

Summary
The adult mammalian intestine is composed of two connected structures, the absorptive villi and the crypts, which house progenitor cells. Mouse crypts develop postnatally and are the architectural unit of the stem cell niche, yet the pathways that drive their formation are not known. Here, we combine transcriptomic, quantitative morphometric and genetic analyses to identify mechanisms of crypt development. We uncover the upregulation of a contractility gene network at the earliest stage of crypt formation, which drives myosin II-dependent apical constriction and invagination of the crypt progenitor cells. Subsequently, hinges form, compartmentalizing crypts from villi. Hinges contain basally constricted cells, and this cell shape change was inhibited by increased hemidesmosomal adhesion in Rac1 null mice. Loss of hinges resulted in reduced villar spacing, revealing an unexpected role for crypts in tissue architecture and physiology. These studies provide a framework for studying crypt morphogenesis and identify essential regulators of niche formation.
Graphical abstract
Sumigray et al. uncover cell biological regulators of stem cell niche morphogenesis in the intestine. Using transcriptomic and genetic approaches, they find two key steps in crypt genesis: an initial apical constriction that is required for invagination and a subsequent compartmentalization of crypts, which promote villar spacing and absorptive activity.
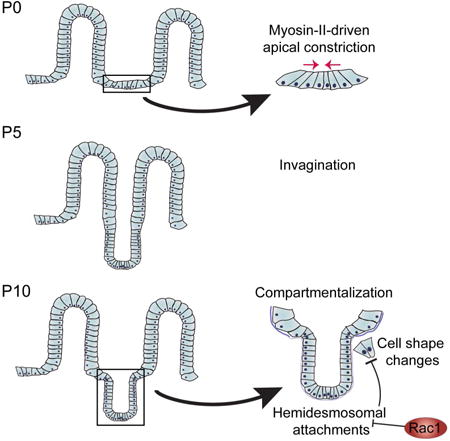
Introduction
The mammalian intestinal epithelium is arranged in a series of finger-like projections into the lumen called villi, and invaginations into the mesenchyme called crypts. The villi are composed of terminally differentiated cells, including absorptive enterocytes, goblet cells, and enteroendocrine cells, whereas the crypt contains stem and transit amplifying cells. The actively cycling adult intestinal stem cells, also known as crypt base columnar cells, sit at the base of the crypt between the terminally differentiated Paneth cells (Clevers, 2013; Tan and Barker, 2014). Crypt base columnar stem cells generate transit-amplifying cells that undergo 4-5 divisions as they move up the crypt axis (Marshman et al., 2002). Cells then exit the crypt compartment and simultaneously undergo differentiation as they enter the villus. Whether crypt exit and differentiation onset are necessarily linked is unknown.
Villus formation occurs during embryogenesis. In mice, this is driven by the formation of mesenchymal cell clusters that induce overlying epithelial cells to form villi; in chick, contraction of the underlying smooth muscle drives villus morphogenesis (Shyer et al., 2015; Shyer et al., 2013; Walton et al., 2012). In either case, formation of the villi also results in the establishment of intervillar domains that contain progenitors. This is due to compartmentalization of signals, such as Shh and Bmp4, which repress progenitor fates in the villi (Walton et al., 2012; Walton et al., 2016). Later events then transform the intervillar region from flat sheets of epithelial cells into cup-like crypts. However, the cell biological mechanisms driving crypt formation have not been reported.
Although the crypt is the architectural unit of the intestinal stem cell niche, the function of this structure in stem cell establishment/maintenance and in organ physiology remains unknown. Possible functions include increasing the area available for the number of progenitor cells needed to fuel the rapid turnover of the intestinal epithelium, compartmentalizing signals between villi and crypts, and protecting stem cells from soluble signals in the lumen. Data supporting this last role has been reported for the colon, where a metabolite generated by the lumenal microbiota suppresses stem cell proliferation (Kaiko et al., 2016). However, because the mechanisms underlying crypt morphogenesis have not been defined, these hypotheses have not yet been directly tested, and we therefore lack insight into the functions of this niche architecture.
To understand how crypt morphogenesis occurs, we combined quantitative morphometric and RNA-seq analyses to identify architectural changes and molecular regulators of crypt morphogenesis. These analyses led us to identify two distinct pathways involved in crypt formation. First, myosin II-mediated apical constriction is required for the earliest stage of crypt invagination. Subsequently, we demonstrate that a hinge region forms between crypts and villi to morphologically compartmentalize them. The formation of this region requires the small GTPase Rac1, which acts to suppress hemidesmosomal integrins in nascent crypts. In the absence of Rac1, remodeling of the basal surface of cells, which is required for hinge cell formation, does not occur. Finally, our data demonstrate that crypt-villus compartmentalization is required for proper villar spacing and mesoscale patterning of the small intestine.
Results
To analyze postnatal crypt morphogenesis, we first needed to label this heterogeneous intervillar cell population. Prior studies focused on profiling adult Lgr5-positive stem cells, which constitute a minority of cells within the crypt compartment. Thus, we searched for markers that labeled the entire developing crypt throughout its morphogenesis. We found that the hyaluronic acid receptor CD44v6 was robustly expressed in crypt progenitor units throughout the formation of the crypt, consistent with previous reports that it is expressed both embryonically (Shyer et al., 2015) and in adults (Alho and Underhill, 1989)(Fig 1A,B). In both tissue sections and epithelial whole mounts, CD44v6 marked discrete cell populations between the villi. Even at postnatal day 0 (P0), there were clear intervillar units before any overt crypt morphogenesis had occurred (Fig 1B, inset). Therefore, we define crypt progenitor units by their CD44v6 expression. Notably, proliferation is confined to the CD44v6 population already before birth (Crosnier et al., 2006; Shyer et al., 2015).
Figure 1. Intestinal crypts form postnatally.
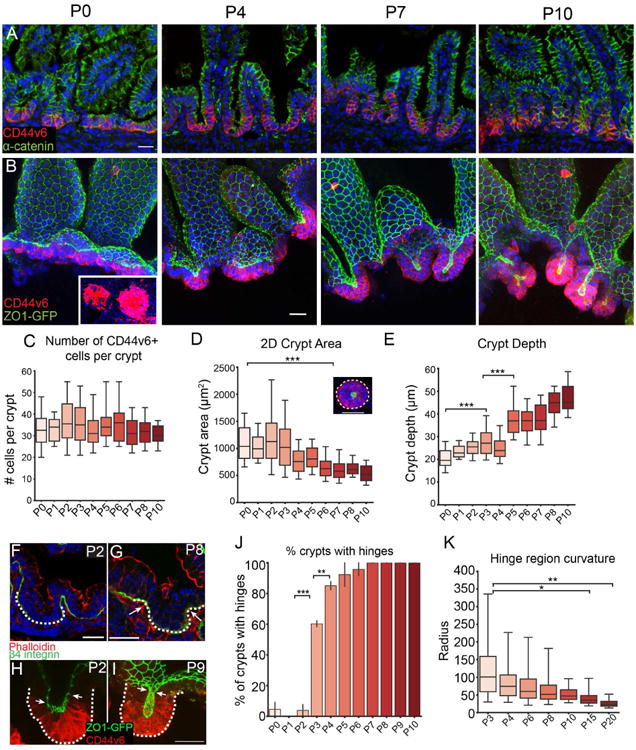
(A,B) Time course of crypt development in tissue sections (A) or in epithelial whole mounts (B). CD44v6 (red) stains intervillar cells in both, α-catenin (green) marks cell-cell adhesions in (A) and ZO1-GFP marks tight junctions in (B). Inset, CD44v6 staining of crypts as viewed from the bottom to reveal distinct morphological units. Images in B are maximum intensity projections. (C) Quantification of number of CD44v6+ cells per crypt. (D) 2D area of crypts at indicated stages. ANOVA, p < 0.05; P0 vs. P6 Tukey HSD, p < 0.001. Inset, representative image of a crypt with its 2D area outlined. (E) Depth of crypts at indicated stages. ANOVA, p < 0.05; P0 vs. P3 Tukey HSD, p < 0.001; P3 vs. P5 Tukey HSD, p < 0.001. (F,G) Basement membrane stained with b4-integrin (green) at P2 (F) and P8 (G) to visualize hinges. The crypt region is marked by a dotted line. Arrows in (G) point to hinge regions. (H,I) Projection of a crypt prior to invagination (H) and after (I), with the crypt marked by a dotted line. The boundary between CD44v6+ crypts and CD44v6- villi are marked by white arrows. ZO1-GFP (green), CD44v6 (red). (J) Quantification of the percentage of crypts with distinguishable hinge regions. ANOVA, p < 0.05; P2 vs. P3 Tukey HSD, p < 0.001; P3 vs. P4 Tukey HSD, p < 0.01. (K) Measurement of the curvature in the hinge region using the ThreepointROI plugin for ImageJ. ANOVA, p < 0.05; P3 vs. P15 Tukey HSD, p < 0.05; P3 vs. P20 Tukey HSD, p < 0.01. All scale bars are 20 μm. For each measurement, a one-way ANOVA was performed, followed by Tukey's HSD test for each pair. *, p < .05, **, p < 0.01, ***, p < .001. For all box and whisker plots, the box boundaries are the 25th and 75th percentiles, and the whiskers are the 5th and 95th percentiles. For all bar plots, the error bars are the SEM. Total number of mice analyzed were 2-4 mice per stage, and more than 10 crypts per mouse. See also Figure S1.
Epithelial whole mounts from ZO1-GFP mice, which marks the apical tight junctions of the epithelial cells (Foote et al., 2013; Huebner et al., 2014), allowed us to quantitate both cell number and morphometry. Because intestinal morphogenesis occurs in a wave from anterior to posterior, we confined all analyses to the medial third of the small intestine. From P0 to P10, the CD44v6-positive compartment reorganized from a flat sheet of cells to a cup-like invagination (Fig 1A,B). Notably, the architecture of the developing crypt was preserved in epithelial whole-mounts in which supporting mesenchymal structures were removed. While this does not rule out roles for surrounding tissue in crypt morphogenesis, it demonstrates that the maintenance of tissue structure is autonomous to the epithelium. Despite their change in appearance, the mean number of cells within these morphogenetic units was constant over the first 10 postnatal days (Fig 1C), thus demonstrating that proliferation does not drive this reorganization. While these cells are still actively proliferating, they give rise to differentiated cells rather than expanding the transit amplifying cell population. During initial morphogenesis (i.e., up to P10), the nascent crypt maintains just above 30 cells (± 9 cells, standard deviation, SD). However, from about P15 onward it undergoes dramatic elongation and expansion of the transit amplifying cell population (Fig S1).
Although the number of cells within each crypt did not change, there was a decrease in the 2D crypt area, as measured by rotating 3D reconstructions of crypts to view from the bottom, then manually outlining the perimeter of the CD44v6-positive region (Fig 1D). The CD44v6-positive area was just above 1 mm2 from P0-P3, and then decreased to approximately half that size by P6. Concomitantly, crypt depth, measured as the distance from the crypt mouth (ZO1-labeled tight junctions of most luminal CD44v6+ cell) to the crypt base (basal side of most distal CD44v6+ cell) increased as it invaginated into the underlying mesenchyme (Fig 1E).
As crypts formed via invagination, we noted the formation of a pronounced border between crypts and villi. We term this region the “hinge”, as the crypt-villus axis contains a clear boundary in adults. This is a three-dimensional structure that forms a rim or ridge at the top of the crypts. This results in a clear plateau between crypts and villi (Fig 2A, rightmost image). When cells were still arranged in a flat sheet (P0-P2), there was no obvious hinge region when viewing either basal (Fig 1F) or apical membranes (Fig 1H). However, a bend with a broad curvature appeared by P3 in approximately 60% of crypts (Fig 1J). We measured the radius of the inflection zone and found that the radius continued to decrease over development, as a sharper angle formed between the developing crypt and villus compartments (Fig 1K). By P7, 100% of crypts examined had defined hinge regions when examining either basal or apical markers (Fig 1G,I) and thus had morphologically compartmentalized their crypts and villi (Fig 1J). Crypt hinges have not been morphometrically characterized before, and the cell biological basis/machinery for their formation is currently unknown.
Figure 2. Identification of core crypt-enriched and villar-enriched genes in the postnatal intestine.
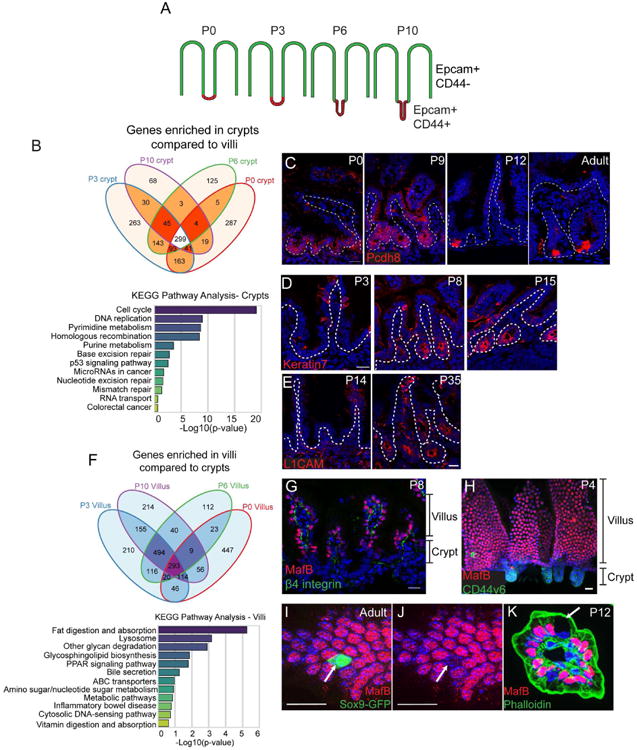
(A) Schematic for FACs sorting strategy. Stages of CD1 mice are listed above each intestinal image. Epcam marks all epithelial cells, while CD44 marks intervillar cells. (B) Venn diagram illustrating the number of genes enriched in crypts compared to villi (at least 2-fold, p < .05) at each stage. A total of 299 genes were enriched in crypts compared to villi at all stages examined. Below is a graph of KEGG pathway terms for genes enriched in crypts versus villi at all stages examined. (C) Staining of the crypt-enriched marker Pcdh8 (PAPC, red) in the medial small intestine at indicated stages. Pcdh8 is broadly expressed in crypts from P0-P9, then becomes enriched in cells at the base of the crypt by P12. The dotted line marks the basement membrane. (D) Keratin 7 (red) staining in P3, P8, and P15 crypts showing its induction at late stages of crypt morphogenesis. (E) L1CAM (red) staining in P14 and P35 crypts showing its induction after morphogenesis is complete. (F) Venn diagram illustrating the number of genes enriched in villi compared to crypts (at least 2-fold, p < .05) at each stage. Below is a graph of KEGG pathway terms for genes enriched in villi compared to crypts at all stages examined. (G) MafB (red) and β4 integrin (green) localization in an intestinal tissue section. MafB is exclusively expressed in cells in the villi. (H) Whole mount staining of MafB (red) and CD44v6 (green). (I-J) MafB (red) staining of a Sox9-GFP (green) intestine. The arrow indicates a Sox9-GFP-positive villar cell that is negative for MafB staining. (K) MafB (red) and phalloidin (green) staining of a villus in cross-section. The arrow indicates a Goblet cell, which does not express MafB. All scale bars are 20 μm. For RNA-seq, 5-10 littermates were pooled for each RNA-seq sample. See also Figures S2 and S3.
Dynamic transcriptome analysis of developing crypts and villi
To identify molecular regulators of the crypt morphogenetic program we characterized above, we developed a FACS isolation protocol to purify developing crypt and corresponding villar cells. Epcam marked all epithelial cells, while CD44 marked the intervillar cells. We sorted Epcam+/CD44- cells and Epcam+/CD44+ cells from the medial small intestine of wild type mice at four stages - P0, P3, P6, and P10 (Fig 2A, Fig S2A-B). qPCR analysis verified that the stem cell marker Ascl2 was enriched in CD44+/Epcam+ cells, whereas the enterocyte marker Lactase was enriched in CD44-/Epcam+ cells (Fig S2C). We performed RNA-seq analysis on the two cell populations at each stage. Principal component analysis revealed that biological replicates clustered together and that the variance between populations was greatest by developmental stage rather than cell compartment (Fig S2D). Interestingly, however, P0 and P10 populations were more similar to each other than either was to P3 or P6. Principal components 2 and 3 segregated crypts from villi more clearly (Fig S2D,E).
We obtained core gene signatures of the crypt and villi (genes expressed and enriched at all time points). There was a core set of 299 genes that were enriched in crypts at all stages examined (Fig 2B). Pathway analysis revealed that developing crypts were enriched for genes regulating the cell cycle, DNA replication, and other processes associated with actively proliferating cells (Fig 2B, Fig S2F). Additionally, the adult intestinal stem cell markers Lgr5, Smoc2, Olfm4, Msi1, Axin2, and Ascl2 were enriched in crypts at all stages. Interestingly, the core set of 299 crypt-enriched genes shared only 64 genes with a previously published adult intestinal stem cell signature (Munoz et al., 2012)(data not shown). This likely reflects the heterogeneity of the crypt pool versus the purified Lgr5-positive cells.
We used antibody staining to validate our RNA-seq data and to identify novel crypt-enriched markers, including the protocadherin Pcdh8 (PAPC). In adults, Pcdh8 has been reported to be an intestinal stem cell marker (Merlos-Suarez et al., 2011). However, Pcdh8 protein was broadly enriched in the crypt compartment throughout crypt morphogenesis and was not restricted to the crypt base until initial crypt morphogenesis had completed (Fig 2C). These data highlight the difference in cellular composition of postnatal crypts compared to adult crypts and suggest a maturation process of adult stem cells from a more homogeneous population of neonatal progenitors.
We also identified genes that were specifically enriched at later stages of crypt development, such as Krt7 and L1cam. Consistent with transcriptome data, keratin 7 protein was absent from early neonatal crypts, though it was expressed in goblet cells in the villi. However, its expression was highly upregulated in crypts at later time points, beginning to be expressed by P8, with all crypts labeled by P15 (Fig 2D). In contrast, while L1CAM levels began to increase by RNAseq at P10, it was still difficult to detect by immunofluorescence. However, at later stages, L1CAM marked crypts (Fig 2E). These results demonstrate the dynamics of crypt marker expression during development and reveal that the entire structure, and not only stem cells, undergoes maturational changes in gene expression.
In addition to crypt-enriched genes, we identified genes whose expression was upregulated in villi at all stages. As shown in the Venn diagram in Fig 2F, 293 genes were enriched in villi (≥ 2-fold compared to crypts, p < 0.05) at all stages examined. Villi were enriched in genes related to metabolism, transport, and digestion (Fig 2F, Fig S2G). Krt20, a known structural component of villi (Zhou et al., 2003), was also upregulated in our dataset (data not shown). Importantly, we found that the transcription factors Maf and Mafb were enriched in villi at all stages examined. We confirmed this by antibody staining and found that MafB was nuclear in the villus, but was not detectable in crypt cells (Fig 2G,H). In addition, MafB was specific for enterocytes, as enteroendocrine cells marked by Sox9-GFP and Goblet cells identified by morphology were negative for nuclear MafB staining (Fig 2I-K). There are currently no known transcriptional markers that are specific for the enterocyte lineage and present at all developmental stages. Maf and MafB have been shown to be necessary for the differentiation of several tissues and cell types (Lopez-Pajares et al., 2015; Miyai et al., 2016) (Artner et al., 2007). Its expression pattern in the differentiated cells of the small intestine suggests that it may play a similar role in enterocyte differentiation. In either case, it serves as a useful new marker for this cell population.
Stem cell marker expression is dynamic in postnatal crypts
In addition to compartment-specific genes, we were very interested in dynamic patterns of transcription that might reveal stage-specific regulators of crypt morphogenesis or differentiation/maturation pathways. We thus performed fuzzy c-means clustering and examined clusters containing genes whose levels increased as crypts developed (Fig S3A). KEGG pathway analysis revealed enrichment for a number of signaling pathways during crypt development, including Wnt, Hippo and PI3K/AKT (Fig S3B). Consistent with this, we saw an increase in Wnt signaling over crypt morphogenesis using a Tcf/Lef-H2B-GFP reporter mouse (Fig S3C,D) and an Axin2-CreER reporter mouse line (Fig S3E). In addition, mRNA expression levels of the stem cell markers Lgr5 and Sox9 (both putative Wnt targets) became more highly expressed as crypts developed (Lgr5, 3.6-fold; Sox9, 2.0-fold). However, while Lgr5 mRNA levels increased, the number of Lgr5-GFP-labeled cells decreased over crypt development, becoming restricted to the crypt base by P13 (Fig S3G). Quantification showed that the percentage of Lgr5-positive cells in each crypt remained constant from P0 to P6, then decreased by P10 (Fig S3F). These data are consistent with postnatal intestinal stem cells undergoing a maturation process to become adult intestinal stem cells, though specific profiling of these cells will be needed to confirm this. Importantly, these data demonstrate that dynamic transcriptional changes occur concomitant with morphological development. We therefore sought to better understand the cell biological basis of crypt formation.
Crypt cells undergo myosin II-dependent apical constriction
Crypt formation involves epithelial invagination, a common morphogenetic event that has diverse underlying mechanisms in different tissues (Pearl et al., 2017). To identify candidates that may be involved in the initiation of crypt formation and invagination, we examined genes that were enriched in P0 crypts compared to villi and compared to later stage crypts. Interestingly, we found that a large subset of myosin II-associated contractility genes (Zaidel-Bar et al., 2015) was enriched in crypts compared to villi (Fig 3A and Table S1), including the regulatory myosin light chain Myl9, the myosin heavy chain gene Myh9, the Rho GEF Ect2, and the actin nucleators Diaph2 and Diaph3. Several of these, including the myosin heavy chain genes, were also enriched in P0 crypts compared to P3 crypts (Myh9 3.7× enriched, Myh14 8× enriched at P0; Table S2). To examine whether there was evidence for myosin II-based contractility in the initiation of crypt formation, we examined the apical surfaces of intervillar cells from neonatal ZO1-GFP intestines. We found that these cells were apically constricted compared to villar cells (Fig 3B). We quantified the apical surface area of crypt and villar cells in 3D using MorphoGraphX software (Barbier de Reuille et al., 2015; de Reuille et al., 2014) and found that crypt cell apical area (18.4 ±0.5 μm2, SD) was smaller than villar cell apical area at P0 (49.5 ±1.1 μm2). Furthermore, the apical area of crypt cells decreased by approximately 3-fold between P0 and P1 (P1: 6.2 ± 0.1 μm2; t-test, p < 0.001), (Fig 3C). When we examined depth-coded images of an invaginating crypt, it was clear that the apical cell areas at the base of the crypt were smallest, and as the cells moved up and out of the crypt, their apical areas became larger and more regular in shape (Fig 3D).
Figure 3. Intervillar cells are apically constricted.
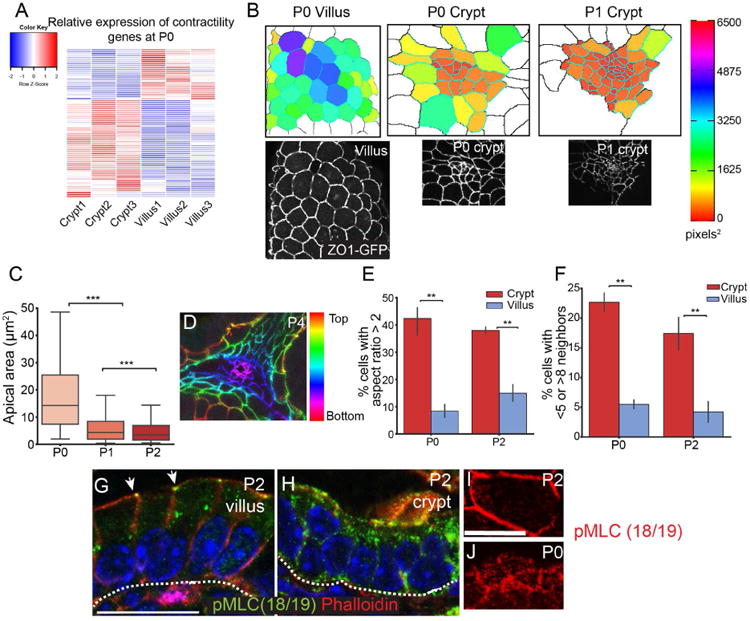
(A) Heatmap of myosin II-associated contractility gene expression in P0 crypts and villi, based on HT-seq gene count data. (B) Images of apical regions of developing crypts and villi, as indicated. The lower panels are epifluorescence images of ZO1-GFP mice, as indicated. The upper panels are segmented images in which cells are colored by their apical area. (C) Quantification of apical area of crypt cells between P0 and P2. The box boundaries are the 25th and 75th percentiles, and the whiskers are the 5th and 95th percentiles. ANOVA, p < 0.05; P0 vs. P1 Tukey HSD, p < 0.001; P1 vs. P2 Tukey HSD, p < 0.001. (D) Depth-coded maximum projection of an invaginating crypt. ZO1-GFP is pseudo-colored by z-position. (E) Percentage of cells in the crypt or villus that have an aspect ratio greater than 2. (F) Percentage of cells in the crypt or villus that have fewer than 5 or more than 8 neighbors. For bar plots, the error bars are the SEM. T-test; ***, p < .001, **, p < .01, *, p < .05. (G,H) phospho-myosin light chain (Thr18/Ser19) staining in villi (G) and crypts (H). pMLC is primarily localized to the zonula adherens in villi (arrows). Scale bar, 20 μm. (I, J) En face view of a villar cell (I) and crypt cells (J) stained for pMLC (red). Scale bar, 20 μm. For apical constriction measurements, 2-3 mice were analyzed per stage, and more than 100 crypt cells per mouse were measured. For panels E-F, 3 mice were analyzed per stage, and more than 25 cells per mouse were measured. See also Tables S1 and S2.
In addition to the constricted apical surfaces, we observed several parameters that suggested that crypt cells may be undergoing dynamic rearrangements prior to invagination. Villar cells were generally hexagonal in shape with an aspect ratio (length of long axis/short axis) slightly over 1. However, there was a much larger range of aspect ratios in crypt cells, with 40% of cells having an aspect ratio over 2.0 (compared to 10-15% in villar cells) (Fig 3E). Because villar cells were stereotypically hexagonal, they had 5-8 neighbors, with approximately 5% of cells falling outside that range. In contrast, crypt cells had irregular shapes and number of neighbors, with over 20% of cells having fewer than 5 or more than 8 neighbors at P0 (Fig 3F), consistent with the cells in the intervillar spaces undergoing dynamic rearrangements.
Apical constrictions that occur in Drosophila gastrulation and Xenopus neural tube closure are driven by actomyosin contractility (Martin et al., 2009; Rolo et al., 2009). The upregulation of contractility genes, which converge on type II non-muscle myosins, suggested that this early morphogenetic change may be myosin II-dependent and required for subsequent crypt invagination. This prompted us to examine whether myosin activation was differentially regulated in distinct intestinal compartments. We examined pMLC(18/19) localization in postnatal intestines and found that it was restricted to the zonula adherens in villi (Fig 3G, arrowheads). In contrast, pMLC was more uniform across the apical surface and down the lateral membranes of postnatal intervillar cells (Fig 3H). When observed en face, intervillar cells had pMLC-positive puncta in the apicomedial region of the cells (Fig 3J), while there was little detectable pMLC in the apicomedial region of villar cells (Fig 3I). These data are consistent with myosin activity being differentially regulated in crypts and villi during crypt morphogenesis. Furthermore, the apicomedial localization of pMLC suggested that there may be an active contractile network at the apical side of intervillar cells.
To determine whether crypt cell apical constriction is myosin II-dependent, we examined the localization of the three type II myosins in the mouse intestine. Myosin heavy chain IIC (MyoIIC) was highly enriched at the apical surface of intestinal epithelial cells (Fig 4A). When observed en face, MyoIIC was localized to the apical cortex and in a medial meshwork (Fig 4B), similar to the medial myosin network observed in gastrulating Drosophila cells (Martin et al., 2009). In contrast to MyoIIC, myosin heavy chain IIA (MyoIIA) primarily localized to lateral junctions (Fig 4C), whereas myosin IIB was not detectable in the intestinal epithelium by antibody staining, consistent with our RNA-Seq data (Fig 4D and data not shown). Due to its apical enrichment, we first examined the intestinal crypt architecture of MyoIIC -/- mice (MyoIIC KO). However, consistent with the lack of overt phenotype of these null mice (Ma et al., 2010), the intestines appeared grossly normal (Fig 4E,F). Interestingly, myosin IIA relocalized to the apical surface of MyoIIC KO intestinal epithelial cells, suggesting that in the intestine, myosin IIA can compensate for the loss of myosin IIC (Fig 4G,H). Therefore, to determine whether type II myosins function in crypt morphogenesis, and particularly in intervillar cell apical constriction, we generated double myosin IIA/myosin IIC knockouts (MyoIIA/C dKO) using the Villin-CreER transgene and injecting with tamoxifen at P0. The apical area of MyoIIA/C dKO crypt cells was significantly larger than control crypt cells at P5 (6.0 ± 0.3 μm2 in control vs. 17.8 ± 1.0 μm2 in MyoIIA/C dKO; p < .001), and was not statistically different from that of the apical areas of P0 control crypt cells (18.4 ± 0.5 μm2 in P0 control)(Fig 3C and Fig 4K-M). These data demonstrate that type II myosins are required for the apical constriction of intestinal crypt cells. In addition to loss of apical constriction, both the invagination (Fig 4I,J,N) and the change in 2D area of the crypt (Fig 4O) were inhibited in the MyoIIA/C dKO intestine. These changes were not secondary to changes in number of crypt progenitor cells, as cell number was comparable to control crypts (Fig 4P). Furthermore, proliferation rates were normal, and we did not observe an increase in crypt cell apoptosis (data not shown). Together, these data demonstrate that type II myosins are required for the initiation of crypt invagination by inducing apical constriction. This process is likely to be highly regulated, and the large number of contractility related genes in the P0 crypt data set should allow for a candidate-based approach to identifying the pathways responsible for the activation of contractility. Notably, once established, the small apical surface area of adult crypts was maintained in a myosin-independent manner, as ablation of myosins IIA/C in adult animals did not affect cell shape (Fig S4A-D).
Figure 4. Myosin-II mediated contractility is required for crypt invagination.
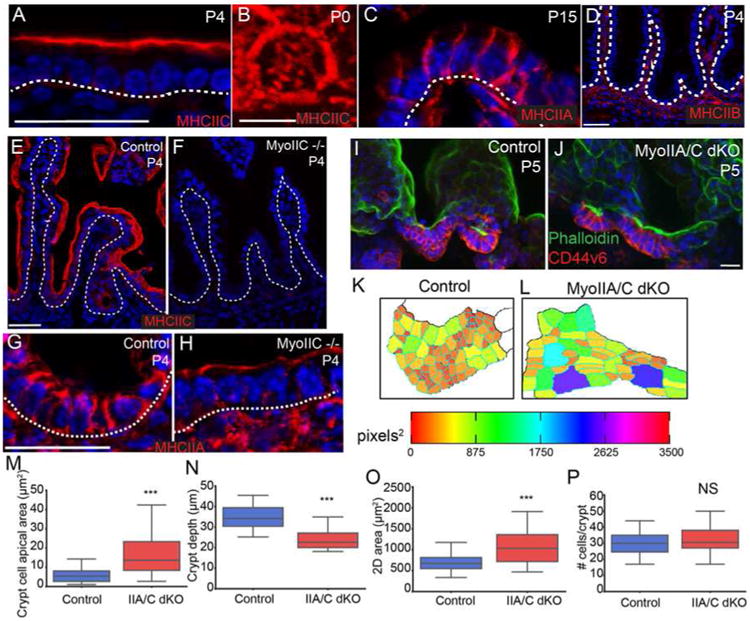
(A) Myosin IIC localization (red) at the apical side of intestinal epithelial cells. The basement membrane is marked with a dotted line. (B) En face view of myosin IIC localization in the apicomedial region of an intestinal epithelial cell. (C) Myosin IIA localization (red) to the lateral membranes of intestinal epithelial cells. The basement membrane is marked with a dotted line. (D) Myosin IIB is not expressed in the intestinal epithelium. Basement membrane is marked with a dotted line. (E,F) Staining of myosin IIC (red) in control and myoIIC -/- intestines, confirming loss of protein. The basement membrane is marked by a dotted line. (G,H) Staining of myosin IIA (red) in control and MyoIIC -/- intestines. In the absence of myosin IIC, myosin IIA localizes to the apical region of intestinal epithelial cells. Basement membrane is marked by a dotted line. (I,J) Maximum intensity projections of epithelial sheets from control (I) and MyoIIA/IIC double KO intestines (J) at P5. Epithelial sheets are stained for phalloidin (green) and CD44v6 (red). (K,L) Segmented images of apical regions of developing crypts are color coded by apical area for control and MyoIIA/IIC double KO intestines. (M) Quantification of apical area of crypt cells in control and MyoIIA/IIC double KO P5 intestines. (N) Quantification of crypt depth in control and MyoIIA/IIC double KO P5 intestines. (O) Quantification of 2D crypt area in control and MyoIIA/IIC double KO P5 intestines. (P) Quantification of the number of CD44v6+ cells per crypt. There is no difference in crypt cell number in the MyoIIA/IIC double KO compared to controls. For box and whisker plots, the boxes mark the 25th to 75th percentile, and the whiskers mark the 5th and 95th percentiles. ***, p < .001 by student's t-test. NS, not significant. All scale bars are 20 μm, with the exception of (B), where it is 5 μm. Total number of mice analyzed were three mice per genotype, and more than 20 cells per mouse. See also Figure S4.
Rac1 signaling is essential for compartmentalization of crypts and villi
Having established the utility of our transcriptomic dataset in identifying pathways important for crypt development, we further mined this resource. We found that the crypt transcriptomic signature was enriched for mediators of Rac1 signaling, which regulates many aspects of cell physiology - including cell migration and cytoskeletal dynamics. As shown in Fig 5A, many Rac1 signaling pathway genes, though not Rac1 itself, were enriched in crypts compared to villi at various stages (yellow nodes). These results are consistent with previous FRET data showing that Rac1 activity is high at the base of adult crypts (Johnsson et al., 2014). We generated intestinal epithelial-specific Rac1 conditional knockout mice by crossing a Rac1 floxed mouse to the Villin-CreER transgenic line and induced recombination at P0 (Chrostek et al., 2006; el Marjou et al., 2004). We observed loss of Rac1 protein by P5 (Fig S5A,B), but intestines at this stage had a normal morphology, demonstrating that Rac1 is not required for initial crypt invagination (Fig S5C-F). Rac1 protein remained absent at P20 (Fig 5B), but at this stage we observed a dramatic phenotype. Whole mount imaging of the epithelium revealed that loss of Rac1 resulted in expansion of the crypts at the expense of villi (Fig 5C). In many areas, the intestine looked similar to the colon, with deep crypts and no villar projections into the lumen. There was considerable variability in the severity of this phenotype, even within the same mouse. Scanning electron microscopy (SEM) demonstrated that rather than the normally well-separated villi of control intestines, the Rac1 mutant intestine had a rather flat surface with short villi that were tightly apposed to each other (Fig 5D-E).
Figure 5. Loss of Rac1 causes dramatic architectural changes in the small intestine.
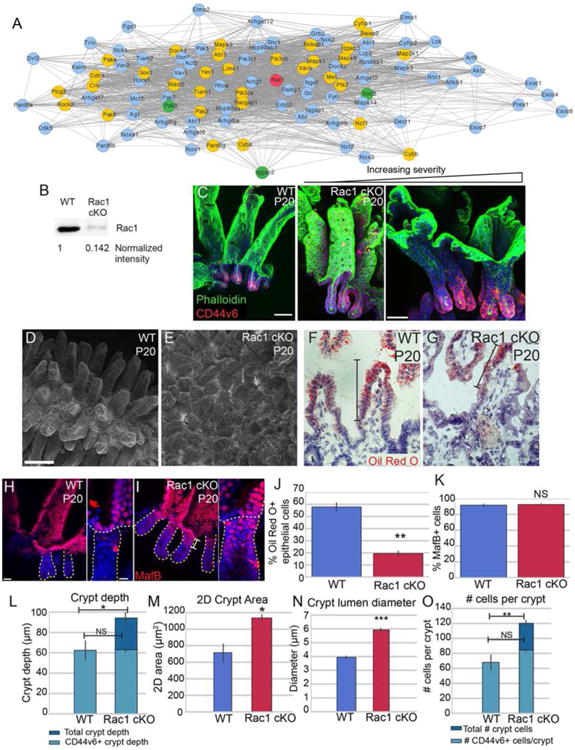
(A) STRING network of Rac1-interacting proteins. Rac1 is marked in red. Gold nodes are genes that were identified by RNA-seq to be enriched in crypts over villi at one or more stage. Green nodes are genes identified by RNA-seq to be enriched in villi over crypts at one or more stage. (B) Western blot of isolated intestinal epithelium from P20 VilCreER; Rac1 fl/fl (Rac1 cKO) or control littermates injected with tamoxifen at P0. The amount of Rac1 was normalized to total protein. (C) Maximum intensity projections of P20 control or Rac1 cKO intestinal epithelial sheets stained for phalloidin (green) and CD44v6 (red). The first panel is from a WT intestine, and the remaining two panels are from Rac1 cKO intestines, illustrating the variation in the severity of the phenotype. Scale bar, 50 μm. (D-E) Scanning electron microscopy of P20 WT (D) and Rac1 cKO (E) intestines. Scale bar, 100 μm. (F-G) Oil Red O and Hematoxylin staining of P20 WT (F) and Rac1 cKO (G) intestinal sections after 30 min oil gavage. Brackets mark the region of the villus that contains Oil Red O-positive puncta. Scale bar, 20 μm. The saturation level of Oil Red O staining was adjusted for both images in Photoshop. (H-I) Epithelial whole mount staining of MafB (red) in WT (H) and Rac1 cKO (I) P20 intestines. Insets show MafB staining in the uppermost region of a single crypt. Individual crypts are marked by a dotted line. Scale bar, 25 μm. Scale bar in insets is 10 μm. (J) Bar plot of the percentage of epithelial cells that were positive for Oil Red O staining. (K) Bar plot of the percentage of CD44v6-negative epithelial cells that stained positive for MafB. (L) Quantification of crypt depth in WT and Rac1 cKO P20 intestines. Note that in WT crypts, all cells are CD44v6+. (M) Quantification of 2D crypt area in WT and Rac1 cKO P20 intestines. (N) Quantification of the lumen diameter of crypts in WT and Rac1 cKO P20 intestines. (O) Quantification of number of cells per crypt. Note that in WT crypts, all cells are CD44v6+. For bar plots, the error bars are the SEM. ***, p < .001, **, p < .01, *, p < .05, NS = not significant. Total number of mice analyzed were 3-5 mice per genotype, and at least 15 crypts per mouse. See also Figure S5.
To test the functional relevance of these changes in tissue architecture, we examined the ability of control and Rac1 cKO intestines to absorb nutrients from the lumen. We gavaged P20 mice with oil, then sacrificed 30 min later. By staining with Oil Red O, we could examine the regions of villi occupied by enterocytes that had taken up the lipid (Fig 5F-G). While 57.9% (± 4.7 SD) of WT epithelial cells absorbed oil, only 19.5% (± 3.0) of Rac1 cKO epithelial cells absorbed oil (Fig 5J). This was not due to a difference in the number of enterocytes present, as quantification of MafB-positive/CD44v6-negative cells revealed similar numbers in WT and Rac1 cKO intestines (Fig 5K). Furthermore, Rac1 cKO enterocytes properly expressed and localized proteins involved in lipid and cholesterol transport (Fig S6N-Q). These data suggest that proper villar spacing is important for maximal surface area exposure to luminal contents.
While the length of Rac1 cKO crypts was longer than WT crypts (measured from crypt mouth to crypt base), the CD44v6+ region was comparable in depth between WT and Rac1 cKOs (Fig 5L). Changes to crypt depth were at the expense of villi, as villar length was shorter in Rac1 cKO intestines, while the length of the crypt-villus axis was comparable to WT intestines (Fig S6A-B). In addition, the 2D area of Rac1 cKO crypts was larger than that of WT crypts, as was the lumen diameter (Fig 5M-N). The defects were not secondary to changes in proliferation or migration, both of which were unchanged in the mutant background (Fig S6C-H). Thus, Rac1 plays an integral role in regulating small intestinal tissue structure, which is essential for optimal intestinal function.
While the Rac1 cKO intestines maintained the proper number of CD44v6+ cells at the bottom of the crypt, they also contained a large population of CD44v6- cells in the crypt neck and mouth (Fig 5C,O). These cells were non-proliferative based on Ki67 staining and EdU incorporation (Fig S6D-G), and they expressed the enterocyte marker MafB, demonstrating that they had exited the cell cycle and were at least partially differentiated (Fig 5H-I). Importantly, these data demonstrate that residence within a crypt and/or insulation from the intestinal lumen is not sufficient for a cell to maintain its proliferative status and that differentiation is not obligately bound to crypt exit.
Because proliferation and migration were normal in Rac1 cKO intestines, we examined whether loss of Rac1 affected cell shapes. In WT intestines, we found that the hinge region that separates villi from crypt was occupied by cells with a distinct morphology. They were wedge-shaped, with an expanded apical domain and a constricted basal footprint (Fig 6A, arrows and insets; Fig S7G, J, K). These cells formed shortly after hinge formation began, at P6. Initial hinge formation from P3-P5 resulted from a bending of the tissue without observable changes in cell shape, though we do not rule out that small changes may underlie this. We refer to the wedge-shaped cells at the crypt/villus boundaries as hinge cells. Hinge cells were basally constricted (Fig 6C) and apically expanded (Fig 6D) compared to cells in the crypt neck. Currently, these cells are defined by their morphology rather than specific markers. That said, they normally exist at or near the boundary between CD44v6-positive crypt cells and MafB-positive villar cells (Fig 6F, Fig S6K).
Figure 6. Loss of Rac1 increases hemidesmosomal attachments and inhibits hinge cell formation.
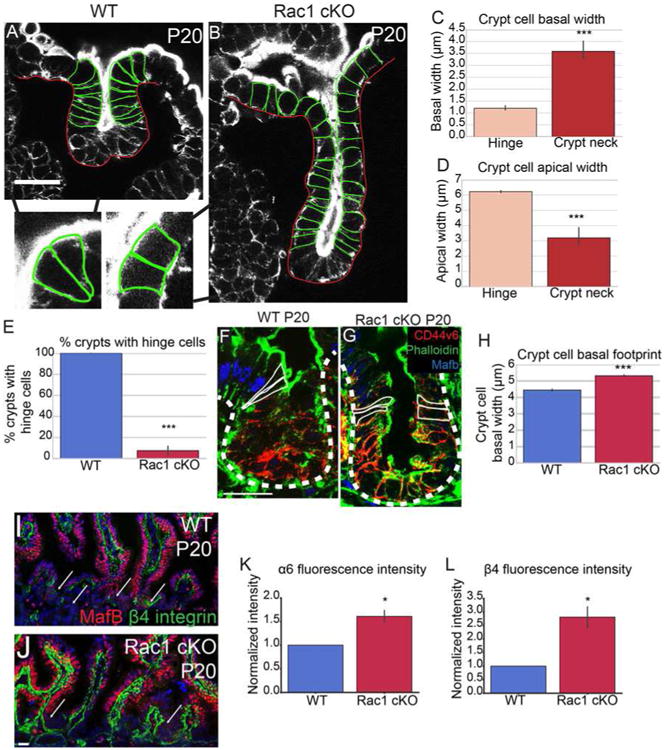
(A-B) Phalloidin staining of P20 WT (A) or Rac1 cKO (B) crypts. Yellow arrowheads in A mark “hinge cells”, the cells at the mouth of the crypt that are basally constricted and apically expanded. Insets show zoomed in regions of hinge cells with the cell boundaries outlined. (C) Quantification of basal width of hinge cells and cells within the crypt neck. (D) Quantification of the apical width of hinge cells and cells within the crypt neck. (E) Quantification of the percent of WT or Rac1 cKO P20 crypts that contain identifiable hinge cells. (F-G) MafB (blue), Phalloidin (green) and CD44v6 (red) staining in P20 crypts. Cells within the hinge region are outlined in white. (H) Quantification of the basal footprint of cells in P20 WT and Rac1 cKO crypts. (I-J) MafB (red) and β4 integrin staining (green) in tissue sections from P20 WT and Rac1 cKO intestines. Arrows point to crypts. (K-L) Normalized fluorescence intensity of α6 integrin (K) and β4 integrin (L) in P20 WT and Rac1 cKO crypts. All scale bars are 20 μm. For all bar plots, the error bars are the SEM. ***, p < .001, **, p < .01, *, p < .05. Total number of mice analyzed were 3-5 mice per genotype, at least 23 crypts per mouse or least 100 cells per mouse. See also Figures S6 and S7.
In stark contrast to WT intestines in which almost every crypt had clear basally constricted hinge cells, these cells were absent in the Rac1 cKO intestine (Fig 6E; Fig S7I-K). We did not observe hinge cells at either the crypt/villus boundary (Fig 6B and inset) or at the CD44v6/MafB boundary (Fig 6G). Rather, the cells across the crypt-villus axis were columnar, with roughly equal apical widths and basal widths. Consistent with this observation, Rac1 mutant mice did not form the acute hinges characteristic of control intestines (Fig 5C, 6B, S5I,J). That said, initiation of hinge formation at P5, which occurs before hinge cells are observed, appeared normal (Fig S5E-H). These data demonstrate that Rac1 affects tissue morphology in part by regulating hinge cell formation. Together with the SEM data (Fig 5D-E), these findings suggest that the hinge functions to maintain the organization of the intestinal epithelium, proper spacing between villi, and compartmentalization between crypts and villi.
Hinge cell formation requires Rac1-dependent suppression of hemidesmosomal integrins
We next turned to understand the cell biological basis for Rac1's role in hinge cell formation. During our examination of hinge cells, we noted that the basal footprints of all crypt cells in the Rac1 mutant intestine were notably larger than in WT controls (Fig 6B,H). The changes in cell shape and the loss of hinge cells also occurred when we induced recombination for a short time (48 hours, Fig S6I-J), demonstrating that they are not secondary to the changes in tissue morphology. The increase in basal footprint suggested that cells in Rac1 cKO crypts had increased attachments to the extracellular matrix (ECM), and those attachments may preclude the cell shape changes required to form a hinge. When we isolated epithelial sheets for whole-mount imaging, we found that it was much more difficult to release the Rac1 cKO epithelium from the underlying mesenchyme, further suggesting that cell-ECM adhesion was increased. This prompted us to examine attachments between the cells and the ECM. The α6 integrin and β4 integrin subunits are the only hemidesmosome-associated integrins (Walko et al., 2015). Immunofluorescence analysis revealed that in WT intestines, the hemidesmosomal localization of both of these integrins was low in crypts and high in villi (Fig 6I). In contrast, the intensity of integrin staining was high in both crypts and villi of Rac1 cKO intestines (Fig 6J-L). We did not see this significant upregulation of α1 integrin (data not shown), suggesting that loss of Rac1 specifically increases hemidesmosomal integrins.
In contrast to the small intestine, the colon does not have villi. Consistent with the idea that Rac1 plays a rather specific role in the compartmentalization of crypts and villi in the small intestine, we did not observe any significant changes in colon architecture in Rac1 mutants (Fig S7A-D). This was despite the fact that loss of Rac1 resulted in an increase in hemidesmosomal integrin levels (Fig S7E) and increased basal footprint size (Fig S7F). Colons lack the apically expanded/basally constricted hinge cells seen in the small intestine (Fig S7G-K), a function that may be replaced by Goblet cells, which are apically expanded and represent a significant fraction of cells in the colon.
To determine whether the increased hemidesmosomal attachments were responsible for the increased basal footprint and the lack of hinge cells in the small intestine, we turned to cultured 3D organoids. Remarkably, we found that Rac1 cKO organoids recapitulated not only the increased basal footprint phenotype (Fig 7C), but also the absence of hinge cells (at both the Cd44v6 positive region, Fig 7A,B, and at the “top” of the crypt, Fig S6L) and the increase in α6β4 integrin levels (Fig 7D-H). Similar to Rac1 cKO intestines, Rac1 cKO organoids contained crypt-like evaginations with CD44v6 staining restricted to the base of the crypt (Fig 7I-J).
Figure 7. Inhibition of α6 integrin binding to laminin rescues hinge cell formation.
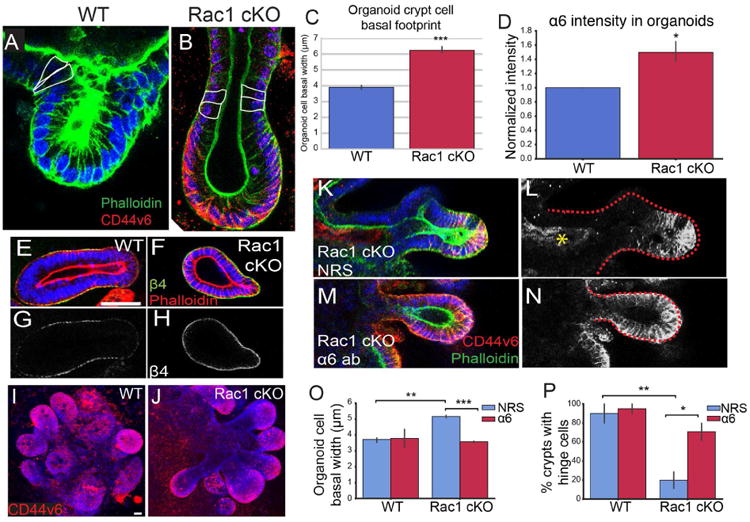
(A-B) Phalloidin (green) staining of WT (A) and Rac1 cKO (B) organoids. In (B), CD44v6 is labeled in red. Cells within the hinge region are outlined in white. (C) Quantification of the basal footprint of cells in P20 WT and Rac1 cKO organoids. (D) Normalized fluorescence intensity of α6 integrin in WT and Rac1 cKO organoids. (E-F) β4 integrin (green) and phalloidin (red) staining in WT (E) and Rac1 cKO (F) organoids. (G-H) β4 integrin staining of the images in E-F. (I-J) Maximum intensity projection of CD44v6 staining (red) in WT (I) and Rac1 cKO organoids (J). (K-M) CD44v6 (red) and phalloidin (green) staining in Rac1 cKO organoids treated with normal rat serum (K) or α6 integrin blocking antibody (M). (L,N) are gray-scale images of CD44v6 in (K, M). The red dotted line marks the boundary of the crypt region. The yellow asterisk in (L) marks non-specific staining due to NRS treatment. (O) Quantification of the basal width of WT or Rac1 cKO cells in organoids treated with normal rat serum (NRS) or α6 integrin blocking antibody. (P) Quantification of the percent of crypts with hinge cells in organoids treated with normal rat serum (NRS) or α6 integrin blocking antibody. All scale bars are 20 μm. For all bar plots, the error bars are the SEM. ***, p < .001, **, p < .01, *, p < .05. Organoids from 3-5 mice per genotype were analyzed, and at least 5 crypts per sample, or 164 cells per sample.
We next aimed to address whether the increased hemidesmosomal integrin levels functionally contributed to the morphology and cell shape defects in Rac1-null organoids. Upon plating epithelial sheets, we incubated the tissues with an antibody that blocks binding of α6 integrin to laminin (α6 blocking antibody) or with normal rat serum (NRS). After allowing the organoids to grow for one week in the presence of antibodies, we examined organoid cell morphology and crypt structure. Inhibition of α6 binding to laminin resulted in CD44v6 staining throughout the Rac1 cKO crypt, in contrast to serum-treated organoids in which CD44v6 was expressed only in the base of the evagination (Fig 7K-N). Importantly, decreasing α6 binding to laminin also rescued crypt cell basal footprint to WT sizes (Fig 7O). Finally, we examined the presence of hinge cells in organoids and found that the majority of WT organoids contained morphologically distinguishable hinge cells, and treatment with the α6 blocking antibody did not affect their morphology (Fig 7P). In contrast, only approximately 20% of Rac1 cKO organoids contained identifiable hinge cells. Treatment with the α6 blocking antibody rescued the formation of hinge cells in approximately 65% of organoids (Fig 7P). These data demonstrate that changes in intestinal morphology in Rac1 cKO intestines were at least partially due to increased hemidesmosomal attachment through α6/β4 integrin. Based on our data, we suggest the following model. In the WT intestine, Rac1 negatively regulates hemidesmosomal integrins to allow for dynamic cell shape changes. These cell shape changes are important for the formation of the hinge cell, which is required for the proper compartmentalization of the crypt and villus (Fig S6R). In the absence of Rac1, the negative regulation of hemidesmosomal integrins is lost. The increased adhesions inhibit the dynamic cell shape changes required to form the hinge cell, resulting in loss of crypt/villus compartmentalization and decreased exposure to lumenal contents.
Discussion
Neither the mechanisms driving crypt formation nor the functions for this anatomical compartment are known. This work identified two discrete pathways essential for early crypt morphogenesis and defined a role for compartmentalization of crypts and villi in normal intestinal physiology.
While intervillar cells are specified around e15.5 days of development in the mouse (Shyer et al., 2015), we did not observe overt morphogenetic changes for crypt development until shortly after birth. Although the trigger for crypt morphogenesis is unknown, the transient upregulation of a contractility network is likely an initiating step in crypt development, as we show that myosin II-dependent apical constriction of cells is required for their subsequent invagination. Understanding the signals that promote the transcriptional upregulation of this contractility program will provide insight into the developmental pathways driving crypt formation.
As invagination proceeds, a previously underappreciated event that must occur is the formation of hinges, which morphologically separate the crypt from the villi. The formation of a hinge appears to be initially driven by tissue bending, followed by the appearance of basally constricted and apically expanded cells (which we term hinge cells) that cover the hinge region. In the absence of Rac1, initial bending occurs, but hinge cells do not form, and hinges are subsequently lost or are very broad. Mechanistically, this is due to increased hemidesmosomal adhesion in Rac1 mutant crypts. Upregulation of hemidesmosomal integrins alters cell shapes, increasing the crypt cell basal footprint. An attractive hypothesis is that increased adhesion prevents the decrease in basal attachment and cell shape changes needed to maintain the hinge. Supporting this, hemidesmosomal integrin blocking antibodies were able to partially rescue Rac1 cell biological phenotypes in organoid cultures. That said, we cannot rule out additional roles for Rac1 in tissue morphology and physiology. Specifically, whether the apical expansion and the basal constriction are linked and both controlled by Rac1 or whether they are separable remains unknown. Notably, similar basal constrictions have been noted during the formation of the midbrain-hindbrain boundary in zebrafish, suggesting this may be a conserved morphogenetic mechanism (Gutzman et al., 2008).
While previous studies have demonstrated that integrins promote Rac1 activation (Cruz-Monserrate and O'Connor, 2008; Mettouchi et al., 2001; Price et al., 1998), control of hemidesmosomal integrin expression by Rac1 has not been reported. Whether this is due to transcriptional effects, signaling, or mechanical changes remains to be tested. Rac1 could negatively regulate hemidesmosomal attachments through its antagonistic relationship with the related small GTPase RhoA (Chauhan et al., 2011; Comunale et al., 2007). Alternatively, Rac1 can regulate several transcriptional and signaling pathways, including NF-κB and JNK activation (Arbibe et al., 2000; Olson et al., 1995; Sulciner et al., 1996). Notably, in Drosophila, loss of JNK signaling results in ectopic β-integrin accumulation (Llense and Martin-Blanco, 2008).
Through their role in compartmentalizing crypts and villi, hinge cells are important for the proper spacing and patterning of villi. By creating an acute hinge region, the hinge cells allow for separation between villi, as seen by SEM. In contrast, lack of hinge cells in the Rac1 cKO resulted in broad villi with no clear separation between them, generating an epithelium that looked more colon-like. This phenotype is consistent with a previous report in which a dominant-negative Rac1 was expressed mosaicly in the intestine (Stappenbeck and Gordon, 2000). One potential role for this villar spacing may be to maximize the exposed surface area of each villus, allowing for absorption of nutrients down the entire villus length. We found evidence for this, as fewer cells absorbed oil in the Rac1 cKO intestine, suggesting that cells on tightly apposed villi could not access the lumenal contents.
The functions of crypt architecture remain an important outstanding question. It is clear from this work that exiting the crypt is not essential for exiting the cell cycle or for enterocyte differentiation, even though these events normally occur as cells leave the crypt. However, due to the lethality of the loss of type II myosins, we have not been able to extend these studies to determine whether loss of crypt morphology results in long-term changes in stem cell maintenance or function. The architecture of crypts likely provides multiple functions, from protection of stem cells from luminal contents, to the ability to package the required number of transit amplifying cells. However, our data suggest that crypt morphology is more generally important for tissue architecture. Further analysis of crypt development mechanisms will allow us to more specifically disrupt architecture to finally address these questions.
Finally, our transcriptomic approach has identified new core crypt and villar markers, many of which are likely to play important roles in the physiology of these tissues in addition to their utility as markers for discrete cell populations. For example, MafB is a transcriptional regulator that specifically marks enterocytes and may provide a foothold into understanding the transcriptional regulation of enterocyte development, which has remained largely unexplored. The ability to track changes in transcripts over the crypt morphogenesis process also revealed dynamic changes in many pathways. This includes not only signaling pathways and stem cell markers as discussed in the text, but a large number of genes that specify proteins for cell-ECM adhesion, many of which are likely involved in the remodeling of the basement membrane that must occur for crypt invagination.
STAR Methods
Contact for Reagent and Resource sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Terry Lechler (terry.lechler@duke.edu).
Experimental Model and Subject Details
Mice
All animal work was approved by Duke University's Institutional Animal Care and Use Committee. Mouse strains used in this study were: CD1 (Charles River), MHCIIA fl/fl (Jacobelli et al., 2010) MMRRC #032096, MHCIIC -/- (Ma et al., 2010) MMRRC #030138, Rac1 fl/fl (gift from Cord Brakebusch, University of Copenhagen (Chrostek et al., 2006)), VillinCreER (gift from Sylvie Robine, Institut Curie (el Marjou et al., 2004)), Lgr5-EGFP-IRES-CreER (Jackson Laboratories (Barker et al., 2007)), TCF/Lef1-Histone2b-eGFP (Jackson Laboratories (Ferrer-Vaquer et al., 2010)), Sox9-EGFP (MMRRC, stock #011019-UCD (Gong et al., 2003)), and ZO1-GFP (Foote et al., 2013; Huebner et al., 2014). Mice were genotyped by PCR and both males and females were analyzed. Mice were maintained in a barrier facility with 12 hour light/dark cycles.
Organoid culture
Organoids were cultured as described previously (Miyoshi and Stappenbeck, 2013) using conditioned media collected from L-WRN cells (gift from Thaddeus Stappenbeck, Washington University in St. Louis). Intestinal fragments were flushed with cold PBS, and the luminal surface was scraped with a glass coverslip to remove villi. The remaining tissue fragments were incubated in 2 mM EDTA in PBS for 30 min at 4°C with gentle rocking. Sheets were lightly shaken to release remaining villi. The PBS was replaced, and the tissue was vigorously shaken to release crypts. The cells were collected, allowed to settle, and washed once in cold PBS before plating in 1× Matrigel (Corning) on glass coverslips. Media was changed every other day and supplemented with 10 μM Jagged (Genscript).
For α6 blocking experiments, 0.25 μg of α6 antibody (Millipore, MAB1378) or Normal Rat Serum was added to the media upon plating. Organoids were allowed to grow for 7 days before analysis. Organoids were washed once in PBS-T and fixed in 4% PFA for 10 min. The organoids were released from the Matrigel by scraping with a pipette tip. For staining, the samples were washed in PBS-T and incubated in blocking buffer for 30 min. Organoids were incubated in primary antibody for 45 minutes, washed three times in PBS-T, followed by incubation with secondary antibody for 30 min. Organoids were imaged on a Leica SP8 upright confocal microscope using a 20×/0.7 HC PL Apo-multiImm objective or 40×/1.3 HC Plan APO CS2 oil immersion objective.
Method Details
Tissue preparation
For all quantifications, the medial region of the intestine was used because crypt development proceeds in a wave down the length of the intestine from duodenum to ileum. The medial small intestine was isolated and embedded in Optimal Cutting Temperature prior to freezing, and samples were stored at -80°C. A cryostat was used to cut 8-μm thick sections. In all cases, 2-5 mice per stage or genotype were analyzed, as detailed in each figure legend.
Intestinal epithelial sheet preparation and whole mount imaging
For whole mount imaging, the medial small intestine was isolated and cut into ∼2 cm segments. The lumen was flushed with cold Hanks Buffered Saline Solution containing Ca2+ and Mg2+ (HBSS). The intestinal tube was then cut longitudinally to expose the epithelium. These sheets were placed in 30 mM EDTA in HBSS. Intestinal segments were incubated at 37°C for 20 minutes, then were vigorously shaken to release the epithelium from the mesenchyme. Epithelial sheets were collected, washed twice in HBSS, and fixed in 4% PFA overnight at 4°C. Note, Rac1 cKO intestinal epithelial sheets were isolated in HBSS lacking Ca2+ and Mg2+.
For staining, sheets were washed three times in PBS-T containing 0.2% Triton X-100. They were blocked in 5% Normal Goat Serum, 5% Normal Donkey Serum, and 3% BSA in PBS-T for 30 min at room temperature. Epithelial sheets were then incubated in primary antibodies for 1 h. After washing, sheets were incubated in secondary antibody for 30 min. Following washes in PBS-T, sheets were mounted on slides in a region contained by VALAP. A solution of 90% glycerol in PBS plus 2.5 mg/ml p-Phenylenediamine was used to mount the slides, which were then coverslipped with 22 × 22 mm no 1.5 glass coverslips. Images were collected at room temperature using a Zeiss 780 upright confocal with a 20×/0.8 Plan-Apochromat objective or 63×/1.4 oil immersion Plan-Apochromat objective. Immersol 518F immersion oil was used for the 63× objective.
Immunofluorescence
For immunofluorescent staining, tissue sections were fixed for 8 min in 4% PFA or 2 min in ice-cold acetone (MHCIIA, MHCIIC, Keratin7). Sections were washed in PBS-T containing 0.2% Triton X-100, then incubated in blocking buffer (3% BSA, 5% NGS, 5% NDS in PBS-T) for 15 min. Sections were incubated in primary antibody diluted in blocking buffer for 15 min – 1h at room temperature. After washing in PBS-T, sections were incubated in secondary antibodies for 10 min. Sections were washed, then mounted in 90% glycerol in PBS plus 2.5 mg/ml p-Phenylenediamine (Sigma-Aldrich). The following primary antibodies were used: Rat anti Mouse CD44v6 (Santa Cruz and BioRad), Rabbit anti mouse Pcdh8 (gift from O. Pourquié, Harvard Medical School; (Chal et al., 2017)), rabbit anti MafB (Sigma-Aldrich), rat anti Beta 4 integrin (BD Biosciences), mouse anti keratin 7 (Abcam), rabbit anti MHCIIC (Biolegend), rabbit anti MHCIIA (Biolegend), rabbit anti phospho-MLC (18/19) (Cell Signaling Technology), rat anti alpha 6 integrin (BD Biosciences), rabbit anti alpha-catenin (Sigma-Aldrich), rabbit anti Ki67 (Abcam), mouse anti NPC1L1 (Santa Cruz), and goat anti CD36 (R&D Systems). Tissue sections were imaged on a Zeiss AxioImager Z1 microscope with Apotome.2 attachment, Plan-APOCHROMAT 20×/0.8 objective or Plan-NEOFLUAR 40×/1.3 oil objective, Axiocam 506 mono camera, and Zen software (Zeiss).
Image analysis
Quantifications of number of cells per crypt, 2D area, crypt depth, hinge measurements, aspect ratio, number of neighbors, crypt lumen diameter, basal footprint, and apical cell area were performed on Z-stacks of epithelial whole mount images. Number of cells per crypt, 2D crypt area, crypt depth, and hinge measurements were analyzed using Fiji/ImageJ. The ThreepointROI plugin was used to measure hinge region curvature. The 2D crypt area was measured by rotating 3D reconstructions of crypts to view from the bottom, then manually outlining the perimeter of the CD44v6-positive region. The area of the drawn shape was considered the 2D crypt area. Crypt depth was measured by rotating 3D reconstructions of crypts to view the entire length of the crypt-villus axis. A line was then drawn from the apical side of the uppermost CD44v6-positive cells to the basal side of CD44v6-positive cells at the base of the crypt. The length of the line was considered the crypt depth. Number of neighbors was quantified by thresholding and watershedding the region of interest, then analyzing the resulting image using the neighbor analysis tool in the BioVoxxel Toolbox. Statistical and graphical analysis was performed in Python using the matplotlib, seaborn, pandas, numpy and scipy packages. When multiple comparisons were made, ANOVA tests were performed, followed by Tukey HSD tests for each pairwise comparison. P < 0.05 was considered statistically significant. Apical cell area was quantified in 3D using the MorphoGraphX software. Images were segmented computationally and then manually validated (Barbier de Reuille et al., 2015; de Reuille et al., 2014). Imaris (Bitplane) was used for volume rendering of cells. Cells were manually outlined in serial slices from which surfaces were created.
FACS sorting
Litters of mice aged P0, P3, P6, or P10 were dissected, and their intestinal epithelium was isolated as described above. Upon washing of epithelial sheets in cold HBSS (containing Ca2+ and Mg2+), sheets were incubated in Epcam-PE/Cy7 (1:100; eBioscience) and APC-CD44 (1:25, BioLegend) for 1 h at 4°C with rotation. Sheets were washed in cold HBSS, then incubated in 0.8 mg/ml dispase II in HBSS at 37°C for 6-10 minutes with frequent shaking. A total of 1 ml FBS and 2 μl DNase I were added to each tube, and the cell solutions were passed through 70 μm cell strainers, followed by 40 μm cell strainers. Cells were collected by centrifugation at 2400 rpm for 5 min at 4°C. Cell pellets were washed with HBSS containing 10% FBS and spun at 2400 rpm for 5 min at 4°C. Pellets were then resuspended in DMEM, high glucose, containing Pen/Strep, B-27, 5% FBS, and 10 mM HEPES. Prior to FACs sorting, cells were run through a Celltrics 30 μm filter, and 1 ul Propidium Iodide was added to the cell suspension. Cells were collected into the same media as above using a BD DiVa cell sorter. Cells were pelleted, media was removed and cell pellets were stored at -80°C until RNA extraction.
RNA isolation and RNA sequencing
RNA was extracted using a Qiagen RNAeasy Mini kit following the manufacturer's protocols. Eluted RNA was precipitated overnight with glycogen, ammonium acetate and cold ethanol at -20°C. The RNA was spun, washed, and resuspended in RNAse-free water.
RNA samples were evaluated for concentration by Qubit (Thermo Fisher) and for integrity using an Agilent 2100 Bioanalyzer. Samples with a RIN value > 7.0 were used for RNA-seq analysis. RNA sequencing was performed using an Illumina HiSeq 2500 instrument with single-end 50 bp reads. Reads were mapped to the mm10 genome using STAR (Dobin et al., 2013; Dobin and Gingeras, 2015), and gene counts were analyzed using HTSeq (Anders et al., 2015). HTSeq counts were input into DESeq2 (Love et al., 2014) to calculate differential expression across stages and cell types. Genes were considered to be enriched in a population if they were at least two-fold more highly expressed than the other population with p < 0.05. Principal component analysis was performed in R using the DESeq2 package. Fuzzy c-means clustering was performed using the MFuzz package (Kumar and Futschik, 2007) in R with an m value of 1.75, and genes with membership scores 0.7 and above were included in KEGG and GO Term analysis. All KEGG pathway and GO Term analysis was performed using DAVID Bioinformatics Resources.
qPCR
For qPCR analysis, RNA was isolated from FACs-sorted cells using a Qiagen RNAeasy Mini kit. cDNA was synthesized using a First Strand cDNA synthesis kit with Superscript II (Thermo Fisher). qPCR was performed using Genecopoeia All-in-One qPCR Mix and a Roche LightCycler 2.0. Fold change expression was calculated using the 2-ΔΔCt method when primer efficiency had been calculated to be 2.0. When primer efficiency was not 2.0, the efficiency correction method was used. Ascl2 primers were as follows: Forward 5′ – AAGCACACCTTGACTGGTACG -3′ and Reverse 5′ –AAGTGGACGTTTGCACCTTCA-3′. Lactase primers were as follows: Forward 5′- GAGACCCAGAACTCAATGACACC- 3′ and Reverse 5′-GGTCAGAGCGGTTCACAAAGT-3′. GAPDH primers were as follows: Forward 5′ – AGGTCGGTGTGAACGGATTTG-3′ and Reverse 5′ – TGTAGACCATGTAGTTGAGGTCA- 3′.
Scanning electron microscopy
For scanning electron microscopy, a 1-2 cm fragment of the medial small intestine was flushed with cold HBSS, cut longitudinally, and fixed at 4°C overnight in 2% glutaraldehyde (Electron Microscopy Sciences), 4% PFA (Electron Microscopy Sciences), 1 mM CaCl2, and 0.05 M sodium cacodylate, pH 7.4. The fixative was removed, and samples were washed twice in 1× PBS, 10 min each wash. Samples were incubated in 1% OsO4 for 1 h at RT, protected from light. Samples were rinsed 3 times in PBS, 10 min each wash. Samples were then dehydrated through a series of EtOH incubations (2 × 30%, 2 × 50%, 2 × 70%, 2 × 90%, 3 × 100%) for 10 min each. After EtOH incubation, samples were incubated in Tetramethylsilane, and the tetramethysilane was replaced three times, 10 min each. Upon drying, samples were sputter coated in gold and imaged on a FEI XL30 ESEM.
Oil gavage and Oil Red O staining
P20 mice were gavaged with 40 μl filter-sterilized corn oil, then sacrificed 30 min later. The intestines were removed, and flushed with cold PBS prior to embedding in OCT. 8-μm thick sections were fixed in 4% PFA in PBS for 30 min. Slides were then washed three times for 30 sec each in dH2O, and incubated in a freshly made Oil Red O solution (60 mg Oil Red O, 7.2 mL triethyl phosphate, 12.8 mL dH2O, filtered through a 0.45 μm syringe filter) for 30 min. Samples were washed three times for 30 sec each in dH2O, followed by 5 min in tap water. Sections were placed in Meyer's Hematoxylin for 3 min, then washed for 2-3 min in dH2O until blue.
Western blotting
Western blotting was quantified using the Odyssey infrared imaging system (LI-COR Biosciences) for the Rac1 antibody (1:1000, BD Biosciences) and IRDye 800 Donkey anti-mouse IgG (1:50,000, LI-COR Biosciences). Samples were normalized according to the manufacturer's protocol. Nitrocellulose was stained with LI-COR Biosciences REVERT Total Protein Stain and fluorescence levels were determined by quantitating the levels across the entire lane. Total protein was normalized to the lane with the highest signal. Normalized intensity of Rac1 bands were calculated by dividing the Rac1 band intensity by the normalized total protein value.
Quantification and Statistical Analysis
Statistical parameters including the exact value of n, the definition of center, dispersion and precision measures (meant ± SEM) and statistical significance are reported in the Figure Legends. Data were judged to be statistically significant when p < 0.05 by two-tailed Student's t-test or pairwise Tukey HSD test when multiple comparisons were made. In figures, asterisks denote statistical significance calculated by the noted method (NS = not significant' *, p < 0.05; **, p < 0.01, ***, p < 0.001). Statistical analysis was performed in Python using the scipy stats package. Tukey HSD tests were performed in R.
Data and Software Availability
Raw data files for the RNA sequencing analysis have been deposited in the NCBI Gene Expression Omnibus under accession number GEO: GSE109054.
Supplementary Material
Highlights.
-Identification of genes associated with crypt formation and villar differentiation
-Myosin II-dependent apical constriction is required for initial crypt invagination
-Hinges compartmentalize crypts and villi and pattern the intestine
-Rac1 controls hinge formation through negative regulation of a6/b4 integrins
Acknowledgments
We thank Julie Underwood for care of the mice; Michel Bagnat, Brigid Hogan and members of the Lechler Lab for comments on the manuscript. This work was supported by the NIH (NIAMS – R01-AR055926, R01-AR067203 and NIGMS – R01-GM111336).
Footnotes
Author Contributions: Conceptualization, K.D.S. and T.L.; Methodology, K.D.S. and T.L., Investigation, K.D.S., M.T. and T.L., Writing – Original Draft, K.D.S. and T.L., Writing – Review and Editing, K.D.S. and T.L., Visualization, K.D.S., Supervision, T.L., Funding Acquisition, T.L.
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alho AM, Underhill CB. The hyaluronate receptor is preferentially expressed on proliferating epithelial cells. J Cell Biol. 1989;108:1557–1565. doi: 10.1083/jcb.108.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbibe L, Mira JP, Teusch N, Kline L, Guha M, Mackman N, Godowski PJ, Ulevitch RJ, Knaus UG. Toll-like receptor 2-mediated NF-kappa B activation requires a Rac1-dependent pathway. Nat Immunol. 2000;1:533–540. doi: 10.1038/82797. [DOI] [PubMed] [Google Scholar]
- Artner I, Blanchi B, Raum JC, Guo M, Kaneko T, Cordes S, Sieweke M, Stein R. MafB is required for islet beta cell maturation. Proc Natl Acad Sci U S A. 2007;104:3853–3858. doi: 10.1073/pnas.0700013104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier de Reuille P, Routier-Kierzkowska AL, Kierzkowski D, Bassel GW, Schupbach T, Tauriello G, Bajpai N, Strauss S, Weber A, Kiss A, et al. MorphoGraphX: A platform for quantifying morphogenesis in 4D. Elife. 2015;4:05864. doi: 10.7554/eLife.05864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003–1007. doi: 10.1038/nature06196. [DOI] [PubMed] [Google Scholar]
- Chal J, Guillot C, Pourquie O. PAPC couples the segmentation clock to somite morphogenesis by regulating N-cadherin-dependent adhesion. Development. 2017;144:664–676. doi: 10.1242/dev.143974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan BK, Lou M, Zheng Y, Lang RA. Balanced Rac1 and RhoA activities regulate cell shape and drive invagination morphogenesis in epithelia. Proc Natl Acad Sci U S A. 2011;108:18289–18294. doi: 10.1073/pnas.1108993108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrostek A, Wu X, Quondamatteo F, Hu R, Sanecka A, Niemann C, Langbein L, Haase I, Brakebusch C. Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol Cell Biol. 2006;26:6957–6970. doi: 10.1128/MCB.00075-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. The intestinal crypt, a prototype stem cell compartment. Cell. 2013;154:274–284. doi: 10.1016/j.cell.2013.07.004. [DOI] [PubMed] [Google Scholar]
- Comunale F, Causeret M, Favard C, Cau J, Taulet N, Charrasse S, Gauthier-Rouviere C. Rac1 and RhoA GTPases have antagonistic functions during N-cadherin-dependent cell-cell contact formation in C2C12 myoblasts. Biol Cell. 2007;99:503–517. doi: 10.1042/BC20070011. [DOI] [PubMed] [Google Scholar]
- Crosnier C, Stamataki D, Lewis J. Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet. 2006;7:349–359. doi: 10.1038/nrg1840. [DOI] [PubMed] [Google Scholar]
- Cruz-Monserrate Z, O'Connor KL. Integrin alpha 6 beta 4 promotes migration, invasion through Tiam1 upregulation, and subsequent Rac activation. Neoplasia. 2008;10:408–417. doi: 10.1593/neo.07868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Reuille PB, Robinson S, Smith RS. Quantifying cell shape and gene expression in the shoot apical meristem using MorphoGraphX. Methods Mol Biol. 2014;1080:121–134. doi: 10.1007/978-1-62703-643-6_10. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Gingeras TR. Mapping RNA-seq Reads with STAR. Curr Protoc Bioinformatics. 2015;51:11 14 11–19. doi: 10.1002/0471250953.bi1114s51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- Ferrer-Vaquer A, Piliszek A, Tian G, Aho RJ, Dufort D, Hadjantonakis AK. A sensitive and bright single-cell resolution live imaging reporter of Wnt/ss-catenin signaling in the mouse. BMC Dev Biol. 2010;10:121. doi: 10.1186/1471-213X-10-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foote HP, Sumigray KD, Lechler T. FRAP analysis reveals stabilization of adhesion structures in the epidermis compared to cultured keratinocytes. PLoS One. 2013;8:e71491. doi: 10.1371/journal.pone.0071491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Zheng C, Doughty ML, Losos K, Didkovsky N, Schambra UB, Nowak NJ, Joyner A, Leblanc G, Hatten ME, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- Gutzman JH, Graeden EG, Lowery LA, Holley HS, Sive H. Formation of the zebrafish midbrain-hindbrain boundary constriction requires laminin-dependent basal constriction. Mech Dev. 2008;125:974–983. doi: 10.1016/j.mod.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner RJ, Lechler T, Ewald AJ. Developmental stratification of the mammary epithelium occurs through symmetry-breaking vertical divisions of apically positioned luminal cells. Development. 2014;141:1085–1094. doi: 10.1242/dev.103333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobelli J, Friedman RS, Conti MA, Lennon-Dumenil AM, Piel M, Sorensen CM, Adelstein RS, Krummel MF. Confinement-optimized three-dimensional T cell amoeboid motility is modulated via myosin IIA-regulated adhesions. Nat Immunol. 2010;11:953–961. doi: 10.1038/ni.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnsson AK, Dai Y, Nobis M, Baker MJ, McGhee EJ, Walker S, Schwarz JP, Kadir S, Morton JP, Myant KB, et al. The Rac-FRET mouse reveals tight spatiotemporal control of Rac activity in primary cells and tissues. Cell Rep. 2014;6:1153–1164. doi: 10.1016/j.celrep.2014.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, Stappenbeck TS. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell. 2016;165:1708–1720. doi: 10.1016/j.cell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar L, Futschik ME. Mfuzz: a software package for soft clustering of microarray data. Bioinformation. 2007;2:5–7. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llense F, Martin-Blanco E. JNK signaling controls border cell cluster integrity and collective cell migration. Curr Biol. 2008;18:538–544. doi: 10.1016/j.cub.2008.03.029. [DOI] [PubMed] [Google Scholar]
- Lopez-Pajares V, Qu K, Zhang J, Webster DE, Barajas BC, Siprashvili Z, Zarnegar BJ, Boxer LD, Rios EJ, Tao S, et al. A LncRNA-MAF:MAFB transcription factor network regulates epidermal differentiation. Dev Cell. 2015;32:693–706. doi: 10.1016/j.devcel.2015.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Jana SS, Conti MA, Kawamoto S, Claycomb WC, Adelstein RS. Ablation of nonmuscle myosin II-B and II-C reveals a role for nonmuscle myosin II in cardiac myocyte karyokinesis. Mol Biol Cell. 2010;21:3952–3962. doi: 10.1091/mbc.E10-04-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshman E, Booth C, Potten CS. The intestinal epithelial stem cell. Bioessays. 2002;24:91–98. doi: 10.1002/bies.10028. [DOI] [PubMed] [Google Scholar]
- Martin AC, Kaschube M, Wieschaus EF. Pulsed contractions of an actin-myosin network drive apical constriction. Nature. 2009;457:495–499. doi: 10.1038/nature07522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlos-Suarez A, Barriga FM, Jung P, Iglesias M, Cespedes MV, Rossell D, Sevillano M, Hernando-Momblona X, da Silva-Diz V, Munoz P, et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011;8:511–524. doi: 10.1016/j.stem.2011.02.020. [DOI] [PubMed] [Google Scholar]
- Mettouchi A, Klein S, Guo W, Lopez-Lago M, Lemichez E, Westwick JK, Giancotti FG. Integrin-specific activation of Rac controls progression through the G(1) phase of the cell cycle. Mol Cell. 2001;8:115–127. doi: 10.1016/s1097-2765(01)00285-4. [DOI] [PubMed] [Google Scholar]
- Miyai M, Hamada M, Moriguchi T, Hiruma J, Kamitani-Kawamoto A, Watanabe H, Hara-Chikuma M, Takahashi K, Takahashi S, Kataoka K. Transcription Factor MafB Coordinates Epidermal Keratinocyte Differentiation. J Invest Dermatol. 2016;136:1848–1857. doi: 10.1016/j.jid.2016.05.088. [DOI] [PubMed] [Google Scholar]
- Miyoshi H, Stappenbeck TS. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc. 2013;8:2471–2482. doi: 10.1038/nprot.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz J, Stange DE, Schepers AG, van de Wetering M, Koo BK, Itzkovitz S, Volckmann R, Kung KS, Koster J, Radulescu S, et al. The Lgr5 intestinal stem cell signature: robust expression of proposed quiescent ‘+4’ cell markers. EMBO J. 2012;31:3079–3091. doi: 10.1038/emboj.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–1272. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- Pearl EJ, Li J, Green JB. Cellular systems for epithelial invagination. Philos Trans R Soc Lond B Biol Sci. 2017;372 doi: 10.1098/rstb.2015.0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price LS, Leng J, Schwartz MA, Bokoch GM. Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell. 1998;9:1863–1871. doi: 10.1091/mbc.9.7.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolo A, Skoglund P, Keller R. Morphogenetic movements driving neural tube closure in Xenopus require myosin IIB. Dev Biol. 2009;327:327–338. doi: 10.1016/j.ydbio.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyer AE, Huycke TR, Lee C, Mahadevan L, Tabin CJ. Bending gradients: how the intestinal stem cell gets its home. Cell. 2015;161:569–580. doi: 10.1016/j.cell.2015.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyer AE, Tallinen T, Nerurkar NL, Wei Z, Gil ES, Kaplan DL, Tabin CJ, Mahadevan L. Villification: how the gut gets its villi. Science. 2013;342:212–218. doi: 10.1126/science.1238842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappenbeck TS, Gordon JI. Rac1 mutations produce aberrant epithelial differentiation in the developing and adult mouse small intestine. Development. 2000;127:2629–2642. doi: 10.1242/dev.127.12.2629. [DOI] [PubMed] [Google Scholar]
- Sulciner DJ, Irani K, Yu ZX, Ferrans VJ, Goldschmidt-Clermont P, Finkel T. rac1 regulates a cytokine-stimulated, redox-dependent pathway necessary for NF-kappaB activation. Mol Cell Biol. 1996;16:7115–7121. doi: 10.1128/mcb.16.12.7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan DW, Barker N. Intestinal stem cells and their defining niche. Curr Top Dev Biol. 2014;107:77–107. doi: 10.1016/B978-0-12-416022-4.00003-2. [DOI] [PubMed] [Google Scholar]
- Walko G, Castanon MJ, Wiche G. Molecular architecture and function of the hemidesmosome. Cell Tissue Res. 2015;360:529–544. doi: 10.1007/s00441-015-2216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton KD, Kolterud A, Czerwinski MJ, Bell MJ, Prakash A, Kushwaha J, Grosse AS, Schnell S, Gumucio DL. Hedgehog-responsive mesenchymal clusters direct patterning and emergence of intestinal villi. Proc Natl Acad Sci U S A. 2012;109:15817–15822. doi: 10.1073/pnas.1205669109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton KD, Whidden M, Kolterud A, Shoffner SK, Czerwinski MJ, Kushwaha J, Parmar N, Chandhrasekhar D, Freddo AM, Schnell S, et al. Villification in the mouse: Bmp signals control intestinal villus patterning. Development. 2016;143:427–436. doi: 10.1242/dev.130112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Zhenhuan G, Luxenburg C. The contractome--a systems view of actomyosin contractility in non-muscle cells. J Cell Sci. 2015;128:2209–2217. doi: 10.1242/jcs.170068. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Toivola DM, Feng N, Greenberg HB, Franke WW, Omary MB. Keratin 20 helps maintain intermediate filament organization in intestinal epithelia. Mol Biol Cell. 2003;14:2959–2971. doi: 10.1091/mbc.E03-02-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.