

Summary
A primary cause of disease progression in type 2 diabetes (T2D) is β cell dysfunction due to inflammatory stress and insulin resistance. However, preventing β cell exhaustion under diabetic conditions is a major therapeutic challenge. Here we identify the vitamin D receptor (VDR) as a key modulator of inflammation and β cell survival. Alternative recognition of an acetylated lysine in VDR by bromodomain proteins BRD7 and BRD9 directs association to PBAF and BAF chromatin remodeling complexes, respectively. Mechanistically, ligand promotes VDR association with PBAF to effect genome-wide changes in chromatin accessibility and enhancer landscape resulting in an anti-inflammatory response. Importantly, pharmacological inhibition of BRD9 promotes PBAF-VDR association to restore β cell function and ameliorate hyperglycemia in murine T2D models. These studies reveal an unrecognized VDR-dependent transcriptional program underpinning β cell survival and identifies the VDR:PBAF/BAF association as a potential therapeutic target for T2D.
eTOC
Modulation of a ligand-dependent switch between VDR-associated chromatin remodeling complexes enhances vitamin D response in β cells and curbs T2D progression.
Introduction
Already at epidemic proportions, the incidence of type 2 diabetes (T2D) is expected to continue largely unabated over the coming decades. Initiated by insulin resistance, β cell dysfunction is characterized by defective insulin secretion, ER stress, eventual β cell loss and disease progression (Ashcroft and Rorsman, 2012; Donath et al., 2013; Halban et al., 2014; Weyer et al., 1999). Though many therapeutic approaches are deployed to combat hyperglycemia, few (if any) treatments directly target β cell pathogenesis. Thus, long-term control of disease progression remains a persistent challenge.
Though the molecular underpinnings of obesity-induced β cell dysfunction are poorly understood, progress is being made. Increasing evidence links inflammation and specifically, the innate immune response of pancreatic islets to metabolic stress, to T2D progression (Donath and Shoelson, 2011; Fernandez-Real and Pickup, 2012; Imai et al., 2013). In particular, inflammatory stress associated with increased interleukin 1β (IL1β) and other cytokines reduces insulin secretion and leads to β cell damage and loss of identity (Herder et al., 2015). Blocking IL1β signaling by antagonists has shown modest β cell functional improvement, although the long term efficacy remains to be determined (Donath et al., 2013).
While vitamin D is widely known for its role in bone homeostasis, interest in its anti-inflammatory potential continues to grow. In addition, epidemiological and human genetic studies linking vitamin D and the VDR to both type 1 and 2 diabetes (Takiishi et al., 2010), suggest a potential therapeutic role in diabetes (Baeke et al., 2010; Bouillon et al., 2008; Cantorna et al., 2004). Indeed, vitamin D supplements are claimed to reverse many diabetic phenotypes in T1D rodent models by suppressing auto-immunity (Del Pino-Montes et al., 2004; Mathieu et al., 1994). However, a therapeutic benefit in type 2 diabetic patients (or rodents) has yet to be established (Takiishi et al., 2010). Distinct from the general role in suppressing bone-derived innate and adaptive immune cells, the function of VDR in β cells remains unclear. Moreover, although VDR has been shown to be involved in anti-inflammatory responses in various contexts (Ding et al., 2013; Sherman et al., 2014), how VDR responds to signals and initiates the downstream transcriptional cascade at the chromatin level remains largely a mystery.
Bromodomain-containing proteins 7 (BRD7) and 9 (BRD9) are closely related members of a diverse nuclear and cytoplasmic family of proteins that recognize acetylated lysines (Filippakopoulos and Knapp, 2014). Interestingly, BRD9 has recently been identified as a core component of the SWI/SNF (BAF) complex, whereas BRD7 belongs to an alternative chromatin remodeling complex PBAF (Kadoch et al., 2013; Kaeser et al., 2008). However, their specific roles within and beyond chromatin remodeling remain uncharacterized.
Here we describe two opposing gene expression networks as upstream modulators of β cell stress, and identify the balance between BAF-BRD9 and PBAF-BRD7 as a pivotal determinant of the VDR-driven anti-inflammatory and pro-survival response. In the absence of ligand, BRD9 recruits the suppressive BAF complex to VDR. In a dual regulatory mechanism, inhibition of the VDR-BRD9 interaction in combination with ligand activation of VDR cooperate to dismiss the BAF-BRD9 complex and shift the balance to the activating PBAF-BRD7 complex to induce a coordinated transcriptional response. Notably, pharmacologically potentiated VDR signaling by a synthetic ligand in combination with a BRD9 inhibitor is able to partially restore β cell function and glucose homeostasis in various T2D mouse models. Our results reveal an unexpected epigenetic regulation of VDR transcriptional response via the post-translational modification of VDR and a balance between its bromodomain readers, and demonstrate the potential of enhanced VDR signaling in diabetes therapeutics.
Results
VDR is required for β cell maintenance in vitro
To explore the mechanisms regulating β cell survival and expansion, a genomic CRISPR knock-out screen was performed in human induced pluripotent stem (iPS) cell-derived β-like cells (Yoshihara et al., 2016). The incorporation of an inducible Cas9 expression system (Wang et al., 2014) into β-like cells carrying a human insulin promoter-driven GFP reporter (INS-GFP) allowed genes essential for β-like cell maintenance to be identified (enrichment of sgRNAs in GFP− compared to GFP+ cells, Figure 1A). Gene ontology analysis of the enriched genes reveals categories related to chromatin modification/remodeling, cell cycle and transcription (Figure 1B). Notably, this unbiased genomic screen identified VDR as one of the most enriched gene targets, with 6 of the 7 sgRNAs targeting VDR found only in the GFP− cells (Figure 1C, D). Consistent with a potential role for VDR in β cells, published data sets confirm pancreatic VDR expression in both β cells and α cells (Benner et al., 2014; DiGruccio et al., 2016; Xin et al., 2016). Further interrogation of a single cell transcriptional dataset revealed VDR to be one of the most highly expressed transcription factors in mouse islet cells, particularly in β cells (Figure S1A, B).
Figure 1. VDR is essential for β cell homeostasis.

(A) Schematic representation of the genome-wide CRISPR loss-of-function screen in human iPSC-derived β-like cells.
(B) Gene ontology analysis of targets compromising β-like cell function (p<0.01, 156 total genes).
(C) p-value rank order plot of genes enriched in the loss-of-function screen, VDR is labeled in red.
(D) Distribution of normalized reads of individual sgRNAs in GFP sorted cells; VDR sgRNAs are labeled in red. X-axis location indicates sgRNA were found only in INS-GFP− cells.
(E) Immunohistochemistry staining of pro-insulin in VDR+/− or VDR−/− islets (scale bar: 100μm, each panel represents an individual mouse).
(F) Serum proinsulin/insulin ratio in VDR+/+ and VDR−/− male mice measured by ELISA (n=3, mean±S.E.M, * p<0.05).
To explore the functional significance of VDR expression in β cells, iPS cell lines harboring inducible shRNA knockdowns of VDR were differentiated into β-like cells and then challenged with IL1β. Consistent with VDR playing a role in cell survival during stress, increased cytokine-induced cell death was seen in the VDR-depleted β-like cells (Figure S2A). Furthermore, in the rat β cell line INS1, the expressions of key β cell genes were reduced in cytokine and palmitic acid treated cells upon VDR knockdown (Figure S2B). Moreover, whole body VDR knock-out (KO) mice display increased islet staining for proinsulin, a marker of ER stress and β cell dysfunction, and an increased proinsulin:insulin ratio in the serum (Figure 1E, F, S2C, D)(Bouillon et al., 2008; Hasnain et al., 2014; Hasnain et al., 2016). While the reduced adiposity of the VDR KO mice limits their utility in evaluating the contribution of β cell-specific defects to altered glucose homeostasis (Narvaez et al., 2009), the above findings combined with reduced levels of insulin and the survival factor MAFA in islets from VDR KO mice (Figure S2E), indicate that β cell function is compromised upon loss of VDR.
Given these findings, we next asked whether VDR activation can protect β cells against inflammatory damage. Isolated mouse islets were cultured with IL1β in the presence or absence of the synthetic VDR ligand, calcipotriol (Cal), and the transcriptomic changes determined by RNA-Seq. Exposure to IL1β for 48 hours induced widespread transcriptional changes, with 704 and 718 genes up- and down-regulated, respectively (p<0.05, Figure S2F). These transcriptional changes were largely maintained, with some even amplified, upon prolonged exposure (1090 and 989 genes up- and down-regulated after 96 hours, Figure S2F). Notably, co-treatment with Cal protected islets from IL1β-induced damage, with more pronounced effects evident at the later time point (Figure S2F–J). Gene ontology of the IL1β-suppressed genes whose expression was restored upon Cal treatment are mainly associated with β cell function, whereas the IL1β-induced genes repressed by Cal are associated with inflammation (Figure S2H, J). These findings support the notion that VDR is able to protect against IL1β-induced β cell dysfunction, in part by counteracting the inflammatory response and maintaining β cell functionality.
VDR shuttles between BRD9 and BRD7
To dissect the molecular underpinning of VDR function, we characterized the VDR interactome in human β-like cells. INS-GFP iPS cells with inducible HA-tagged VDR were differentiated into β-like cells, and VDR expression induced to coincide with the peak expression of insulin, as measured by GFP expression. VDR was subsequently immunoprecipitated from cells grown in the presence or absence of Cal 24 hours after VDR induction, and associating proteins identified by mass spectrometry. Consistent with previous studies, multiple transcription elongation factors and components of the mediator and FACT complexes were found to co-immunoprecipitate with VDR (Figure 2A). In addition, novel VDR-interacting proteins were identified, most notably the bromodomain-containing factor 9 (BRD9), and its paralog, BRD7 (Figure 2A). BRD9 was recently identified as a core component of the BAF complex, whereas BRD7 is found in the PBAF complex. Consistent with this, we showed that BRD9 selectively associates with the BAF but not the PBAF complex, as previously reported (Figure S3A, (Hodges et al., 2016)). Interestingly, BRD7 was also identified in our CRISPR screen (p=0.002) (Figure 1B, S3B).
Figure 2. Bromodomain proteins BRD7 and BRD9 bind acetylated VDR.

(A) Heat map of VDR interacting proteins in human β-like cells in the absence or presence of ligand Cal. Spectral counts normalized to VDR.
(B) Western blot of BRD9 in immunoprecipitates (IP) of endogenous VDR from 293T cells 1h after indicated treatments.
(C) Western blot of ectopically expressed VDR in IP of 293T cells expressing Flag-tagged BRD9, 1h after indicated treatments.
(D) In vitro interaction of purified full-length HA-VDR with the bromodomain of BRD9 fused to GST.
(E) Alignment of T-box sequences from related nuclear receptors showing conservation of the lysine acetylated in VDR.
(F) Crystal structure of the DNA binding domains of VDR (red) bound to DNA (blue), highlighting the sidechain of K91 (green). Image is derived from the VDR crystal structure PDB ID 1kb2 (Shaffer and Gewirth, 2002).
(G) Western blot of endogenous BRD9 in IP from 293T cells expressing wild type, K91R, or K91A HA-tagged VDR.
(H) Western blot of ectopically expressed VDR in IP from 293T cells expressing Flag-tagged BRD7, 1h after indicated treatments.
(I) Western blot of ectopically expressed Flag-BRD7 in IP from 293T cells expressing wild type, K91R, or K91A HA-tagged VDR.
(J) Reported increases in hydrogen-deuterium exchange upon addition of 1,25 (OH)2 vitamin D (Zhang et al., 2011), mapped onto the crystal structure of the DNA binding domain of VDR.
(K) Western blot of endogenous PCAF in IP from INS1 cells expressing HA-VDR, 1h after indicated treatments.
(L) Western blot of ectopically expressed HA-VDR in IP from 293T cells over-expressing wild type, or an enzymatically dead PCAF (D608A).
See also Figure S3.
To verify these novel VDR interactions, we first confirmed that endogenous BRD9 co-immunoprecipitates with HA-tagged VDR as well as endogenous VDR, and conversely, that HA-tagged VDR can be immunoprecipitated with Flag-tagged BRD9 in 293T cells (Figure 2B and 2C, S3C). Somewhat surprisingly, the strength of this interaction was markedly decreased in the presence of VDR ligand as measured by proteomic and Western blot analyses (Figure 2A–C, S3C–E). Furthermore, a selective inhibitor of the bromodomain of BRD9 (iBRD9) (Theodoulou et al., 2016), also reduced the interaction between VDR and BRD9, suggesting that BRD9 directly binds to VDR via its bromodomain (Figure 2B and 2C). Consistent with this, we show that the bromodomain of BRD9 is sufficient for the interaction with VDR (purified recombinant His-tagged VDR binds the GST-tagged bromodomain of BRD9, Figure 2D and S3F). Since bromodomains bind acetylated lysines, we hypothesized that acetylation of VDR was required for the interaction. Indeed, mass spectrometry clearly showed the presence of an acetylated lysine in a VDR peptide containing lysine 91 (K91Ac) (Figure S3G). Interestingly, K91 is located in the T-box domain (Lee et al., 1994; Quack et al., 1998) juxtaposed to the DNA-binding zinc fingers, a domain highly conserved in closely related nuclear receptors including RARα and PXR (Figure 2E). The structure of the VDR DNA-binding domain bound to direct repeat DNA elements (Orlov et al., 2012; Shaffer and Gewirth, 2002) reveals the side-chain of K91 protruding away from the DNA double helix, suggesting it may serve as a docking site for the interaction with BRD9 (Figure 2F). To confirm the importance of K91Ac in the VDR:BRD9 interaction, we mutated K91 to alanine (K91A) or arginine (K91R). As predicted, mutating K91 significantly reduced the interaction with BRD9, as well as the total acetylation level of VDR (Figure 2G and S3H).
Similarly, we confirmed that HA-tagged VDR was able to bind Flag-tagged BRD7 (Figure 2H and S3I). However, in contrast with BRD9, the interaction of VDR with BRD7 progressively increased in the presence of Cal and Cal+iBRD9, suggestive of competitive BRD9/BRD7 binding (Figure 2H). Indeed, we showed that VDR acetylation at the same site, K91, promoted the interaction with BRD7 (Figure 2I), supporting the competitive binding hypothesis. The notion of a ligand-induced shift in the association of VDR from BRD9 to BRD7 is supported by previous findings that revealed increased conformational flexibility in the DNA-binding domain and T box regions upon binding of 1,25(OH)2 vitamin D (Zhang et al., 2011) (Figure 2J).
As the T-box region has been previously implicated in the interaction of nuclear receptors with PCAF (KAT2b) (Blanco et al., 1998), we examined the interaction between VDR and PCAF. PCAF binding increased with Cal and/or iBRD9 treatment (Figure 2K), as seen for BRD7 but the reverse of that seen for BRD9 binding. As a lysine acetyltransferase, we explored whether PCAF directly acetylates VDR. VDR acetylation indeed increased when expressed together with PCAF, but not with the enzymatically-dead PCAF D608A mutant (Jin et al., 2014) (Figure 2L). Taken together, the above findings suggest an unrecognized regulatory mechanism for VDR whereby T-box acetylation partitions recruitment between BRD9 or BRD7, depending on the absence or presence of VDR ligand, respectively.
BRD9 dissociation promotes VDR-mediated transcription in β cells
As BRD9 and BRD7 are components of the SWI/SNF (BAF) and PBAF complexes, respectively, they are in a position to mediate the alternative recruitment of VDR. To explore the idea of complex exchange, we added VDR ligand (Cal) to favor BRD7-PBAF association, iBRD9 to block re-association to BAF, as well as both compounds together. Proteomic analyses of INS1 cells expressing HA-tagged VDR confirmed the ligand-dependent association of VDR with its heterodimeric partner RXR as well as components of the basal transcriptional machinery (Figure 3A). VDR association proteins common to the BAF and PBAF complexes similarly increased in the presence of Cal or iBRD9 (e.g. SMARCA1, SMARCA2, SMARCA4, Figure 3A–B, S4A–E). In addition, we find significantly enhanced interactions with key components of the PBAF complex including PBRM1 (BAF180) and ARID2 (but not BAF-specific components such as ARID1A and ARID1B) (Figure 3A–B, S4A–E). These findings suggest that the activation state of VDRK91Ac (i.e. ± Cal or iBRD9) is conditional, dependent on its association with the BAF or PBAF complexes.
Figure 3. BRD9 represses VDR activation upon acute cytokine stress in β cells.

(A) Heat map of VDR interactome in INS1 cells expressing HA-tagged VDR treated with vehicle, Cal, iBRD9, or Cal+iBRD9. Spectral counts normalized to VDR.
(B) Western blots of immunoprecipitates from INS1 cells expressing HA-tagged VDR with indicated treatments.
(C) Expression of Nfkbia in INS1 cells 30 minutes after exposure to IL1βand indicated co-treatments.
(D) Western bolt of NFKBIA (IκBα) in INS1 cells 30 minutes after exposure to IL1β with and without Cal, iBRD9, or Cal+iBRD9.
(E) Time course of Nfkbia expression in INS1 cells in response to IL1β with indicated treatments.
(F) Nfkbia levels in IL1β-stressed INS1 cells with siRNA knock down of PBRM1 or PCAF.
n=3, mean±S.E.M., *P<0.05
See also Figure S4.
Based on the above findings, we posited that BRD9 attenuates VDR activity. To explore the functional relevance of this molecular toggle in β cells, we determined the effects of Cal and iBRD9 upon IL1β-induced stress in INS1 cells (Hahn et al., 1997; Riachy et al., 2006). Within 30 min of exposure, IL1β induced the expression of IκBα (aka Nfkbia), a key negative feedback early response gene (Verma et al., 1995). Interestingly, this anti-inflammatory modulator was increased with both Cal and Cal+iBRD9 treatments (Figure 3C), with parallel changes seen at the protein level (Figure 3D). Furthermore, the induction of Nfkbia upon Cal treatment was largely abrogated in cells expressing the VDR dominant negative mutant (K91R) (Figure S4F). Beyond an acute effect, the iBRD9-Cal combination led to sustained Nfkbia induction (>24 hrs, Figure 3E) portending a possible sustained anti-inflammatory effect (see below). Moreover, knockdown of the PBAF component PBRM1 or PCAF significantly compromised the activation of Nfkbia (Figure 3F). These findings support BRD9 as a regulator of unliganded VDR, whereby inhibition of BRD9 binding allows increased association of VDR with the PBAF complex and PCAF to prolong transcriptional activation.
Enhanced activation of VDR promotes a β cell stress response
Transcriptional changes induced by the addition of Cal and iBRD9 were used to explore mechanistic transitions in cytokine-stressed β cells. 1 hour exposure of IL1β alone was sufficient to trigger rapid and widespread changes in INS1 cells, with 499 and 496 genes significantly induced and repressed, respectively (fold change >1.5) (Figure 4A). Notably, co-treatment with Cal+iBRD9 partially reversed the cytokine-induced inflammatory response, abrogating ~25% of the IL1β-induced transcriptional changes (141 up- and 127 down-regulated genes, Figure 4A and B). Within this regulated network, gene ontology (GO) analyses of these Cal+iBRD9 responsive genes identified β cell function (Figure 4C), along with cytokine response and NF-κB signaling (Figure 4D) in the IL1β-repressed and induced gene sets, respectively. Thus, the Cal+iBRD9 combination significantly protects against cytokine-induced transcriptional changes by reducing the pro-inflammatory response and preventing the loss in functionality of β cells.
Figure 4. Cal and iBRD9 cooperate to reverse inflammation-driven transcriptional changes.

(A) IL1β-induced transcriptional changes in INS1 cells, 1 hour after treatment (fold change>1.5) Black bars represent genes rescued by co-treatment with Cal+iBRD9.
(B) Heat map showing relative expression of the subset of rescued genes in A.
(C and D) Gene ontology categories enriched in the subset of IL1β repressed (C) and induced (D) genes whose expression is restored with Cal+iBRD9 treatment.
(E) Percentage of peaks with significant changes in chromatin accessibility (fold change>4).
(F) Chromatin accessibility at sites that significantly increased in response to Cal+iBRD9.
(G) Transcription factors (TF) motifs enriched in chromatin regions showing increased accessibility upon Cal+iBRD9 treatment.
(H) Relative accessibility at VDR response elements (VDREs) 1 hour after indicated treatments.
(I) Browser tracks showing ATAC-seq data 1 hour after indicated treatments at indicated loci.
Our proteomics data indicated that inhibiting the interaction with BRD9 redirects VDR equilibrium towards the PBAF complex (Figure 3A and C). Consistent with this, we show that Cal+iBRD9 treatment reduced ARID1B binding – a component of the BAF complex, and increased BAF180 binding – a hallmark of the PBAF complex, at multiple target gene promoters (Figure S4G–L). Given the established roles of the BAF and PBAF complexes in chromatin remodeling, we examined the impact of this VDR-shifted equilibrium on genome-wide chromatin accessibility. Notably, transposase-based sequencing (ATAC-Seq) (Buenrostro et al., 2013) revealed chromatin remodeling upon Cal and iBRD9 treatment. IL1β-induced stress led to an overall reduction in chromatin accessibility in INS1 cells one hour after treatment (Figure 4E). In contrast, Cal and iBRD9 as single agents antagonized a large portion of these changes, resulting a significant increase (>4 fold) in chromatin accessibility at ~4% (1935 and 1703 peaks) of all ATAC-Seq peaks in IL1β treated cells (Figure 4E and F). The combined Cal+iBRD9 treatment further increased accessibility and resulted in global increases (>4 fold) at >3 times more peaks (~15%, 7932 out of 51397 peaks) (Figure 4E and F). Motif analyses of Cal+iBRD9-induced ATAC-Seq peaks identified Vitamin D response elements (VDREs) as the top-ranking motif together with CTCF (Figure 4G), implicating a direct role of VDR in counteracting the effects of IL1β. Indeed, chromatin accessibility was markedly increased at identified VDREs upon Cal+iBRD9 treatment (Figure 4H). In particular, chromatin accessibility at Nfkbia and Nfkbiz was significantly increased upon Cal+iBRD9 treatment (Figure 4I), consistent with the sustained Nfkbia expression (Figure 3I). In addition, the reduction in chromatin accessibility induced by IL1β stress at key genes involved in β cell functionality including Cckar was reversed by Cal+iBRD9 treatment (Figure 4I). These findings support a model in which dissociating BRD9 from VDR by ligand or inhibitor results in selected chromatin accessibility by stabilizing “hyper-open” regions at VDR target genes.
Activation of VDR reverses cytokine-induced changes in enhancer landscape
To better understand how VDR shuttling between BAF and PBAF complexes impacts chromatin states, a genome-wide enhancer landscape was generated using H3K27Ac as a mark of active enhancers (Rada-Iglesias et al., 2011). Short-term IL1β exposure (1 hour) significantly altered the enhancer landscape, with widespread decreases and increases in H3K27 acetylation (Figure 5A and B, respectively). As was observed for chromatin accessibility (Figure 4E), Cal and iBRD9 as single agents were both able to partially reverse the impact of IL1β (Figure 5A and B). Examination of the sites where Cal+iBRD9 treatment increased H3K27 acetylation revealed VDRE as the most enriched motif (Figure 5C), implicating a direct role for VDR in reversing the IL1β-induced enhancer changes. Furthermore, an interrogation of the Cal+iBRD9 rescued gene set (Figure 4B) revealed that the VDRE-proximal decreases (at repressed genes, Figure 5D) and increases (at activated genes, Figure 5E) in H3K27 acetylation induced by IL1β were partially restored with Cal+iBRD9 treatment (Figure 5D–F).
Figure 5. VDR directly antagonizes global enhancer dynamics induced by IL1.

β
(A and B) H3K27Ac peak intensities at sites repressed (A) or induced (B) by IL1β (>1.5 fold) 1 hour after indicated treatments in INS1 cells.
(C) TF binding motifs enriched at sites of Cal-induced increases in H3K27Ac.
(D and E) H3K27Ac density at accessible VDREs (within ATAC-Seq peaks) in IL1β repressed (D) or induced (E) genes rescued by Cal+iBRD9.
(F) Browser tracks showing H3K27Ac at Camk4 and Cckar after indicated treatments.
(G and H) H3K27Ac peaks synergistically enhanced by iBRD9 (IL1β+Cal+iBRD9 vs. IL1β+Cal, 6 hours after treatment) and associated GO categories (H).
(I) Browser tracks showing H3K27Ac at Nfkbia and Camk4 loci 6 hours after IL1β+Cal and IL1β+Cal+iBRD9 treatments.
(J and K) H3K27Ac peaks synergistically repressed by iBRD9 (IL1β+Cal+iBRD9 vs. IL1β+Cal 6 hours after treatment) and associated GO categories (K).
(L) Browser tracks showing H3K27Ac at Ccl28 and Txnip loci 6 hours after indicated treatments.
Given the sustained hyper-activation of Nfkbia expression observed with the combined Cal+iBRD9 treatment (Figure 3E), we next determined the synergistic effect on epigenomic changes after a prolonged IL1β stress (6 hours). At this later time point, increased H3K27Ac with Cal+iBRD9 treatment was seen at 945 peaks compared to Cal alone, of which 149 (16%) contained VDREs (Figure 5G). The genes associated with these VDRE+ peaks are mainly related to β cell function (e.g. Camk4) and anti-inflammatory responses (e.g. Nfkbia, Figure 5H and I). Concomitantly, Cal+iBRD9 treatment reduced H3K27Ac at 1232 peaks, of which 130 (10.5%) contain VDREs (Figure 5J), with the VDRE+ gene set mainly related to the inflammatory response (Figure 5K and L). Together, these data indicate a synergistic action of Cal+iBRD9 on H3K27Ac levels that affects both transcriptional activation and repression. The less pervasive changes in H3K27Ac levels compared to those in chromatin accessibility are consistent with alterations in enhancer elements being secondary to changes in chromatin accessibility. Combined with the marked changes in chromatin accessibility (Figure 4E–I), these data identify a balance between PBAF- and BAF-modulated VDR transcription whereby VDR activation in combination with BRD9 inhibition shifts VDR towards the PBAF complex, resulting in reshaping of the enhancer landscape and antagonism of cytokine-induced transcriptomic changes.
VDR-PBAF complex reverses β cell dysfunction to reduce hyperglycemia
The improvements in markers of β cell survival and function with Cal+iBRD9 treatment suggest that this approach may have therapeutic utility. To explore this possibility, we investigated the benefits of Cal+iBRD9 treatment in a dietary stress mouse model. B6.BKS(D)-Leprdb/J (hereafter referred to as db/db (B6)) mice have markedly increased levels of cytokines that lead to both β cell dysfunction and insulin resistance (Chan et al., 2013). To assess the potential for VDR-targeted therapies to ameliorate dietary-induced inflammatory stress, mice were injected with vehicle, Cal alone (60μg/kg i.p.), iBRD9 alone (10mg/kg i.p.), or the combination (Cal: 60μg/kg i.p. + iBRD9:10mg/kg i.p.) 3 times a week for 9 weeks. Vehicle-treated db/db (B6) mice developed pronounced hyperglycemia, with fasted blood glucose levels ~400mg/dl (Figure 6A). Cal and iBRD9 as single agents achieved modest improvements in blood glucose levels after 4–6 weeks treatment. Notably, mice receiving the Cal+iBRD9 combination showed a significant reduction in blood glucose after 4 weeks treatment, with the beneficial effects progressively increasing with treatment duration (Figure 6A). These improvements in glucose homeostasis were achieved without significant changes in body weight (Figure 6B), liver or epididymal white adipose tissue (eWAT) weights, metabolic rate, or serum calcium level (Figure S5 A–E). Intraperitoneal glucose tolerance tests (IPGTT) were performed to evaluate the functionality of the β cells independent of the incretin effect. While mice treated with Cal alone showed improved glucose clearance compared to vehicle-treated mice, more significant improvements were achieved with Cal+iBRD9 (Figure 6C), suggesting improved insulin secretion with the combination therapy. Consistent with this, random fed serum insulin levels were significantly increased by Cal+iBRD9 treatment, and increased insulin secretion was observed during an intraperitoneal glucose challenge (Figure 6D, E). These findings indicate that activation of VDR combined with the dismissal of the BAF complex is able to improve β cell function and thereby glucose homeostasis in an inflammation-driven diabetes model.
Figure 6. Cal+iBRD9 treatment rescues β cell dysfunction in diabetic mice.

(A) Overnight fasted blood glucose levels in db/db (B6) mice during indicated treatments (n=6–11).
(B) Body weights of mice in A (n=6).
(C) IPGTT of db/db (B6) mice after 9 weeks of indicated treatments (n=6).
(D) Fasting serum insulin levels in 9 week-treated db/db (B6) mice as described in A (n=4–6).
(E) Serum insulin levels in vehicle and Cal+iBRD9 treated db/db (B6) mice in response to a glucose challenge.
(F) Overnight fasted blood glucose levels in db/db (BKS) mice during treatment (n=8).
(G) Fasting serum insulin levels in 12 week-treated db/db (BSK) mice as described in F (n=5).
(H) Overnight fasted blood glucose levels in STZ-treated C57/B6 mice during indicated treatments (n=8–10).
(G) IPGTT of STZ mice after 7 weeks treatment with vehicle or Cal+iBRD9 (n=5).
Data reported as mean±S.E.M, *P<0.05.
See also Figure S5.
Mice lacking the leptin receptor on a BKS genetic background (C57BLKS-Leprdb, hereafter db/db (BKS)) develop a more severe diabetic phenotype than db/db(B6) mice (Garris and Garris, 2004). Chronic hyperglycemia in db/db (BKS) progresses to β cell atrophy, hypoinsulinemia and β cell failure, resembling the progression of patients with genetic defects in β cell function. In these genetically predisposed mice, treatment with Cal alone (60μg/kg i.p. 3 times/week) provided only limited improvements in glucose management (Figure 6F). In contrast, 3 weeks co-treatment with Cal+iBRD9 was able to reduce the fasting blood glucose levels, and continued treatment maintained these levels for ~10 weeks without any body weight changes (Figure 6F, S5F). Consistent with improved glucose management, Cal+iBRD9 treatment partially reversed the hallmark hypoinsulinemia associated with the db/db (BKS) model (Figure 6G).
Finally, we explored the therapeutic utility of targeting VDR in a β cell damage model, designed to mimic the severe β cell loss observed in the final stages of T2D patients. B6/J mice treated with low doses of streptozotocin (STZ) progressively lose glucose control. Interestingly, Cal and iBRD9 as single agents provided limited to no benefit in this β cell damage model. Notably, the combination Cal+iBRD9 treatment provided extended improvements in glucose control (Figure 6H) and increased glucose tolerance and serum insulin levels (Figure 6I, Figure S6D). In combination, in vivo application of VDR ligand and iBRD9 results in enhanced β cell function and insulin secretion in each of the three mouse models representing different stages of T2D progression, strongly supporting the potential therapeutic value of this synergetic strategy.
Targeting the VDR-BRD9 axis rescues β cell dysfunction in mouse and human islets
To understand how VDR activation improves glucose homeostasis in each of the three mouse models, we compared the molecular and structural changes induced in islets. The marked induction of Cyp24A1, a classic VDR target gene, confirmed VDR activation in islets as well as in the intestine of the Cal treated db/db (B6) mice (Figure S6A). In addition, Ins2 mRNA levels trended upwards in Cal-treated islets, consistent with the increase seen in serum insulin levels (Figure S6B, 6D). Transcriptomic analysis revealed the upregulation of multiple key transcription factors including Nkx6-1 and Nr4a1 (Nur77) (Figure 7A), whose repression have been shown to correlate with β cell dysfunction (Guo et al., 2013; Talchai et al., 2012). Furthermore, multiple cytokine and chemokine genes were downregulated, further supporting the conclusion that activation of VDR reduced the pro-inflammatory response in islets (Figure 7A). Gene ontology of the upregulated genes identified pathways involved in β cell function including OXPHOS, secretion and β cell proliferation enriched in the Cal-treated islets (Figure 7B). In addition, we found reduced expression of gluconeogenesis genes including Pepck, Fbp1 and G6pase in the livers of Cal-treated mice, most likely linked to enhanced levels of serum insulin (Figure S6C). Thus, the benefits of VDR activation in the inflammation-driven db/db (B6) model are multifactorial, including reduced inflammation, improved β cell function and decreased hepatic gluconeogenesis.
Figure 7. VDR reverses β cell stress in vivo.

(A and B) Volcano plot (A) and GO analysis (B) of Cal-induced transcriptional changes in islets from db/db (B6) mice.
(C) H&E staining of pancreata from db/db (BKS) mice treated with vehicle or Cal+iBRD9. (Scale bar: 50μm)
(D) Islet area, expressed as a percentage of total pancreas, in db/db (BKS) mice described in C (vehicle n=6, Cal+iBRD9 n=15).
(E) Islet size in treated db/db (BKS) mice described in C.
(F and G) Immunohistochemistry and quantification (G) of Ki67 in vehicle and Cal+iBRD9 treated pancreas sections from db/db (BKS) mice. (Scale bar: 50μm)
(H) Immunofluorescence staining of insulin (red) in pancreas sections from vehicle, Cal and Cal+iBRD9 treated db/db (BKS) mice. (Scale bar: 50μm)
(I) Immunofluorescence staining of insulin (red) and glucagon (green) in pancreas sections from vehicle and Cal+iBRD9 treated low dose STZ mice. (Scale bar: 50μm).
(J) Relative β and α cell numbers in pancreas sections in I (n=3)
(K) Transmission electron microscopic images of healthy human islets treated with vehicle, IL1β+IFNγ, IL1β+IFNγ+Cal, or IL1β+IFNγ+Cal+iBRD9 for 24 hours. Magnified images of insulin granules and ER are shown in the upper and lower right panels, respectively. (Scale bar: 1μm).
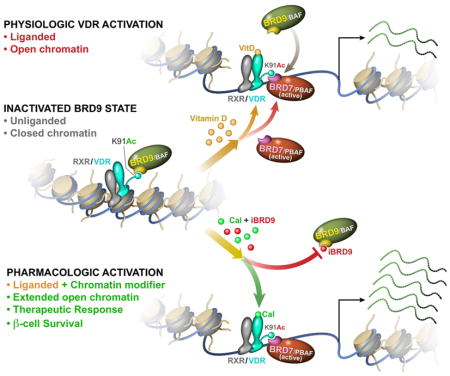
(L) Schematic model depicting the synergy between Cal and iBRD9 in directing the association of VDR with alternate chromatin remodeling complexes and the consequential effects on transcription.
Data reported as mean±S.E.M., *P<0.05, **P<0.01.
Distinct from diet-induced T2D models, β cell intrinsic defects in db/db (BKS) mice severely compromise β cell proliferation and are considered the major cause for the phenotype severity (Puff et al., 2011). Notably, 12 weeks of Cal+iBRD9 treatment led to ~70% increase in islet area in the pancreas, as well as a significant increase (~100%) in the size of individual islets in db/db (BKS) mice (Figure 7C–E). Consistent with these observations, the number of proliferating cells measured by Ki67 labeling more than doubled after Cal+iBRD9 treatment (Figure 7F and G). Moreover, based on immunologic stains, intracellular insulin levels were also rescued by Cal or Cal+iBRD9 treatment (Figure 7H), suggesting that dysfunction within the β cell (Guo et al., 2013) is reversed upon VDR activation.
As a common model of phase III or end stage diabetes, low-dose STZ treatment induces islet loss and α cell expansion (Li et al., 2000). Cal+iBRD9 treatment was protective against progressive β cell death, with increased insulin and decreased glucagon staining (Figure 7I) seen after 7 weeks of treatment (quantified in Figure 7J). These changes were accompanied by an increase in the level of NKX6-1, a marker for β cell function (Figure S6E), further supporting that the β cell dysfunction caused by multiple layers of stress is ameliorated upon enhanced VDR activation. The above findings indicate that Cal+iBRD9 treatment improves glucose homeostasis in multiple diabetic mouse models through the reversal of β cell dysfunction and increased insulin secretion.
In addition to inflammation, ER stress, IAPP amyloid deposition, lipo- and gluco-toxicity all contribute to islet dysfunction in human T2D patients (Mukherjee and Soto, 2017; Papa, 2012). To test whether vitamin D signaling can modulate the damage attributed to additional stressor, we treated islets from aged (15 months) transgenic mice carrying a copy of human islet amyloid polypeptide driven by rat insulin II promoter (Janson et al., 1996) with Cal or Cal+iBRD9 for 72 hr ex vivo. Multiple inflammatory and stress markers were reduced upon Cal or Cal+iBRD9 treatment (Figure S6F). In addition, Thapsigargin or Tunicamycin-induced ER stress in INS1 cells was attenuated by Cal treatment (Figure S6G). Finally, ER stress markers Calnexin and PERK were reduced in db/db (BKS) mice treated with Cal or Cal+iBRD9 (Figure S6H), suggesting that VDR activation can reduce ER stress in highly dysfunctional β cells in vivo.
While the significant improvements seen in the diabetic models are consistent with the effects observed in β cell lines upon Cal + iBRD9 treatment, efficacy in related tissues such as macrophages or the intestines, or off target effects cannot be excluded (Hohmann et al., 2016). In support of the specificity of iBRD9, structurally-unrelated BRD9 inhibitors (Hohmann et al., 2016; Martin et al., 2016) induced similar anti-inflammatory effects on NF-κB signaling (Figure S6I). In addition, the maximum plasma iBRD9 concentration (~750nM, Figure S6J) is sufficient to activate BRD9 (IC50=50nM) while sparing other bromodomain members such as BRD4 (IC50=5μM) (Theodoulou et al., 2016). Collectively, these data support that the therapeutic efficacy of iBRD9 in vivo is on-target.
As islet allogenic transplants are a medium-term therapy for select patients, we explored the ability of VDR activation to protect isolated human islets. Healthy human islets were challenged with an inflammatory stress (Arnush et al., 1998) (IL1β+IFNγ for 24 hrs) with or without activation of VDR. Transmission electron microscopy confirmed that IL1β and IFNγ caused severe islet damage including loss of dense insulin granules and dilation of the ER, as previously reported (Ravelli et al., 2013; Song et al., 2008) (Figure 7K upper and lower inserts respectively). Activation of VDR alone or in combination with iBRD9 rescued the damage caused by IL1β and IFNγ, most notably at the level of the insulin granules (upper inserts, bottom panels) and ER morphology (lower inserts, bottom panel) (Figure 7K). Additionally, Cal increased survival and insulin secretion capacity in long term ex vivo human islet cultures (Figure S7A and S7B). While these findings will require further validation in additional human samples, they support the conclusion from the mouse models that enhanced vitamin D signaling has significant beneficial effects on islets during cytokine stress.
Discussion
Here we identify VDR as a critical factor in β cell survival and function. Initially identified in an unbiased CRISPR knockout screen, genetic loss-of-function and ex vivo gain-of-function studies support a protective role for VDR in β cells and the modulation of islet stress. In exploring the regulatory mechanisms of VDR-driven transcriptional action, we demonstrate the shuttling of VDR between competing chromatin remodeling complexes (Figure 7L). Specifically, we describe a novel acetylated lysine in VDR that facilitates its recognition by the highly related bromodomain reader proteins, BRD9 and BRD7, respective components of the BAF and PBAF complexes. We show that VDR shuttles between BAF and PBAF complexes in a ligand-dependent fashion. Furthermore, we show that a highly selective BRD9 inhibitor, iBRD9, cooperates with VDR ligand to favor PBAF complex binding and significantly potentiates VDR signaling. Mechanistically, binding of the PBAF complex enhances chromatin accessibility at consensus VDR binding elements to modulate the expression of key inflammatory response genes including the inhibitor of NFκB, Nfkbia.
Despite many shared components, the BAF and PBAF complexes are distinct assemblies with functional differences (Hodges et al., 2016). While the PBAF complex has been shown to be a superior transcriptional activator for nuclear receptors compared to the BAF complex in vitro (Lemon et al., 2001; Rafati et al., 2011), the ability of these two complexes to mediate ligand-dependent signaling was not previously known. Here we describe a novel mechanism in which acetylation at K91 in VDR is recognized by the BRD7/9 subfamily of bromodomain proteins that facilitate the alternative association of the PBAF and BAF complexes in a ligand-dependent fashion. Fortuitously, the switching of VDR’s partner from BAF-BRD9 to PBAF-BRD7 can also be pharmacologically manipulated by targeting the “reader” of VDR acetylation, thereby providing an additional level of nuclear receptor signaling regulation.
Pro-inflammatory cytokines such as IL1β and TNFα are detrimental to β cell and islet function (Nackiewicz et al., 2014), and are the ongoing targets of multiple clinical trials (Imai et al., 2013). However, use of selective inhibitors of inflammation to treat T2D have produced only modest improvements (Donath, 2016). In contrast, exploiting VDR as a negative modulator of NFκB signaling in β cells is highly effective, and as we show the combination of a VDR activator with a BRD9 inhibitor results in remarkably prolonged suppression of the damaging effects of inflammatory insults.
While multiple therapies aimed at improving β cell function are in the clinic or under development, chronic stress-induced damage to β cells continues to pose a serious challenge for long term therapeutic efficacy (Zhou et al., 2016). By focusing on the VDR shuttle between BAF and PBAF complexes, we uncover an unusual therapeutic strategy to suppress inflammation via sustained receptor activation in β cells. This strategy demonstrates the power of genomic reprogramming, tackling the problem at the genome-wide level. As this is a natural way in which β cells normally limit chronic inflammation, exploiting the power of the VDR, its acetylation and the safety of its ligands offer a very different approach in the treatment of T2D.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-BRD9 | Bethyl | Cat# A303-781A RRID:AB_11218396 |
| Rabbit polyclonal anti-VDR | Santa Cruz | Cat# sc-1008 RRID:AB_632070 |
| Mouse monoclonal anti-VDR | Santa Cruz | Cat# sc-13133 RRID:AB_628040 |
| Mouse monoclonal anti-FLAG | Sigma | Cat# P2983 RRID:AB_439685 |
| Rat polyclonal anti HA | Roche | Cat# 11867423001 RRID:AB_390918 |
| Rabbit polyclonal anti-lysine acetylation | Cell Signaling | Cat# 9441S RRID:AB_331805 |
| Rabbit polyclonal anti-GST | Cell Signaling | Cat# 2622 RRID:AB_331670 |
| Rabbit polyclonal anti-PCAF | Cell Signaling | Cat# 3378S RRID:AB_2128409 |
| Rabbit polyclonal anti-SMARCC1/2 | Cell Signaling | Cat# 11956 |
| Rabbit polyclonal anti-BRG1 | Cell Signaling | Cat# 3508S RRID:AB_2193944 |
| Rabbit polyclonal anti-PBRM1 | Bethyl | Cat# A301-591A RRID:AB_1078808 |
| Rabbit polyclonal anti-PHF10 | Genetex | Cat# GTX116314 RRID:AB_11176023 |
| Rabbit polyclonal anti-ARID1B | Bethyl | Cat# A301-040A RRID:AB_817895 |
| Rabbit polyclonal anti-HDAC3 | Cell Signaling | Cat# 2632 RRID:AB_331545 |
| Rabbit polyclonal anti-IKBa | Cell Signaling | Cat# 4814 RRID:AB_390781 |
| Rabbit polyclonal anti-p65 | Cell Signaling | Cat# 8242 RRID:AB_10859369 |
| Rabbit polyclonal anti-IKKb | Cell Signaling | Cat# 8943 RRID:AB_11024092 |
| Mouse monoclonal anti-a Tubulin | EMD | Cat# CP06-100UG RRID:AB_212802 |
| Rabbit polyclonal anti-H3K27Ac | Abcam | Cat# ab4729 RRID:AB_2118291 |
| Rabbit polyclonal anti-Nkx6-1 | Cell Signaling | Cat# 54551 |
| Mouse monoclonal anti-glucagon | Abcam | Cat# ab82270 RRID:AB_1658481 |
| Guinea pig polyclonal anti-insulin | Abcam | Cat# ab7842 RRID:AB_306130 |
| Mouse monoclonal anti-proinsulin | R&D | Cat# MAB13361 RRID:AB_2126534 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab16667 RRID:AB_302459 |
| Rabbit polyclonal anti-Calnexin | Cell Signaling | Cat# 2433S RRID:AB_2243887 |
| Rabbit monoclonal anti-PERK | Cell Signaling | Cat# 3192 RRID:AB_2095847 |
| Rabbit polyclonal anti-MAFA | Novus | Cat# NB400-137 RRID:AB_10002142 |
| Rabbit monocolonal anti-phospho-PERK | Cell Signaling | Cat# 3179S RRID:AB_2095853 |
| Biological Samples | ||
| Human islets | See Table S1 | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Calcipotriol | Tocris | Cat#2700 |
| i-BRD9 | R&D | Cat#5591 |
| IL-1b (mouse) | Peprotech | Cat#211-11b |
| IL-1b (human) | Peprotech | Cat#200-01b |
| IL-23 | Peprotech | Cat#200-23 |
| IL-24 | Peprotech | Cat#200-35 |
| IFN-g | Peprotech | Cat#315-05 |
| Streptozotocin | Sigma | S0130 |
| Activin | R&D | 338-AC |
| Wnt3a | R&D | 5036-WN |
| Dorsomophin | Biovision | 1656-d |
| Retinoic acid | Sigma | R2625 |
| SB431542 | Sigma | S4317 |
| B27 | Gibco | 17504-044 |
| Forskolin | Sigma | F3886 |
| Dexmethasone | Sigma | D4904 |
| Alk5 inhibitor II | Calbiochem | 616452 |
| Nicotinamide | Sigma | 72340 |
| Triiodo-L-thyronine sodium salt (T3) | Sigma | T6397 |
| Doxycycline | Cayman | 14422-10 |
| SAHA | Cayman | 10009929-100 |
| 2-Hydroxypropyl-β Cyclodextrin | Sigma | H-107 |
| Collagenase P | Roche | 11213873001 |
| Histopaque-1077 | Sigma | H8889 |
| Dynabeads protein A | Life technologies | 10002D |
| Dynabeads protein G | Life technologies | 10004D |
| Recombinant full length His-VDR (SF9 origin) | Life Technologies | 12025H08B25 |
| Recombinnat BRD9 bromodomain GST tagged | BPS biosciences | 31091 |
| EDTA free protease inhibitor cocktail | Roche | 11836170001 |
| Dynabeads Co-Immunoprecipitation Kit | Life Technologies | 14321D |
| Critical Commercial Assays | ||
| Mouse insulin ELISA | Millipore | EZRMI-13K |
| Human insulin ELISA | Millipore | EZHI-14K |
| Vectastain ABC kit | Vector lab | PK-6101 |
| Annexin V | Ebioscience | 88-8088-74 |
| Mouse proinsulin ELISA | Mercodia | NC9088881 |
| Deposited Data | ||
| RNA-seq, ChIP-seq and ATAC-seq | This study | |
| Experimental Models: Cell Lines | ||
| INS-1 | AddexBio | C0018007 |
| HEK-293T | ATCC | CRL-11268 |
| HUVEC-iPS | Yoshihara et. al., 2016 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.129S4-Vdrtm1Mbd/J | The Jackson Laboratory | Stock No. 006133 |
| Mouse: B6.BKS(D)-Leprdb/J | The Jackson Laboratory | Stock No. 000697 |
| Mouse: BKS.Cg-Dock7m +/+ Leprdb/J | The Jackson Laboratory | Stock No. 000642 |
| Mouse: C57BL6/J | The Jackson Laboratory | Stock No. 000664 |
| Mouse: FVB/N-Tg(Ins2-IAPP)RHFSoel/J | The Jackson Laboratory | Stock No. 008232 |
| Oligonucleotides | ||
| qPCR primers: See Table S2 | See Table S2 | N/A |
| siRNA: VDR | GE Healthcare | L-097753-02-0005 |
| siRNA: PCAF | GE Healthcare | L-085879-02-0005 |
| siRNA: BAF180 | GE Healthcare | L-088113-02-0005 |
| siRNA: non-targeting pool | GE Healthcare | D-001810-10-05 |
| Recombinant DNA | ||
| Lentiviral sgRNA library | Wang et. al., 2014; | Addgene: 51048 |
| Plasmid: pCW-Cas9 | Wang et. al., 2014; | Addgene: 50661 |
| Plasmid: human insulin promoter-GFP | Yoshihara et al. 2016 | N/A |
| Plasmid: Lv-VDR-HA | This study | N/A |
| Plasmid: Lv-BRD9-Flag | This study | N/A |
| Plasmid: LV-VDR-K91R-HA | This study | N/A |
| Plasmid: Lv-VDR-K91A-HA | This study | N/A |
| Plasmid: Lv-BRD7-Flag | This study | N/A |
| Plasmid: pMSCVpuro-Gcn5(D608A) (PCAF) | Jin et. al., 2014 | Addgene: 63707 |
| Plasmid: pMSCVpuro-Gcn5 (PCAF) | Jin et. al., 2014 | Addgene: 63705 |
| Plasmid: pCMV-p300-myc | N/A | Addgene: 30489 |
| Software and Algorithms | ||
| Homer | N/A | http://homer.ucsd.edu/homer/ |
| CASAVA-1.8.2 | Illumina | https://www.illumina.com/ |
| Metascape | N/A | metascape.org |
| DAVID | N/A | https://david.ncifcrf.gov/ |
| MAGECK | N/A | https://sourceforge.net/projects/mageck/ |
| STAR | N/A | https://github.com/alexdobin/STAR |
| Fiji | N/A | https://fiji.sc/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact Ronald M. Evans (evans@salk.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Differentiation of hiPSC to human β-like cells
As previously described (Yoshihara et al., 2016), INS-GFP hiPSCs (derived from HUVECs) were cultured to 90% confluency in complete mTeSR Media, at which time the media was changed to 100 ng/ml Activin (R&D Systems, 338-AC), 25 ng/ml Wnt3a (R&D Systems, 5036-WN) in differentiation media (800 ml DMEM/F12, 13.28 g BSA, 10 ml Glutamax, 560 mg NaHCO3, 330 mg thiamine, 100 mg reduced glutathione, 3300 mg vitamin C, 14 μg selenium, 10 ml NEAA, 2 ml trace element B, 1 ml trace element C, 7 μl β-ME, 2 ml DLC, 2 ml GABA, 2 ml LiCl, 129.7 μg pipecolic acid, insulin 2 mg up to 1,000 ml) for 2 days and then 100 ng/ml activin in differentiation media for another 2 days. Subsequently, media was replaced with differentiation media with 1 μM dorsomorphin (Biovision, 1656-5), 2 μM retinoic acid (Sigma, R2625-1G), 10 μM SB431542 (Sigma, S4317-5MG), and 1% B27 supplement (GIBCO, 17504-044) for 7 days. Media was then replaced with 10 μM Forskolin (Sigma, F6886-25MG), 10 μM dexamethasone (Sigma, D4902-100MG), 10 μM Alk5 inhibitor II (Calbiochem, 616452 or Enzo, ALX-270-445), 10 mM nicotinamide (Sigma, 72340-100G), 1uM 3,3′,5-Triiodo-L-thyronine sodium salt (T3), and 1% B27 supplement 10 days. At days 21, the expression of INS-GFP were confirmed regularly by fluorescence microscopy. Stem cell culture were supported by the Stem Cell Core Facility of the Salk Institute with funding from the Helmsley Charitable Trust (K. Diffenderfer).
Mice
Animals were maintained in a specific pathogen-free animal facility on a 12-hr light-dark cycle at an ambient temperature of 23°C. Water and food were provided ad lib. All animal experiments used age-matched male mice. VDR KO and control litter mice were maintained on a VDR KO rescue diet containing 21% calcium, 0.67% phosphorus, and 20% lactose supplemented with 4.4 U vitamin D per gram diet. All procedures involving animals were performed in accordance with protocols approved by the IACUC and Animal Resources Department of the Salk Institute for Biological Studies.
T2D animal model treatment
For db/db (B6), mice were grouped based on glucose levels at age of 5 weeks. Treatments were started from 6 weeks of age and animals were treated with vehicle (30% 2-Hydroxypropyl-β Cyclodextrin, Sigma, H-107) or drugs 3 times a week. Cal (Tocris, 60ug/kg) and iBRD9 (R&D, 10mg/kg) were dissolved in 30% β-cyclodextran and injected intraperitoneally 3 times a week. For db/db (BKS), treatment started from the age of 10 weeks. Cal (Tocris, 30ug/kg) and iBRD9 (R&D, 5mg/kg) were dissolved in 30% β-cyclodextran and injected intraperitoneally 3 times a week.
Low-dose STZ induction
8 week old C57Bl6 male mice were injected intraperitoneally with streptozotocin (Sigma, S0130) at the dose of 50mg/kg every day for 5 consecutive days. Blood glucose was measured 1 week after the final injection and mice were separated into different treatment groups to ensure all groups have similar starting glucose levels.
Isolation of Pancreatic Islets
Mouse pancreatic islets were isolated as previously described (Yoshihara et al., 2010) with slight modification. Briefly, 0.5 mg/ml collagenase P (Roche REF11213873001, diluted in HBSS buffer, GIBCO, 14170-112) was injected through the common bile duct, and the perfused pancreas dissected and incubated at 37°C for 21 min. Digested exocrine cells and intact islets were separated via centrifugation (900 × g for 15 min) over Histopaque-1077 (Sigma, H8889), and intact islets were manually selected. Human islets were provided by the Integrated Islets Distribution Program under an approved protocol (Table S1).
METHOD DETAILS
CRISPR screening
“Drop off” genome-wide CRISPR screening was performed using a protocol similar to that described previously (Wang et al., 2014). We transduced a Tet-ON inducible rtTA and subsequently a genomic lentiviral sgRNA library into an INS (human insulin promoter)-GFP iPS cell lines as reported in previous work (Yoshihara et al., 2016). By selecting with 3 different markers (puromycin for rtTA, blasticidin for sgRNA library and neomycin for insulin-GFP), we obtained iPS cell lines that contain both vectors and maintained at least 5X107 cells for every passage to guarantee the coverage of library. Cells were subjected to standard differentiation protocols and the rtTA was induced after 2 weeks of differentiation, concurrent to expression of INS-GFP. Doxycycline (1μg/ml) was then added for 1 week. Subsequently, the cells were FACS-sorted by GFP expression and both GFP+ and GFP- cells from the same batch were collected, extracted for genomic DNA and libraries were prepared for high throughput sequencing. The sgRNA sequences were amplified with primers described previously(Wang et al., 2014) and sequenced by HiSeq 2500. The sequences were aligned and analyzed by MAGeCK (https://sourceforge.net/p/mageck/wiki/Home/). FACS was supported by the Flow Cytometry Core Facility of the Salk Institute with funding from NIH-NCI CCSG: P30 014195 (C. O’Connor).
RNA-Seq library generation
Total RNA was isolated from tissue or cell pellets using the RNA mini kit (Qiagen). Sequencing libraries were prepared from 100–500ng total RNA using the TruSeq RNA Sample Preparation Kit v2 (Illumina) according to the manufacturer’s protocol. Briefly, mRNA was purified, fragmented, and used for first-, then second-strand cDNA synthesis followed by adenylation of 3′ ends. Samples were ligated to unique adapters and subjected to PCR amplification. Libraries were then validated using the 2100 BioAnalyzer (Agilent), normalized, and pooled for sequencing. RNA-Seq libraries prepared from two biological replicates for each experimental condition were sequenced on the Illumina HiSeq 2500 using bar-coded multiplexing and a 100bp read length.
High-throughput sequencing and analysis
Image analysis and base calling were performed with Illumina CASAVA-1.8.2. This yielded a median of 29.9M usable reads per sample. Short read sequences were mapped to a UCSC mm10 reference sequence using the RNA-seq aligner STAR. Known splice junctions from mm10 were supplied to the aligner and de novo junction discovery was also permitted. Differential gene expression analysis, statistical testing and annotation were performed using Cuffdiff 2. Transcript expression was calculated as gene-level relative abundance in fragments per kilobase of exon model per million mapped fragments and employed correction for transcript abundance bias. RNA-Seq results for genes of interest were also explored visually using the UCSC Genome Browser.
Gene Expression Analysis by qPCR
Samples were run in triplicate and expression was normalized to the levels of the housekeeping controls Rplp0 (36b4) for human and mouse. Primers used are listed in Table S2. Samples were analyzed by qPCR, using SYBR Green dye (Bio-rad). Statistical comparisons were made using Student’s t test. Error bars are mean ± SEM.
Metabolic Cage Analyses
Metabolic cage analyses were conducted with a Comprehensive Lab Animal Monitoring System (Columbus Instruments). CO2 production, O2 consumption, RER, and ambulatory counts by x-peak were determined for 3 consecutive days and nights, with at least 24 hr adaptation before data recording.
Histology (H&E staining, Immunohistochemistry and immunofluorescence)
H&E staining was performed by Pacific Pathology (San Diego) and UCSD pathology core.
For immunohistochemistry, FFPE tissue sections were FFPE tissues sections were deparrafinized and antigen retrieval were performed using Vector Antigen Unmasking buffer (H3301). VectaStain ABC kit and ImmPACT DAB substrate (Vector Lab) were used to develop signal. Antibody used: Pro-insulin (R&D, MAB13361, 1:100). For immunofluorescence staining, FFPE tissues sections were deparrafinized and antigen retrieval were performed using Vector Antigen Unmasking buffer (H3301). Antibody used: insulin (Abcam, ab7842, 1:100), MAFA (Novus, NB400-137, 1:50), NKX6-1 (Cell Signaling, 54551, 1:100), Glucagon (abcam, ab82270, 1:100). DAPI-containing mounting media (VECTASHIELD mounting medium for fluorescence) was used for nuclear staining. Immunostaining was visualized by ZEISS 780 confocal microscopy analysis.
Immunoprecipitation
Briefly, cells are lysed using ice-cold lysis buffer (HEPES 20mM pH8, EDTA 0.2mM, NaCl 0.3 mM, NP40 0.5%, 15% glycerol) for 15 min, and after centrifuge the supernatant was collected. For HA- or Flag-tagged protein, immunoprecipitations were performed using HA-magnetic beads (Pierce, PI88836), or Flag-magnetic beads (Sigma, M8823) for 1.5 hr at 4C. For other immunoprecipitations, primary antibodies were incubated with lysate for 2 hr followed by Protein A magnetic beads (Life Technologies, 10001D). After 3 washes with wash buffer (HEPES 10mM pH8, EDTA 0.2 mM, NaCl 0.3 mM, NP40 0.1%, 15% glycerol), the beads were boiled with NuPage LDS sample buffer and the lysate were stored at −20°C. For mass-spec samples detecting VDR acetylation, HA-tagged VDR was transfected together with a plasmid expressing p300 and HDAC inhibitor TSA (1μM) were added in all buffers. For immunoprecipitation with endogenous VDR, antibody was first crosslinked with dynabeads using Dynabeads immunoprecipitation kit (Life technologies, 14321D).
Mass-spec
Samples were precipitated by methanol/chloroform. Dried pellets were dissolved in 8 M urea/100 mM TEAB, pH 8.5. Proteins were reduced with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich) and alkylated with 10 mM chloroacetamide (Sigma-Aldrich). Proteins were digested overnight at 37°C in 2 M urea/100 mM TEAB, pH 8.5, with trypsin (Promega). Digestion was quenched with formic acid, 5 % final concentration.
The digested samples were analyzed on a Fusion Orbitrap tribrid mass spectrometer (Thermo). The digest was injected directly onto a 30cm, 75um ID column packed with BEH 1.7um C18 resin (Waters). Samples were separated at a flow rate of 200nl/min on a nLC 1000 (Thermo). Buffer A and B were 0.1% formic acid in water and acetonitrile, respectively. A gradient of 1–30%B over 90min, an increase to 40%B over 30min, an increase to 90%B over another 10min and held at 90%B for a final 10min of washing was used for 140min total run time. Column was re-equilibrated with 20ul of buffer A prior to the injection of sample. Peptides were eluted directly from the tip of the column and nanosprayed directly into the mass spectrometer by application of 2.5kV voltage at the back of the column. The Orbitrap Fusion was operated in a data dependent mode. Full MS1 scans were collected in the Orbitrap at 120K resolution with a mass range of 400 to 1600 m/z and an AGC target of 5e5. The cycle time was set to 3sec, and within this 3sec the most abundant ions per scan were selected for CID MS/MS in the ion trap with an AGC target of 1e4 and minimum intensity of 5000. Maximum fill times were set to 50ms and 100ms for MS and MS/MS scans respectively. Quadrupole isolation at 1.6m/z was used, monoisotopic precursor selection was enabled and dynamic exclusion was used with exclusion duration of 5 sec.
Protein and peptide identification were done with Integrated Proteomics Pipeline – IP2 (Integrated Proteomics Applications). Tandem mass spectra were extracted from raw files using RawConverter(He et al., 2015) and searched with ProLuCID(Xu et al., 2015) against rat UniProt database. The search space included all fully-tryptic and half-tryptic peptide candidates. Carbamidomethylation on cysteine was considered as a static modification. Acetylation (42.010565) was considered as a differential modification on Lysine and the N-terminus. Data was searched with 50 ppm precursor ion tolerance and 600 ppm fragment ion tolerance. Data was filtered to 10 ppm precursor ion tolerance post search. Identified proteins were filtered using DTASelect (Tabb et al., 2002) and utilizing a target-decoy database search strategy to control the false discovery rate to 1% at the protein level. Mass-spec was performed by Mass Spectrometry Core of the Salk Institute with funding from NIH-NCI CCSG: P30 014195 and the Helmsley Center for Genomic Medicine (J. Moresco, J. Diedrich and W. Low).
siRNA knock-down assay
INS1 cells were cultured at 37°C in 5% CO2 in air in RPMI-1640 (Sigma Aldrich) supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) Antibiotic-Antimycotic (Gibco) 10 mM HEPES, 2 mM glutamax, 1 mM sodium pyruvate, and 50 μM β-mercaptoethanol (RPMI for INS1 medium). INS1 cells were transfected with Lipofectamine RNAiMAX (Invitrogen) for 24 hr, and cytokines were added for 24 hr or 1 hour.
Intraperitoneal Glucose or Insulin Tolerance Tests
Intraperitoneal GTTs were performed on overnight fasted mice. Blood glucose values were assessed before and at 0, 15, 30, 60, and 120 min after i.p. administration of 0.5 g/kg of glucose. Serum insulin levels were assessed before and at 0, 15, and 60 min after the i.p. administration of glucose using a rat/mouse Insulin ELISA kit (Millipore). IP-ITT assays were performed on mice after a 6 hr fast with an injection of 2 U/kg of insulin (Humalin R, Eli Lilly).
shRNA knockdown
shRNA constructs for human VDR, as well as control shRNA, were purchased from Transomics. Lentiviral shRNA were produced in 293T cells and polybrene (6μg/ml) was used in transduction. Puromycin selection performed for 1 week at concentration of 1ug/ml.
FACS analysis
Annexin V staining were performed per manufacturers’ instructions (Ebioscience, 88-8008-74, BD, 561126). FACS analysis were performed using BD FACSCantoII.
ELISA
ELISA were performed per manufacturer’s instructions: mouse insulin (Millipore, EZRMI-13K), mouse proinsulin (Mercodia, NC9088881).
Chromatin Immunoprecipitation (ChIP)
INS1 cells were treated with Cal (100 nM), iBRD9 (10uM) and/or IL1β with conditions specified in the text. Cells were then harvested for the ChIP assay. The experimental procedure for ChIP was as previously described (Barish et al., 2010). Briefly, after fixation, nuclei from were isolated, lysed, and sheared with a Diagenode Bioruptor to yield DNA fragment sizes of 200–1,000 bp followed by immunoprecipitation. Primers used are described in Table S2.
ChIP-Seq Data Analysis
Briefly, short DNA reads were aligned against the mouse mm10 or rat rn4 reference genome using the Illumina CASAVA-1.8.2. Reads were aligned using the Bowtie aligner, allowing up to two mismatches in the read. Only tags that map uniquely to the genome were considered for further analysis. Subsequent peak calling and motif analysis were conducted using HOMER, a software suite for ChIP-seq analysis. The methods for HOMER, which are described below, have been implemented and are freely available at http://homer.ucsd.edu. One tag from each unique position was considered to eliminate peaks resulting from clonal amplification of fragments during the ChIP-seq protocol. Peaks were identified by searching for clusters of tags within a sliding 200 bp window, requiring adjacent clusters to be at least 1 kb away from each other. The threshold for the number of tags that determine a valid peak was selected for an FDR < 0.001, as empirically determined by repeating the peak finding procedure using randomized tag positions. Peaks are required to have at least 4-fold more tags (normalized to total count) than input or immunoglobulin G control samples and 4-fold more tags relative to the local background region (10 kb) to avoid identifying regions with genomic duplications or nonlocalized binding. Differential H3K27Ac peaks are called by Homer using a threshold of 1.5 fold change and p value< 0.0001. Peaks are annotated to gene products by identifying the nearest RefSeq transcriptional start site. Visualization of ChIP-seq results was achieved by uploading custom tracks onto the University of California, Santa Cruz genome browser. Gene ontology analysis were performed using Metascape (metascape.org) or DAVID (https://david.ncifcrf.gov/).
ATAC-Seq
INS1 cells were pretreated with vehicle, Cal, iBRD9, or iBRD9 16 hrs before IL1β were added for 1 hr and 5X104 cells were harvested to prepare ATAC-seq libraries. Reads were aligned by Bowtie to rn4 and peaks were called by Homer using default settings. Differential peaks were called by Homer using default settings (Fold change >4, p value <0.0001).
Transmission electronic microscopy (TEM)
Islets in suspension were pelleted in 2% low melting point agarose and subsequently fixed in 2.5% glutaraldehyde with 2% paraformaldehyde in 0.15M cacodylate buffer containing 2mM calcium chloride, pH 7.4, for one hour at 4°C. The pellet was trimmed down to contain the minimal amount of agarose possible, washed in buffer, and secondarily fixed in 1% osmium tetroxide/0.3% potassium ferrocyanide in buffer. Subsequently, the pellet was washed in water and en bloc stained with 2% uranyl acetate followed by a graded dehydration in ethanol (35%, 50%, 70%, 90%, 100%, 100%). Samples were then rapidly infiltrated in Spurr’s resin using a Pelco BioWave microwave processing unit (Ted Pella, Redding, CA), embedded in Pelco Pyramid tip mold (Ted Pella, Redding, CA), and cured at 60°C overnight. 70nm ultrathin sections were then cut on a Leica UC7 ultramicrotome (Leica, Vienna) and Islets were examined on a Libra120 (Zeiss, Oberkochen, Germany) at 120V. TEM was supported by the Waitt Advanced Biophotonics Core Facility of the Salk Institute with funding from NIH-NCI CCSG: P30 014195, NINDS Neuroscience Core Grant: NS072031 and the Waitt Foundation (S. Dunn).
LC-MS Analysis
An internal standard of d4-succinate was added to all samples prior to analysis. 500 μL of methanol containing 100 pmol of d4-succinate was added to 50 uL plasma samples and vortexed for 1 min. The samples were then centrifuged at 17.000 x g for 10 min at 4 oC. After drying the supernatant via SpeedVac, the extract was dissolved in H2O (50 μL) and 10 μL was subjected to liquid chromatography mass spectrometry (LC-MS). Compound i-BRD9 was measured using a TSQ Quantiva mass spectrometer (Thermo) and Dionex Ultimate 3000 (Thermo) fitted with a Gemini C18 HPLC column (5 μm; 4.6 mm × 50 mm, Phenomenex). LC solvents used were: solvent A, 0.1 % formic acid in water; solvent B, 0.1 % formic acid in acetonitrile. Separations took place over 16 min with a flow rate of 0.4 mL/min and consisted of the following steps: 5 to 50 % solvent B over 9 min, 50 to 80 % B over 1 min, 80 % B for 1 min, 80 to 5% B over 0.1 min, and 5% B for 5 min. MS analyses were performed using electrospray ionization (ESI) in positive ion mode for i-BRD9 and negative ion mode for d4-succinate. Positive and negative mode source parameters were as follows: spray voltage 3.5 kV, ion transfer tube temperature of 325 °C, and a vaporizer temperature of 275 °C. Multiple reaction monitoring transitions for target compound and internal standard are shown in (Table S3).
QUANTIFICATION AND STATISTICAL ANALYSES
All statistical details of experiments are included in the Figure legends or specific Methods section. Band intensities were quantified with Fiji and normalized with internal control.
DATA AND SOFTWARE AVAILABILITY
RNA-Seq data reported in this paper have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database, Accession No. SRP103617. ChIP-Seq and ATAC-Seq data have been deposited in the SRA database, Accession No. SRP103615.
Supplementary Material
A. Ranking of highly expressed transcription factors by average RPKM in single-cell RNA seq. Average RPKM was shown. VDR is showed in red.
B. Heat map of single cell expression value of key α cell and β cell markers, together with VDR.
A. FACS analysis of Annexin-V+ population percentage, which indicate apoptotic cells, in control and VDR knockdown human β-like cells.
B. Heat map of ratio of gene expression level by qPCR between siVDR and siCtrl INS1 cells under different stressing condition.
C. Serum insulin level of VDR+/+ and VDR−/− mice (n=3, error bars show S.E.M., *P<0.05).
D. Serum proinsulin level of VDR+/+ and VDR−/− mice (n=3, error bars show S.E.M.).
E. Staining of MAFA and Insulin in VDR WT and VDR KO islets.
F. Heat map of mouse islets treated with vehicle, IL1β, or IL1β+Cal for 48 or 96 hours, showing the genes induced by IL1b are partially rescued by Cal treatment.
G. Venn diagram showing the overlap of between genes significantly suppressed by IL1b and genes rescued by Cal at 96 hours.
H. Gene ontology categories of the 169 genes suppressed by IL1b and rescued by Cal at 96 hours.
I. Venn diagram showing the overlap of between genes significantly induced by IL1b and genes rescued by Cal at 96 hours.
J. Gene ontology categories of the 332 genes induced by IL1b and rescued by Cal at 96 hours.
A. Immunoprecipitation of endogenous BAF/PBAF components in 293T cells shows that BRD9 interacts with BAF component BRG1 and ARID1B, but not interact with PBAF component ARID2.
B. Scatter plot of the p-value of all screening genes enriched in the drop-out screening, VDR is labeled in red. Several epigenetic modifiers, including BRD7, are labeled in green.
C. Western blot of endogenous BRD9 in immunoprecipitates from 293T cells expressing HA-tagged VDR treated with vehicle (DMSO), Cal, the BRD9 inhibitor iBRD9, or Cal+iBRD9.
D. Quantification of relative signal intensity corresponding to Figure S3C (n=2, error bars show S.E.M., **P<0.01).
E. Quantification of relative signal intensity corresponding to Figure 2C (n=2, error bars show S.E.M., *P<0.05, **P<0.01). Western blotting of full length his-VDR showed the SF9 (insect) produced recombinant protein is acetylated.
F. Mass-spec results suggest the lysine 91 on human VDR is acetylated.
G. Quantification of relative signal intensity corresponding to Figure 2G (n=2, error bars show S.E.M., *P<0.05, **P<0.01).
H. Quantification of relative signal intensity corresponding to Figure 2H (n=2, error bars show S.E.M., **P<0.01).
I. Quantification of relative signal intensity corresponding to Figure 2I (n=2, error bars show S.E.M., **P<0.01).
A–E. Quantification of relative signal intensity corresponding to Figure 3B (n=2, error bars show S.E.M., **P<0.01).
F. INS-1 cells expression VDR-WT or VDR-K91R were treated with Cal and exposed to IL1β for 1hr, Nfkbia level were measured by qPCR (n=3, error bars show S.E.M., *P<0.05)
G–I. ChIP-qPCR of ARID1B, a BAF specific component, show that addition of iBRD9 reduce BAF complex binding at key VDR target loci Nfkbia, Nfkbiz, and Camk4.
J–L. ChIP-qPCR of BAF180, a PBAF specific component, show that addition of iBRD9 increase PBAF complex binding at key VDR target loci Nfkbia, Nfkbiz, and Camk4.
A. Liver weight of db (B6) mice treated with Cal or iBRD9 (n=6, error bars show S.E.M.).
B. Epididymal white adipose tissue (eWAT) weight of db/db (B6) mice treated with Cal or iBRD9 (n=6, error bars show S.E.M.).
C. O2 consumption of mice treated with vehicle or Cal+iBRD9.
D. CO2 production of mice treated with vehicle or Cal+iBRD9.
E. Serum calcium level of mice in all treatment group (n=10, error bars show S.D).
F. Whole body weight of db (BKS) mice treated with Cal or Cal+iBRD9.
A. Target gene of Cal Cyp24A1 expression in multiple tissues, indicating islets is targeted by Cal directly (n=3, error bars show S.E.M., *P<0.05).
B. Islets from db (B6) treated with Cal show increased expression of Ins2 (n=3, error bars show S.E.M., *P<0.05).
C. Liver expression of key gluconeogenesis genes are repressed in Cal treated db (B6) mice, suggesting higher level of serum insulin suppressed gluconeogenesis from liver.
D. Low dose STZ mice treated with Cal or iBRD9 showed increased level of insulin (n=8–10, error bars show S.E.M.).
E. Immunofluorescence staining of NKX6-1 (red), a key transcription factor for β cell identity and function, in vehicle and Cal+iBRD9 treated low dose STZ pancreas sections. (Scale bar: 50μm).
F. Heatmap of qPCR results of multiple inflammatory genes expression in islets from aged h-IAPP mice treated with Cal or Cal+iBRD9. Islets were collected from 15 months old mice and cultured ex vivo in ultra-low attachment plates. Cal or Cal+iBRD9 were treated for 72 hours.
G. Western blotting of ER stress marker p-PERK in INS-1 cells treated with Thapsigargin (0.25μM) or Tunicamycin (5μg/ml) overnight. Cal was pretreated 1 hour.
H. Immunohistochemistry staining of ER stress marker Calnexin and PERK with pancreatic tissue section in db/db (BKS) mice.
I. qPCR of Nfkbia expression in INS-1 cells exposed to IL1β and Cal and multiple BRD9 inhibitors. (n=3, error bars show S.E.M., *P<0.05, **P<0.01).
J. In vivo pharmacokinetic study of iBRD9 (n=3, error bars show S.E.M.).
A. Cell number of ex vivo cultured human islets treated with vehicle or Calcipotriol for 4 weeks (n=3, error bars show S.E.M., *P<0.05).
B. Insulin secretion assay of ex vivo cultured human islets treated with vehicle or Calcipotriol for 1 week. (n=3, error bars show S.E.M., **P<0.01).
Highlights.
BRD9 and BRD7 bromodomains recognize VDR K91Ac
VDR ligand switches association from BRD9/BAF (inactive) to BRD7/PBAF (active)
Inhibiting BRD9 enhances Vitamin D response
Enhanced VDR signaling reduces β cell failure and curbs T2D progression
Acknowledgments
We thank D. Hargreaves and C. Benner for discussion, L. Chuong, H. Juguilon and B. Collins for technical support, E. Ong and C. Brondos for administrative support, and all Evans lab members for discussion. R.M.E. is an Investigator of the Howard Hughes Medical Institute at the Salk Institute and March of Dimes Chair in Molecular and Developmental Biology, and is supported by NIH (DK057978, HL088093, HL105278, and ES010337), NIEHS (P42ES010337), the Glenn Foundation for Medical Research, the Leona M. and Harry B. Helmsley Charitable Trust (#2017PG-MED001), Ipsen/Biomeasure, California Institute for Regenerative Medicine, The Ellison Medical Foundation, and by a gift from Steven and Lisa Altman. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This study was supported by Salk Cores (see online methods).
Footnotes
Declaration of interest
Z.W., R.T.Y, M.H.S, A.A, M.D., and R.M.E are co-inventors of inventions related to vitamin D and bromodomain technologies and may be entitled to royalties.
Author Contributions
Conceptualization, Z. W., E.Y., A.R.A, R.T.Y, M.D., and R.M.E.; Methodology, Z. W., E.Y., A.R.A, R.T.Y, M.D. and R.M.E.; Investigation, Z.W., E.Y., N.H., T.H., Y.W., B.R. G.E., A.M.P, and Y.D.; Data Curation, Z.W., N. H., C.L, and R.T.Y.; Resources, W.F., N.D., M.H.S., X.Z. and N.H.; Writing – Original Draft, Z.W.; Writing – Review & Editing, Z.W., E.Y., A.R.A, R.T.Y, M.D. and R.M.E.; Funding Acquisition, R.M.E.; Supervision, M.D. and R.M.E.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arnush M, Heitmeier MR, Scarim AL, Marino MH, Manning PT, Corbett JA. IL-1 produced and released endogenously within human islets inhibits beta cell function. The Journal of clinical investigation. 1998;102:516–526. doi: 10.1172/JCI844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Diabetes mellitus and the beta cell: the last ten years. Cell. 2012;148:1160–1171. doi: 10.1016/j.cell.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. Vitamin D: modulator of the immune system. Current opinion in pharmacology. 2010;10:482–496. doi: 10.1016/j.coph.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Benner C, van der Meulen T, Caceres E, Tigyi K, Donaldson CJ, Huising MO. The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non-coding RNA and protein-coding gene expression. BMC genomics. 2014;15:620. doi: 10.1186/1471-2164-15-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco JC, Minucci S, Lu J, Yang XJ, Walker KK, Chen H, Evans RM, Nakatani Y, Ozato K. The histone acetylase PCAF is a nuclear receptor coactivator. Genes & development. 1998;12:1638–1651. doi: 10.1101/gad.12.11.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, Lieben L, Mathieu C, Demay M. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocrine reviews. 2008;29:726–776. doi: 10.1210/er.2008-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantorna MT, Zhu Y, Froicu M, Wittke A. Vitamin D status, 1,25-dihydroxyvitamin D3, and the immune system. Am J Clin Nutr. 2004;80:1717S–1720S. doi: 10.1093/ajcn/80.6.1717S. [DOI] [PubMed] [Google Scholar]
- Chan JY, Luzuriaga J, Bensellam M, Biden TJ, Laybutt DR. Failure of the adaptive unfolded protein response in islets of obese mice is linked with abnormalities in beta-cell gene expression and progression to diabetes. Diabetes. 2013;62:1557–1568. doi: 10.2337/db12-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Pino-Montes J, Benito GE, Fernandez-Salazar MP, Covenas R, Calvo JJ, Bouillon R, Quesada JM. Calcitriol improves streptozotocin-induced diabetes and recovers bone mineral density in diabetic rats. Calcif Tissue Int. 2004;75:526–532. doi: 10.1007/s00223-004-0118-9. [DOI] [PubMed] [Google Scholar]
- DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, van der Meulen T, Huising MO. Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab. 2016;5:449–458. doi: 10.1016/j.molmet.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding N, Yu RT, Subramaniam N, Sherman MH, Wilson C, Rao R, Leblanc M, Coulter S, He M, Scott C, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153:601–613. doi: 10.1016/j.cell.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donath MY. Multiple benefits of targeting inflammation in the treatment of type 2 diabetes. Diabetologia. 2016;59:679–682. doi: 10.1007/s00125-016-3873-z. [DOI] [PubMed] [Google Scholar]
- Donath MY, Dalmas E, Sauter NS, Boni-Schnetzler M. Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity. Cell metabolism. 2013;17:860–872. doi: 10.1016/j.cmet.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nature reviews. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- Fernandez-Real JM, Pickup JC. Innate immunity, insulin resistance and type 2 diabetes. Diabetologia. 2012;55:273–278. doi: 10.1007/s00125-011-2387-y. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nature reviews Drug discovery. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Garris DR, Garris BL. Cytochemical analysis of pancreatic islet hypercytolipidemia following diabetes (db/db) and obese (ob/ob) mutation expression: influence of genomic background. Pathobiology. 2004;71:231–240. doi: 10.1159/000080056. [DOI] [PubMed] [Google Scholar]
- Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, Robertson RP, Powers AC, Stein R. Inactivation of specific beta cell transcription factors in type 2 diabetes. The Journal of clinical investigation. 2013;123:3305–3316. doi: 10.1172/JCI65390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn HJ, Kuttler B, Mathieu C, Bouillon R. 1,25-Dihydroxyvitamin D3 reduces MHC antigen expression on pancreatic beta-cells in vitro. Transplantation proceedings. 1997;29:2156–2157. doi: 10.1016/s0041-1345(97)00273-x. [DOI] [PubMed] [Google Scholar]
- Halban PA, Polonsky KS, Bowden DW, Hawkins MA, Ling C, Mather KJ, Powers AC, Rhodes CJ, Sussel L, Weir GC. beta-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes care. 2014;37:1751–1758. doi: 10.2337/dc14-0396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, Das I, Wang R, Chen AC, Loudovaris T, et al. Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nature medicine. 2014;20:1417–1426. doi: 10.1038/nm.3705. [DOI] [PubMed] [Google Scholar]
- Hasnain SZ, Prins JB, McGuckin MA. Oxidative and endoplasmic reticulum stress in beta-cell dysfunction in diabetes. J Mol Endocrinol. 2016;56:R33–54. doi: 10.1530/JME-15-0232. [DOI] [PubMed] [Google Scholar]
- He L, Diedrich J, Chu YY, Yates JR., 3rd Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Analytical chemistry. 2015;87:11361–11367. doi: 10.1021/acs.analchem.5b02721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herder C, Dalmas E, Boni-Schnetzler M, Donath MY. The IL-1 Pathway in Type 2 Diabetes and Cardiovascular Complications. Trends in endocrinology and metabolism: TEM. 2015;26:551–563. doi: 10.1016/j.tem.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Hodges C, Kirkland JG, Crabtree GR. The Many Roles of BAF (mSWI/SNF) and PBAF Complexes in Cancer. Cold Spring Harbor perspectives in medicine. 2016:6. doi: 10.1101/cshperspect.a026930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AF, Martin LJ, Minder JL, Roe JS, Shi J, Steurer S, Bader G, McConnell D, Pearson M, Gerstberger T, et al. Sensitivity and engineered resistance of myeloid leukemia cells to BRD9 inhibition. Nature chemical biology. 2016;12:672–679. doi: 10.1038/nchembio.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Dobrian AD, Morris MA, Nadler JL. Islet inflammation: a unifying target for diabetes treatment? Trends in endocrinology and metabolism: TEM. 2013;24:351–360. doi: 10.1016/j.tem.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, Kreutter DK, Butler PC. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:7283–7288. doi: 10.1073/pnas.93.14.7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q, Zhuang L, Lai B, Wang C, Li W, Dolan B, Lu Y, Wang Z, Zhao K, Peng W, et al. Gcn5 and PCAF negatively regulate interferon-beta production through HAT-independent inhibition of TBK1. EMBO reports. 2014;15:1192–1201. doi: 10.15252/embr.201438990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nature genetics. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser MD, Aslanian A, Dong MQ, Yates JR, 3rd, Emerson BM. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. The Journal of biological chemistry. 2008;283:32254–32263. doi: 10.1074/jbc.M806061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MS, Sem DS, Kliewer SA, Provencal J, Evans RM, Wright PE. NMR assignments and secondary structure of the retinoid X receptor alpha DNA-binding domain. Evidence for the novel C-terminal helix. European journal of biochemistry/FEBS. 1994;224:639–650. doi: 10.1111/j.1432-1033.1994.00639.x. [DOI] [PubMed] [Google Scholar]
- Lemon B, Inouye C, King DS, Tjian R. Selectivity of chromatin-remodelling cofactors for ligand-activated transcription. Nature. 2001;414:924–928. doi: 10.1038/414924a. [DOI] [PubMed] [Google Scholar]
- Li Z, Karlsson FA, Sandler S. Islet loss and alpha cell expansion in type 1 diabetes induced by multiple low-dose streptozotocin administration in mice. The Journal of endocrinology. 2000;165:93–99. doi: 10.1677/joe.0.1650093. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Koegl M, Bader G, Cockcroft XL, Fedorov O, Fiegen D, Gerstberger T, Hofmann MH, Hohmann AF, Kessler D, et al. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. Journal of medicinal chemistry. 2016;59:4462–4475. doi: 10.1021/acs.jmedchem.5b01865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu C, Waer M, Laureys J, Rutgeerts O, Bouillon R. Prevention of autoimmune diabetes in NOD mice by 1,25 dihydroxyvitamin D3. Diabetologia. 1994;37:552–558. doi: 10.1007/BF00403372. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Soto C. Prion-Like Protein Aggregates and Type 2 Diabetes. Cold Spring Harbor perspectives in medicine. 2017;7 doi: 10.1101/cshperspect.a024315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nackiewicz D, Dan M, He W, Kim R, Salmi A, Rutti S, Westwell-Roper C, Cunningham A, Speck M, Schuster-Klein C, et al. TLR2/6 and TLR4-activated macrophages contribute to islet inflammation and impair beta cell insulin gene expression via IL-1 and IL-6. Diabetologia. 2014;57:1645–1654. doi: 10.1007/s00125-014-3249-1. [DOI] [PubMed] [Google Scholar]
- Narvaez CJ, Matthews D, Broun E, Chan M, Welsh J. Lean phenotype and resistance to diet-induced obesity in vitamin D receptor knockout mice correlates with induction of uncoupling protein-1 in white adipose tissue. Endocrinology. 2009;150:651–661. doi: 10.1210/en.2008-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlov I, Rochel N, Moras D, Klaholz BP. Structure of the full human RXR/VDR nuclear receptor heterodimer complex with its DR3 target DNA. The EMBO journal. 2012;31:291–300. doi: 10.1038/emboj.2011.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa FR. Endoplasmic reticulum stress, pancreatic beta-cell degeneration, and diabetes. Cold Spring Harbor perspectives in medicine. 2012;2:a007666. doi: 10.1101/cshperspect.a007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puff R, Dames P, Weise M, Goke B, Seissler J, Parhofer KG, Lechner A. Reduced proliferation and a high apoptotic frequency of pancreatic beta cells contribute to genetically-determined diabetes susceptibility of db/db BKS mice. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2011;43:306–311. doi: 10.1055/s-0031-1271817. [DOI] [PubMed] [Google Scholar]
- Quack M, Szafranski K, Rouvinen J, Carlberg C. The role of the T-box for the function of the vitamin D receptor on different types of response elements. Nucleic acids research. 1998;26:5372–5378. doi: 10.1093/nar/26.23.5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470:279–283. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS biology. 2011;9:e1001206. doi: 10.1371/journal.pbio.1001206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli RB, Kalicharan RD, Avramut MC, Sjollema KA, Pronk JW, Dijk F, Koster AJ, Visser JT, Faas FG, Giepmans BN. Destruction of tissue, cells and organelles in type 1 diabetic rats presented at macromolecular resolution. Scientific reports. 2013;3:1804. doi: 10.1038/srep01804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riachy R, Vandewalle B, Moerman E, Belaich S, Lukowiak B, Gmyr V, Muharram G, Kerr Conte J, Pattou F. 1,25-Dihydroxyvitamin D3 protects human pancreatic islets against cytokine-induced apoptosis via down-regulation of the Fas receptor. Apoptosis. 2006;11:151–159. doi: 10.1007/s10495-006-3558-z. [DOI] [PubMed] [Google Scholar]
- Shaffer PL, Gewirth DT. Structural basis of VDR-DNA interactions on direct repeat response elements. The EMBO journal. 2002;21:2242–2252. doi: 10.1093/emboj/21.9.2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, Collisson EA, Connor F, Van Dyke T, Kozlov S, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Scheuner D, Ron D, Pennathur S, Kaufman RJ. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. The Journal of clinical investigation. 2008;118:3378–3389. doi: 10.1172/JCI34587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH, Yates JR., 3rd DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. Journal of proteome research. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiishi T, Gysemans C, Bouillon R, Mathieu C. Vitamin D and diabetes. Endocrinology and metabolism clinics of North America. 2010;39:419–446. doi: 10.1016/j.ecl.2010.02.013. table of contents. [DOI] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell. 2012;150:1223–1234. doi: 10.1016/j.cell.2012.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoulou NH, Bamborough P, Bannister AJ, Becher I, Bit RA, Che KH, Chung CW, Dittmann A, Drewes G, Drewry DH, et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. Journal of medicinal chemistry. 2016;59:1425–1439. doi: 10.1021/acs.jmedchem.5b00256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes & development. 1995;9:2723–2735. doi: 10.1101/gad.9.22.2723. [DOI] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science (New York, NY. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. The Journal of clinical investigation. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y, Kim J, Ni M, Wei Y, Okamoto H, Lee J, Adler C, Cavino K, Murphy AJ, Yancopoulos GD, et al. Use of the Fluidigm C1 platform for RNA sequencing of single mouse pancreatic islet cells. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:3293–3298. doi: 10.1073/pnas.1602306113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. Journal of proteomics. 2015;129:16–24. doi: 10.1016/j.jprot.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara E, Fujimoto S, Inagaki N, Okawa K, Masaki S, Yodoi J, Masutani H. Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nature communications. 2010;1:127. doi: 10.1038/ncomms1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara E, Wei Z, Lin CS, Fang S, Ahmadian M, Kida Y, Tseng T, Dai Y, Yu RT, Liddle C, et al. ERRgamma Is Required for the Metabolic Maturation of Therapeutically Functional Glucose-Responsive beta Cells. Cell metabolism. 2016;23:622–634. doi: 10.1016/j.cmet.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Chalmers MJ, Stayrook KR, Burris LL, Wang Y, Busby SA, Pascal BD, Garcia-Ordonez RD, Bruning JB, Istrate MA, et al. DNA binding alters coactivator interaction surfaces of the intact VDR-RXR complex. Nature structural & molecular biology. 2011;18:556–563. doi: 10.1038/nsmb.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Pedersen HK, Dawed AY, Pearson ER. Pharmacogenomics in diabetes mellitus: insights into drug action and drug discovery. Nature reviews. 2016;12:337–346. doi: 10.1038/nrendo.2016.51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. Ranking of highly expressed transcription factors by average RPKM in single-cell RNA seq. Average RPKM was shown. VDR is showed in red.
B. Heat map of single cell expression value of key α cell and β cell markers, together with VDR.
A. FACS analysis of Annexin-V+ population percentage, which indicate apoptotic cells, in control and VDR knockdown human β-like cells.
B. Heat map of ratio of gene expression level by qPCR between siVDR and siCtrl INS1 cells under different stressing condition.
C. Serum insulin level of VDR+/+ and VDR−/− mice (n=3, error bars show S.E.M., *P<0.05).
D. Serum proinsulin level of VDR+/+ and VDR−/− mice (n=3, error bars show S.E.M.).
E. Staining of MAFA and Insulin in VDR WT and VDR KO islets.
F. Heat map of mouse islets treated with vehicle, IL1β, or IL1β+Cal for 48 or 96 hours, showing the genes induced by IL1b are partially rescued by Cal treatment.
G. Venn diagram showing the overlap of between genes significantly suppressed by IL1b and genes rescued by Cal at 96 hours.
H. Gene ontology categories of the 169 genes suppressed by IL1b and rescued by Cal at 96 hours.
I. Venn diagram showing the overlap of between genes significantly induced by IL1b and genes rescued by Cal at 96 hours.
J. Gene ontology categories of the 332 genes induced by IL1b and rescued by Cal at 96 hours.
A. Immunoprecipitation of endogenous BAF/PBAF components in 293T cells shows that BRD9 interacts with BAF component BRG1 and ARID1B, but not interact with PBAF component ARID2.
B. Scatter plot of the p-value of all screening genes enriched in the drop-out screening, VDR is labeled in red. Several epigenetic modifiers, including BRD7, are labeled in green.
C. Western blot of endogenous BRD9 in immunoprecipitates from 293T cells expressing HA-tagged VDR treated with vehicle (DMSO), Cal, the BRD9 inhibitor iBRD9, or Cal+iBRD9.
D. Quantification of relative signal intensity corresponding to Figure S3C (n=2, error bars show S.E.M., **P<0.01).
E. Quantification of relative signal intensity corresponding to Figure 2C (n=2, error bars show S.E.M., *P<0.05, **P<0.01). Western blotting of full length his-VDR showed the SF9 (insect) produced recombinant protein is acetylated.
F. Mass-spec results suggest the lysine 91 on human VDR is acetylated.
G. Quantification of relative signal intensity corresponding to Figure 2G (n=2, error bars show S.E.M., *P<0.05, **P<0.01).
H. Quantification of relative signal intensity corresponding to Figure 2H (n=2, error bars show S.E.M., **P<0.01).
I. Quantification of relative signal intensity corresponding to Figure 2I (n=2, error bars show S.E.M., **P<0.01).
A–E. Quantification of relative signal intensity corresponding to Figure 3B (n=2, error bars show S.E.M., **P<0.01).
F. INS-1 cells expression VDR-WT or VDR-K91R were treated with Cal and exposed to IL1β for 1hr, Nfkbia level were measured by qPCR (n=3, error bars show S.E.M., *P<0.05)
G–I. ChIP-qPCR of ARID1B, a BAF specific component, show that addition of iBRD9 reduce BAF complex binding at key VDR target loci Nfkbia, Nfkbiz, and Camk4.
J–L. ChIP-qPCR of BAF180, a PBAF specific component, show that addition of iBRD9 increase PBAF complex binding at key VDR target loci Nfkbia, Nfkbiz, and Camk4.
A. Liver weight of db (B6) mice treated with Cal or iBRD9 (n=6, error bars show S.E.M.).
B. Epididymal white adipose tissue (eWAT) weight of db/db (B6) mice treated with Cal or iBRD9 (n=6, error bars show S.E.M.).
C. O2 consumption of mice treated with vehicle or Cal+iBRD9.
D. CO2 production of mice treated with vehicle or Cal+iBRD9.
E. Serum calcium level of mice in all treatment group (n=10, error bars show S.D).
F. Whole body weight of db (BKS) mice treated with Cal or Cal+iBRD9.
A. Target gene of Cal Cyp24A1 expression in multiple tissues, indicating islets is targeted by Cal directly (n=3, error bars show S.E.M., *P<0.05).
B. Islets from db (B6) treated with Cal show increased expression of Ins2 (n=3, error bars show S.E.M., *P<0.05).
C. Liver expression of key gluconeogenesis genes are repressed in Cal treated db (B6) mice, suggesting higher level of serum insulin suppressed gluconeogenesis from liver.
D. Low dose STZ mice treated with Cal or iBRD9 showed increased level of insulin (n=8–10, error bars show S.E.M.).
E. Immunofluorescence staining of NKX6-1 (red), a key transcription factor for β cell identity and function, in vehicle and Cal+iBRD9 treated low dose STZ pancreas sections. (Scale bar: 50μm).
F. Heatmap of qPCR results of multiple inflammatory genes expression in islets from aged h-IAPP mice treated with Cal or Cal+iBRD9. Islets were collected from 15 months old mice and cultured ex vivo in ultra-low attachment plates. Cal or Cal+iBRD9 were treated for 72 hours.
G. Western blotting of ER stress marker p-PERK in INS-1 cells treated with Thapsigargin (0.25μM) or Tunicamycin (5μg/ml) overnight. Cal was pretreated 1 hour.
H. Immunohistochemistry staining of ER stress marker Calnexin and PERK with pancreatic tissue section in db/db (BKS) mice.
I. qPCR of Nfkbia expression in INS-1 cells exposed to IL1β and Cal and multiple BRD9 inhibitors. (n=3, error bars show S.E.M., *P<0.05, **P<0.01).
J. In vivo pharmacokinetic study of iBRD9 (n=3, error bars show S.E.M.).
A. Cell number of ex vivo cultured human islets treated with vehicle or Calcipotriol for 4 weeks (n=3, error bars show S.E.M., *P<0.05).
B. Insulin secretion assay of ex vivo cultured human islets treated with vehicle or Calcipotriol for 1 week. (n=3, error bars show S.E.M., **P<0.01).