with APCs pulsed with the SL9 9 mer alone versus the GNP pepmix (Figure 4). As shown in Figure 4A, it was not possible to force Gag-SL9 specificity in any of the HIV-seronegative donor-derived products (n = 3; Figure 4A). However, from the same donors, we produced HNA-T and CB-T products specific for other Gag/Nef/Pol epitopes (Figure 4B). In contrast, and as expected, we could successfully expand Gag-SL9-specific T cells from an HIV+ donor using the same methodology (Figure 4C).
Figure 4.

HLA-A02-Restricted HIV-Specific T Cells Derived from Virus-Naive Donors Do Not Recognize Gag SL9, Despite Forced Expression Manufacturing Methods
(A) No SL9-specific HNA-T or CB-T products were generated using forced expression methods with SL9 9-mer peptide. (B) In comparison, GNP-specific HNA-Ts and CB-Ts were generated from the same donors as a control. (C) SL9-specific T cells were produced using our manufacturing methods from an HIV-positive donor (OM9), determined by IFNγ ELISPOT. (D) SL9-specific T cells show low functional avidity on IFNγ ELISPOT, stimulating with progressive dilutions of SL9 9-mer peptide. In this figure, error bars represent the SD.
Because HIV-specific T cells derived from HLA A2+ naive donors did not recognize the expected SL9 Gag epitope and instead recognized other epitopes spanning HIV Gag, Nef, and Pol, we considered whether the avidities of these T cell receptors for their respective peptide/MHC complexes might differ depending on the donor source. The functional avidity was determined by the EC50, or the concentration of peptide used to still provide one-half the maximum magnitude of IFNγ response. Functional avidity for HPA donor OM9 Gag SL9-specific T cells revealed a low functional avidity (EC50 = 3.72 ng/mL; Figure 4D).
To compare this functional avidity to the HIV-negative products, we used limiting dilutions of peptides to test the functional avidity of two HIV epitopes (one Gag and one Pol epitope) recognized by both HNA-T and CB-T products. As shown in Figure S3A, for T cells recognizing the Gag epitope HLVWASREL, the mean peptide concentration needed to induce a half-maximum IFNγ response (EC50) was 0.1 ng/mL in the CB-T-derived donor product compared to an EC50 = 0.009 ng/mL observed with the seronegative adult donor product. Additionally, naive donor-derived T cells that recognized the Pol epitope KLVGKLNWA showed similar avidity irrespective of the donor source: 0.01 ng/mL (CB-T donor) versus 0.03 ng/mL (adult seronegative donor; Figure S3B), suggesting that HIV-naive donor-derived HNA-Ts and CB-Ts may have higher functional avidity for their cognate epitope compared to HPA-T products.
Discussion
This is the first description of a GMP-compliant, reproducible platform for generating HIV-specific T cells from HIV-seronegative, naive T cells derived from adult or cord blood donors. These HNA-Ts and CB-Ts demonstrated HIV specificity against epitopes spanning the breadth of Gag, Nef, and Pol. Assessing functionality of these products, HNA-Ts and CB-Ts suppressed active HIV infection in vitro and lysed autologous LCL and PHA blast targets pulsed with HIV pepmix. We also observed a cytokine response dominated by production of IFNγ and TNF-α in response to HIV pepmix stimulation, suggestive of a TH1 skewed response associated with intracellular pathogens, such as HIV. IL-8 was also detected, normally involved in innate immune responses, such as neutrophil recruitment, and is not unexpected, based on the diverse phenotypes of these HNA-T and CB-T products.
Hanley et al.25, 26 previously demonstrated that CMV-specific T cells can be generated from cord blood, an obligatory source of naive T cells. It was shown that HLA-A2+ CMV-specific T cells derived from naive cord blood recognized atypical epitopes LQT and MLN, whereas CMV-specific T cells generated from CMV+ donors recognize the typical NLV epitope. That study demonstrated in vivo that CMV-specific T cells recognizing atypical epitopes were protective, with the presence of these T cells correlating with an absence of CMV reactivation. Extending this model to HIV, we compared the epitope recognition breadths of HNA-Ts, CB-Ts, and HPA-Ts together with known HIV+ epitopes from the Los Alamos National Database (LAND).
Interestingly, we were unable to generate Gag-SL9-specific T cells from HLA-A2+ HIV-seronegative donors, suggesting that naive donor-derived T cells recognize a different epitope repertoire from HIV+ donors. This finding is similar to that of a study of 13 HIV-uninfected individuals where a Gag vaccine did not produce Gag-SL9 responses37 and even forced expression failed to produce Gag-SL9-specific T cells from healthy adult or cord donors. Nevertheless, our approach was able to produce SL9-specific T cells from the HIV+ individuals. Other investigators have also succeeded in generating SL9-specific T cells from healthy cord blood, suggesting the type of APC used for manufacturing may play a role in determining T cell specificity.38 Gag-SL9-specific T cells are commonly identified in chronically infected HIV+ individuals, but not during acute infection, demonstrating the HIV epitope repertoire and resulting T cell responses change over the course of HIV infection.39, 40, 41 In chronic HIV infection, HIV+ individuals may be unable to control viral load due to the accumulation of escape mutations in targeted epitopes,11, 12 T cell exhaustion,42, 43, 44 or the presence of CTLs that recognize immunodominant epitopes, such as Gag SL9 (SLYNTVATL), a response negatively associated with viral load in HIV progression but limited to chronic infection.41, 45, 46, 47 This has significant implications for developing non-exhausted T cell products that target epitopes associated with multiple stages of HIV infection and progression. Importantly, HNA-Ts and CB-Ts display a wide breadth of specificity across Gag, Nef, and Pol antigens, offering extensive coverage, and may be critical for preventing immune escape.
Despite this advantage, one concern with the infusion of HNA-T and CB-T products is the presence of a notable CD4+ population with the potential to be infected by the recipient’s virus. To address this concern, we are currently investigating two strategies to render HIV-specific T cells resistant to HIV infection: gene modification and selection of donors naturally resistant to HIV. Gene modification approaches have shown promise in the HIV field. Several groups have shown chimeric antigen receptors (CARs) can target conserved HIV epitopes.48 Other groups have employed CRISPR technology and zinc finger nucleases (ZFNs) to target HIV co-receptor CCR5 through disruption of the host genome to prevent viral entry.49, 50 Applying these gene modification strategies to our HIV-specific T cells could produce a potent cell product with the desired bi-functionality—cytotoxicity and resistance to infection.
We are also exploring generating HIV-specific T cells from HIV-negative homozygous Delta32 CCR5 donors, who possess natural resistance to R5-tropic HIV strains.51 The National Marrow Donor Program (NMDP) and the German Cord Blood Bank (DKMS) have typed homozygous Delta32 CCR5 cord units, providing a platform for rapid availability and clinical translation.52
Lastly, it is important to note that demonstrating the persistence of these HIV-specific T cells will be critical to produce durable, long-term anti-HIV immunity, especially if ART interruption is considered. The use of other virus-specific T cells against EBV, CMV, and adenovirus in a post-transplant setting has demonstrated durability during this period of immune suppression, with few patients experiencing recurring viral infection or progression.53 Translating this to the HIV setting, it is critical that HIV-specific T cells persist to produce a durable long-term anti-HIV response, particularly during immune reconstitution post-HSCT, where low levels of residual reservoir virus may rebound if left unchecked.
Ultimately, we envision multiple platforms for using HIV-specific T cells in the allogeneic setting. First, HIV+ individuals with hematologic malignancies who receive an allogeneic-HSCT could receive HIV-specific T cells from the same stem cell transplant donor to help control viral rebound during immune reconstitution. Second, HIV-specific T cells derived from HIV-seronegative sources could be used in combination with latency-reversing agents to “shock and kill” infected cells and reactivate latent infection for subsequent targeting by T cells.54, 55, 56 The broad epitope recognition of HNA-Ts and CB-Ts may be critical in this setting, as they can recognize multiple HIV epitopes, reducing the chance of immune escape. These potential applications for HIV-specific T cells from HIV-seronegative donors set the stage for future cell therapy trials to validate the efficacy of adoptive T cell therapy in HIV. Currently, we are also exploring safety and efficacy of autologous HIV-specific T cells generated using the same approach in HIV+ individuals (NCT02208167 and NCT03212989).
In summary, we show that HIV-specific T cells can be generated from the naive T cell compartment of HIV-seronegative adults and cord blood. These products have wide epitope recognition, suppress HIV in vitro, and demonstrate cytolytic abilities. This has important implications for HIV+ individuals undergoing stem cell transplantation for malignant disease where the HSCT donor can serve both as a source of hematopoietic stem cells and for the generation of HIV-specific T cells.
Materials and Methods
Isolation of PBMCs
PBMCs were isolated from HIV-negative and HIV+ donors on ART with acute or chronic HIV infection (University of North Carolina, Chapel Hill, NC; University of Texas MD Anderson Cancer Center, Houston, TX; and The George Washington University, Washington, DC). All donations were obtained under informed consent approved at each institution. PBMCs were diluted 1:4 to 1:2 (blood: 1× PBS) and layered on top of 10–15 mL of Lymphocyte Separation Medium (MP Biomedicals, CA). Blood was spun for 30 min at 600 rpm at room temperature (RT). PBMCs were harvested from the lymphocyte layer and washed three times with 1× PBS prior to counting.
Generation of Dendritic Cells
PBMCs were plated for 2-hr adherence at 37°C, after which non-adherent cells were washed off and frozen. Dendritic cells (DCs) isolated from plastic adherence of PBMCs were fed with IL-4 (1,000 U/mL) and granulocyte macrophage colony-stimulating factor (GM-CSF) (800 U/mL) for 6 days, added on day 1 and day 4. DCs were matured on day 7 with IL-4 (1,000 U/mL), GM-CSF (800 U/mL), IL-6 (100 ng/mL), TNF-α (10 ng/mL), IL-1b (10 ng/mL; all R&D Systems, MN), and Prostaglandin E1 (1 mg/mL; Sigma-Aldrich, MO). DCs were harvested 24–48 hr after maturation for stimulation 1.
Generation of PHAbs for HNA-T Manufacturing
To generate PHA blasts (PHAbs), PBMCs were stimulated with PHA-P (5 mg/mL; Sigma-Aldrich, MO) in the presence of IL-2 on day 1. PHAbs were fed with IL-2 on day 3 and every 2 or 3 days thereafter with IL-2. PHAbs were used as APCs in stimulations (stims) 2 and 3 for the HNA-T and HPA-T manufacturing protocols.
Generation of HIV-Specific T Cells from the HNA-Ts Compartment
A magnetic-activated cell sorting (MACS) column (Miltenyi Biotech, Germany) was used to positively select naive T cells from adult HIV-negative PBMCs selecting for CD3+CD45RA+CCR7+CD62L+. The negatively selected fraction was flushed through the magnetic column, and both the naive and non-naive T cell compartments were stimulated with antigen-coated DCs in stim 1 and subsequently expanded according to our established protocol.32, 33 Irradiated PHAbs were used as APCs in stims 2 and 3. APCs were pulsed with HIV Gag, Nef, and Pol pepmixes. These were chosen as they are more conserved compared to Env and allow targeting of multiple stages of HIV infection, as Nef is expressed early whereas Gag and Pol are expressed later in infection.57 These overlapping HIV peptide libraries consisted of 15 mers, overlapping by 11 amino acids (JPT, Germany), based on consensus sequences of HIV-1. For stim 3, modified K562 cells were added, expressing co-stimulatory molecules 41-BBL, CD80, CD83, and CD86 (gift of Dr. Clio Rooney, Baylor, TX) to aid in expansion. The ratios of cells cultured were as follows: stim 1 (1:10; T:DC); stim 2 (1:4; T:PHAb); and stim 3 (1:1:4; T:PHAb:K562).
Generation of Autologous LCLs for CB-T Manufacturing
Autologous LCL lines were generated from cord blood mononuclear cells (CBMCs) to serve as APCs for the generation of CB-Ts. 5e6 PBMCs were pelleted and resuspended in 200 μL of live B95-8 EBV (produced by infected marmoset cells). This B95-8-CBMC mixture was then resuspended in 2 mL of cRPMI containing cyclosporine A (1 μg/mL). On a 96-well plate, 5 wells were plated at 200 μL each of the viral cell suspension. The remaining 1 mL was diluted to 2 mL with cRPMI containing cyclosporine and plated in 10 wells of 200 μL each. The cells were monitored weekly, and as the wells became confluent, cells were expanded into 24-well plates, T25 flasks, and ultimately T75 flasks. The generation of autologous LCL lines required from 1 to 2 months.
Generation of CB-Ts
The protocol for the generation of CB-Ts is similar to that of the HNA-Ts,32, 33 with several key differences: (1) K562s were not used to expand CB-Ts in the 3rd stimulation; (2) at stim 1, IL-7, IL-12, and either IL-15 or IL-21 was used to assist in proliferation and expansion of T cells; and (3) autologous LCLs replaced PHA blasts as the APCs for the second and third stimulation at a LCL:T cells ratio of 1:1 (Figure S1).
IFNγ ELISPOT Assay and Epitope Mapping
No peptide or the irrelevant peptide, actin (JPT, Germany), was used as negative control. Staphylococcus enterotoxin B (SEB) was used as a positive control (Sigma-Aldrich, MO). T cells were plated at 1e5/well on IFNγ-coated ELISPOT plates (Millipore, NJ). Positive responses are defined as having more than double the spot-forming cells (SFCs) obtained in the negative controls, with a minimum of 50 IFNγ SFC/1e5 cells/well. For epitope mapping, the 15-mer peptides overlapping by 11 amino acids spanning the consensus region of the Gag, Nef, and Pol antigens were pooled and used according to previously published matrices.32 Using the matrices, cross-reactive pools were analyzed for common 15-mer epitopes, and these 15-mer epitopes were then individually tested on ELISPOT to confirm epitope specificity.
HLA Epitope Specificity: IFNγ ELISPOT
Based on epitope-mapping ELISPOTs, expanded HIV-specific T cells were tested for HLA specificity to Gag, Nef, Pol, or individual peptides. For HLA blocking, 1 × 105 cells/well were treated with monoclonal mouse anti-human HLA class I or HLA class II antibody (Dako, Agilent, CA) in a 96-well round-bottom plate for 1 hr at 37°C. Treated cells were transferred to ELISPOT plate, stimulated with peptide, and developed as previously described.
Flow Cytometry Phenotyping and Exhaustion Panels
Flow for phenotyping and exhaustion panels were run on the MACSQUANT Analyzer (Miltenyi Biotech, Germany) with analysis done with FlowJo software (FlowJo, OR). For the phenotyping, the following antibodies were used: anti-CD3; CD4; CD8; CD45RA; CD45RO; CD56; CD16; and CD62L (Miltenyi Biotech, Germany). For exhaustion phenotyping, the following antibodies were used: anti-CD3; PD-1; LAG-3; TIM-3; KLRG1; and CD57. For isotype controls, the recommended isotype for each of the previous antibodies was purchased and used. 1e6 cells were stained per condition, incubated for 30 min at 4°C, washed twice with fluorescence-activated cell sorting (FACS) buffer (2%–5% FBS/1× PBS), and run on the MACSQUANT.
Multiplex Assay
To assess polyfunctionality of T cell products, a multiplex assay was run using the Bio-plex Pro Human 17-plex Cytokine Assay kit (Bio-Rad, CA). HPA-T, HNA-T, and CB-T products were thawed overnight with IL-2 (50 U/mL) on day 1. On day 2, T cells were washed and plated at 1e6 cells/well with 1 μl of corresponding pepmix: actin; GNP; or SEB. On day 3, supernatants were harvested from the wells and plated on the multiplex plate. The multiplex protocol provided by Bio-Rad was followed for the 17-plex kit, and the plate was analyzed for concentrations of cytokines, based on the standard curves produced.
Viral Inhibition Assay
CD8-depleted PBMCs were activated in IL-2 (50 U/mL) and PHA (2 μg/mL) before being infected with HIV laboratory strain SF162. Infected target cells were co-cultured for 5 days with expanded HIV-specific T cells or unexpanded CD8 T cells that were isolated using magnetic beads (Miltenyi Biotech, Germany) added at 1:2 E:T and 20:1 E:T. The following conditions were used as controls: uninfected CD8-depleted PBMCs; infected CD8-depleted PBMCs alone; antiretrovirals (ARVs); ARVs + expanded HIV-specific T cells; and expanded CMV- and EBV-specific T cells. Day 5 supernatants were measured for HIV-1 gag p24 concentration by ELISA (ABL, Rockville, MD). The ARVs used in combination were indinavir and raltegravir (Selleck Chemicals, TX).
Cytotoxicity Assay
The cytolytic activity of HPA-T products was determined with a chromium-51-release assay. Autologous PHAb targets were pulsed with nothing (negative control) or Gag/Nef/Pol pepmix and incubated with chromium 51 for 1 hr. Targets were then washed 3 times and co-cultured with autologous HPA-Ts at E:Ts of 40:1, 20:1, 10:1, and 5:1. Targets alone were plated as a spontaneous release control. Targets mixed with 1% Triton X (Sigma-Aldrich, MO) were plated as a maximum release control. Targets were co-cultured with effectors for 4 or 5 hr at 37°C. Plates were spun, and supernatant was collected onto a Luma plate (PerkinElmer, MA). Plate was left overnight, and Cr51 release was measured the next morning in a MicroBeta2 counter. Specific lysis % was measured as (experimental release − spontaneous release)/(maximum release − spontaneous release) × 100.
Functional Avidity
Functional avidity of HPA-T, HNA-T, and CB-T products were tested using limiting dilutions of individual peptides (NIH AIDS Reagent Program and GenScript, NJ). Once our T cell products were confirmed as specific against individual 15-mer peptides (AIDS Reagent Program), the individual 9-/10-mer peptides within the 15-mer sequence were ordered (GenScript, NJ) to determine the exact epitope responsible. T cells were plated on IFNγ ELISPOT at 1e5 cells/well with limiting dilutions of peptide to determine the EC50. ELISPOTs were sent out for analysis for unbiased spot counting (Zellnet, NJ).
HLA Typing
Samples were sent for high-resolution HLA typing (Kashi Clinical Laboratories, OR).
Statistical Analysis
Two-way ANOVA with Holm-Sidak correction was used to determine statistical significance of IFNγ release on ELISPOT in response to HIV antigens compared to the negative control as well as significance of HIV suppression in viral inhibition assay. Means, medians, and ranges were provided where applicable.
Author Contributions
S.P. contributed to the writing of this manuscript and formal data analysis. S.P., E.C., and S.A. conducted the experiments. S.P., C.R.C., R.B.J., and C.M.B. contributed to the protocol development and experimental plans. C.R.C., R.B.J., E.J.S., D.M.M., R.F.A., and C.M.B. provided expertise on experimental approaches and edited the manuscript. C.M.B. supervised the experiments, provided funding acquisition, and contributed to the writing and editing of the manuscript.
Conflicts of Interest
The authors have no conflicts of interest.
Acknowledgments
This work was supported by grants from the BELIEVE Martin Delaney Collaboratory (NIAID award UM1AI26617; co-funded by NIDA, NIMH, and NINDS), P01 CA148600, and R01 HL132791-0. The following materials were supplied by the NIH AIDS Reagent Program: Gag; Nef; and Pol 15-mer peptide sets and HIV SF162 virus. We would also like to thank Zabrina Brumme from Simon Fraser University for her HIV epitope database analysis and expertise.
Footnotes
Supplemental Information includes three figures and can be found with this article online at https://doi.org/10.1016/j.ymthe.2018.04.009.
Supplemental Information
References
- 1.Hütter G., Ganepola S. Eradication of HIV by transplantation of CCR5-deficient hematopoietic stem cells. Sci. World J. 2011;11:1068–1076. doi: 10.1100/tsw.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hütter G., Nowak D., Mossner M., Ganepola S., Müssig A., Allers K., Schneider T., Hofmann J., Kücherer C., Blau O. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 3.Henrich T.J., Hanhauser E., Marty F.M., Sirignano M.N., Keating S., Lee T.H., Robles Y.P., Davis B.T., Li J.Z., Heisey A. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: report of 2 cases. Ann. Intern. Med. 2014;161:319–327. doi: 10.7326/M14-1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henrich T.J., Hu Z., Li J.Z., Sciaranghella G., Busch M.P., Keating S.M., Gallien S., Lin N.H., Giguel F.F., Lavoie L. Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J. Infect. Dis. 2013;207:1694–1702. doi: 10.1093/infdis/jit086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borrow P., Lewicki H., Hahn B.H., Shaw G.M., Oldstone M.B. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koup R.A., Safrit J.T., Cao Y., Andrews C.A., McLeod G., Borkowsky W., Farthing C., Ho D.D. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ndhlovu Z.M., Kamya P., Mewalal N., Kløverpris H.N., Nkosi T., Pretorius K., Laher F., Ogunshola F., Chopera D., Shekhar K. Magnitude and kinetics of CD8+ T cell activation during hyperacute HIV infection impact viral set point. Immunity. 2015;43:591–604. doi: 10.1016/j.immuni.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmitz J.E., Kuroda M.J., Santra S., Sasseville V.G., Simon M.A., Lifton M.A., Racz P., Tenner-Racz K., Dalesandro M., Scallon B.J. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 9.Fellay J., Shianna K.V., Ge D., Colombo S., Ledergerber B., Weale M., Zhang K., Gumbs C., Castagna A., Cossarizza A. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pereyra F., Jia X., McLaren P.J., Telenti A., de Bakker P.I., Walker B.D., Ripke S., Brumme C.J., Pulit S.L., Carrington M., International HIV Controllers Study The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557. doi: 10.1126/science.1195271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goonetilleke N., Liu M.K., Salazar-Gonzalez J.F., Ferrari G., Giorgi E., Ganusov V.V., Keele B.F., Learn G.H., Turnbull E.L., Salazar M.G., CHAVI Clinical Core B The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 2009;206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu M.K., Hawkins N., Ritchie A.J., Ganusov V.V., Whale V., Brackenridge S., Li H., Pavlicek J.W., Cai F., Rose-Abrahams M., CHAVI Core B Vertical T cell immunodominance and epitope entropy determine HIV-1 escape. J. Clin. Invest. 2013;123:380–393. doi: 10.1172/JCI65330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang O.O., Kalams S.A., Rosenzweig M., Trocha A., Jones N., Koziel M., Walker B.D., Johnson R.P. Efficient lysis of human immunodeficiency virus type 1-infected cells by cytotoxic T lymphocytes. J. Virol. 1996;70:5799–5806. doi: 10.1128/jvi.70.9.5799-5806.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leen A.M., Myers G.D., Sili U., Huls M.H., Weiss H., Leung K.S., Carrum G., Krance R.A., Chang C.C., Molldrem J.J. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat. Med. 2006;12:1160–1166. doi: 10.1038/nm1475. [DOI] [PubMed] [Google Scholar]
- 15.Gerdemann U., Katari U.L., Papadopoulou A., Keirnan J.M., Craddock J.A., Liu H., Martinez C.A., Kennedy-Nasser A., Leung K.S., Gottschalk S.M. Safety and clinical efficacy of rapidly-generated trivirus-directed T cells as treatment for adenovirus, EBV, and CMV infections after allogeneic hematopoietic stem cell transplant. Mol. Ther. 2013;21:2113–2121. doi: 10.1038/mt.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gerdemann, U., Vera, J.F., Rooney, C.M., and Leen, A.M. (2011). Generation of multivirus-specific T cells to prevent/treat viral infections after allogeneic hematopoietic stem cell transplant. J. Vis. Exp. (51), 2736. [DOI] [PMC free article] [PubMed]
- 17.Hanley P.J., Melenhorst J.J., Nikiforow S., Scheinberg P., Blaney J.W., Demmler-Harrison G., Cruz C.R., Lam S., Krance R.A., Leung K.S. CMV-specific T cells generated from naïve T cells recognize atypical epitopes and may be protective in vivo. Sci. Transl. Med. 2015;7:285ra63. doi: 10.1126/scitranslmed.aaa2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garcia J.V., Miller A.D. Downregulation of cell surface CD4 by nef. Res. Virol. 1992;143:52–55. doi: 10.1016/s0923-2516(06)80080-4. [DOI] [PubMed] [Google Scholar]
- 19.Koenig S., Conley A.J., Brewah Y.A., Jones G.M., Leath S., Boots L.J., Davey V., Pantaleo G., Demarest J.F., Carter C. Transfer of HIV-1-specific cytotoxic T lymphocytes to an AIDS patient leads to selection for mutant HIV variants and subsequent disease progression. Nat. Med. 1995;1:330–336. doi: 10.1038/nm0495-330. [DOI] [PubMed] [Google Scholar]
- 20.Kamphorst A.O., Ahmed R. CD4 T-cell immunotherapy for chronic viral infections and cancer. Immunotherapy. 2013;5:975–987. doi: 10.2217/imt.13.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rerks-Ngarm S., Pitisuttithum P., Nitayaphan S., Kaewkungwal J., Chiu J., Paris R., Premsri N., Namwat C., de Souza M., Adams E., MOPH-TAVEG Investigators Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 22.Day T.A., Kublin J.G. Lessons learned from HIV vaccine clinical efficacy trials. Curr. HIV Res. 2013;11:441–449. doi: 10.2174/1570162x113116660051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim J.H., Excler J.L., Michael N.L. Lessons from the RV144 Thai phase III HIV-1 vaccine trial and the search for correlates of protection. Annu. Rev. Med. 2015;66:423–437. doi: 10.1146/annurev-med-052912-123749. [DOI] [PubMed] [Google Scholar]
- 24.Berglund S., Magalhaes I., Gaballa A., Vanherberghen B., Uhlin M. Advances in umbilical cord blood cell therapy: the present and the future. Expert Opin. Biol. Ther. 2017;17:691–699. doi: 10.1080/14712598.2017.1316713. [DOI] [PubMed] [Google Scholar]
- 25.Hanley P.J., Bollard C.M., Brunstein C.G. Adoptive immunotherapy with the use of regulatory T cells and virus-specific T cells derived from cord blood. Cytotherapy. 2015;17:749–755. doi: 10.1016/j.jcyt.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanley P.J., Cruz C.R., Savoldo B., Leen A.M., Stanojevic M., Khalil M., Decker W., Molldrem J.J., Liu H., Gee A.P. Functionally active virus-specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T-cell populations in cord blood and will target a range of viral epitopes. Blood. 2009;114:1958–1967. doi: 10.1182/blood-2009-03-213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Micklethwaite K.P., Savoldo B., Hanley P.J., Leen A.M., Demmler-Harrison G.J., Cooper L.J., Liu H., Gee A.P., Shpall E.J., Rooney C.M. Derivation of human T lymphocytes from cord blood and peripheral blood with antiviral and antileukemic specificity from a single culture as protection against infection and relapse after stem cell transplantation. Blood. 2010;115:2695–2703. doi: 10.1182/blood-2009-09-242263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barker J.N., Kurtzberg J., Ballen K., Boo M., Brunstein C., Cutler C., Horwitz M., Milano F., Olson A., Spellman S. Optimal practices in unrelated donor cord blood transplantation for hematologic malignancies. Biol. Blood Marrow Transplant. 2017;23:882–896. doi: 10.1016/j.bbmt.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altfeld M., Allen T.M., Kalife E.T., Frahm N., Addo M.M., Mothe B.R., Rathod A., Reyor L.L., Harlow J., Yu X.G. The majority of currently circulating human immunodeficiency virus type 1 clade B viruses fail to prime cytotoxic T-lymphocyte responses against an otherwise immunodominant HLA-A2-restricted epitope: implications for vaccine design. J. Virol. 2005;79:5000–5005. doi: 10.1128/JVI.79.8.5000-5005.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altfeld M.A., Livingston B., Reshamwala N., Nguyen P.T., Addo M.M., Shea A., Newman M., Fikes J., Sidney J., Wentworth P. Identification of novel HLA-A2-restricted human immunodeficiency virus type 1-specific cytotoxic T-lymphocyte epitopes predicted by the HLA-A2 supertype peptide-binding motif. J. Virol. 2001;75:1301–1311. doi: 10.1128/JVI.75.3.1301-1311.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitt-Haendle M., Bachmann O., Harrer E., Schmidt B., Bäuerle M., Harrer T. Recognition patterns of HLA-A2-restricted human immunodeficiency virus-1-specific cytotoxic T-lymphocytes in a cohort of HIV-1-infected individuals. Viral Immunol. 2005;18:627–636. doi: 10.1089/vim.2005.18.627. [DOI] [PubMed] [Google Scholar]
- 32.Patel S., Lam S., Cruz C.R., Wright K., Cochran C., Ambinder R.F., Bollard C.M. Functionally active HIV-specific T cells that target Gag and Nef can be expanded from virus-naïve donors and target a range of viral epitopes: implications for a cure strategy after allogeneic hematopoietic stem cell transplantation. Biol. Blood Marrow Transplant. 2016;22:536–541. doi: 10.1016/j.bbmt.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lam S., Sung J., Cruz C., Castillo-Caro P., Ngo M., Garrido C., Kuruc J., Archin N., Rooney C., Margolis D., Bollard C. Broadly-specific cytotoxic T cells targeting multiple HIV antigens are expanded from HIV+ patients: implications for immunotherapy. Mol. Ther. 2015;23:387–395. doi: 10.1038/mt.2014.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sung J.A., Lam S., Garrido C., Archin N., Rooney C.M., Bollard C.M., Margolis D.M. Expanded cytotoxic T-cell lymphocytes target the latent HIV reservoir. J. Infect. Dis. 2015;212:258–263. doi: 10.1093/infdis/jiv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.HIV Molecular Immunology Database. (2017). CTL/CD8+ epitope summary. https://www.hiv.lanl.gov/content/immunology/tables/ctl_summary.html.
- 36.Carlson J.M., Listgarten J., Pfeifer N., Tan V., Kadie C., Walker B.D., Ndung’u T., Shapiro R., Frater J., Brumme Z.L. Widespread impact of HLA restriction on immune control and escape pathways of HIV-1. J. Virol. 2012;86:5230–5243. doi: 10.1128/JVI.06728-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrari G., Neal W., Ottinger J., Jones A.M., Edwards B.H., Goepfert P., Betts M.R., Koup R.A., Buchbinder S., McElrath M.J. Absence of immunodominant anti-Gag p17 (SL9) responses among Gag CTL-positive, HIV-uninfected vaccine recipients expressing the HLA-A*0201 allele. J. Immunol. 2004;173:2126–2133. doi: 10.4049/jimmunol.173.3.2126. [DOI] [PubMed] [Google Scholar]
- 38.Colleton B.A., Huang X.L., Melhem N.M., Fan Z., Borowski L., Rappocciolo G., Rinaldo C.R. Primary human immunodeficiency virus type 1-specific CD8+ T-cell responses induced by myeloid dendritic cells. J. Virol. 2009;83:6288–6299. doi: 10.1128/JVI.02611-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altfeld M., Rosenberg E.S., Shankarappa R., Mukherjee J.S., Hecht F.M., Eldridge R.L., Addo M.M., Poon S.H., Phillips M.N., Robbins G.K. Cellular immune responses and viral diversity in individuals treated during acute and early HIV-1 infection. J. Exp. Med. 2001;193:169–180. doi: 10.1084/jem.193.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaul R., Dong T., Plummer F.A., Kimani J., Rostron T., Kiama P., Njagi E., Irungu E., Farah B., Oyugi J. CD8(+) lymphocytes respond to different HIV epitopes in seronegative and infected subjects. J. Clin. Invest. 2001;107:1303–1310. doi: 10.1172/JCI12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goulder P.J., Altfeld M.A., Rosenberg E.S., Nguyen T., Tang Y., Eldridge R.L., Addo M.M., He S., Mukherjee J.S., Phillips M.N. Substantial differences in specificity of HIV-specific cytotoxic T cells in acute and chronic HIV infection. J. Exp. Med. 2001;193:181–194. doi: 10.1084/jem.193.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones R.B., Ndhlovu L.C., Barbour J.D., Sheth P.M., Jha A.R., Long B.R., Wong J.C., Satkunarajah M., Schweneker M., Chapman J.M. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008;205:2763–2779. doi: 10.1084/jem.20081398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Petrovas C., Casazza J.P., Brenchley J.M., Price D.A., Gostick E., Adams W.C., Precopio M.L., Schacker T., Roederer M., Douek D.C., Koup R.A. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trautmann L., Janbazian L., Chomont N., Said E.A., Gimmig S., Bessette B., Boulassel M.R., Delwart E., Sepulveda H., Balderas R.S. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 45.Borrow P., Lewicki H., Wei X., Horwitz M.S., Peffer N., Meyers H., Nelson J.A., Gairin J.E., Hahn B.H., Oldstone M.B., Shaw G.M. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 46.Goulder P.J., Sewell A.K., Lalloo D.G., Price D.A., Whelan J.A., Evans J., Taylor G.P., Luzzi G., Giangrande P., Phillips R.E., McMichael A.J. Patterns of immunodominance in HIV-1-specific cytotoxic T lymphocyte responses in two human histocompatibility leukocyte antigens (HLA)-identical siblings with HLA-A*0201 are influenced by epitope mutation. J. Exp. Med. 1997;185:1423–1433. doi: 10.1084/jem.185.8.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deng K., Pertea M., Rongvaux A., Wang L., Durand C.M., Ghiaur G., Lai J., McHugh H.L., Hao H., Zhang H. Broad CTL response is required to clear latent HIV-1 due to dominance of escape mutations. Nature. 2015;517:381–385. doi: 10.1038/nature14053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ali A., Kitchen S.G., Chen I.S., Ng H.L., Zack J.A., Yang O.O. HIV-1-specific chimeric antigen receptors based on broadly neutralizing antibodies. J. Virol. 2016;90:6999–7006. doi: 10.1128/JVI.00805-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li C., Guan X., Du T., Jin W., Wu B., Liu Y., Wang P., Hu B., Griffin G.E., Shattock R.J., Hu Q. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J. Gen. Virol. 2015;96:2381–2393. doi: 10.1099/vir.0.000139. [DOI] [PubMed] [Google Scholar]
- 50.Perez E.E., Wang J., Miller J.C., Jouvenot Y., Kim K.A., Liu O., Wang N., Lee G., Bartsevich V.V., Lee Y.L. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allers K., Schneider T. CCR5Δ32 mutation and HIV infection: basis for curative HIV therapy. Curr. Opin. Virol. 2015;14:24–29. doi: 10.1016/j.coviro.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 52.Petz L.D., Redei I., Bryson Y., Regan D., Kurtzberg J., Shpall E., Gutman J., Querol S., Clark P., Tonai R. Hematopoietic cell transplantation with cord blood for cure of HIV infections. Biol. Blood Marrow Transplant. 2013;19:393–397. doi: 10.1016/j.bbmt.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Leen A.M., Bollard C.M., Mendizabal A.M., Shpall E.J., Szabolcs P., Antin J.H., Kapoor N., Pai S.Y., Rowley S.D., Kebriaei P. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121:5113–5123. doi: 10.1182/blood-2013-02-486324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marsden M.D., Loy B.A., Wu X., Ramirez C.M., Schrier A.J., Murray D., Shimizu A., Ryckbosch S.M., Near K.E., Chun T.W. In vivo activation of latent HIV with a synthetic bryostatin analog effects both latent cell “kick” and “kill” in strategy for virus eradication. PLoS Pathog. 2017;13:e1006575. doi: 10.1371/journal.ppat.1006575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walker-Sperling V.E., Pohlmeyer C.W., Tarwater P.M., Blankson J.N. The effect of latency reversal agents on primary CD8+ T cells: implications for shock and kill strategies for human immunodeficiency virus eradication. EBioMedicine. 2016;8:217–229. doi: 10.1016/j.ebiom.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Archin N.M., Liberty A.L., Kashuba A.D., Choudhary S.K., Kuruc J.D., Crooks A.M., Parker D.C., Anderson E.M., Kearney M.F., Strain M.C. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487:482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Y. HIV-1 gene expression: lessons from provirus and non-integrated DNA. Retrovirology. 2004;1:13. doi: 10.1186/1742-4690-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.