

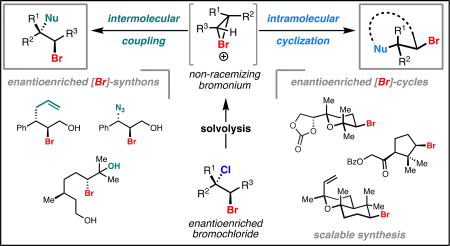
Abstract
Herein we report that under mild solvolytic conditions, enantioenriched bromochlorides can be ionized, stereospecifically cyclized to an array of complex bromocyclic scaffolds, or intermolecularly trapped by exogenous nucleophiles. Mechanistic investigations support an ionic mechanism wherein the bromochloride serves as an enantioenriched bromonium surrogate. Several natural product-relevant motifs are accessed in enantioenriched form for the first time with high levels of stereocontrol, and this technology is applied to the scalable synthesis of a polycyclic brominated natural product. An array of nucleophiles including olefins, alkynes, heterocycles, and epoxides are competent traps in the bromonium-induced cyclizations, leading to the formation of enantioenriched mono-, bi-, and tricyclic products. This strategy is further amenable to intermolecular coupling between cinnamyl bromochlorides and a diverse set of commercially available nucleophiles. Collectively this work demonstrates that enantioenriched bromonium chlorides are confiugurationally stable under solvolytic conditions in the presence of a variety of functional groups.
TOC graphic
BACKGROUND
The number of known halogenated natural products has increased dramatically since the early 1970’s; to date over 2,000 secondary metabolites containing either a chlorine- or bromine-bearing stereocenter have been isolated and structurally characterized.1 Of these, approximately 500 contain a brominated five-, six-, or seven-membered cyclic scaffold that is commonly embedded within a larger polycyclic framework (1–9, Figure 1A).1 Promising biological activity has been demonstrated for brominated cyclic terpenes, with noteworthy examples including dactylone (1) as a cancer preventative agent,2 callophycoic acid G (6) as an antibiotic against resistant Staphylococci and Enterococci strains,3 and thyrsiferyl 23-acetate (7) which exhibits sub-nanomolar activity against a P-388 leukemia cell line.4
Figure 1.

(A) Representative bromocyclic natural products. (B) Putative biosynthetic pathway for the biosynthesis of two related natural products involving several discrete bromocyclization steps. (C) Our previous work on a stereospecific bromopolyene cyclization and that accomplished in this report.
Biosynthetically, these compounds are thought to arise from the carbocationic cyclization of an isoprenoid initiated by an electrophilic source of bromine (Br+) (Figure 1B) followed by discrete steps including: cyclohexane formation (Figure 1B, path a), decalin formation (path a,b), and/or heteroatom trapping (e.g. paths b, c, d).5 Recapitulating this reactivity with high levels of chemo-, diastereo-, and enantioselectivity in a laboratory setting has long eluded chemists. The first report of a racemic bromopolyene cyclization was published more than 50 years ago by van Tamelen and Hessler.6 Since this seminal report, a variety of reactive brominating reagents have been used to initiate polyene cyclizations, albeit in generally low yields.7 In fact, a high yielding protocol did not emerge until the popularization of more reactive sulfonium-based brominating agents by the Snyder laboratoy.8 The work recently published by Yamamoto and co-workers stands as the only example of a catalytic enantioselective bromopolyene cyclization.9a While selectivities for this reaction are generally impressive, the scope of this reaction is limited to cyclizations that terminate in an arene or a phenol, significantly limiting its potential utility in the context of natural product synthesis. Although some formal bromopolyene cyclizations (multistep cyclization sequences or cyclizations from a bromohydrin derivative) provide access to natural product-relevant scaffolds, these reactions are generally limited by narrow substrate scopes, moderate selectivities, or the use of stoichiometric toxic heavy metals.10
To address these shortcomings, we anticipated that enantioenriched vicinal dihalides could be deployed under solvolytic conditions to effect bromopolyene cyclizations across a broad range of substrates. Recently our laboratory disclosed a stereospecific solvolytic cyclization of an enantioenriched bromochloride en route to the synthesis of four distinct members of the brominated chamigrene sesquiterpenes, including the highly strained aplydactone (2) (Figure 1A).11 We hypothesized this solvolytic approach could be a useful platform for the generation of an enantiopure bromonium ion (10, Figure 1C) which could then be captured in an intra- or intermolecular sense. Herein, we describe our investigation into mechanistic aspects of this cyclization, a full exploration of the substrate scope with various enantioenriched bromochloride motifs in conjunction with a range of pendant nucleophiles, application to a scalable enantioselective synthesis, and an extension of this solvolytic reaction manifold to intermolecular couplings.
DEVELOPMENT AND MECHANISM OF A SOLVOLYTIC CYCLIZATION
The bromonium ion, a reactive intermediate universally taught in undergraduate organic chemistry, has a rich history in chemical synthesis. This intermediate can be readily accessed from olefins in racemic fashion. However, forming an enantiopure bromonium ion has remained a longstanding challenge for synthetic chemists. Studies on the nature and reactivity of racemic bromonium ions date back to the pioneering work by Winstein in the 1940’s.12 It wasn’t until 2010 when Denmark and co-workers investigated the configurational stability of enantiopure bromonium ions;13 in that report, the stereochemical integrity of a bromonium ion was observed to be highly dependent on the nature of the trapping nucleophile and presence of alkene in ionizing media. However, the Braddock group has demonstrated that a configurationally stable enantiopure bromonium ion can be generated and captured in the presence of an alkene nucleophile and an oxophilic Lewis acid in nitromethane.10d Together, these reports highlight a rather tenuous understanding of the configurational stability of a bromonium ion in varied reaction settings. With our recent total synthesis of aplydactone notwithstanding, there have been no investigations into the stereochemical nature of a bromonium ion derived from an enantioenriched dihalide.
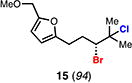
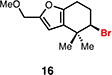
Our laboratory previously disclosed a chemo-, regio-, and enantioselective Schiff base-catalyzed dihalogenation reaction of allylic alcohols, providing access to highly enantioenriched bromochlorides, dibromides, and dichlorides.14 We initially hypothesized that subjecting these species to ionizing conditions could facilitate the generation of an enantioenriched halonium ion that could be captured intramolecularly for productive cyclization (Figure 1C). To test our hypothesis we first subjected enantioenriched dibrominated furan alcohol 11 to common ionizing conditions of 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP)15 hopeful that an enantioenriched bromonium ion 12 could be generated and captured by the nucleophilic furan, ultimately leading to carbocycle 13 (Table 1A). Unfortunately, facile dibromide racemization (i.e. 11 to ent-11) was found to occur at room temperature (entry 1), with only trace racemic cyclization product 13 being observed after 96 hours (entry 2). Fortunately, subjecting the analogous enantioenriched bromochloroalcohol 14 to the ionizing conditions led to no observed racemization of the starting material, with slow equilibration to the isomeric bromochloride over extended reaction times (entries 3 and 4). When the temperature of the reaction was elevated to 50 °C cyclization was observed (10%) with no loss in enantiopurity of the cyclized product or starting material (entry 5). We next sought to improve the yield of the solvolytic cyclization and hypothesized that the presence of the alcohol used to direct the dihalogenation was impeding ionization and/or subsequent cyclization. Indeed, when deoxygenated furyl bromochloride 15 was subjected to the solvolysis at room temperature facile equilibration of the bromochloride regioisomers was observed with concomitant production of bromocycle 16 in synthetically viable yields (40%) and high enantiospecificity (entry 6).
Table 1.
Timepoint analysis for the solvolytic cyclization of enantioenriched dihalides.

|
(A) aPrepared as a >20:1 mixture of constitutional bromochloride isomers.
1.5 equiv. K2CO3, 0.05 M in HFIP.
Based on 1H NMR analysis using 1,4-dinitrobenzene as an internal standard.
According to chiral HPLC analysis.
Ratio of constitutional isomers according to 1H NMR analysis.
HPLC integration was complicated by the presence of the corresponding constitutional isomer.
(B) Proposed mechanism for equilibration of bromochloride constitutional isomers and racemization of dibromides.
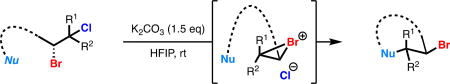
A mechanistic rationale explaining the observed trends (Table 1B) involves the initial formation of bromonium intermediate 18 from all dihalide starting materials (11, 14, 15). In the case of bromochlorides 14 and 15, recapture by chloride anion could lead to the observed mixture of constitutional isomers (X = Cl, 17 and 19). In the case of dibromide 11, such ionization and recapture by bromide anion would lead to dibromide racemization (via 18 or 20). Bromochlorides 14 and 15 thus serve as non-racemizing, enantioenriched bromonium surrogates under these conditions.
Seeking to better understand the mechanism of bromochloride solvolysis and to distinguish it from the closely related dyotropic rearrangement of dihalides,16 we computationally investigated the interconversion between isomeric bromochlorides of 2-methyl-2-butene (Figure 2). We hypothesized that solvent dielectric plays an important role in stabilizing intermediates or transition states that have high charge separation. Calculations were performed at the M06-2X/6-31G+(d) level of theory with the IEFPCM implicit solvation model using either toluene or 2,2,2-trifluoroethanol (TFE) as solvent (see Supporting information and references therein for details). In the nonpolar solvent model with implicit toluene, a dyotropic rearrangement transition state structure was identified, but no ground state structure corresponding to an ion pair was found. Conversely, in the polar solvent model with implicit TFE, two transition state structures corresponding to ionization of each isomer were found, along with an intermediate bromonium chloride ion pair ground state. Overall, implicit polar solvation lowers the calculated barrier to rearrangement by 8 kcal/mol. Inclusion of a single explicit HFIP molecule hydrogen-bonded to chloride further lowers this barrier by an additional 5.7 kcal/mol (see Supporting Information). These results suggest that both solvent dielectric and hydrogen bonding play a role in activating vicinal bromochlorides towards ionization under our solvolytic conditions, and establish the bromonium chloride tight ion pair as a viable intermediate in the mechanism of these solvolytic reactions.
Figure 2.

Computational investigation of the interconversion of bromochloride constitutional isomers at the M06-2X/6-31G+(d) level of theory; uncorrected electronic energies are listed in kcal/mol (PCM = polarizable continuum model).
SUBSTRATE SCOPE AND APPLICATIONS
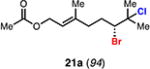
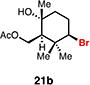
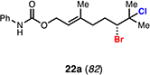
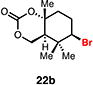
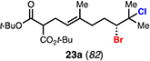
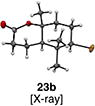
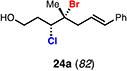
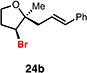
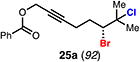
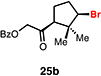
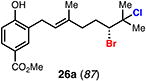
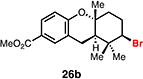
With optimal conditions in hand, a wide variety of substrates were synthesized and investigated. We discovered that the conditions for solvolysis were readily applicable to a diverse set of bromochlorides synthesized from the corresponding trisubstituted allylic alcohol. The varied nature of nucleophiles used to trap the bromonium ions generated from the corresponding bromochlorides is shown in Table 2 and includes trisubstituted olefins, free alcohols, alkynes, carbamates, esters, phenols, and a furan heterocycle. As previously described in Table 1, furyl bromochloride 15 is efficiently cyclized with perfect enantiospecificity. Enantioenriched bromochlorides derived from geraniol are effectively cyclized with various terminating groups (21a, 22a, 23a, and 26a) and were isolated as single diastereomers. The cyclization of 21a was performed on gram-scale and proceeded with 96% enantiospecificity, producing the simple bromo-cyclohexane motif which we utilized in our recently disclosed total synthesis of the chamigrene sesquiterpenes.11 The bicyclic bromolactone 23b derived from di-tert-butyl malonate 23a, whose absolute stereochemistry was determined by X-ray crystallography, confirmed that the cyclization proceeded with complete retention of the bromide stereocenter. Cyclization of internal bromochloride 24a proceeded in good yield and high enantiospecificity (91% es), but was isolated as a mixture of epimeric tertiary ethers. This result is particularly intriguing as it suggests that the intermediate bromonium exists as, or in equilibrium with an open β-bromo carbocation; non-stereoselective nucleophilic attack then accounts for the formation of both diastereomers in 24b. At this time we cannot rule out competing participation of the styrene, or subtle hydrogen bonding effects with the free alcohol, as a rationale for the lower levels of diastereoselectivity. It is worth noting that accessing and effectively capturing the bromonium from bromochloride 24a would be difficult or impossible with other protocols given the polyunsaturated nature of the substrate. Cyclization of alkynyl bromochloride 25a interestingly provided cyclopentyl bromide 25b. To our knowledge, this is the only example of a bromonium-induced cyclization affording a bromocyclopentane- a motif found in bromocyclic natural products (e.g. 5, Figure 1A).
Table 2.
Substrate Scope of the Solvolytic Cyclization.
| |||
|---|---|---|---|
| Starting material (%ee)a | Product | %Yield (dr)b | es (%)c |
|
|
54%d | 99 |
|
|
|
96 |
|
|
40%f | 96 |
|
|
67g | 98 |
|
|
59% | 91 |
| (3 : 1 dr) | |||
|
|
36% | >99 |
|
|
64% | 98 |
|
|
30d | >99 |
| (2.3 : 1 dr)h | |||
Based on chiral HPLC analysis of the corresponding bromochloroalcohol.
Yields shown are of the major isolated diastereomer; d.r. given is for combined diastereomer content unless otherwise noted.
Based on chiral HPLC analysis of the cyclization product or derivative; es (enantiospecificity) = ((%ee s/m)/(%ee product)) × 100%.
Reaction conducted at 4 °C.
See ref. 10.
After subsequent hydrolysis step; see SI.
After subsequent decarboxylation step.
dr shown for two isolated diastereomers.
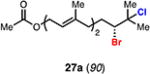
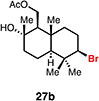
Expanding on successful cyclizations with geraniol-derived bromochlorides, we subjected bromochlorofarnesyl acetate 27a to the solvolytic cyclization conditions. We were able to isolate the bicyclic bromodecalin 27b and a corresponding diastereomer in a combined 30% yield. Also isolated were small amounts (ca. 5%) of additional minor diastereomer content and significant amounts of partially cyclized material (ca. 40%). Mechanistically, these results implicate a stepwise polyolefin cyclization being operative under these reaction conditions; however, we cannot rule out a concerted cyclization at this time. This bromodecalin has been previously used in various racemic natural product syntheses17 but has never been accessed in enantioenriched form.
We were particularly intrigued by trying to access bromopyran 29 (Scheme 1), a motif that is found in close to 100 natural products (e.g. 7, Figure 1), however synthetic technologies enabling enantio- and diastereoselective access to this motif are largely deficient.18 To date, only one approach utilizing an epoxide opening kinetic resolution with a bromohydrin has been successful in accessing this motif with regio- and enantiocontrol.19 We were confident that we could access this challenging motif using our dihalide solvolysis, and to test this strategy, we constructed epoxidized bromochlorogeraniol derivative 28 and subjected this substrate to standard solvolysis conditions. Unfortunately, the reaction produced a complex mixture of products. However, when this crude mixture was treated with wet HCl in ether, we observed formation of 29 as the major reaction product. Our working hypothesis is that when substrate 28 is subjected to the solvolysis conditions oxonium 30 is initially formed and nonselectively trapped, either intramolecularly via the carbamate or intermolecularly by adventitious water, leading to a mixture of bromopyran 29 and the corresponding 7-membered bromoether (i.e. from the trapping of 30 at C-6). We believe that subjecting the crude mixture to acidic aqueous ether leads to reformation of 30, but is selectively trapped by water at C-7 to 30a under these conditions leading to near-exclusive formation of pyran 29.
Scheme 1.

Solvolytic epoxide opening cascade for the synthesis of a brominated pyrana
aReagents and conditions: (i) K2CO3 (1.5 equiv), 1,1,1,3,3,3-hexafluoroisopropanol (0.05 M), rt; (ii) HCl (concentrated aq., 5 equiv), wet Et2O, rt, 34% over two steps.
While we previously demonstrated the utility of this solvolytic methodology in the total synthesis of the brominated chamigrene sesquiterpenes, we sought to demonstrate that this methodology could be applied towards the scalable synthesis of a brominated polycyclic natural product (Scheme 2). We targeted the bicyclic, brominated ether 3420 which we believed could be accessed in a straightforward fashion from bromolactone ent-23b. Solvolysis of di-tert-butyl malonate bromochloride ent-23a on 5.7 gram-scale proceeded smoothly, and a subsequent decarboxylation step on the crude material cleanly afforded bicyclic bromolactone ent-23b in 75% yield as a single isolated diasteromer, with complete enantiospecificity, over two steps. Our strategy called for conversion of lactone ent-23b to allylic diol 33, which required conversion to the acyclic Weinreb amide 31, and was then converted to methyl ketone 32 on gram-scale. This intermediate proved to be unstable, where upon exposure to acidic media or silica gel the material was observed to rapidly dehydrate to a cyclic enol ether. Bypassing a purification step, crude keto alcohol 32 was treated with vinylmagnesium chloride on gram-scale to afford diol 33 in 73% yield over two steps.
Scheme 2.

Application of the dihalide solvolysis to a scalable, enantioselective synthesis of a brominated natural producta
aReagents and conditions: (a) K2CO3 (1.5 equiv), 1,1,1,3,3,3-hexafluoroisopropanol (0.05 M), rt; (b) trifluoroacetic acid, CH2Cl2, rt; PhMe, 110 °C, 75% over two steps; (c) trimethylaluminum (3.0 equiv), N,O-dimethylhydroxylamine hydrochloride (3.0 equiv), CH2Cl2, 0 °C to rt; (d) methyllithium lithium bromide complex (3.5 equiv), THF, −78 °C; (e) vinylmagnesium chloride, Et2O, 0 °C, 73% over two steps; (f) BF3· OEt2 (1.5 equiv), CH2Cl2, 4 °C, 50% (6:1 dr).
Treatment of diol 33 with boron trifluoride diethyl etherate readily afforded target natural product 34 as a mixture of diastereomers (ca. 4:1 dr at C8) on small scale. However, attempts to scale this reaction gave inconsistent results. Specifically, variable amounts of the natural product and its epimer were produced, in addition to an isomeric byproduct. These findings prompted us to investigate the nature of this cyclization and after extensive experimentation on this system (see Supporting Information for details), we were able to develop a mechanistic hypothesis consistent with our data. Treating 33 with a slight excess of boron trifluoride diethyl etherate led to rapid (<5 mins) conversion of the starting material to bicyclic ether 34 and its corresponding C8-epimer (ca. 1:1 dr). Over time, the diastereomeric ratio of the desired natural product increased with slight erosion in overall yield, which was concomitant with formation of an isomeric, bicyclic tetrahydrofuran. This suggests that 34 and its epimer are initially formed with low diastereoselectivity (ca. 1:1 dr) and rapidly and reversibly interconvert as the reaction progresses. Each diastereomer is then irreversibly funneled to undesired byproduct at a different rate, giving rise to the enrichment in the desired epimer of 34. Allowing this reaction to run for extended periods resulted in exclusive formation of the undesired isomer, suggesting it is the thermodynamic product in this setting. The consequence of this mechanistic scenario is that the reaction must be run for an amount of time such that good levels of diastereoselectivity are obtained while the amount of isomeric byproduct formed is minimized, thereby maximizing the yield of desired 34. Allowing the cyclization to run for a period of 1 hour at 4 °C proved to be optimal conditions on scale, and we obtained the natural product in 50% yield and 6:1 dr. Having produced a total amount of 233 mg, we have demonstrated the use of this this methodology toward the scalable enantioselective synthesis of a brominated natural product.
EXTENSION TO INTERMOLECULAR COUPLING
Having demonstrated the utility of this approach in an intramolecular sense, as well as its utility in total synthesis, we set out to see if this methodology could be extended to intermolecular solvolytic couplings. Given the extensive use of alkyl halides as versatile building blocks in chemical synthesis, we believed that developing a strategy for providing access to a diverse set of highly enantioenriched anti-functionalized alkyl bromides would be an enabling technology.
After investigating an array of enantioenriched dihalides, we identified cinnamyl bromochloride 35 as an optimal substrate which was observed to engage with a diverse set of exogenous nucleophiles in an intermolecular coupling reaction. Our solvolysis conditions employed in the intramolecular cyclization were general for intermolecular coupling with some minor modifications. Running the reactions at a concentration of 0.1 M (cyclizations are run at 0.05 M) at room temperature and employing 5 equivalents of a commercially available nucleophile proved optimal. Under these conditions, a variety of carbon–heteroatom bond formations can be effected and the scope of this reaction is highlighted in Table 3.
Table 3.
Scope of the intermolecular solvolytic coupling.

|
Isolated as a 4:1 mixture of thiocyanate and isothiocyanate.
Triethylsilyl protected 35 was used in this reaction.
Isolated as a 12:1 mixture of diastereomers.
Isolated as a 2.5:1 mixture of ortho,para- to ortho,ortho-isomers.
Isolated as a 9:1 mixture of diastereomers.
HPLC integration was complicated by the presence of a trace impurity.
Simple nucleophiles can be readily deployed to effect carbon–nitrogen, carbon–fluorine, carbon–sulfur, and carbon–oxygen bond formation in good yield, high levels of diastereoselectivity, and with excellent enantiospecificity (all >90%; 37–42). Triethylsilane can be employed to effect stereospecific reduction of the bromochloride (36). This methodology can be extended to carbon–carbon formation using allyltrimethylsilane as nucleophile (43 and 46). Triethylsilyl-protected cinnamyl bromochloride can be allylated in excellent yield producing the product (43) in 12:1 dr with near perfect enantiospecificity. Simple arenes also serve as competent nucleophiles in this reaction for productive carbon–carbon bond formation (44). Deoxygenated cinnamyl bromochlorides were observed to be considerably more reactive than their alcohol-bearing counterpart and underwent clean reactivity with several distinct nucleophiles (45–47). A current limitation of this methodology is the requirement of an arene to stabilize the bromonium resulting from the solvolysis of the bromochloride. Currently, most alkyl-derived bromochlorides do not engage in the solvolytic coupling, however citronellyl bromochloride 48 can be diastereospecifically converted to its corresponding bromohydrin (49), a common motif found in halogenated natural products (e.g. 8), under these conditions. We are actively engaged in further exploring the substrate scope of this reaction, specifically with regard to non-benzylic substrates.
CONCLUSION
We have demonstrated that under mild solvolytic conditions enantioenriched bromochlorides can be efficiently cyclized to a diverse array of natural product-relevant motifs, and coupled to exogenous nucelophiles in an intermolecular sense, with high levels of enantiospecificity. Bromochlorides were demonstrated to be superior to their dibromide counterparts, which were observed to rapidly and competitively racemize under the cyclization conditions. A variety of enantioenriched anti-functionalized brominated compounds can be constructed from their corresponding bromochloride precursor, which we anticipate will serve as valuable building blocks in chemical synthesis. This solvolytic approach provides scaffolds that have seen use in racemic total syntheses and are herein accessed in highly enantioenriched form for the first time.
Supplementary Material
Acknowledgments
We are grateful to Dr. A. Oliver (University of Notre Dame) for X-ray crystallographic analysis and Dr. S. Lynch (Stanford University) for assistance with NMR spectroscopy.
Funding Sources
This work was supported by Stanford University, the National Institutes of Health (R01 GM114061), and the National Science Foundation (DGE-114747 GRF to AJB).
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interests
References
- 1.(a) Gribble GW. Naturally Occurring Organohalogen Compounds – A Comprehensive Survey. Springer-Verlag; Wien: 1996. [PubMed] [Google Scholar]; (b) Gribble GW. Naturally Occurring Organohalogen Compounds – A Comprehensive Update. Springer-Verlag; Wien: 2010. [Google Scholar]; (c) Wang BG, Gloer JB, Ji NY, Zhao JC. Chem. Rev. 2013;113:3632–3685. doi: 10.1021/cr9002215. [DOI] [PubMed] [Google Scholar]; (d) Brito I, Dias T, Diaz-Marrero AR, Darias J, Cueto M. Tetrahedron. 2006;62:9655–9660. [Google Scholar]; (e) Brito I, Dias T, Diaz-Marrero AR, Darias J, Cueto M. Tetrahedron. 2007;63:3908. [Google Scholar]
- 2.(a) Federov SN, Reshetnyak AP, Shchedrin AP, Il’in SG, Struchkov YT, Stonik VA, Elyakov GB. Dokl. Akad. Nauk SSSR. 1989;305:877–879. [Google Scholar]; (b) Lyakhova EG, Federov SN, Shubina LK, Radchenko OS, Kalinovsky AI, Dvitrenok PS, Stonki VA. Russ. Chem. Bull. Int. Ed. 2003;52:970–974. [Google Scholar]; (c) Federov SN, Shubina LK, Bode AM, Stonik VA, Dong Z. Cancer Res. 2007;67:5194–5920. doi: 10.1158/0008-5472.CAN-06-3723. [DOI] [PubMed] [Google Scholar]
- 3.Lane AL, Stout EP, Hay ME, Prusak AC, Hardcastle K, Fairchild CR, Franzblau SG, Le Roch K, Prudhomme J, Aalbersberg W, Kubanek J. J. Org. Chem. 2007;72:7343–7351. doi: 10.1021/jo071210y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Suzuki T, Suzuki M, Furusaki A, Matsumoto T, Kato A, Imanaka Y, Kurosawa E. Tetrahedron Lett. 1985;26:1329–1332. [Google Scholar]; (b) Suzuki T, Takeda S, Suzuki M, Kurosawa E, Kato A, Imanaka Y. Chem. Lett. 1987:361–364. [Google Scholar]
- 5.(a) Carter-Franklin JN, Butler AJ. J. Am. Chem. Soc. 2004;126:15060–15066. doi: 10.1021/ja047925p. [DOI] [PubMed] [Google Scholar]; (b) Fukuzawa A, Miyamoto M, Kumagai Y, Abiko A, Takaya Y, Masamune T. Chem. Lett. 1985:1259–1262. [Google Scholar]; (c) Findlay JA, Li G. Can. J. Chem. 2002;80:1697–1707. [Google Scholar]
- 6.van Tamelen EE, Hessler EJ. J. Chem. Soc. Chem. Comm. 1966:411–413. [Google Scholar]
- 7.(a) Wolinsky LE, Faulkner DJ. J. Org. Chem. 1976;41:597–600. [Google Scholar]; (b) Kato T, Ichinose I, Kamoshida A, Kitahara Y. J. Chem. Soc. Chem. Comm. 1976:518–519. [Google Scholar]; (c) Shieh HM, Prest-wich GD. Tetrahedron Lett. 1982;23:4643–4646. [Google Scholar]; (d) Kato T, Mochizuki M, Hirano T, Fujiwara S, Uyehara T. J. Chem. Soc. Chem. Commun. 1984:1077–1078. [Google Scholar]
- 8.Snyder SA, Treitler DS. Angew. Chem. Int. Ed. 2009;48:7899–7903. doi: 10.1002/anie.200903834.Snyder SA, Treitler DS, Brucks AP. J. Am. Chem. Soc. 2010;132:14303–14314. doi: 10.1021/ja106813s.For additional bromonium-in-duced cyclizations using BDSB see: Snyder SA, Treitler DS, Brucks AP. J. Am. Chem. Soc. 2011;133:15898–15901. doi: 10.1021/ja2069449.Snyder SA, Brucks AP, Treitler DS. J. Am. Chem. Soc. 2012;134:17714–17721. doi: 10.1021/ja3076988.
- 9.Samanta RC, Yamamoto H. J. Am. Chem. Soc. 2017;139:1460–1463. doi: 10.1021/jacs.6b13193.Lower levels of enantioselectivity have been achieved in an earlier report of a direct bromopolyene cyclization: Sakakura A, Ukai A, Ishihara K. Nature. 2007;445:900–903. doi: 10.1038/nature05553.
- 10.(a) Couladouros EA, Vidali VP. Chem. Eur. J. 2004;10:3822–3835. doi: 10.1002/chem.200400407. [DOI] [PubMed] [Google Scholar]; (b) Snyder SA, Treitler DS, Schall A. Tetrahedron. 2010;66:4796–4804. [Google Scholar]; (c) Braddock DC, Marklew JS, Thomas AJF. Chem. Commun. 2011;47:9051–9053. doi: 10.1039/c1cc13619d. [DOI] [PubMed] [Google Scholar]; (d) Braddock DC, Marklew JS, Foote KM, White AJP. Chirality. 2013;25:692–700. doi: 10.1002/chir.22194. [DOI] [PubMed] [Google Scholar]
- 11.Burckle AJ, Vasilev VH, Burns NZ. Angew. Chem. Int. Ed. 2016;55:11476–11479. doi: 10.1002/anie.201605722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.“The Role of Neighboring Groups in Replacement Reactions” is a series of >20 reports by Winstein and co-workers, many of which contain seminal studies on the mechanism of reactions in which a bromonium ion is generated from a dihalide, a halohy-drin, or a derivative thereof. For a summary of key findings, see part XIII of this series: Winstein S, Grunwald E. J. Am. Chem. Soc. 1948;70:828–837.
- 13.Denmark SE, Burk MT, Hoover AJ. J. Am. Chem. Soc. 2010;132:1232–1233. doi: 10.1021/ja909965h. [DOI] [PubMed] [Google Scholar]
- 14.(a) Hu DX, Seidl FJ, Bucher CB, Burns NZ. J. Am. Chem. Soc. 2015;137:3795–3798. doi: 10.1021/jacs.5b01384. [DOI] [PubMed] [Google Scholar]; (b) Hu DX, Shibuya GM, Burns NZ. J. Am. Chem. Soc. 2013;135:12960–12963. doi: 10.1021/ja4083182. [DOI] [PubMed] [Google Scholar]; (c) Landry ML, Hu DX, McKenna GM, Burns NZ. J. Am. Chem. Soc. 2016;138:5150–5158. doi: 10.1021/jacs.6b01643. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bucher CB, Deans RM, Burns NZ. J. Am. Chem. Soc. 2015;137:12784–12787. doi: 10.1021/jacs.5b08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) For a classic solvolysis study using HFIP see: Schadt FL, Bentley TW, Schleyer PvR. J. Am. Chem. Soc. 1976;98:7667–7674.For additional physical studies of HFIP see Schadt FL, Schleyer PvR. Tetrahedron Lett. 1974;27:2355–2338.Ammer J, Mayer H. J. Phys. Org. Chem. 2013;26:59–63.For recent synthetic studies employing HFIP as a solvent or co-solvent see: Chen G, Gong W, Zhuang Z, Andrä MS, Chen Y-Q, Hong X, Yang Y-F, Liu T, Houk KN, Yu J-Q. Science. 2016;353:1023–1027. doi: 10.1126/science.aaf4434.Gray EE, Nielsen MK, Choquette KA, Kalow JA, Graham TJA, Doyle AG. J. Am. Chem. Soc. 2016;138:10802–10805. doi: 10.1021/jacs.6b06770.Adams AM, Du Bois JD. Chem. Sci. 2014;5:656–659.
- 16.(a) Cresswell AJ, Eey ST-C, Denmark SE. Angew. Chem. Int. Ed. 2015;54:15642–15682. doi: 10.1002/anie.201507152. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fernández I, Cossío FP, Sierra MA. Chem. Rev. 2009;109:6687–6711. doi: 10.1021/cr900209c. [DOI] [PubMed] [Google Scholar]; (c) Zou J-W, Yu C-H. J. Phys. Chem. A. 2004;108:5649–5654. [Google Scholar]; (d) Fernández I, Sierra MA, Cossío FP. Chem. Eur. J. 2006;12:6323–30. doi: 10.1002/chem.200501517. [DOI] [PubMed] [Google Scholar]
- 17.For use of structural variants of this bromodecalin in total synthesis see: reference 10b; Tadahiro K, Ishi K, Ichinose I, Nalai Y, Kumagai T. J. Chem. Soc. Chem. Comm. 1980;23:1106–1108.Murai A, Abiko A, Masamune T. Tetrahedron Lett. 1984;25:4955–4958.Yamaguchi Y, Uyehara T, Kato T. Tetrahedron Lett. 1985;26:343–346.Nishizawa M, Takenaka H, Hayashi Y. J. Org. Chem. 1986;51:806–813.Fujiwara S, Takeda K, Uyehara T, Kato T. Chem. Lett. 1986;10:1763–1766.
- 18.Close to 100 natural products containing the bromopyran motif highlighted in Scheme 2 have been isolated and structurally characterized, of which almost 30 belong to the thyrsenol or thyrsiferol family. For further reading see ref’s 1a, b. For isolation and structural characterization of 7 see ref. 4. For synthetic studies see: Gonzalez IC, Forsyth CJ. J. Am. Chem. Soc. 2000;122:9099–9108.Hashimoto M, Kan T, Nozaki K, Yanagiya M, Shirahama H, Matsumoto T. J. Org. Chem. 1990;55:5088–5107.Kan T, Hashimoto M, Yanagiya M, Shirahama H. Tetrahedron Lett. 1988;29:5417–5418.
- 19.McDonald FE, Wei X. Org. Lett. 2002;4:593–595. doi: 10.1021/ol0171968.Nishiguchi GA, Little RD. J. Org. Chem. 2005;70:5249–5256. doi: 10.1021/jo050465d.(c) For a comprehensive overview of approaches to the synthesis of bromopyrans see Supporting Information.
- 20.For isolation see: Faulkner DJ. Phytochemistry. 1976;15:1993–1994. For synthetic studies see: Hoye TR, Kurth MJ. J. Org. Chem. 1979;44:3461–3467.Kato T, Ishii K, Ichinose I, Nakai Y, Kumagai T. J. Chem. Soc. Chem. Comm. 1980:1106–1108.Ref 5a. For discussion on the characterization of this natural product and determination of absolute stereochemistry, see the Supporting Information
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.