

Abstract
Preterm preeclampsia is associated with the failure of trophoblast invasion, placental hypoxic/ischemic injury and the release of toxic substances, which promote the terminal pathway of preeclampsia. In term preeclampsia, factors yet unknown trigger the placenta to induce the terminal pathway. The contribution of the villous trophoblast to these pathologic events has not been fully elucidated. Here we aimed to study how stress and signaling pathways influence trophoblastic functions in various subforms of preeclampsia. Tissue microarrays (TMAs) were constructed from placentas obtained from pregnant women in the following groups: 1-2) preterm preeclampsia with (n=8) or without (n=7) HELLP syndrome; 3) late-onset preeclampsia (n=8); 4-5) preterm (n=5) and term (n=9) controls. TMA slides were stained for phosphorylated Akt-1, ERK1/2, JNK, and p38 kinases, and trophoblastic immunostainings were semi-quantitatively evaluated. BeWo cells were kept in various stress conditions, and the expression of FLT1, GCM1, LEP, and PGF was profiled by qRT-PCR, while Akt-1, ERK1/2, JNK, and p38 kinase activities were measured with phospho-kinase immunoassays. We found that: 1) Placental LEP and FLT1 expression was up-regulated in preterm preeclampsia with or without HELLP syndrome compared to controls; 2) Mean pp38 immunoscore was higher in preterm preeclampsia, especially in cases with HELLP syndrome, than in controls. 3) Mean pERK1/2 immunoscore was higher in preterm preeclampsia with HELLP syndrome than in controls. 4) In BeWo cells, ischemia up-regulated LEP expression, and it increased JNK and decreased ERK1/2 activity. 5) Hypoxia up-regulated FLT1 and down-regulated PGF expression, and it increased ERK1/2, JNK and p38 activity. 6) IL-1β treatment down-regulated PGF expression, and it increased JNK and p38 activity. 7) The p38 signaling pathway had the most impact on LEP, FLT1 and PGF expression. In conclusion, hypoxic and ischemic stress, along with unknown factors, activates trophoblastic p38 signaling, which has a key role in villous trophoblastic functional changes in preterm preeclampsia. The activation of ERK1/2 signaling may induce additional trophoblastic functional changes in HELLP syndrome, while distinct mechanisms may promote late-onset preeclampsia.
Keywords: anti-angiogenic factor, hypertension, mitogen activated protein kinase, oxidative stress, pregnancy, signal transduction, syncytiotrophoblast
INTRODUCTION
Preeclampsia, diagnosed by new-onset hypertension and proteinuria after 20 weeks of gestation, is a major cause of maternal, perinatal and neonatal morbidity and mortality [1]. It is a syndrome consisting of various subforms among which preterm preeclampsia develops before, while term preeclampsia develops after the 37th week of gestation. Another sub-classification has suggested the 34th week to differentiate between early-onset and late-onset preeclampsia [2]. A growing body of evidence has shown that late-onset or term preeclampsia is often mild, while preterm or early-onset preeclampsia is more severe and is often associated with HELLP (Hemolysis, Elevated Liver enzymes, and Low Platelet count) syndrome and intrauterine growth restriction (IUGR) [1–4]. Although the pathogenesis of preeclampsia is not completely understood, it is evident that the placenta plays an important role, since the only effective cure of preeclampsia is the delivery of the placenta [1, 4].
The pathogenesis of preeclampsia may differ in its subforms based on histopathological [5, 6] and transcriptomic [7, 8] evidence. Preterm preeclampsia is frequently associated with the failure of trophoblast invasion and consequent abnormal remodeling of the uterine spiral arteries [9, 10] resulting in hypoxic/ischemic placental injury [11–17]. Subsequently, the stressed placenta releases increased amounts of anti-angiogenic factors [e.g. soluble fms-like tyrosine kinase 1 (sFlt-1/sVEGFR1), soluble endoglin (sEng)] [18–20], pro-inflammatory molecules [e.g. leptin, corticotropin-releasing hormone (CRH)] [7, 15, 21, 22] and syncytiotrophoblast debris [15, 23]. The release of toxic placental substances and the decreased availability of pro-angiogenic molecules [e.g. placenta growth factor (PGF)] then promotes the terminal pathway of preeclampsia, including maternal systemic anti-angiogenic and pro-inflammatory states, generalized endothelial dysfunction, leukocyte activation, hypertension and proteinuria [4, 15, 17, 18, 20, 24–28]. Although placental histopathological and transcriptomic changes are less prevalent in late-onset preeclampsia [4–6], it has recently been suggested that normal placental growth may also induce syncytiotrophoblastic stress and increased production of sFlt-1 late in pregnancy, promoting the progression of the terminal pathway [29].
Studies focusing on placental injury or the placental dysregulation of genes in preeclampsia have revealed a complex interplay between hypoxia and ischemia in the activation or inhibition of various signaling pathways, leading to altered gene expression and placental functions [11–17, 30]. Our recent microarray study [7] has shown the differential placental expression of a set of genes in preterm preeclampsia and HELLP syndrome, which are induced by villous trophoblast differentiation [7, 31, 32]. This finding suggested an important role of the syncytiotrophoblast, the terminally differentiated villous trophoblast that covers the placenta towards maternal blood, in preeclampsia pathogenesis [7]. In fact, defects in villous trophoblast differentiation and syncytialization, characteristic transcriptomic, biochemical and morphological changes governed by cAMP [31, 33, 34], were hypothesized to promote the development of preeclampsia [35]. Since several genes that are induced by cAMP-signaling and trophoblast differentiation (e.g. CGB, CRH, LEP) have increased expression in preeclampsia [7, 8, 36], while the placental expression of the syncytialization-inducer GCM1 is down-regulated in early-onset preeclampsia [37], emerging evidence suggests that villous trophoblast syncytialization rather than differentiation is altered in early-onset preeclampsia. However, the contribution of various signaling pathways in the dysregulation of villous trophoblast physiology and the pathogenesis of various preeclampsia subforms has not been elucidated to date.
This study targeted how stress and signaling pathways influence villous trophoblastic functions in distinct subforms of preeclampsia by the investigation of kinases involved in the regulation of cell survival, proliferation, differentiation, apoptosis, and the release of inflammatory mediators and anti-angiogenic factors [15, 17]. In addition, selected genes were examined, which are highly expressed by the villous trophoblast and have been implicated in the placental pathogenesis of preeclampsia [8, 37–39]. In accord, the aims of the current study were: 1) to examine the in vivo changes in villous trophoblastic signaling pathways in different subforms of preeclampsia by the high-throughput analysis of immunostaining signatures of the selected protein kinases; 2) to determine the in vitro effect of cAMP signaling on the trophoblastic expression of the selected genes; and 3) to model the alterations in villous trophoblastic signaling pathways and gene expression signatures in preeclampsia in vitro by applying various stress conditions to differentiating BeWo cells.
MATERIALS AND METHODS
The collection and use of human biological materials for research purposes were approved by the Health Science Board (Budapest, Hungary) and the Institutional Review Boards of Wayne State University (Detroit, MI, USA) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD, NIH, DHHS, Bethesda, MD, USA). Placentas were histopathologically examined and used for microarray and tissue microarray (TMA) studies [40]. Immunostained TMAs were digitally scanned and used for virtual microscopic evaluations [41]. Primary trophoblasts isolated from normal term placentas [38] and BeWo cells were used for functional studies. Total RNA and protein lysates were isolated and analyzed with quantitative real-time reverse transcription PCR (qRT-PCR) and phospho-kinase assays, respectively. Demographic, clinical and immunoscore data were analyzed using SPSS v.12.0 (SPSS Inc., Chicago, IL, USA). All other data were analyzed in the R statistical language (www.r-project.org). All methods are described in detail in the Supplementary Information; additional data are shown in Suppl. Tables 1–4 and Suppl. Figs. 1–4.
RESULTS
Demographic, clinical and histopathological data
Demographic and clinical characteristics are displayed in Suppl. Table 1. Peak systolic and diastolic blood pressures were higher in all patient groups than in controls. Proteinuria was detected in all cases, but not in controls. Term and preterm controls were matched to cases within two weeks of gestational age; however, the median gestational age of term controls was slightly higher than that of cases with late-onset preeclampsia. Birth weight was significantly lower in late-onset preeclampsia, and it tended to be lower in preterm preeclampsia with or without HELLP syndrome than in respective controls.
Placental gene expression changes in preterm preeclampsia with or without HELLP syndrome
First, we revisited our placental microarray data [7] for potential target genes of signaling pathways altered in the villous trophoblast. Out of the selected four genes, we found two to be differentially expressed in our microarray data (Suppl. Fig. 1). Placental LEP expression was 108.9-fold (q=6.4×10−5) and 56.9-fold (q=0.002) up-regulated in preterm preeclampsia without or with HELLP syndrome compared to preterm controls, respectively. Similarly, placental FLT1 expression was 3.6-fold (q=0.085) and 4.3-fold (q=0.04) up-regulated in preterm preeclampsia without or with HELLP syndrome compared to preterm controls, respectively. The magnitude of changes for GCM1 (1.9-fold) and PGF (1.8-fold) were higher in preterm preeclampsia with HELLP syndrome than without this severe syndrome; however, the differential expression of these genes did not reach statistical significance according to the criteria set by our study [7]. Nevertheless, the placental down-regulation of these genes in preeclampsia has been shown by recent studies [37–39].
Changes in trophoblastic signaling pathways in preeclampsia and HELLP syndrome
Next, we looked for alterations in trophoblastic signaling pathways by evaluating phosphorylated Akt-1, ERK1/2, JNK, and p38 immunostainings of the villous trophoblast in various subforms of preeclampsia. In accord with the findings of a parallel study on placental signaling pathway alterations in preeclampsia [30], the most impact of preeclampsia was observed on the p38 signaling pathway. Phosphorylated p38 immunoscore was higher in all preterm preeclampsia cases (mean±SD: 2.07±0.39, p<0.001) than in preterm controls (1.19±0.51), while it was not changed in late-onset preeclampsia (1.60±0.39, p=0.5) compared to term controls (1.33±0.53) (Fig. 1). Among cases of preterm preeclampsia, the mean pp38 immunoscore was increased in larger extent in cases with HELLP syndrome (2.15±0.43, p=0.004) than in those who did not have HELLP syndrome (1.98±0.37, p=0.01) (Fig. 1).
Fig. 1. Villous trophoblastic pp38 immunostaining is increased in preterm preeclampsia associated with or without HELLP syndrome.
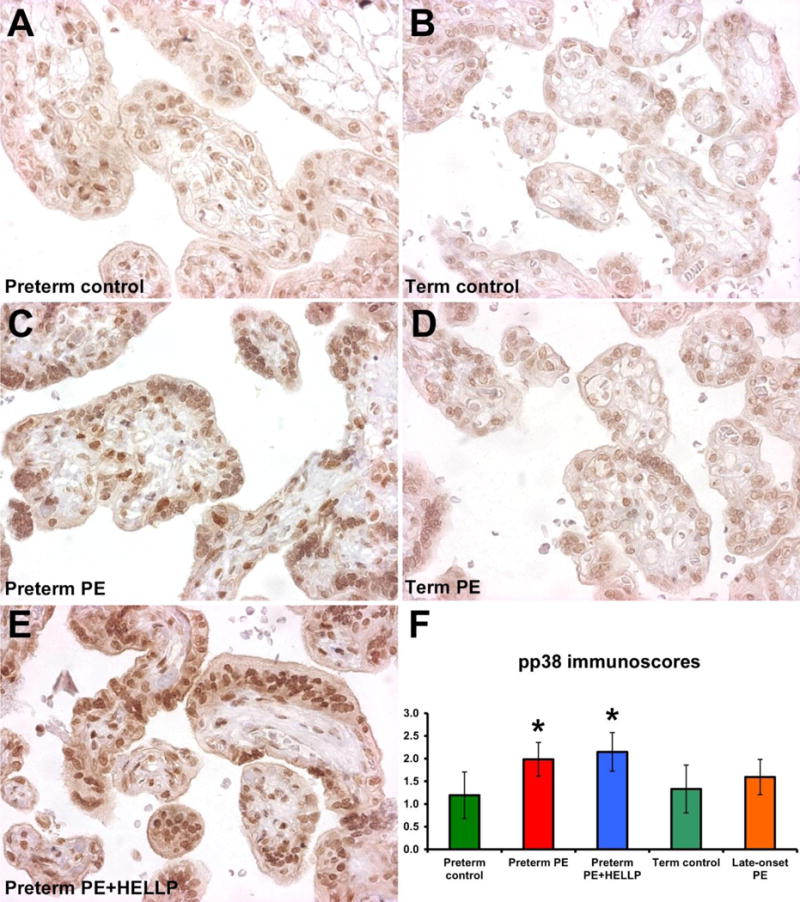
A-E) Phosphorylated p38 (pp38) immunostaining was detected mostly in the nuclei of villous trophoblasts. A,C,E) Villous trophoblastic nuclear immunostaining was stronger in preterm preeclampsia with or without HELLP syndrome than in preterm controls. B,D) There was no difference in villous trophoblastic nuclear immunostaining between term controls and late-onset preeclampsia cases. Representative images for the groups are shown in 400× magnifications. F) Villous trophoblastic pp38 immunoscores were higher in preterm preeclampsia (mean+SD: 1.98+0.37, p=0.01) and in preterm preeclampsia with HELLP syndrome (2.15+0.43, p=0.004) than in preterm controls (1.19+0.51). Mean pp38 immunoscores were not different between late-onset preeclampsia (1.60+0.39, p=0.5) and term controls (1.33+0.53). PE, preeclampsia; HELLP, HELLP syndrome; PE+HELLP, preeclampsia with HELLP syndrome. Stars denote significant changes.
Similar, but less intense changes were observed in the ERK1/2 signaling pathway (Suppl. Fig. 2A). The mean pERK1/2 immunoscore tended to be higher in all preterm preeclampsia cases (1.23±0.56, p=0.09) than in preterm controls (0.72±0.50), while it was not changed in late-onset preeclampsia (0.46±0.54, p=0.8) compared to term controls (0.53±0.52). Among cases of preterm preeclampsia, the mean pERK1/2 immunoscore was significantly higher in cases associated with HELLP syndrome (1.33±0.38, p=0.03), while the increase in mean pERK1/2 immunoscore did not reach statistical significance in cases who did not have HELLP syndrome (1.11±0.73, p=0.3).
There was no change in pJNK immunoscores in preterm or term preeclampsia although it tended to be higher in late-onset preeclampsia (1.80±0.4, p=0.14) compared to term controls (1.51±0.37) (Suppl. Fig. 2B). We did not observe difference between pAkt-1 immunoscores in any of the comparisons (Suppl. Fig. 2C). These results suggest that mitogen-activated protein kinase (MAPK) signaling pathways are activated in the villous trophoblast in preterm but not in late-onset preeclampsia, and that these changes are exaggerated in preterm preeclampsia cases associated with HELLP syndrome.
Trophoblast differentiation affects angiogenic/anti-angiogenic gene expression balance
Next, we modeled in vitro how the alterations in signaling pathways may impact villous trophoblastic expression of selected genes. To optimize our assays, we differentiated primary villous trophoblasts obtained from normal term placentas, and observed that days 2 and 3 represented the peak expression for the selected genes (Suppl. Fig. 3A). The expression of FLT1 modestly changed, while PGF expression had large increase by day 3, suggesting that differentiation affects angiogenic/anti-angiogenic gene expression balance in the trophoblast, which may have impact on the development of preeclampsia.
Because of the limitations in obtaining enough primary trophoblast cells for the experiments, we looked for the gene expression changes in the trophoblast-like BeWo cells, which syncytialize similarly to primary trophoblasts after cAMP-induced differentiation. When we differentiated BeWo cells with the cAMP-analogue Forskolin, we observed a plateau in the expression of the selected genes between days 2 and 3 of differentiation (Suppl. Fig. 3B). Similar to primary trophoblasts, we detected a remarkable increase in PGF and LEP expression during differentiation, while FLT1 expression changed only modestly. Since a relatively high and stable expression of these genes was needed for further experiments with BeWo cells, we chose to use the day 2 to 3 time-window in differentiation.
Trophoblastic hypoxia and ischemia mimic gene expression changes in preterm preeclampsia
To study the changes in selected genes’ expression in stress conditions relevant to preeclampsia in the placenta, we differentiated BeWo cells, and then applied three different stress conditions (Fig. 2A): 1) In ischemia, LEP expression increased by 2.1-fold (p=0.04), in accord with that LEP was the most up-regulated placental gene in preterm preeclampsia and HELLP syndrome in our study [7]. We did not see a difference in FLT1 or PGF expression in this condition. 2) In hypoxia, the expression of FLT1 increased by 2.5-fold (p=0.056), while PGF was down-regulated by 1.4-fold (p=0.04), consistent with the increased placental FLT1 expression in preterm preeclampsia [7]. 3) After IL-1β treatment that mimicked pro-inflammatory changes in the placenta, we observed the down-regulation of PGF by 1.6-fold (p=0.01); however, there was no significant change in the expression of FLT1 or LEP. These findings suggest that the combination of ischemia and hypoxia along with unknown factors may promote the functional changes in the villous trophoblast in preterm preeclampsia and HELLP syndrome.
Fig. 2. Gene expression and kinase activity changes in BeWo cells under various stress conditions.
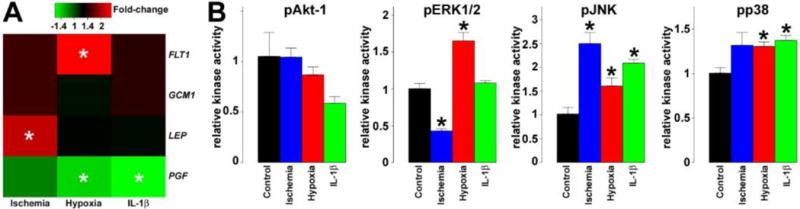
A) Heatmap representing qRT-PCR data reveals various effects of stress conditions on FLT1, GCM1, LEP and PGF expression in differentiating BeWo cells. The color bar depicts fold-changes relative to differentiating BeWo cells in normoxic conditions. In ischemia, LEP expression increased by 2.1-fold (p=0.04). In hypoxia, the expression of FLT1 increased by 2.5-fold (p=0.056), while PGF was down-regulated by 1.4-fold (p=0.04). After IL-1β treatment, PGF expression was decreased by 1.6-fold (p=0.01). B) Bar-charts representing kinase activity assay data reveal various effects of stress conditions on pAkt-1, pERK1/2, pJNK and pp38 activities in differentiating BeWo cells (n=3). In ischemia, JNK had a 2.5-fold increased activity (q=0.03), ERK1/2 had a 2.4-fold decreased activity (q=0.02), while there was a 1.3-fold, marginally significant increase in p38 activity (q=0.17). In hypoxia, the activity pERK1/2 (1.7-fold, q=0.03), pJNK (1.6-fold, q=0.1) and pp38 (1.3-fold, q=0.06) increased significantly. After IL-1β treatment, pJNK had a 2.1-fold increased activity (q=0.03), while pp38 activity was increased by 1.4-fold (q=0.04). All experiments were run in triplicate. Stars denote significant changes.
Hypoxia and ischemia mimic kinase pathway changes in preterm preeclampsia
To investigate the possible changes in the activities of kinases that are stimulated by oxidative stress and may promote the observed gene expression changes, we treated BeWo cells in the same way as for the qRT-PCR experiments, and kinase activity assays were run on protein lysates (Fig. 2B): 1) In ischemia, JNK had a 2.5-fold increased activity (q=0.03), ERK1/2 had a 2.4-fold decreased activity (q=0.02), while there was a 1.3-fold, marginally significant increase in p38 activity (q=0.17). 2) In hypoxia, there was a 1.7-fold increase in ERK1/2 activity (q=0.03), a 1.6-fold increase in JNK activity (q=0.1), and a 1.3-fold increase in p38 activity (q=0.06). 3) After IL-1β treatment, JNK had a 2.1-fold increased activity (q=0.03), while p38 activity was increased by 1.4-fold (q=0.04). These findings suggest that trophoblastic p38 and JNK kinases may be activated in all three stress conditions, while the effect of ischemia and hypoxia may be opposing on ERK1/2 activation in the trophoblast.
p38 signaling regulates gene expression changes in hypoxic and ischemic BeWo cells
Finally, we tested how the inhibition of kinase pathways may change gene expression patterns in stress conditions applied to BeWo cells compared to normoxic control BeWo cells. As depicted in Fig. 3 and Suppl. Fig. 4, in normoxia, the ERK1/2 inhibitor decreased the expression of FLT1, LEP and PGF, in line with the role of ERK1/2 in supporting trophoblast differentiation and the expression of trophoblastic genes [42]. In hypoxia, the p38 and JNK inhibitors had the most effect on gene expression compared to normoxic control cells, in line with the role of p38 and JNK kinases in signaling environmental stress in the placenta [22, 43, 44]. In ischemia, the p38 and Akt-1 inhibitors had the most effect on gene expression compared to normoxic control cells, while the p38 inhibitor impacted mostly gene expression after IL-1β treatment compared to normoxic control cells. Overall, p38 signaling had the most impact on LEP, FLT1 and PGF expression in the applied stress conditions.
Fig. 3. Gene expression changes in BeWo cells treated with kinase inhibitors under various stress conditions.
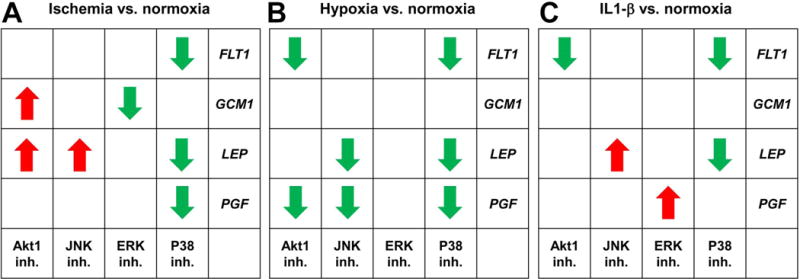
The tables represent altered effects of kinase inhibitors on gene expression in differentiating BeWo cells in a given stress condition compared to control, normoxic conditions. The actual qRT-PCR data is presented in Suppl. Fig. 4. Red arrows depict increase in expression so that up-regulation trends observed in normoxia become significant in the current stress condition or the significance of down-regulation observed in normoxia is lost in the current stress condition. Green arrows represent decrease in expression so that down-regulation trends observed in normoxia become significant in the current stress condition or the significance of up-regulation observed in normoxia is lost in the current stress condition.
DISCUSSION
Principal findings of this study
1) Trophoblastic pp38 immunoscore was higher in preterm preeclampsia, especially in cases with HELLP syndrome, than in controls, while it was not changed in late-onset preeclampsia; 2) pERK1/2 immunoscore was higher in preterm preeclampsia with HELLP syndrome than in controls while it was not changed in other patient groups; 3) Placental LEP and FLT1 expression was up-regulated in preterm preeclampsia with or without HELLP syndrome compared to controls; 4) In BeWo cells, ischemia up-regulated LEP expression, and it increased JNK and decreased ERK1/2 activity; 5) Hypoxia up-regulated FLT1 and down-regulated PGF expression, and it increased ERK1/2, JNK and p38 activity; 6) IL-1β treatment down-regulated PGF expression, and it increased JNK and p38 activity; and 7) The p38 signaling pathway had the most impact on LEP, FLT1 and PGF expression in BeWo cells under stress conditions compared with control normoxic BeWo cells.
Villous trophoblastic signaling pathways affected in preterm preeclampsia and HELLP syndrome
The emerging data suggest a complex interplay between hypoxia and ischemia in the activation and inhibition of signaling pathways in the placenta, leading to altered functions and gene expression, and the pathogenesis of preeclampsia [11–17]. First, the increased placental production and release of anti-angiogenic sFlt-1 and sEng in hypoxia was found [13, 14]. Subsequently, in vitro and in vivo evidence have suggested that oxidative stress may play a dominant role in the pathogenesis of preeclampsia by activating the MAPK and NFκB signaling pathways and inhibiting the PI3 kinase/Akt pathway in the placenta [15, 17, 22]. These signaling pathways strongly impact cell survival or apoptosis, cell proliferation or differentiation, and the release of inflammatory mediators and anti-angiogenic factors, all relevant for the pathogenesis of preeclampsia [15, 17]. Among MAPK kinases, ERK1/2 can be activated by mitogenic signals and reactive oxygen species (ROS), whereas JNK and p38 signaling are initiated largely by ROS and pro-inflammatory conditions [42, 45]. When activated by ROS, the ERK1/2 pathway promotes cell survival and proliferation, while JNK and p38 pathways are inducers of apoptosis [15, 45]. These findings are particularly significant since trophoblastic apoptosis and aponecrosis have been implicated as key pathologic events in preeclampsia [41, 46, 47].
Studies to date only focused on the examination of altered signaling in the whole placenta in preeclampsia. Our study is the first to explore the contribution of the villous trophoblast to placental stress signaling in distinct subforms of preeclampsia and HELLP syndrome. Among the characteristic alterations, the most significant change was found for the activated pp38 kinase, which had a higher mean immunoscore in the villous trophoblast in preterm preeclampsia compared to controls, while there was no change in the pp38 immunoscore in late-onset preeclampsia. Of note, no change was observed in the activated pJNK kinase immunoscores in the disease groups (Fig. 4). These findings are in accord with the observations on placental stress pathways in preeclampsia by a parallel study [30], and suggest that preterm but not late-onset preeclampsia is associated with villous trophoblastic stress. Moreover, these results are consistent with the more extensive placental pathology [4–6, 9, 10] and transcriptomic changes [7, 8], and the earlier and larger increase in anti-angiogenic protein release from the placenta [4] in preterm than in late-onset preeclampsia. Of interest, among patients with preterm preeclampsia, the pp38 immunoscore was more robustly increased in cases with HELLP syndrome, suggestive for a heightened stress response in the villous trophoblast (Fig. 4). This phenomenon is in accord with the findings of our microarray study using the same placentas, which showed a more severe placental pathology and pronounced inflammatory gene expression signature in cases with HELLP syndrome [7].
Fig. 4. Summary figure.
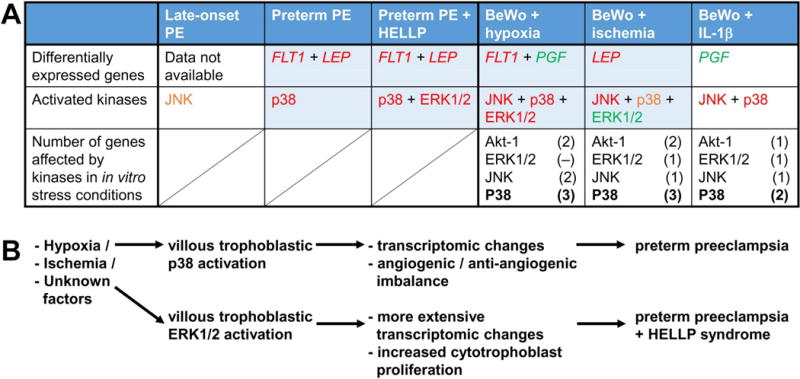
A) The table summarizes differential expression among selected genes and activation among selected kinases in placentas in the disease groups and in BeWo cells in the applied stress conditions. Red letters denote significant increase, orange letters denote marginal increase, while green letters denote significant decrease. Data in boxes highlighted with blue suggest that trophoblastic ischemia and hypoxia may lead to the signaling and transcriptomic changes observed in preterm preeclampsia. Treatments with kinase inhibitors revealed the key role of p38 signaling in impacting trophoblastic gene expression unique to various stress conditions. B) The activation of trophoblastic p38 signaling may be key in placental transcriptomic changes and consequent angiogenic/anti-angiogenic imbalance in preterm preeclampsia. The trophoblastic activation of ERK1/2 signaling may drive the more extensive transcriptomic changes and placental inflammation, and the increased cytotrophoblast proliferation in preterm preeclampsia with HELLP syndrome compared to preterm preeclampsia alone.
Patients with HELLP syndrome had a significant increase in the pERK1/2 immunoscore compared to those without HELLP syndrome (Fig. 4), suggestive for an additional stimulus that could facilitate the activation of the villous trophoblastic ERK1/2 signaling. Of note, the activation of ERK1/2 signaling promotes cell proliferation [15, 45], and it is pivotal for the regulation of cytotrophoblast proliferation and differentiation [42]. In accord with our findings, cytotrophoblast proliferation was found to be increased in placentas from patients with HELLP syndrome but not in placentas from women with preeclampsia as evaluated by Ki67 immunostainings [48, 49], suggestive for an ERK1/2 signaling mediated imbalance in cytotrophoblast proliferation and differentiation in HELLP syndrome.
Hypoxia and ischemia alters gene expression in BeWo cells similar to those found in preeclampsia
Next, we investigated which stress stimuli may activate a similar response in BeWo cells as in the villous trophoblast in preeclampsia. We set up an in vitro model in which we induced BeWo cells with Forskolin to syncytialize in order to model the mixture of cytotrophoblasts and the syncytiotrophoblast in the villi, and used a time-window for the stress stimuli when the expression of the four genes was high and stable.
The first experiments revealed that the pro-inflammatory condition mimicked by IL-1β treatment did not increase FLT1 or LEP expression, suggesting that it cannot be the cause of the placental transcriptomic changes in preeclampsia. On the other hand, ischemia up-regulated LEP expression, while hypoxia increased FLT1 expression and decreased PGF expression. Since the placental up-regulation of LEP and FLT1 is a hallmark of preterm preeclampsia [7, 8], our in vitro results suggest that the combination of ischemia and hypoxia may promote the characteristic changes in gene expression and function of the villous trophoblast in preterm preeclampsia and HELLP syndrome. Since neither of these in vitro stress conditions could solely mimic placental findings in preterm preeclampsia, it is possible that the extent of hypoxia or ischemia applied in our experiments, or the use of BeWo cells, might have been suboptimal for modeling villous trophoblastic stress in preeclampsia. Moreover, yet unknown factors in addition to hypoxia or ischemia may also contribute to these transcriptomic and functional changes in the villous trophoblast in preeclampsia. Supporting this latter concept, it has recently been shown that preeclampsia is associated with the differential methylation of LEP and FLT1 in the placenta in preeclampsia, which may promote the increased placental expression of these genes [50, 51].
Signaling pathways affected in BeWo cells by hypoxic, ischemic and pro-inflammatory stimuli
Interestingly, all applied stress conditions could induce the activation of the p38 and JNK pathways, hypoxia and ischemia had an opposing effect on ERK1/2 signaling, while IL-1β did not affect this latter pathway (Fig. 4). Similar to that proposed by the gene expression changes, kinase activity assays also suggest the combination of hypoxia and ischemia to lead to the closest simulation of altered signaling pathways in the villous trophoblast in preterm preeclampsia. Our results may also suggest that a robust hypoxic component besides ischemia may activate ERK1/2 signaling in the villous trophoblast and the development of HELLP syndrome. Nevertheless, we can conclude that either the applied in vitro stress conditions could not entirely mimic those present in vivo, or yet unknown factors in addition to hypoxia or ischemia may also contribute to villous trophoblastic stress in preeclampsia and HELLP syndrome.
The use of various kinase inhibitors revealed that the ERK1/2 pathway had the most impact on gene expression in normoxic conditions in accord with the pivotal role of ERK1/2 in villous trophoblast differentiation [42]. However, in all stress conditions when compared to normoxia, p38 signaling had the most impact on trophoblastic LEP, FLT1 and PGF expression. These findings are in line with the role of p38 kinase in signaling environmental stress in the villous placenta [22], and they underline its pivotal role in the development of the anti-angiogenic and pro-inflammatory states in preeclampsia.
CONCLUSIONS
Hypoxic and ischemic stress, along with unknown factors, activates trophoblastic p38 signaling, which may have a key role in villous trophoblastic functional changes in preterm preeclampsia, associated with or without HELLP syndrome. The activation of ERK1/2 signaling may induce trophoblastic functional changes characteristic for HELLP syndrome, while distinct mechanisms may promote the development of late-onset preeclampsia.
Supplementary Material
Acknowledgments
The experiments, analysis and interpretation of data and/or writing of the manuscript were supported, in part, by: the European Union FP6 grant “Pregenesys 037244” (to N.G.T.); the Perinatology Research Branch, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD, NIH, DHHS); Federal funds from the NICHD under Contract No. HHSN275201300006C; and the Hungarian Academy of Sciences Momentum grant (#LP2014-7/2014 to N.G.T.). The authors thank everyone who made this work possible, including patients, nurses, lab staff, and clinicians. The authors are grateful to Dr. Katalin Éder, Edit Parsch, Dr. Tibor Várkonyi (Semmelweis University), Po Jen Chiang, Jianhua Du and Dr. Theodore Price (Wayne State University) for their helpful technical assistance, and Sara Tipton (Wayne State University) for her critical reading of the manuscript.
Footnotes
AUTHOR CONTRIBUTIONS
SzSz, YX, TF and NGT designed research; RR, IK, ZP, and NGT contributed clinical specimens / analytical tools; SzSz, MM, YX, KK, NM, and TK performed research; SzSz, MM, RR, YX, NM, ZX, GB, TF, PH, JR, ALT, SSH, TC, IK, ZP, and NGT analyzed / interpreted data; and SzSz, MM, RR, YX, KK, NM, ZX, GB, TF, PH, TK, JR, ALT, SSH, TC, IK, ZP, and NGT wrote the manuscript.
DISCLOSURE/CONFLICT OF INTEREST
The authors have no conflicts of interest.
References
- 1.Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005;365(9461):785–799. doi: 10.1016/S0140-6736(05)17987-2. [DOI] [PubMed] [Google Scholar]
- 2.von Dadelszen P, Magee LA, Roberts JM. Subclassification of preeclampsia. Hypertens Pregnancy. 2003;22(2):143–148. doi: 10.1081/PRG-120021060. [DOI] [PubMed] [Google Scholar]
- 3.Weinstein L. Syndrome of hemolysis, elevated liver enzymes, and low platelet count: a severe consequence of hypertension in pregnancy. Am J Obstet Gynecol. 1982;142(2):159–167. doi: 10.1016/s0002-9378(16)32330-4. [DOI] [PubMed] [Google Scholar]
- 4.Than NG, Vaisbuch E, Kim CJ, Mazaki-Tovi S, Erez O, Yeo L, Mittal P, Hupuczi P, Varkonyi T, Hassan SS, Papp Z, Romero R. Early-Onset Preeclampsia and HELLP Syndrome: An Overview. Handbook of growth and growth monitoring in health and disease. 2012:1867–1891. [Google Scholar]
- 5.Moldenhauer JS, Stanek J, Warshak C, Khoury J, Sibai B. The frequency and severity of placental findings in women with preeclampsia are gestational age dependent. Am J Obstet Gynecol. 2003;189(4):1173–1177. doi: 10.1067/s0002-9378(03)00576-3. [DOI] [PubMed] [Google Scholar]
- 6.Ogge G, Chaiworapongsa T, Romero R, Hussein Y, Kusanovic JP, Yeo L, Kim CJ, Hassan SS. Placental lesions associated with maternal underperfusion are more frequent in early-onset than in late-onset preeclampsia. J Perinat Med. 39(6):641–652. doi: 10.1515/JPM.2011.098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varkonyi T, Nagy B, Fule T, Tarca AL, Karaszi K, Schonleber J, Hupuczi P, Mihalik N, Kovalszky I, Rigo J, Jr, Meiri H, Papp Z, Romero R, Than NG. Microarray profiling reveals that placental transcriptomes of early-onset HELLP syndrome and preeclampsia are similar. Placenta. 2011;32(Suppl):S21–29. doi: 10.1016/j.placenta.2010.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleinrouweler CE, van Uitert M, Moerland PD, Ris-Stalpers C, van der Post JA, Afink GB. Differentially expressed genes in the pre-eclamptic placenta: a systematic review and meta-analysis. PLoS ONE. 2013;8(7):e68991. doi: 10.1371/journal.pone.0068991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brosens IA. Morphological changes in the utero-placental bed in pregnancy hypertension. Clin Obstet Gynaecol. 1977;4(3):573–593. [PubMed] [Google Scholar]
- 10.Brosens I, Pijnenborg R, Vercruysse L, Romero R. The "Great Obstetrical Syndromes" are associated with disorders of deep placentation. Am J Obstet Gynecol. 2011;204(3):193–201. doi: 10.1016/j.ajog.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genbacev O, Joslin R, Damsky CH, Polliotti BM, Fisher SJ. Hypoxia alters early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental defects that occur in preeclampsia. J Clin Invest. 1996;97(2):540–550. doi: 10.1172/JCI118447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Redman CW, Sargent IL. Placental debris, oxidative stress and pre-eclampsia. Placenta. 2000;21(7):597–602. doi: 10.1053/plac.2000.0560. [DOI] [PubMed] [Google Scholar]
- 13.Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, Zamudio S, Post M, Caniggia I. Molecular evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab. 2005;90(7):4299–4308. doi: 10.1210/jc.2005-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, Many A, Zamudio S, Caniggia I. Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol. 2006;291(4):R1085–1093. doi: 10.1152/ajpregu.00794.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cindrova-Davies T. Gabor Than Award Lecture 2008: pre-eclampsia – from placental oxidative stress to maternal endothelial dysfunction. Placenta. 2009;30(Suppl A):S55–65. doi: 10.1016/j.placenta.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 16.Burton GJ, Woods AW, Jauniaux E, Kingdom JC. Rheological and physiological consequences of conversion of the maternal spiral arteries for uteroplacental blood flow during human pregnancy. Placenta. 2009;30(6):473–482. doi: 10.1016/j.placenta.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burton GJ, Yung HW, Cindrova-Davies T, Charnock-Jones DS. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta. 2009;30(Suppl A):S43–48. doi: 10.1016/j.placenta.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111(5):649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaiworapongsa T, Romero R, Kim YM, Kim GJ, Kim MR, Espinoza J, Bujold E, Goncalves L, Gomez R, Edwin S, Mazor M. Plasma soluble vascular endothelial growth factor receptor-1 concentration is elevated prior to the clinical diagnosis of pre-eclampsia. J Matern Fetal Neonatal Med. 2005;17(1):3–18. doi: 10.1080/14767050400028816. [DOI] [PubMed] [Google Scholar]
- 20.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, Bdolah Y, Lim KH, Yuan HT, Libermann TA, Stillman IE, Roberts D, D’Amore PA, Epstein FH, Sellke FW, Romero R, Sukhatme VP, Letarte M, Karumanchi SA. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12(6):642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 21.Ng EK, Leung TN, Tsui NB, Lau TK, Panesar NS, Chiu RW, Lo YM. The concentration of circulating corticotropin-releasing hormone mRNA in maternal plasma is increased in preeclampsia. Clin Chem. 2003;49(5):727–731. doi: 10.1373/49.5.727. [DOI] [PubMed] [Google Scholar]
- 22.Cindrova-Davies T, Spasic-Boskovic O, Jauniaux E, Charnock-Jones DS, Burton GJ. Nuclear factor-kappa B, p38, and stress-activated protein kinase mitogen-activated protein kinase signaling pathways regulate proinflammatory cytokines and apoptosis in human placental explants in response to oxidative stress: effects of antioxidant vitamins. Am J Pathol. 2007;170(5):1511–1520. doi: 10.2353/ajpath.2007.061035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crocker I. Gabor Than Award Lecture 2006: pre-eclampsia and villous trophoblast turnover: perspectives and possibilities. Placenta. 2007;28(Suppl A):S4–13. doi: 10.1016/j.placenta.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 24.Sacks GP, Studena K, Sargent K, Redman CW. Normal pregnancy and preeclampsia both produce inflammatory changes in peripheral blood leukocytes akin to those of sepsis. Am J Obstet Gynecol. 1998;179(1):80–86. doi: 10.1016/s0002-9378(98)70254-6. [DOI] [PubMed] [Google Scholar]
- 25.Gervasi MT, Chaiworapongsa T, Pacora P, Naccasha N, Yoon BH, Maymon E, Romero R. Phenotypic and metabolic characteristics of monocytes and granulocytes in preeclampsia. Am J Obstet Gynecol. 2001;185(4):792–797. doi: 10.1067/mob.2001.117311. [DOI] [PubMed] [Google Scholar]
- 26.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308(5728):1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 27.Than NG, Abdul Rahman O, Magenheim R, Nagy B, Fule T, Hargitai B, Sammar M, Hupuczi P, Tarca AL, Szabo G, Kovalszky I, Meiri H, Sziller I, Rigo J, Jr, Romero R, Papp Z. Placental protein 13 (galectin-13) has decreased placental expression but increased shedding and maternal serum concentrations in patients presenting with preterm pre-eclampsia and HELLP syndrome. Virchows Arch. 2008;453(4):387–400. doi: 10.1007/s00428-008-0658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szalai G, Xu Y, Romero R, Chaiworapongsa T, Xu Z, Chiang PJ, Ahn H, Sundell B, Plazyo O, Jiang Y, Olive M, Jacques SM, Qureshi F, Tarca AL, Erez O, Dong Z, Papp Z, Hassan SS, Hernandez-Andrade E, Than NG. In vivo experiments reveal the good, the bad and the ugly faces of sFlt-1 in pregnancy. PLoS ONE. 2014;9(11):e110867. doi: 10.1371/journal.pone.0110867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Redman CW, Sargent IL, Staff AC. IFPA Senior Award Lecture: Making sense of pre-eclampsia Two placental causes of preeclampsia? Placenta. 2014;35:S20–S25. doi: 10.1016/j.placenta.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 30.Yung HW, Atkinson D, Campion-Smith T, Olovsson M, Charnock-Jones DS, Burton GJ. Differential activation of placental unfolded protein response pathways implies heterogeneity in causation of early- and late-onset pre-eclampsia. J Pathol. 2014;234(2):262–276. doi: 10.1002/path.4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aronow BJ, Richardson BD, Handwerger S. Microarray analysis of trophoblast differentiation: gene expression reprogramming in key gene function categories. Physiol Genomics. 2001;6(2):105–116. doi: 10.1152/physiolgenomics.2001.6.2.105. [DOI] [PubMed] [Google Scholar]
- 32.Ge YC, Li JN, Ni XT, Guo CM, Wang WS, Duan T, Sun K. Cross talk between cAMP and p38 MAPK pathways in the induction of leptin by hCG in human placental syncytiotrophoblasts. Reproduction. 2011;142(2):369–375. doi: 10.1530/REP-11-0053. [DOI] [PubMed] [Google Scholar]
- 33.Kudo Y, Boyd CA, Sargent IL, Redman CW, Lee JM, Freeman TC. An analysis using DNA microarray of the time course of gene expression during syncytialization of a human placental cell line (BeWo) Placenta. 2004;25(6):479–488. doi: 10.1016/j.placenta.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Bischof P, Irminger-Finger I. The human cytotrophoblastic cell, a mononuclear chameleon. Int J Biochem Cell Biol. 2005;37(1):1–16. doi: 10.1016/j.biocel.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 35.Huppertz B. Placental origins of preeclampsia: challenging the current hypothesis. Hypertension. 2008;51(4):970–975. doi: 10.1161/HYPERTENSIONAHA.107.107607. [DOI] [PubMed] [Google Scholar]
- 36.Karteris E, Vatish M, Hillhouse EW, Grammatopoulos DK. Preeclampsia is associated with impaired regulation of the placental nitric oxide-cyclic guanosine monophosphate pathway by corticotropin-releasing hormone (CRH) and CRH-related peptides. J Clin Endocrinol Metab. 2005;90(6):3680–3687. doi: 10.1210/jc.2004-2210. [DOI] [PubMed] [Google Scholar]
- 37.Chen CP, Chen CY, Yang YC, Su TH, Chen H. Decreased placental GCM1 (glial cells missing) gene expression in pre-eclampsia. Placenta. 2004;25(5):413–421. doi: 10.1016/j.placenta.2003.10.014. [DOI] [PubMed] [Google Scholar]
- 38.Than NG, Romero R, Xu Y, Erez O, Xu Z, Bhatti G, Leavitt R, Chung TH, El-Azzamy H, LaJeunesse C, Wang B, Balogh A, Szalai G, Land S, Dong Z, Hassan SS, Chaiworapongsa T, Krispin M, Kim CJ, Tarca AL, Papp Z, Bohn H. Evolutionary origins of the placental expression of chromosome 19 cluster galectins and their complex dysregulation in preeclampsia. Placenta. 2014;35:855–865. doi: 10.1016/j.placenta.2014.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Andraweera PH, Dekker GA, Laurence JA, Roberts CT. Placental expression of VEGF family mRNA in adverse pregnancy outcomes. Placenta. 2012;33(6):467–472. doi: 10.1016/j.placenta.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 40.Redline RW. Placental pathology: a systematic approach with clinical correlations. Placenta. 2008;29(Suppl A):S86–91. doi: 10.1016/j.placenta.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Ratts VS, Tao XJ, Webster CB, Swanson PE, Smith SD, Brownbill P, Krajewski S, Reed JC, Tilly JL, Nelson DM. Expression of BCL-2, BAX and BAK in the trophoblast layer of the term human placenta: a unique model of apoptosis within a syncytium. Placenta. 2000;21(4):361–366. doi: 10.1053/plac.1999.0486. [DOI] [PubMed] [Google Scholar]
- 42.Daoud G, Amyot M, Rassart E, Masse A, Simoneau L, Lafond J. ERK1/2 and p38 regulate trophoblasts differentiation in human term placenta. J Physiol. 2005;566(Pt 2):409–423. doi: 10.1113/jphysiol.2005.089326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cindrova-Davies T, Yung HW, Johns J, Spasic-Boskovic O, Korolchuk S, Jauniaux E, Burton GJ, Charnock-Jones DS. Oxidative stress, gene expression, and protein changes induced in the human placenta during labor. Am J Pathol. 2007;171(4):1168–1179. doi: 10.2353/ajpath.2007.070528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li M, Wu ZM, Yang H, Huang SJ. NFkappaB and JNK/MAPK activation mediates the production of major macrophage- or dendritic cell-recruiting chemokine in human first trimester decidual cells in response to proinflammatory stimuli. J Clin Endocrinol Metab. 2011;96(8):2502–2511. doi: 10.1210/jc.2011-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang C, Liang J, Qian J, Jin L, Du M, Li M, Li D. Opposing role of JNK-p38 kinase and ERK1/2 in hydrogen peroxide-induced oxidative damage of human trophoblast-like JEG-3 cells. Int J Clin Exp Pathol. 2014;7(3):959–968. [PMC free article] [PubMed] [Google Scholar]
- 46.Huppertz B, Rote NS, Nelson DM, Reister F, Black S, Hunt JS. Apoptosis: molecular control of placental function–a workshop report. Placenta. 2001;22(Suppl A):S101–103. doi: 10.1053/plac.2001.0645. [DOI] [PubMed] [Google Scholar]
- 47.Longtine MS, Chen B, Odibo AO, Zhong Y, Nelson DM. Villous trophoblast apoptosis is elevated and restricted to cytotrophoblasts in pregnancies complicated by preeclampsia, IUGR, or preeclampsia with IUGR. Placenta. 2012;33(5):352–359. doi: 10.1016/j.placenta.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jeschke U, Schiessl B, Mylonas I, Kunze S, Kuhn C, Schulze S, Friese K, Mayr D. Expression of the proliferation marker Ki-67 and of p53 tumor protein in trophoblastic tissue of preeclamptic, HELLP, and intrauterine growth-restricted pregnancies. Int J Gynecol Pathol. 2006;25(4):354–360. doi: 10.1097/01.pgp.0000225838.29127.6. [DOI] [PubMed] [Google Scholar]
- 49.Prusac IK, Zekic Tomas S, Roje D. Apoptosis, proliferation and Fas ligand expression in placental trophoblast from pregnancies complicated by HELLP syndrome or pre-eclampsia. Acta Obstet Gynecol Scand. 2011;90(10):1157–1163. doi: 10.1111/j.1600-0412.2011.01152.x. [DOI] [PubMed] [Google Scholar]
- 50.Hogg K, Blair JD, von Dadelszen P, Robinson WP. Hypomethylation of the LEP gene in placenta and elevated maternal leptin concentration in early onset pre-eclampsia. Mol Cell Endocrinol. 2013;367(1-2):64–73. doi: 10.1016/j.mce.2012.12.018. [DOI] [PubMed] [Google Scholar]
- 51.Sundrani DP, Reddy US, Joshi AA, Mehendale SS, Chavan-Gautam PM, Hardikar AA, Chandak GR, Joshi SR. Differential placental methylation and expression of VEGF, FLT-1 and KDR genes in human term and preterm preeclampsia. Clin Epigenetics. 2013;5(1):6. doi: 10.1186/1868-7083-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.