

CONSPECTUS
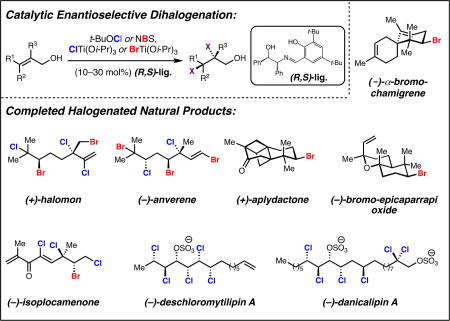
To date, more than 5,000 biogenic halogenated molecules have been characterized. This number continues to increase as chemists explore chloride- and bromide-rich marine environments in search of novel bioactive natural products. Naturally-occurring organohalogens span nearly all biosynthetic structural classes, exhibit a range of unique biological activities, and have been the subject of numerous investigations. Despite the abundance of and interest in halogenated molecules, enantioselective methods capable of forging carbon-halogen bonds in synthetically-relevant contexts remain scarce. Accordingly, syntheses of organohalogens often rely on multistep functional group interconversions to establish carbon-halogen stereocenters. Our group has developed an enantioselective dihalogenation reaction and utilized it in the only reported examples of catalytic enantioselective halogenation in natural product synthesis. In this Account, we describe our laboratory’s development of a method for catalytic, enantioselective dihalogenation and the application of this method to the synthesis of both mono- and polyhalogenated natural products. In the first part, we describe the initial discovery of a TADDOL-mediated dibromination of cinnamyl alcohols. Extension of this reaction to a second-generation system capable of selective bromochlorination, dichlorination, and dibromination is then detailed. This system is capable of controlling the enantioselectivity of dihalide formation, chemoselectivity for polyolefinic substrates, and regioselectivity in the case of bromochlorination. The ability of this method to exert control over regioselectivity of halide delivery permits selective halogenation of electronically non-biased olefins required for total synthesis.
In the second part, we demonstrate how the described dihalogenation has provided efficient access to a host of structurally diverse natural products. The most direct application of this methodology is in the synthesis of naturally-occurring vicinal dihalides. Chiral vicinal bromochlorides represent a class of >175 natural products; syntheses of five members of this class, including its flagship member, (+)-halomon, have been accomplished through use of the catalytic, enantioselective bromochlorination. Likewise, enantioselective dichlorination has provided selective access to two members of the chlorosulfolipids, a class of linear, acyclic polychlorides. Synthesis of chiral monohalides has been achieved through solvolysis of enantioenriched bromochlorides; this approach has resulted in the synthesis of five bromocyclohexane-containing natural products through an enantiospecific bromopolyene cyclization. In reviewing these syntheses, a framework for the synthesis of chiral organohalogens mediated by catalytic, enantioselective dihalogenation has emerged.
The development of a selective dihalogenation method has been highly enabling in the synthesis of halogenated natural products. In this Account, we detail all examples of catalytic, enantioselective halogenation in total synthesis and encourage the further development of synthetically-useful halogenation methodologies.
Graphical abstract
INTRODUCTION
The synthesis of chiral halogenated natural products is a frontier in organic chemistry that motivates and underscores the need for methodological development. In contrast to the ubiquity and complexity of organohalogens found in nature, there are comparatively few synthetic methods capable of rendering these molecules with enantiocontrol.1,2 Prior to reports from our laboratory, there existed a single example of enantioselective halogenation in natural product synthesis by Snyder and co-workers; this dichlorination relied on the use of a super-stoichiometric chiral auxiliary.3 Numerous examples of catalytic, enantioselective methods for halogenation have been described but have yet to find use in natural product synthesis.2 Surveying the structures of natural organohalogens, we anticipated that the development of an enantioselective dihalogenation would be of particular synthetic value. Vicinal dihalides not only represent a large component of naturally-occurring chiral organohalogens but also might serve as progenitors to chiral monohalogenated natural products.1
Controlling the enantioselectivity of alkene dihalogenation has been a long-standing challenge in catalysis and limited progress in this area is attributed to a number of factors.4 First, molecular halogens and their surrogates are highly reactive and often exhibit nonselective background reactivity, even at reduced temperatures. Second, solely controlling the enantioselectivity of halonium formation is not sufficient in generating enantioenriched dihalide products (i.e. 1 to 2, Scheme 1). For dichlorination and dibromination, control over the regioselectivity of halide delivery is required for an enantioselective process as well; in these cases, regioisomeric dihalides are enantiomeric (2 to 3 or 4, Scheme 1). Namely, even if an enantiopure halonium were generated, non-regioselective capture by halide would result in an overall racemic dihalogenation. For interhalogenation (i.e. the addition of two distinct halogen atoms across an olefin), differing courses in regioselectivity of halide delivery produce discrete constitutional isomers. In addition to these unique mechanistic challenges, Denmark has demonstrated that enantioenriched bromonium ions can rapidly racemize in the presence of exogenous olefin (2 to ent-2, Scheme 1).5 Owing to these obstacles, only a single report of a catalytic, enantioselective dihalogenation by Nicolaou and co-workers had been disclosed prior to our work.6 This dichlorination requires use of styrenyl olefins to electronically bias chloride delivery and hence has limited synthetic utility. Cognizant of the value and challenges associated with enantioselective dihalogenation, we endeavored to develop a method capable of accessing dihalide motifs on natural product-relevant scaffolds.
Scheme 1.

Selectivity Challenges Associated with Enantioselective Dihalogenation of Alkenes.
FIRST-GENERATION SYSTEM FOR ENANTIOSELECTIVE DIBROMINATION
Initial efforts toward developing a system for enantioselective dihalogenation were focused on the dibromination of alkenes. Such a reaction was absent from the compendium of organic transformations and we anticipated that chiral dibromides would serve as useful, stereodefined building blocks. Prior to our work there existed a single report claiming a palladium-catalyzed enantioselective dibromination,7a however extensive reinvestigation by Denmark cast serious doubt on the validity of this work.7b The principle challenges associated with dihalogenation guided the design of our methodology. To address the highly reactive nature of commonly employed reagents for dibromination, we decided to formally separate the electrophilic and nucleophilic halogen sources into two individually unreactive partners; ideally, these would only become a mutually reactive source of dihalogen in the presence of a chiral entity (5 and 6, Figure 1). We postulated that an appropriate chiral complex might then be able to exert enantiocontrol over bromonium formation. To temper racemization of the intermediate bromonium ion, we adopted a directing group strategy wherein alcoholic functionality on the substrate could chelate to the chiral complex and provide a persistent stereodefined environment. Additionally, the use of a directing group would render halide transfer an intramolecular process, providing a handle for controlling regioselectivity (7 and 8, Figure 1). Taking inspiration from Sharpless’ work on enantioselective epoxidation,8 we selected allylic alcohols as substrates for the development of our dihalogenation.
Figure 1.

First-Generation Enantioselective Dibromination Design Principles.
The strategy detailed above was reduced to practice through use of dibromomalonates as latent sources of electrophilic bromine (i.e. 5, Figure 1). These electrophiles exhibit little to no background reactivity with olefins and, to the best of our knowledge, have seen only a single use in the halogenation of unactivated alkenes.9 A range of Lewis-acidic metal halides were found to efficiently activate dibromomalonates as sources of electrophilic bromine while serving as competent halide donors in the halogenation of olefins. The dual role of transition metal halides in both bromonium generation and bromide delivery provided a clear entry point for asymmetric induction. Intensive investigations revealed that tartaric acid-derived TADDOL ligands paired with titanium halides were capable of furnishing dihalide products with enantiocontrol (Scheme 2).
Scheme 2.

First-Generation System for Enantioselective Dibromination.
Our dibromination system provided access, for the first time, to enantioenriched vicinal dibromides directly from their corresponding olefins.7,10 With our optimized conditions, a variety of trans cinnamyl alcohols could be dibrominated in fair yield and good enantioselectivity using a stoichiometric amount of chiral ligand 12; use of substoichiometric (20 mol%) ligand was accompanied by a slight erosion in selectivity (9 to 10, Scheme 2). In part, high enantioselectivities are achieved with cinnamyl substrates as they provide an inherent electronic bias for halide delivery to the benzylic carbon. Interaction of the aromatic substituent of the substrate and ligand could additionally provide a context for facial selectivity in halonium formation. Interestingly, application of the dibromination conditions to less-electronically-biased alkyl-substituted allylic alcohols still produced enriched (27–55% ee) product, confirming the ability of this system to exert some external control over the regioselectivity of bromide delivery to bromonium intermediates.
Multiple shortcomings limited the synthetic utility of our first-generation system. First, the poor efficiency of the ligated system compared to background racemic dibromination led to the requirement for high loading of chiral material. Second, synthetically-relevant, non-cinnamyl substrates could only be accessed with low enantioselectivity. Third, slow conversion and modest yields were observed due to the low reactivity of dibromomalonates, necessitating the use of highly polar nitromethane solvent. Use of more reactive sources of electrophilic bromine, such as N-bromosuccinimide (NBS), increased yields and conversion rates but proceeded with reduced enantioselectivity. Encouraged by this increase in reactivity, we began to develop strategies for restoring enantioselectivity to this titanium-mediated dihalogenation. In principle, the lower enantioselectivity observed with NBS could arise from increased racemic background dibromination, decreased enantioselectivity in bromonium generation, and/or decreased regioselectivity in bromide delivery. Whereas background dihalogenation is inherent to the reactivity of NBS, we anticipated that both the enantioselectivity of bromonium formation and regioselectivity of bromide capture could be influenced through a redesign of the chiral ligand.
SECOND-GENERATION SYSTEM FOR ENANTIOSELECTIVE DIHALOGENATION
Development of a second-generation system for dihalogenation was driven by consideration of titanium coordination chemistry. In our initial dibromination, halogenated malonates were presumed to act as neutral, bidentate ligands on titanium. Addition of dianionic bidentate TADDOL, a monodentate alkoxide substrate, and halide could form a six-coordinate octahedral titanium(IV) complex which we hypothesized was responsible for enantioselective bromination (13, Figure 2). We ascribed the high selectivity exerted by this complex to its well-defined, saturated coordination sphere and its low reactivity to the relatively stable dibromomalonate ligand. Use of neutral, monodentate NBS as a source of electrophilic bromine in place of halomalonates would disrupt the octahedral coordination sphere about titanium. We postulated that this might allow for multiple reactive coordination geometries, lowering the selectivity of dihalogenation but increasing reactivity (14, Figure 2). Accordingly, we reasoned that use of NBS in combination with a dianionic, tridentate ligand would restore the octahedral geometry of the titanium complex, improving selectivity (15, Figure 2).
Figure 2.

Rational Design of a Second-Generation Dihalogenation Platform.
The study of olefin bromochlorination provided an ideal context for developing a second-generation system for two reasons. First, bromochlorination adds two distinguishable halogens across an alkene allowing enantioselectivity to be decoupled from regioselectivity, unlike in the case of dibromination. Independent quantification of enantiomeric and regioisomeric ratios could then provide insight into what structural elements of a ligand influenced each of these forms of selectivity. Second, a catalytic, enantioselective bromochlorination would enable access to prevalent bromochlorinated natural product motifs.
Optimization of our second-generation bromochlorination was purposefully conducted on non-cinnamyl allylic alcohols so as to maximize its potential synthetic utility. In accord with our initial hypothesis, use of NBS 19 in place of dibromomalonate 11 as a source of electrophilic bromine provided kinetically faster and higher yielding reactions, but produced dihalides with lower enantioselectivity. Bromochlorination of allylic alcohol 16a with NBS 19, ClTi(i-OPr)3, and the optimal chiral ligand 12 from our first-generation system illustrated this notion, rendering bromochlorides 17a and iso-17a in good combined yield but low enantioselectivity (entry 1, Table 1). Bromochlorides 17a and iso-17a were produced as 1:2 mixture of constitutional isomers, favoring isomer iso-17a which results from Markovnikov delivery of chloride. Following our hypothesis concerning coordination chemistry, we replaced dianionic bidentate TADDOL-derived ligand 12 with dianionic tridentate Schiff base 18 under otherwise identical reaction conditions. Gratifyingly, the combination of ligand 18 and NBS 19 substantially increased the enantioselectivity of the bromochlorination (entry 2, Table 1); control over regioselectivity remained poor for non-cinnamyl substrates such as 16a. It was empirically discovered that use of hexanes as a reaction solvent improved both the regio- and enantioselectivity of this process (entry 3, Table 1). Further optimization revealed that catalyst loading could be reduced at lower temperatures (entry 4, Table 1). Under these fully optimized conditions, isomer 17a which results from contra electronic chloride delivery is produced with >20:1 regioselectivity.
Table 1.
Development of a Second-Generation Dihalogenation
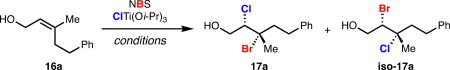
| ||||
|---|---|---|---|---|
|
| ||||
| entry | conditions | 17a + iso-17a (% yield) |
17a : iso-17a (cr) |
17a, iso-17a (% ee) |
| 1 | 50 mol % 12, CH2Cl2, r.t. | 88 | 1 : 2 | 6, 8 |
| 2 | 50 mol % 18, CH2Cl2, r.t. | 67 | 1 : 1 | 63, 6 |
| 3 | 50 mol % 18, hexanes, r.t. | 70 | 8 : 1 | 94, 52 |
| 4 | 10 mol % 18, hexanes, −20 °C | 88 | >20 : 1 | 94, − |
|
| ||||
|
| ||||
Under our optimized conditions, a range of alkyl-substituted allylic alcohols could be selectively bromochlorinated in high yield (16 to 17, Scheme 3).11 A salient feature of this platform is its ability to react chemoselectively with the allylic alcohol functionality of polyolefins (17e–h, Scheme 3). Additionally, this system is capable of overriding inherent substrate bias to provide anti-Markovnikov dihalide products (17a,b,e,f,n, Scheme 3). In the case of non-electronically-biased cis allylic alcohols, exquisite control over regioselectivity of chloride delivery is achieved (17o,p, Scheme 3); regioselectivity is poor in the case of trans disubstituted allylic alcohols. Our second-generation is more selective and higher yielding than our first-generation system for all classes of allylic alcohols and dihalogenations examined. This method represented the first means to access enriched bromochlorides and is directly applicable to the synthesis of natural products.
Scheme 3.

Scope of Catalytic Enantioselective Bromochlorination.
Our proposed catalytic cycle for the bromochlorination of allylic alcohols is depicted in Scheme 4. In the first step of this mechanism, chiral ligand 18, allylic alcohol substrate 16, NBS 19, and halide are assembled on a mutual titanium center with expulsion of three equivalents of isopropanol. In complex 20, coordination of NBS 19 to Lewis-acidic titanium could promote transfer of electrophilic bromine to the bound allylic alcohol, forming putative zwitterion 21; the presence of chiral ligand 18 thus may provide a source of enantiocontrol in bromonium formation. At this time reversible nonselective bromonium formation followed by selective opening with chloride in a dynamic kinetic resolution cannot be ruled out.5 Intramolecular delivery of chloride via transition state structure 22 offers a rationale for regiocontrol in dihalide formation wherein steric interactions between the large ligand and the substrate on titanium may favor opening at the bromonium carbon closer to the allylic alcohol. Alcoholysis liberates bromochloride product 17 and regenerates catalyst 18, completing the catalytic cycle. We observe that reactions conducted with ligand are kinetically faster than those conducted without, suggesting that this dihalogenation is an example of ligand-accelerated catalysis.12 While speculative, this mechanism has proven to be a useful hypothesis for conceptualizing our reaction and has enabled further development.
Scheme 4.

Proposed Catalytic Cycle for Bromochlorination of Allylic Alcohols.
Critical to the selectivity of our bromochlorination is the use of non-polar solvents, such as hexanes. Our working hypothesis is that the insolubility of NBS in hexanes limits unselective, non-catalyzed background halogenation; coordination of NBS to a chiral titanium complex may solubilize this source of electrophilic bromine and impart selectivity over bromonium formation. Given the heterogeneous nature of NBS in hexanes, stir rate is an important factor in these reactions with fast, efficient stirring providing optimal yield and selectivity. Coordination of titanium to allylic alcohol substrates also increases their propensity to go into solution, allowing for a surprisingly wide range of alcohols to be soluble under these conditions. In cases where allylic alcohol substrates are insoluble, carbon tetrachloride or toluene can be used as co-solvents. Despite the known sensitivity of titanium(IV) complexes to water, conducting our bromochlorination with reagent-grade hexanes and in non-flame-dried glassware has a negligible effect on yield and selectivity, which greatly facilitates reaction setup.
EXTENSION TO DICHLORINATION AND DIBROMINATION
Our approach of formally separating electrophilic and nucleophilic halogen sources provided modularity for the extension of this system to other forms of olefin difunctionalization. As our second-generation system was capable of exerting high levels of regiocontrol in halide delivery, we were ideally positioned to reinvestigate the dibromination of non-electronically biased allylic alcohols. Substitution of ClTi(Oi-Pr)3 by BrTi(Oi-Pr)3 directly provided access to enriched dibromides in considerably improved yield and enantioselectivity compared to our first-generation system (Scheme 5, top).13 Non-cinnamyl olefins were competent substrates using these conditions, which feature substantially reduced ligand loadings. Extension of this system to dichlorination by replacement of NBS with N-chlorosuccinimide provided dichlorides in low yield and selectivity. After much investigation, it was found that use of tert-butyl hypochlorite in conjunction with ClTi(Oi-Pr)3 significantly improved the efficiency and selectivity of this process. With these conditions, dichlorination proceeded with yields and selectivities similar to those obtained for dibromination (Scheme 5, bottom).
Scheme 5.

Dibromination and Dichlorination with Second-Generation System.
SYNTHETIC APPLICATIONS OF DIHALOGENATION
All examples of catalytic enantioselective halogenation in natural product synthesis thus far have been conducted by our laboratory and have utilized our above-described dihalogenation reaction. This methodology has enabled the synthesis of twelve halogenated natural products; five of these molecules had never been synthesized before and four had only been made in racemic fashion. All of our syntheses feature enantioselective dihalogenation in an early step, illustrating the practicality and scalability of the method.
VICINAL BROMOCHLORIDE NATURAL PRODUCTS
The most direct application of our methodology is found in the synthesis of chiral vicinal di- and polyhalides. Within all known vicinal dihalide natural products, bromochlorides are most common, representing over 175 naturally occurring molecules.1 In nature, these molecules presumably derive from enantioselective alkene bromochlorination, although no dihalogenation biosynthetic pathways have yet been characterized.14 Previous to our work, no selective synthetic bromochlorination reaction existed.6b The advent of our regio- and enantioselective bromochlorination method thus enabled rapid access to this class of molecules.
The premier example of a bromochlorinated natural product is halomon, a highly oxidized terpene with anticancer activity.14,15 Halomon was selected by the National Cancer Institute for preclinical drug development as it exhibited “one of the most extreme cases of differential cytotoxicity” in their human tumor cell line screen.15 However, efforts in this vein were curtailed explicitly due to lack of material.16 Two prior syntheses had been reported, however both feature nonselective dihalogenation reactions which necessitated multiple HPLC purification steps.17 Recognizing halomon as a high-value target that could prove as a testing ground for our methodology, we embarked on a selective synthesis of this pentahalogenated monoterpene (Scheme 6).18
Scheme 6.

Syntheses of (+)-Halomon, (+)-Bromochloromyrcene, (−)-Anverene, (−)-Isoplocamenone, and (−)-Plocamenone.
Gram-scale bromochlorination of known allylic alcohol 25 proceeded in good yield and 90% ee, demonstrating the robustness of our protocol. Deoxygenation of alcohol 26 to bromochloride 27 using a two-step procedure proceeded in 95% overall yield and illustrated the impressive stability of vicinal dihalides (see below) as well as the high chemoselectivity that can be achieved in the reduction of alkyl sulfonates over alkyl halides. We have found this deoxygenation procedure to be a convenient and reliable way to deoxygenate a range of dihaloalcohols. Formal 1,4-bromohydration of diene 27 provided allylic alcohol 28, which then was subjected to our conditions for dichlorination and yielded pentahalide 29 in high yield and >20:1 d.r. Dichlorination with other reagents produced pentahalide 29 in ca. 1:1 d.r. The synthesis of halomon (+)-30 was completed via activation and elimination of alcohol 29, which served as a further demonstration of the stability of polyhalides.
Syntheses of (+)-bromochloromyrcene, (−)-anverene, (−)-plocamenone, and (−)-isoplocamenone were conducted using a logic similar to that applied to (+)-halomon. Each of these syntheses makes use of a catalytic enantioselective bromochlorination as the initial step and showcases synthetically useful aspects of our method (Scheme 6, bottom).11,18,19 In our synthesis of bromochloromyrcene (+)-33, bromochlorination occurs chemoselectively at the allylic alcohol functionality, even in the presence of a more electron-rich trisubstituted olefin (31 to 32, Scheme 6). The first example of an enantioselective dihalogenation of a homoallylic alcohol is demonstrated in our synthesis of anverene (−)-36 (34 to 35, Scheme 6). Evaluating reaction parameters with this substrate revealed that the addition of titanium tetraisopropoxide led to increased yield, regio-, and enantioselectivity. Our route to isoplocamenone (−)-39 and plocamenone (−)-40 features the bromochlorination of an allylic chloride, generating an enantioenriched trihalide in a single step (37 to 38, Scheme 6). Bromochlorides 32, 35, and 38 can be efficiently translated to their respective natural products. All of these syntheses share the strategy of coupling bromochlorination with subsequent alcohol oxidation and olefination.
VICINAL POLYCHLORIDE NATURAL PRODUCTS
Having demonstrated the synthetic utility of our bromochlorination reaction, we set out to evaluate the applicability of our dichlorination system to natural product synthesis. Among chlorinated natural products, the chlorosulfolipids have garnered particular synthetic attention owing to their complex, acyclic polychlorinated stereoarrays.20 Chlorosulfolipids have been isolated from freshwater algae and toxic mussels in locations ranging from the Adriatic Sea to the Taiwan Strait. Their interest as synthetic targets derives from a limited understanding of their biogenesis, role in biological membranes, and contribution to Adriatic shellfish poisoning.20 Although some members of the chlorosulfolipid family have been synthesized, the carbon-chlorine bonds of these natural products never have been constructed directly through an enantioselective dichlorination. Instead, chlorine-bearing chiral centers were crafted through functional group interconversion or kinetic resolution. Our dihalogenation system has provided direct and scalable access to enantioenriched dichloroalcohols which readily can be elaborated to chlorosulfolipids.
Our synthesis of (–)-deschloromytilipin A commenced with dichloride 41, which was directly accessed on gram-scale and in 80% ee using our dichlorination method (Scheme 7).13 Oxidation and chloroallylation of dichloride 41 provided trichloroalcohol 42 as a single diastereomer. Acryloylation of alcohol 42 followed by ring-closing metathesis and reduction gave enediol 43. Dichlorination with Et4NCl3 and subsequent triflation and silylation yielded pentachloride 44. Copper-catalyzed alkyl−alkyl cross coupling of triflate 44 followed by sulfation completed the first synthesis of (−)-deschloromytilipin A (−)-45 in 11 steps from crotyl alcohol. This work represents the first use of a catalytic, enantioselective dichlorination in the total synthesis of chlorosulfolipid natural products.
Scheme 7.

Synthesis of (−)-Deschloromytilipin A.
The chlorosulfolipid danicalipin A has attracted much synthetic interest owing to its unusual lipid structure and to its ambiguous role in biological membranes.20 Previous syntheses of danicalipin A have installed its key 1,2-dichloroalcohol motif via racemic, diastereoselective, or stereospecific chlorination reactions; we devised a convergent strategy for synthesizing danicalipin A which leveraged our ability to access enantioenriched dichloroalcohols (Scheme 8).21 The key step of this synthesis was a coupling reaction between sensitive α,β-dichloroaldehyde 48 and complex (Z)-chlorovinylboron reagent 54 (Scheme 9). Aldehyde 48 was prepared directly from oxidation of alcohol 47.13 Synthesis of chiral boronic ester 54 commenced from commercially available boronate 49, which upon action of iodine monochloride and subsequent treatment with aqueous hydroxide in the presence of enantiopure diol 50 provided chlorovinylboronic ester 51 as a single olefin stereoisomer. Matteson homologation of boronate 51 with dichloromethyllithium provided 1,3-dichloroallyl boronic ester 52.22 Homologation of boronic ester 52 by the Grignard reagent of alkyl iodide 53 produced fully functionalized boronic ester 54. Allylboronate 54 could be induced to react with aldehyde 48 via an allylborinate according to a procedure reported by Aggarwal, affording gram quantities of chlorohydrin alkene 55 as a single enantiomer and diastereomer.23 Formal hydrochlorination of 55 was effected by bromochlorination with Me4N(Cl2Br) followed by tin-mediated radical debromination. Sulfation completed our synthesis of (−)-danicalipin A (−)-46 in eight linear steps and provides scalable access to either enantiomer of danicalipin A.
Scheme 8.

Comparison of Synthetic Strategies for (−)-Danicalipin A.
Scheme 9.

Synthesis of (−)-Danicalipin A.
A general strategy has emerged throughout our synthetic work on vicinal bromochloride and polychloride natural products. All of our syntheses commence from simple allylic alcohols that were directly subjected to catalytic, enantioselective dihalogenation at an early stage (16 to 24, Scheme 10). The alcohol used to direct dihalogenation is then either leveraged to extend the carbon backbone or is removed (Scheme 10). In the synthesis of natural products with more than one vicinal dihalide motif, this sequence of halogenation followed by alcohol manipulation can be iterated as our system can be used to control the diastereoselectivity of further halogenations. From analysis of our syntheses, we assert that it is strategically valuable to incorporate chiral dihalide functionality early in a synthesis. Such an approach provides a handle for the relay of chiral information throughout a synthesis and underscores the surprising stability of dihalides relative to their monohalide counterparts (see below). The efficiency and robustness of our halogenation methodology permits its application early in a synthesis, allowing simple allylic alcohols to be quickly elaborated into complex chiral organohalogens. The use of alcohols as directing groups in this methodology not only allows for unprecedented chemoselectivity in the halogenation of polyolefinic substrates but also may be exploited synthetically through later functional group interconversion. We anticipate that these tactics will find use in the synthesis of an array of halogenated natural products.
Scheme 10.

Summary of Strategies for Catalytic, Enantioselective Dihalogenation in Synthesis.
The increased stability of dihalides relative to monohalides is underappreciated and, in our opinion, best understood through consideration of reaction mechanism (Figure 3). In an E1 or SN1 manifold, rate-determining ionization of a monohalide produces a cationic intermediate; introduction of an additional electronegative halogen, as in the case of a dihalide, inductively destabilizes this cation and increases both the barrier to its formation as well as the relative energy of the produced cation (Figure 3a). As higher energy species, α-bromocarbocations may gain stabilization through formation of 3-membered, cyclic bromonium ions, however this intermediate should be recognized as higher in energy than the corresponding non-halogenated carbocation in comparison to their respective ground states. Solvolysis of mono- and dibromides in ethanol supports this mechanistic picture, with dibromides being three orders of magnitude slower to ionize than the corresponding monohalide, assuming ionization, and not nucleophilic capture, is rate-determining (Figure 3b).24 Dihalides are also less reactive than monohalides in an SN2 context. The rate-determining displacement of a bromide in this process is disfavored sterically in the case of dihalides owing to the additional, adjacent bromide. SN2 displacement is also disfavored electronically for dihalides; the buildup of partial positive charge on the carbon that is undergoing substitution is inductively destabilized by the presence of the nearby electronegative bromine atom (Figure 3c). These mechanistic considerations provide a basis for the surprising stability of dihalides in comparison to monohalides, a principle we did not fully appreciate at the outset of our work.
Figure 3.

Mechanistic Rationale for Dihalide Stability.
MONOBROMIDE NATURAL PRODUCTS
Having established the value of our dihalogenation system in the synthesis of polyhalide natural products, we began to explore the utility of enantioenriched dihalides in the synthesis of monohalide natural products. The ubiquity of monobrominated cyclohexane motifs in natural products provided a compelling target for our study of dihalide functionalization. These compounds biosynthetically derive from bromonium-initiated carbocationic cyclizations of isoprenoids; enzymes, such as vanadium-dependent bromoperoxidases, render bromonium formation chemo- and enantioselective (56 to 57, Scheme 11, top).25 Bromination reagents developed by Snyder have enabled efficient bromopolyene cyclizations of polyunsaturated substrates in a synthetic context, however an enantioselective variant capable of producing enantioenriched natural product-relevant scaffolds had yet to be reported at the start of our work.26 We envisioned that Nature’s bromopolyene cyclizations could be recapitulated by using enriched dihalides as progenitors of chiral non-racemizing bromonium ions (58 to 57, Scheme 11, top). Application of our dihalogenation system to polyunsaturated isoprenoid substrates seemed ideal, owing to its capability to chemoselectively functionalize polyolefins.
Scheme 11.

A Solvolytic Approach to Monobrominated Natural Products.
Alcohol-directed dihalogenation followed by deoxygenation and ionization of the resulting bromochloride would formally provide a surrogate for haloperoxidase activity. The key challenge of this approach was identifying a means to ionize bromochlorides which, as previously discussed, possess greater stability than monobromides in an SN1 manifold. This strategy was reduced to practice through use of basic hexafluoroisopropanol as a strongly ionizing medium for bromonium generation. Under these conditions a variety of enriched bromochlorides could be ionized and stereospecifically captured, providing access to valuable bromocyclic motifs for use in synthesis. Importantly, subjecting enantioenriched dibromides to these conditions results in rapid racemization. Such an approach has so far resulted in the synthesis of five monobrominated natural products.
We first applied our solvolytic functionalization approach to the brominated chamigrene sesquiterpenes, a class of more than 50 bromocyclohexane-containing natural products.1 Among these molecules is the structurally unique aplydactone and its related congener, dactylone. Aplydactone presents multiple synthetic challenges: it has two fused cyclobutanes, three contiguous quaternary centers, and a neopentyl secondary bromide. Prior reports suggested that aplydactone may arise from dactylone through an intramolecular [2+2] cycloaddition, although initial attempts to verify this hypothesis by Stonik and co-workers was unsuccessful.27 We anticipated that dactylone and its less oxidized isomeric variants, α- and β-bromochamigrene, could be assembled rapidly via our solvolytic strategy. Bromochlorogeranyl acetate 59 was prepared in five steps using the strategies developed during our synthetic work on bromochlorinated natural products (Scheme 11, middle).28 Solvolysis of bromochloride 59 on gram-scale provided key bromocyclohexane 60 with 96% enantiospecificity. Dehydration of alcohol 60 followed by oxidative cleavage yielded ketone 61 which smoothly underwent β-elimination and in situ Diels–Alder cycloaddition with isoprene to provide isomeric spirocycles 63 and 64. Spirocycle 63 was readily elaborated to (−)-α- and β-bromochamigrene (−)-65 and (−)-66 through methylation and dehydration. Spirocycle 64 was transformed in to (−)-dactylone (−)-67 through methylenation and allylic oxidation.29 Exposure of (−)-dactylone (−)-67 to ultraviolet light formed the highly-strained (+)-aplydactone (+)-68. This work constitutes the first enantioselective syntheses of (−)-dactylone and (+)-aplydactone.30
Our synthesis of (−)-bromo-epicaparrapi oxide follows a similar logic to that employed for the chamigrenes (Scheme 11, bottom). Multigram-scale solvolysis of malonate-tethered bromochloride 69 cleanly afforded bicycle 70 after acidic deprotection and thermal decarboxylation.31 Formation of Weinreb amide 71 was followed by treatment with methyllithium and vinylmagnesium chloride to yield diol 73 via ketone 72. Treatment of diol 73 with boron trifluoride diethyl etherate directly produced (−)-bromo-epicaparrapi oxide (−)-74.
Our enantiospecific solvolytic functionalization of bromochlorides has provided efficient access to monobrominated natural products. The mild conditions employed in bromochloride ionization are aptly aligned with their use in the cyclization of complex, sensitive isoprenoids. We have demonstrated the applicability of this chemistry with the synthesis of five naturally-occurring organobromides and anticipate that this novel approach to generating enantioenriched bromonium ions will enable the synthesis of other halogenated natural products.
CONCLUSION
R. B. Woodward once said that “The synthesis of substances occurring in Nature, perhaps in greater measure than activities in any other area of organic chemistry, provides a measure of the conditions and powers of science.”32 This quote aptly applies to chiral halogenated natural products, where synthesis serves as a sensitive barometer for the maturity of selective halogenation methods. In this Account, we endeavored to provide a narrative for the development of our enantioselective dihalogenation and to illustrate its use in the synthesis of both poly- and monohalogenated natural products. Clear trends emerge throughout our work and provide a general strategic outline for the application of our methodology in a synthetic context. There are currently over 2,000 known natural products containing a chlorine- or bromine-bearing stereocenter; twelve of these have been synthesized using a catalytic, enantioselective halogenation. We hope that this work encourages others both to develop synthetically-relevant, enantioselective halogenation methods and to undertake syntheses within the ever-expanding class of natural organohalogens.
Acknowledgments
This work was supported by Stanford University, the National Institutes for Health (R01 GM114061), and the National Science Foundation (DGE-114747 GRF to M.L.L.).
Biographies
Noah Z. Burns received a B.A. in chemistry from Columbia University in 2004 and a Ph.D. from the Scripps Research Institute in 2009 under the direction of Phil Baran. After continued studies at Harvard University as an NIH postdoctoral fellow in the lab of Eric Jacobsen, Noah joined the Stanford Department of Chemistry as an assistant professor in 2012.
Matthew L. Landry received an A.B. in chemistry from Princeton University in 2009. He is currently a student in the Burns group.
Footnotes
The authors declare no competing financial interest.
References
- 1.(a) Gribble GW. Naturally Occurring Organohalogen Compounds–A Comprehensive Survey. Springer-Verlag; Wien, Austria: 1996. [PubMed] [Google Scholar]; (b) Gribble GW. Naturally Occurring Organohalogen Compounds–A Comprehensive Update. Springer-Verlag; Wien Austria: 2010. [Google Scholar]
- 2.(a) Chung W-j, Vanderwal CD. Stereoselective Halogenation in Natural Product Synthesis. Angew. Chem. Int. Ed. 2016;55:4396–4434. doi: 10.1002/anie.201506388. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cheng YA, Yu WZ, Yeung Y-Y. Recent Advances in Asymmetric Intra- and Intermolecular Halofunctionalizations of Alkenes. Org. Biomol. Chem. 2014;12:2333–2343. doi: 10.1039/c3ob42335b. [DOI] [PubMed] [Google Scholar]; (c) Chen J, Zhou L. Recent Progress in the Asymmetric Intermolecular Halogenation of Alkenes. Synthesis. 2014;46:586–595. [Google Scholar]; (d) Tan CK, Yeung Y-Y. Recent Advances in Stereoselective Bromofunctionalization of Alkenes Using N-Bromoamide Reagents. Chem. Commun. 2013;49:7985–7996. doi: 10.1039/c3cc43950j. [DOI] [PubMed] [Google Scholar]
- 3.Snyder SA, Tang Z-Y, Gupta R. Enantioselective Total Synthesis of (−)-Napyradiomycin A1 via Asymmetric Chlorination of an Isolated Olefin. J. Am. Chem. Soc. 2009;131:5744–5745. doi: 10.1021/ja9014716. [DOI] [PubMed] [Google Scholar]
- 4.(a) Denmark SE, Kuester WE, Burk MT. Catalytic, Asymmetric Halofunctionalization of Alkenes–A Critical Perspective. Angew. Chem. Int. Ed. 2012;51:10938–10953. doi: 10.1002/anie.201204347. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cresswell AJ, Eey ST-C, Denmark SE. Catalytic, Stereoselective Dihalogenation of Alkenes: Challenges and Opportunities. Angew. Chem. Int. Ed. 2015;54:15642–15682. doi: 10.1002/anie.201507152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Denmark SE, Burk MT, Hoover AJ. On the Absolute Configurational Stability of Bromonium and Chloronium Ions. J. Am. Chem. Soc. 2010;132:1232–1233. doi: 10.1021/ja909965h. [DOI] [PubMed] [Google Scholar]; (b) Brown RS. Investigation of the Early Steps in Electrophilic Bromination through the Study of the Reaction with Sterically Encumbered Olefins. Acc. Chem. Res. 1997;30:131–137. [Google Scholar]
- 6.(a) Nicolaou KC, Simmons NL, Ying Y, Heretsch PM, Chen JS. Enantioselective Dichlorination of Allylic Alcohols. J. Am. Chem. Soc. 2011;133:8134–8137. doi: 10.1021/ja202555m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Soltanzadeh B, Jaganathan A, Yi Y, Yi H, Staples RJ, Borhan B. Highly Regio- and Enantioselective Vicinal Dihalogenation of Allyl Amides. J. Am. Chem. Soc. 2017;139:2132–2135. doi: 10.1021/jacs.6b09203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) El-Qisairi AK, Qaseer HA, Katsigras G, Lorenzi P, Trivedi U, Tracz S, Hartman A, Miller JA, Henry PM. New Palladium(II)-Catalyzed Asymmetric 1,2-Dibromo Synthesis. Org. Lett. 2003;5:439–441. doi: 10.1021/ol0273093. [DOI] [PubMed] [Google Scholar]; (b) Denmark SD, Carson N. Reinvestigation of a Catalytic, Enantioselective Alkene Dibromination and Chlorohydroxylation. Org. Lett. 2015;17:5728–5731. doi: 10.1021/acs.orglett.5b02650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katsuki T, Sharpless KB. The First Practical Method for Asymmetric Epoxidation. J. Am. Chem. Soc. 1980;102:5974–5976. [Google Scholar]
- 9.Schmidt E, Ascherl A, von Knilling W. Das gleichartige Verhalten von persubstituierten Halogenverbindungen und Halogenyl-acylaminen. Ber. Dtsch. Chem. Ges. 1926;59:1876–1888. [Google Scholar]
- 10.Hu DX, Shibuya GM, Burns NZ. Catalytic Enantioselective Dibromination of Allylic Alcohols. J. Am. Chem. Soc. 2013;135:12960–12963. doi: 10.1021/ja4083182. [DOI] [PubMed] [Google Scholar]
- 11.Hu DX, Seidl FJ, Bucher CB, Burns NZ. Catalytic Chemo-, Regio-, and Enantioselective Bromochlorination of Allylic Alcohols. J. Am. Chem. Soc. 2015;137:3795–3798. doi: 10.1021/jacs.5b01384. [DOI] [PubMed] [Google Scholar]
- 12.Berrisford DJ, Bolm C, Sharpless KB. Ligand-Accelerated Catalysis. Angew. Chem. Int. Ed. Engl. 1995;34:1059–1070. [Google Scholar]
- 13.Landry ML, Hu DX, McKenna GM, Burns NZ. Catalytic Enantioselective Dihalogenation and the Selective Synthesis of (−)-Deschloromytilipin A and (−)-Danicalipin A. J. Am. Chem. Soc. 2016;138:5150–5158. doi: 10.1021/jacs.6b01643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burreson JB, Woolard FX, Moore RE. Evidence for the Biogenesis of Halogenated Myrcenes from the Red Alga Chondrococcus hornemannii. Chem. Lett. 1975;4:1111–1114. [Google Scholar]
- 15.Fuller RW, Cardellina JH, Kato Y, Brinen LS, Clardy J, Snader KM, Boyd MR. A Pentahalogenated Monoterpene from the Red Alga Portieria hornemannii Produces a Novel Cytotoxicity Profile Against a Diverse Panel of Human Tumor Cell Lines. J. Med. Chem. 1992;35:3007–3011. doi: 10.1021/jm00094a012. [DOI] [PubMed] [Google Scholar]
- 16.Fuller RW, Cardellina JH, Jurek J, Scheuer PJ, Alvarado-Lindner B, McGuire M, Gray GN, Steiner JR, Clardy J, Menez E, Shoemaker RH, Newman DJ, Snader KM, Boyd MR. Isolation and Structure/Activity Features of Halomon-Related Antitumor Monoterpenes from the Red Alga Portieria hornemannii. J. Med. Chem. 1994;37:4407–4411. doi: 10.1021/jm00051a019. [DOI] [PubMed] [Google Scholar]
- 17.(a) Schlama T, Baati R, Gouverneur V, Valleix A, Falck JR, Mioskowski C. Total Synthesis of (±)-Halomon by a Johnson-Claisen Rearrangement. Angew. Chem. Int. Ed. 1998;37:2085–2087. doi: 10.1002/(SICI)1521-3773(19980817)37:15<2085::AID-ANIE2085>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; (b) Sotokawa T, Noda T, Pi S, Hirama M. A Three-Step Synthesis of Halomon. Angew. Chem. Int. Ed. 2000;39:3430–3432. doi: 10.1002/1521-3773(20001002)39:19<3430::aid-anie3430>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 18.Bucher CB, Deans RM, Burns NZ. Highly Selective Synthesis of Halomon, Plocamenone, and Isoplocamenone. J. Am. Chem. Soc. 2015;137:12784–12787. doi: 10.1021/jacs.5b08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seidl FJ, Burns NZ. Selective Bromochlorination of a Homoallylic Alcohol for the Total Synthesis of (−)-Anverene. Beilstein J. Org. Chem. 2016;12:1361–1365. doi: 10.3762/bjoc.12.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) Bedke DK, Vanderwal CD. Chlorosulfolipids: Structure, Synthesis, and Biological Relevance. Nat. Prod. Rep. 2011;28:15–25. doi: 10.1039/c0np00044b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nilewski C, Carreira EM. Recent Advances in the Total Synthesis of Chlorosulfolipids. Eur. J. Org. Chem. 2012:1685–1698. [Google Scholar]; (c) Chung W-J, Vanderwal CD. Approaches to the Chemical Synthesis of the Chlorosulfolipids. Acc. Chem. Res. 2014;47:718–728. doi: 10.1021/ar400246w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Yoshimitsu T, Nakatani R, Kobayashi A, Tanaka T. Asymmetric Total Synthesis of (+)-Danicalipin A. Org. Lett. 2011;13:908–911. doi: 10.1021/ol1029518. [DOI] [PubMed] [Google Scholar]; (b) Umezawa T, Shibata M, Kaneko K, Okino T, Matsuda F. Asymmetric Total Synthesis of Danicalipin A and Evaluation of Biological Activity. Org. Lett. 2011;13:904–907. doi: 10.1021/ol102882a. [DOI] [PubMed] [Google Scholar]; (c) Chung W-j, Carlson JS, Vanderwal CD. General Approach to the Synthesis of the Chlorosulfolipids Danicalipin A, Mytilipin A, and Malhamensilipin A in Enantioenriched Form. J. Org. Chem. 2014;79:2226–2241. doi: 10.1021/jo5000829. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Bailey AM, Wolfrum S, Carreira EM. Biological Investigations of (+)-Danicalipin A Enabled Through Synthesis. Angew. Chem. Int. Ed. 2016;55:639–643. doi: 10.1002/anie.201509082. [DOI] [PubMed] [Google Scholar]
- 22.(a) Matteson DS, Majumdar D. α-Chloro Boronic Esters from Homologation of Boronic Esters. J. Am. Chem. Soc. 1980;102:7588–7590. [Google Scholar]; (b) Matteson DS. Boronic Esters in Asymmetric Synthesis. J. Org. Chem. 2013;78:10009–10023. doi: 10.1021/jo4013942. [DOI] [PubMed] [Google Scholar]
- 23.(a) Chen JL-Y, Scott HK, Hesse MJ, Willis CL, Aggarwal VK. Highly Diastereo- and Enantioselective Allylboration of Aldehydes using α-Substituted Allyl/Crotyl Pinacol Boronic Esters via in Situ Generated Borinic Esters. J. Am. Chem. Soc. 2013;135:5316–5319. doi: 10.1021/ja401564z. [DOI] [PubMed] [Google Scholar]; (b) Chen JL-Y, Aggarwal VK. Highly Diastereoselective and Enantiospecific Allylation of Ketones and Imines Using Borinic Esters: Contiguous Quaternary Stereogenic Centers. Angew. Chem. Int. Ed. 2014;53:10992–10996. doi: 10.1002/anie.201407127. [DOI] [PubMed] [Google Scholar]
- 24.Winstein S, Grunwald E. Neighboring Groups and Reactivity. J. Am. Chem. Soc. 1946;68:536–537. doi: 10.1021/ja01205a038. [DOI] [PubMed] [Google Scholar]
- 25.Carter-Franklin JN, Butler AJ. Vanadium Bromoperoxidase-Catalyzed Biosynthesis of Halogenated Marine Natural Products. J. Am. Chem. Soc. 2004;126:15060–15066. doi: 10.1021/ja047925p. [DOI] [PubMed] [Google Scholar]
- 26.(a) Snyder SA, Treitler DS, Brucks AP. Simple Reagents for Direct Halonium-induced Polyene Cyclizations. J. Am. Chem. Soc. 2010;132:14303–14314. doi: 10.1021/ja106813s. [DOI] [PubMed] [Google Scholar]; (b) Snyder SA, Treitler DS, Schall A. A Two-step Mimic for Direct, Asymmetric Bromonium- and Chloronium-induced Polyene Cyclizations. Tetrahedron. 2010;66:4796–4804. [Google Scholar]; (c) Samanta RC, Yamamoto H. Catalytic Asymmetric Bromocyclization of Polyenes. J. Am. Chem. Soc. 2017;139:1460–1463. doi: 10.1021/jacs.6b13193. [DOI] [PubMed] [Google Scholar]; (d) Arnold AM, Pöthig A, Drees M, Gulder T. NXS, Morpholine, and HFIP: The Ideal Combination for Biomimetic Haliranium-Induced Polyene Cyclizations. J. Am. Chem. Soc. ASAP. doi: 10.1021/jacs.8b00113. [DOI] [PubMed] [Google Scholar]
- 27.Fedorov SN, Radchenko OS, Shubina LK, Kalinovsky AI, Gerasimenko AV, Popov DY, Stonik VA. Aplydactone, a New Sesquiterpenoid with an Unprecedented Carbon Skeleton from the Sea Hare Aplysia dactylomela, and Its Cargill-Like Rearrangement. J. Am. Chem. Soc. 2001;123:504–505. doi: 10.1021/ja003254t. [DOI] [PubMed] [Google Scholar]
- 28.Burckle AJ, Vasilev VH, Burns NZ. A Unified Approach for the Enantioselective Synthesis of the Brominated Chamigrene Sesquiterpenes. Angew. Chem. Int. Ed. 2016;55:11476–11479. doi: 10.1002/anie.201605722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan T-H, Tsai C-C, Chien C-T, Cho C-C, Huang P-C. Dichloromethane Activation. Direct Methylenation of Ketones and Aldehydes with CH2Cl2 Promoted by Mg/TiCl4/THF. Org. Lett. 2004;6:4961–4963. doi: 10.1021/ol0478887. [DOI] [PubMed] [Google Scholar]
- 30.(a) Meier R, Trauner D. A Synthesis of (±)-Aplydactone. Angew. Chem. Int. Ed. 2016;55:11251–11255. doi: 10.1002/anie.201604102. [DOI] [PubMed] [Google Scholar]; (b) Shen M, Kretschmer M, Brill ZG, Snyder SA. Strategies for the Total Synthesis of Diverse Bromo-Chamigrenes. Org. Lett. 2016;18:5018–5021. doi: 10.1021/acs.orglett.6b02478. [DOI] [PubMed] [Google Scholar]; (c) Liu C, Chen R, Shen Y, Liang Z, Hua Y, Zhang Y. Total Synthesis of Aplydactone by a Conformationally Controlled C-H Functionalization. Angew. Chem. Int. Ed. 2017;56:8187–8190. doi: 10.1002/anie.201703803. [DOI] [PubMed] [Google Scholar]
- 31.Burckle AJ, Gál B, Seidl FJ, Vasilev VH, Burns NZ. Enantiospecific Solvolytic Functionalization of Bromochlorides. J. Am. Chem. Soc. 2017;139:13562–13569. doi: 10.1021/jacs.7b07792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Todd AR. Perspectives in Organic Chemistry. Interscience Publishers, Inc.; New York: 1956. [Google Scholar]