

SUMMARY
Hemophilia B is an ideal target for gene- and cell-based therapies because of its monogenic nature and broad therapeutic index. Here, we demonstrate the use of cell therapy as a potential long-term cure for hemophilia B in our FIX-deficient mouse model. We show that transplanted, cryopreserved, cadaveric human hepatocytes remain functional for more than a year and secrete FIX at therapeutic levels. Hepatocytes from different sources (companies and donors) perform comparably in curing the bleeding defect. We also generated induced pluripotent stem cells (iPSCs) from two hemophilia B patients and corrected the disease-causing mutations in them by two different approaches (mutation specific and universal). These corrected iPSCs were differentiated into hepatocyte- like cells (HLCs) and transplanted into hemophilic mice. We demonstrate these iPSC-HLCs to be viable and functional in mouse models for 9–12 months. This study aims to establish the use of cells from autologous and heterologous sources to treat hemophilia B.
In Brief
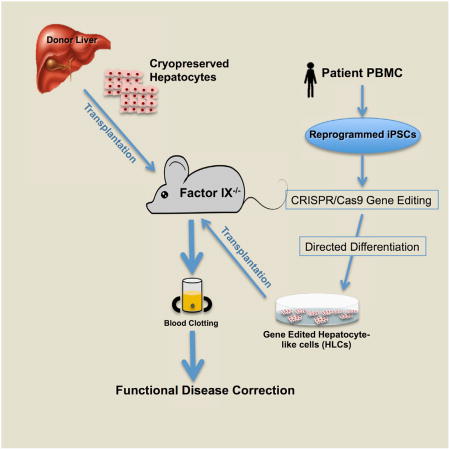
Ramaswamy et al. show that hepatocytes transplanted into a mouse model can alleviate symptoms of hemophilia B. Induced pluripotent cells from patients with hemophilia B can be gene-corrected and converted to hepatocyte-like cells for cell therapy. This provides evidence for potential treatment of monogenic diseases of the liver using cell therapy.
INTRODUCTION
Hemophilia B is an X-linked congenital clotting disorder caused by systemic lack of clotting factor IX and affects 1 in 30,000 male births (Stonebraker et al., 2012). It is clinically categorized as mild (5%–40% activity), moderate (1%–5% activity), or severe (<1% activity) based on the extent of factor IX (FIX) activity seen in patients (Blanchette et al., 2014). Patients suffer from recurrent bleeds in soft tissues, joints, and muscles, leading to chronic joint inflammation, crippling arthropathy, and physical disability in addition to the risk of life-threatening bleeds. Recombinant human Factor IX supplements administered intravenously on a prophylactic basis are currently used to manage the disease. In addition to being expensive, the need for frequent intravenous administration reduces compliance and increases the susceptibility of patients to blood-borne infections (Hepatitis C virus [HCV], Hepatitis B virus [HBV], HIV, etc.) (Knight et al., 2006).
Being a monogenic disorder with a broad therapeutic window and excellent animal models, hemophilia B is an ideal candidate for gene and/or cell therapy. The normal circulating levels of FIX are reported to be in the range of 5 µg/mL, and a 3- to 5-fold increase in its levels in severely affected patients (3%–5% of 5 µg/mL) can significantly improve the quality of life of patients. Over the years, gene therapy with viral vectors, like adeno-associated viral (AAV) vectors, has emerged as a potential long-term therapeutic option. However, despite the recent success, gene therapy with viral vectors is still challenged by problems with low transient expression, random integration, possible tissue damage, and immunogenicity (Nathwani et al., 2011, 2014; Nienhuis et al., 2017). Being the natural site of FIX synthesis, liver transplantation is a long-term therapeutic option and has been shown to be effective (Delorme et al., 1990; Gibas et al., 1988; Merion et al., 1988). Expression of FIX in its native site, the liver (an immune-privileged site), is also envisaged to promote accurate post-translational modifications, immune tolerance, and circulatory access (Knolle and Gerken, 2000; Arruda and Samelson-Jones, 2016). However, an acute shortage of donor livers and the need for long-term immunosuppression prevent more widespread adoption.
In this study, we developed a quadruple knockout mouse model of hemophilia B that allows the engraftment and expansion of human hepatocytes. These mice are derived from the crossing of transgenic FIX−/−-deficient mice (Wang et al., 1997) with Rag2−/− IL2rγ−/− Fah−/− mice (Bissig et al., 2007). The deficiency in Rag2−/− and IL2rγ−/− renders these animals immune-deficient (B, T, and natural killer [NK] cell-deficient) and permissive to the engraftment of human cells. A genetic deletion of fumarylacetoacetate hydrolase (FAH) in the recipient allows the engraftment of human hepatocytes and selectively promotes their expansion. As has been reported, the compound 2-(2-nitro-4-trifluoro-methylbenzoyl)-1,3-cyclohexanedione (NTBC) blocks the enzyme hydroxyphenylpyruvate dioxygenase upstream of FAH, preventing the accumulation of hepatotoxic metabolites. However, when NTBC is withdrawn, the resident mouse hepatocytes undergo toxic death, creating a regenerative niche that supports expansion of the transplanted cells. We reasoned that, in this model, the transplanted cells could expand more and help us maximize therapeutic efficacies.
Here we examined the potential of cadaveric cryopreserved human hepatocytes to persist and retain long-term functionality in this quadruple knockout (KO) mouse model. We show that hepatocyte transplantation can restore clotting function to normal wild-type (WT) levels and is at least 10-fold higher than the levels that would be needed for a significant improvement in treating the disease. We generated iPSCs from two severe hemophilia B patients and employed the CRISPR-Cas9 system to correct disease-causing mutations in the patient’s genome. We adopted two approaches: a universal correction approach where a full-length human FIX cDNA was knocked into exon 1 of the resident FIX gene and a more precise point correction at the mutated base pair to restore the WT sequence. We then differentiated these genetically corrected induced pluripotent stem cell (iPSC) lines into hepatocyte-like cells (HLCs) through an optimized in vitro differentiation protocol. The differentiated iPSC-HLCs were transplanted into our quadruple KO mouse model. Mice transplanted with iPSC-HLCs showed expression of human Albumin (hAlb) suggesting successful engraftment and expansion of the transplanted iPSCs. We also confirmed the presence of FIX in the gene-corrected transplanted hepatocytes 6–9 months after transplantation. These studies thus provide proof of concept for the potential use of autologous and heterologous human hepatocytes in the treatment of hemophilia B and other monogenic diseases of the liver.
RESULTS
Engraftment and Expansion of Human Hepatocytes in a Quadruple Knockout Mouse Model of Hemophilia B
We have previously reported the generation of a mouse model of hemophilia B where a gene-targeting strategy was used to disrupt the FIX gene, as a result of which a 2-kb fragment of the FIX gene (with the C-terminal 164 amino acids and the 3′ UTR) was deleted, leading to complete loss of the factor IX gene product (Wang et al., 1997). For transplantation and expansion of human hepatocytes, we have previously generated an immune-deficient mouse by crossing the fumarylacetoacetate hydrolase Fah−/− mouse with the recombination activating gene 2 KO (Rag2−/−) and interleukin 2 receptor gamma (IL2Rγ−/−) KO mouse lines (Bissig et al., 2007). These triple KO mice allow more efficient engraftment of human cells by virtue of NTBC-based selection for the transplanted cells. We crossed these two transgenic lines to derive a quadruple KO Rag2−/− IL2rγ−/− Fah−/− FIX−/− mouse line (Figure S1A shows the breeding scheme employed). The resulting line exhibited prolonged clotting times and showed less than 10% of WTFIX activity in a one-stage FIX activity test (a physiologically relevant assay to measure clotting activity) (Figure 1A). This clotting defect could be alleviated by intravenous administration of purified FIX protein, confirming that the clotting defect was indeed caused by lack of FIX protein (Figure 1A). We also confirmed the absence of circulating FIX protein in these mice by a western blot (Figure 1B) and their ability to support the engraftment and expansion of human hepatocytes. Animals receiving cryopreserved human hepatocytes (hHeps) (Triangle Research laboratories [TRL] donor HUM4037), showed a strong presence of circulating hAlb in their sera, as detected by an ELISA (Figure 1C). In contrast, sera from PBS-transplanted and untransplanted parental mice (WT, FIX−/−, and FRGFIX−/− mice) lacked any hAlb (Figure 1C). Additionally, we collected liver tissue from the transplanted mice at experimental endpoints and found hAlb- and Fah-positive clusters of functional hHeps in the livers of Fah-deficient recipient mice (Figure 1D).
Figure 1. Transplantation of Cadaveric Hepatocytes from Multiple Donors Can Alleviate the Clotting Defect for Up to a Year, If Not Longer.

(A) The newly derived quadruple KO FRGFIX−/− mice are hemophilic and exhibit poor clotting in a one-stage FIX activity test. As can be seen, these mice behave similarly as the previously reported FIX−/− mice. Administration of commercially available rhFIX protein alleviates the clotting deficiency and reduces the clotting time.
(B) The quadruple KO mice lack any circulating FIX protein, as detected by western blotting. Serum from WT and the parental FRG−/− strain shows a strong signal for FIX protein, which is completely absent from the FIX−/− and quadruple KO mice.
(C) Similar to the parental FRG−/− mice, the quadruple KO mice allow for robust engraftment of cadaveric hHeps. 35 days after transplantation, one can detect a strong and specific signal for hAlb in the sera of hHeps-transplanted (TRL HUM4037) animals. On the other hand, PBS-transplanted animals, untransplanted quadruple KO animals, WT, and FIX−/− animals show no hAlb in their sera. We used culture supernatant from hHep cultures as a positive control.
(D) Similar to the parental FRG−/− mice, the quadruple KO mice allow for robust engraftment of cadaveric hHep. At the end of the transplantation study, in the livers of hepatocyte-transplanted animals, we detected clusters of hAlb- and Fah-positive hepatocytes. The two stains also show strong co-localization with each other, further confirming their human origins. These clusters are conspicuously absent in the control PBS-transplanted animals. Infecting the transplanted (donor) hepatocytes with an Alb-Luc lentiviral vector before transplantation also allowed us to track the engraftment by luciferase-based Intravital imaging systems (IVIS).
(E) PBS-transplanted quadruple KO animals did not show any hAlb in their serum, as detected by an ELISA.
(F) The animals transplanted with cadaveric hepatocytes (TRL HUM4037), on the other hand, showed strong and sustained expression of hAlb in the mouse serum for up to a year from transplantation (if not longer).
(G) PBS transplanted quadruple KO animals did not show any hFIX in their serum as detected by an ELISA assay.
(H) The animals transplanted with cadaveric hepatocytes (TRL HUM4037), however, showed strong expression of hFIX in the mouse serum for up to a year from transplantation (if not longer). In general, we find FIX to be expressed at a lower level compared with hAlb. This is a direct function of their expression levels and the affinities of the respective antibodies.
(I) Attesting to the low FIX levels, the clotting efficiency in these PBS-transplanted control mice also remained at the baseline in the one-stage FIX activity assay 6–8 months after transplantation.
(J) In the hepatocyte-transplanted animals, the secreted FIX protein was duly modified and functional because it resulted in a dramatic improvement in clotting efficiency (from 20% at 30 days to ~120% at 10–12 months).
(K and L) Examining the tissue sections from the PBS-transplanted animals at clinical or experimental endpoints also did not identify any transplanted cells (K).
On the other hand, animals receiving the cadaveric hepatocytes showed clusters of FIX- and Fah-positive cells (L).
Heterologous, Cryopreserved, Cadaveric hHeps Restore Clotting
To examine the utility and efficiency of cell therapy, commercially available, cryopreserved, cadaveric hHeps were intrasplenically transplanted in quadruple KO mice following 24–48 hr of in vitro culture. NTBC was immediately withdrawn, and after 2.5 weeks, mice were put back on NTBC for 10 days, and the cycle was repeated. Animals were bled at the end of every cycle, and circulating levels of hAlb and human FIX (hFIX) were determined by a sandwich ELISA. The cycles of NTBC withdrawal help provide a selective advantage to the donor cells without compromising the recipient’s health. As can be seen, untransplanted or PBS-transplanted animals show no hAlb (Figure 1E), hFIX (Figure 1G), or clotting activity in their serum (Figure 1I). They also lack expression of FIX or FAH in the liver (Figure 1K). On the other hand, animals transplanted with cadaveric hepatocytes show a regular and sustained increase in the circulating levels of hAlb (0–150 µg/mL of Albumin) (Figure 1F), hFIX (5–10 µg/mL) (Figure 1H), therapeutic levels of clotting activity (25%–125% of the normal level) (Figure 1J), and clusters of FIX- or Fah-positive cells in their liver (Figure 1L). The levels of FIX protein seen in an ELISA are equivalent to or higher than the normal (WT) circulating levels (2–5 µg/mL). Because of the broad therapeutic and tolerance range for FIX, even in cases where the animals exhibited higher FIX levels (>5 µg/mL), there were no adverse consequences or thrombotic events. We also tested hepatocytes from different vendors (Xenotech, Celsis, Triangle Research Laboratories, Lonza, etc.), donors (different ages, genders, and causes of death), and preparations and found no significant effect on the transplantability or engraftment potential of these cells (Figures S1B–S1D and data not shown). We observed that, depending on the number of transplanted cells, most animals reached their peak engraftment levels (as detected by hAlb) in 3–6 months and seemed to sustain this level for the rest of the study (more than 1 year), if not longer. We further confirmed these observations by staining the liver sections for FIX and Fah proteins at the experimental endpoints and observed significant regions of the mouse liver to be humanized by the transplanted cells. Depending on the initial number of transplanted cells, anywhere from 10%–90% of the mouse liver can be humanized by this transplantation and selection approach. They also remain healthy, functional, and non-tumorigenic for the duration of the entire study. We have tested hepatocytes from multiple donors and sources and have not seen any adverse reactions in the more than 40 animals we have tested so far. We conclude that cadaveric hHeps from heterologous sources produce sustained levels of circulating FIX that can almost completely abolish the clotting defect in our hemophilic mice for up to a year after transplantation (if not longer).
Generation of iPSCs and Identification of the Disease-Causing Mutations in Two Hemophilia B Patients
To examine the potential use of autologous patient-derived iPSCs in developing cell therapy-based approaches, we obtained peripheral blood samples from two de-identified severe (<1% FIX activity) hemophilia B patients (see Figure S2 for an overall schematic). Peripheral blood derived mononuclear cells (PBMCs) were cultured under two conditions: conditions that supported the viability and expansion of adherent late endothelial progenitor cells (EPCs) and conditions that supported the proliferation and expansion of non-adherent mononuclear cells. After 2 weeks of in vitro culture, the resulting adherent and non-adherent cells were reprogrammed into iPSCs using non-integrating Sendai viruses (Figure S3A). The resulting iPSC lines were validated for their pluripotency by the expression of various markers (transcript and protein levels) (Figures S3B, S3C, S3F, and S3G). They were also confirmed to have a stable karyotype and pluripotency potential, as assessed by a teratoma formation assay (Figures S3D, S3H, and S3E, and S3I).
The FIX gene is spread over 32 kb of genomic sequence, and the FIX locus-specific mutation database (FIXdb) lists more than 3,000 pathogenic mutations, with mutations affecting 73% of the total 461 residues (Goodeve, 2015). To identify the disease-causing mutations in these two patients, we PCR-amplified and cloned the exonic sequences and sequenced multiple bacterial clones by Sanger sequencing (data not shown). By aligning sequences from two normal individuals (NBD3 and NBD4), the consensus human sequence, and the sequences from the two patients (HB1 and HB2), we identified an A→C transition in exon 2 (GAA→GAC, glutamine to aspartate) of the HB1 patient and a G→T transversion in the last residue of exon 5 for the HB2 patient (Figures S4A and S4B). We did not find any additional mutations in any of the other exons or samples, suggesting sufficient power in our analyses to account for sequencing-related technical errors. After identifying these mutations in the original patient sample, we similarly confirmed their presence in the derived iPSC lines.
Correction of the Disease-Causing Mutations Using CRISPR/Cas9
To correct the causative mutations in the patient-derived iPSC lines, we developed two different CRISPR/Cas9-based approaches. Guided by a single-stranded guide RNA (sgRNA), the Cas9 nuclease catalyzes a double-stranded break that then allows the homology directed repair (HDR)/non-homologous end joining (NHEJ) machineries to come into play for gene correction. We used this technology to achieve gene correction by two different approaches: a more universal knockin of the FIX cDNA into exon 1 of the resident FIX gene and a point correction that restores the mis-sense mutation to the original WT sequence.
For the universal correction approach in HB1-derived iPSCs, we designed an sgRNA that targets exon 1 of the human FIX gene (Figure 2A). We also developed a targeting vector with the full-length FIX cDNA flanked by 500- to 700-bp homology arms (sequences in and around FIX exon 1). Co-nucleofection of these components into HB1-derived iPSC lines followed by PCR-based screening of single cell clones identified several iPSC lines that carried the full-length FIX cDNA cassette. Our primers are targeted to the homology arms in the donor DNA and will amplify both targeted and off-target integrations. As can be seen from Figures 2A and 2B, insertion of the FIX cDNA in the targeted locus yields a single 1.9-kb PCR amplicon as opposed to a 350-bp amplicon from the parental clones. Mixed clonal populations or populations with random (off-target) integrations will yield both bands. We found several partial clones (~45 of 120 clones screened; data not shown), suggesting robust cleavage and integration efficiencies. However, there were only 6 iPSC lines (of which three clones were shortlisted for further characterization: 4#21, 4#25, and 4#35) that showed clear clonal integration of the donor template. We further verified these clones by PCR amplification with primers at exon-exon junctions (Figures 2A and 2B). As can be seen, all partial and fully corrected clones showed a single clear 1.3-kb band that extended from the exon 2/3 junction to the exon 7/8 junction (not possible from the endogenous genomic sequence). Finally, to confirm the correction, we PCR-amplified and sequenced the exons from these corrected iPSC lines. As can be seen in Figure 2C, sequences from exon 2 followed exon 1 in the shortlisted universally corrected clones (4#21 and 4#25) as opposed to the intronic sequence. Additionally, as expected, exon 2 in these universally corrected iPSC clones carried the original A→C disease-causing mutation (Figure 2C). Based on these screens, we shortlisted two iPSCs from the HB1 patient, 4#21 and 4#25, and validated them for pluripotency (Figures S5A and S5B, HB1 Cl4#21, and S5E and S5F, HB1 Cl 4#25), teratoma formation (Figures S5D and S5H), and a stable karyotype (Figures S5C and S5G).
Figure 2. CRISPR/Cas9-Based Genomic Correction of HB1 Patient-Derived iPSCs.

(A) In the universal correction approach, we knock in a full-length FIX cDNA into exon 1 of the resident FIX gene to restore full functionality. This removes the need for sequencing the patient’s genome to identify the mutation and the need to custom-design and optimize all genome editing tools. As can be seen in the schematic, 3 sgRNAs were designed and tested for their cleavage efficiency (data not shown). Along with the Cas9 and the guides, we also nucleofected an HDR donor DNA that included the FL-FIX cDNA flanked by homology arms around the cleavage site. The nucleofected cells were identified by co-transfection of a GFP construct, and individual clones were allowed to emerge. We then picked and screened putative clones for the presence of the knockin cassette. As can be seen, the parental WT sequence yields a 350-bp band, whereas the corrected clones yield a 1.9-kb band. Mixed clonal populations will show the presence of both bands. We could further confirm the presence of the FIX cDNA by screening with exon-exon junction primers that will yield a product only from the knockin cDNA cassette.
(B) Multiple iPSc clones were identified based on the presence of a strong 1.9-kb band from the HDR donor. Some of these clones (#24, #27, and #42) were also mixed because they exhibited a varying presence of the parental 350-bp sequence. Finally, as one would expect, all of these clones exhibited a strong 1.3-kb band from the exon-exon junction-specific primers. Based on this PCR profile, we shortlisted HB1 EPC Cl4 #21 and #25 for additional characterization.
(C) We sequenced these shortlisted clones by Sanger sequencing and confirmed the presence of the knockin cassette. As can be seen, although exon 1 leads into intron 1 in the parental sequence (text in black), in the corrected iPSC lines, we detect the FL FIX cDNA. Further, sequencing the endogenous exon 2 from these lines confirmed the presence of the parental A→C mutation. Thus, any FIX to be seen in these cells should be derived from the knockin full-length FIX cassette.
(D) As a minimally invasive approach, we also developed a point correction strategy for the HB1-derived iPSCs. The objective here was to leave a minimal genomic footprint. We designed 4 sgRNAs targeting exon 2 of the FIX gene of the HB1 patient. We cloned them into pX330 or pX450 vectors and tested their efficiency in a Surveyor assay (data not shown). Finally, we nucleofected the shortlisted guides along with Cas9 and the HDR donor template. The donor was designed to restore the mutant base pair (G:C) in the HB1 patient to the WT sequence (A:T). The donor DNA also carried silent point mutations to prevent binding and re-cutting by the guide. We also introduced an XhoI site by making silent, synonymous substitutions to facilitate screening of the corrected clones. All synonymous substitutions were made to incorporate codons with greater or similar usage in mammalian cells.
(E) Genomic DNA was extracted from the isolated iPSC clones, and a PCR-based screening strategy was employed. The genomic sequence flanking exon 2 from these clones was PCR-amplified and screened for the presence of the XhoI site (introduced in the donor DNA). A negative clone like 4#7 gave a single ~1.2-kb band that was not cleaved by XhoI. The donor DNA (positive control) and the corrected iPSC clone (4 #9) both gave a larger PCR product that was successfully cleaved into 2 smaller bands in the presence of XhoI.
(F) We further verified this genomic correction by Sanger sequencing of the exon 2 region from both parental and gene-corrected iPSCs. As can be seen, the corrected clone 4 #9 showed the presence of all the synonymous silent mutations in addition to the XhoI site. The A→C mutation in the parental and uncorrected clone (#7) was also restored to the WT sequence in the gene-corrected clone #9 but not in clone #7.
We also corrected the disease-causing mutation by a more conservative approach that restored the mutated base pair to the WT sequence by deploying a plasmid donor containing the corrective target sequence flanked by homology arms. In this case, we designed sgRNAs that cut in the vicinity of the disease-causing mutation (for both HB1 and HB2) (Figures 2D and S6A). We also designed a targeting vector with homology arms (500–800 bp) that flanked the region around the mutation. The target sequence in the donor vector was engineered to restore the point mutation to the WT sequence. We also introduced some synonymous mutations in the target sequence to facilitate screening (the XhoI site in HB1 and the PstI site in HB2) and to prevent binding and re-cutting of the corrected sequence by the sgRNAs (Figures 2D and S6A). We reasoned that this would ensure better correction efficiencies.
We designed multiple sgRNAs flanking the mutation site in HB1 and introduced an XhoI site to identify corrected clones. PCR amplification of parental iPSC clones results in a 1-kb amplicon that cannot be cleaved by XhoI. Incorporation of the corrective donor DNA (in clone 4#9), however, results in two smaller products when treated with XhoI (Figures 2D and 2E). Any non-specific or off-target integrations should result in 3 bands—cleaved and non-cleaved amplicons. We thus identified one corrected clone, HB1 Cl4#9, which, upon sequencing, confirmed the presence of the silent mutations and the absence of the disease-causing A→C mutation (Figure 2F). Finally, we confirmed the pluripotency (Figures S5I, S5J, and S5L) and karyotypic stability of this iPSC line (Figure S5K).
For our second patient, HB2, we adopted only the point correction approach (Figure S6A). Clonal expansion and screening of the resulting iPSC clones identified two iPSC lines, HB2 EPC Cl# 3A+ and Cl3# B+, which generated a PCR product that could be cleaved by PstI (Figure S6B). We further verified this by Sanger sequencing, which confirmed the presence of all silent mutations introduced in the donor DNA (Figure S6C). We then verified the pluripotency of these shortlisted, corrected iPSc lines by staining for pluripotency markers (Figures S6D and S6G), RNA analysis (Figure S6J), teratoma formation assay (Figures S6E and S6H), and karyotyping (Figures S6F and S6I).
Directed Differentiation of the Corrected iPSCs to HLCs
We developed an in vitro differentiation protocol to generate HLCs from multiple iPSC lines in a robust and reproducible manner (Figure S7). Briefly, our parental (hemophilia B) and gene-corrected iPSCs were differentiated by this optimized protocol from the pluripotent stage through the definitive endoderm, hepatic progenitor state, and hepatoblast state into mature hepatocytes (see the protocol in the Experimental Procedures and Figure S7). We tracked the differentiation process by monitoring the culture supernatant for the presence of hAlb by an ELISA. We also tested for additional hepatocyte and definitive endoderm/hepatoblast/hepatocyte-specific markers like HNF4a, Fah, SOX17, FOXA2, MRP2, etc. at both the transcript and protein levels (see a representative example in Figures S8A and S8B). The in-vitro-differentiated HLCs are also positive for periodic acid-Schiff (PAS) staining (Figure S8C) and are permissive to the expression of reporters driven by the synthetic liver-specific promoter (LSP; a 790-bp synthetic liver-specific promoter consisting of the 475-bp thyroid hormone-binding globulin promoter and two copies of the 96-bp α1-microglobulin enhancer) (Figure S8D; Jacobs et al., 2008). The in-vitro-differentiated HLCs also exhibit many of the morphological features exhibited by hepatocytes, such as a large cytoplasmic-to-nuclear ratio, numerous vacuoles and vesicles, and prominent nucleoli (Figure S8D, arrows). Several cells were found to be multi-nucleated, and the differentiated cells formed sheets of an epithelial nature with localization of ZO-1 and MRP2 on the membrane (Figure S8D and data not shown). These observations confirm our ability to derive mature HLCs that exhibit many of the phenotypes of hepatocytes through our optimized in vitro differentiation protocol.
Having established an optimized differentiation protocol, we compared the corrected lines with the parental lines by staining for a panel of stage-specific markers. As can be seen in Figures 3A and 3E, directed differentiation of the HB1 iPSC lines (parental 4; gene corrected 4#9, 4#21, and 4#25) and HB2 iPSC lines (parental 3 and 6 and the gene-corrected 3#A+ and 3#B+) resulted in strong increases in secreted hAlb in the culture supernatant. These levels were comparable with the levels produced by cadaveric hepatocytes in vitro (Figure 3A, red marker). More importantly, during the differentiation process, the iPSC lines lost pluripotency markers like Oct-4 and Lin28 and gained hepatocyte-specific markers like CYP3A4, GATA4, Alpha fetoprotein (AFP), and Albumin (Figures S9A–S9D). We also tested for additional hepatocyte- and definitive endoderm/hepatoblast/hepatocyte-specific markers like HNF4a, Fah, SOX17, FOXA2, MRP2, etc. (Figures S9A–S9D). The differentiated HLCs also displayed many epithelium-specific markers like ZO1, CK18, etc. (Figures S9A–S9D). We further confirmed the expression of these markers by qPCR analysis. We assayed cells at different stages of the differentiation process by a panel of 30 stage-specific marker genes, including Albumin, AFP, CONN32, TFR, OCT-4, LIN28, NANOG, CK18, CK19, HNF4A, GATA4, FOXA1, FOXA2, Fah, etc. (Figures S9B and S9D).
Figure 3. In-Vitro-Directed Differentiation of the Parental, Gene-Corrected HB001 and HB002 iPSC Lines.

(A) After verifying the correction of their respective mutations, we differentiated the parental and gene-corrected HB1-iPSCs into hepatocytes using our optimized protocol. As can be seen, all 4 lines differentiated equivalently and secreted hAlb into the culture supernatant. These levels of hAlb were comparable with the levels produced by cadaveric hepatocytes (red marker).
(B) All four iPSC lines differentiated comparably and were positive for the standard panel of known hepatocyte markers such as Albumin, AFP, and CYP3A4.
(C) RT-PCR analysis was done to examine the effect of genomic correction on the expression of FIX transcript. As can be seen, although, on day 0, both the parental and the gene-corrected iPSCs showed little to no FIX transcript. However, as differentiation progressed, all gene-corrected iPSCs (4#9, 4#21, and 4#25) exhibited robust upregulation of the FIX transcript. Actin B (ACTB) was used as the internal reference for the RT-PCR.
(D) We verified functional restoration of FIX at the protein level by immunofluorescence staining. As can be seen, on day 0, both parental and gene-corrected iPSCs appear mostly negative for FIX protein; however, at the end of the differentiation, the gene-corrected iPSCs exhibit strong upregulation of FIX protein. We also see marginally weaker levels of expression in the point correction mutant (4#9) compared with the universal correction variants (4#21 and 4#25).
(E) Similarly, we differentiated the parental and gene-corrected HB2-iPSCs into hepatocytes using our optimized protocol. As can be seen, all 4 lines differentiated and produced hAlb in the culture supernatant.
(F) We further confirmed the presence of HLCs by immunofluorescence staining for various hepatocyte markers such as ALB, AFP, and CYP3A4.
(G) RT-PCR analysis was done to examine the effect of genomic correction on the expression of the FIX transcript. As can be seen, although, on day 0, both the parental and the gene-corrected iPSCs showed little to no FIX transcript (HB2 EPC 3 and 6), as differentiation progressed, all gene-corrected iPSCs (3A+ and 3B+) showed robust upregulation of the FIX transcript levels.
(H) We further verified this functional restoration of FIX by immunofluorescence staining for the FIX protein. As can be seen on day 37, although the parental iPSCs appear mostly negative for the FIX protein, the gene-corrected iPSCs exhibited strong upregulation of the FIX protein. Thus, we are able to show that gene correction of the patient-derived iPSCs and their in vitro differentiation is able to restore the expression of FIX protein in these deficient cells.
Finally, we tested these iPSCs for functional correction of the FIX deficiency. Although all lines were previously verified for sequence correction at the genomic level, they were not confirmed to have functional restoration of the FIX protein. Thus, we examined both the parental and gene-corrected iPSC lines for the expression of FIX upon termination of the differentiation protocol. As can be seen in Figures 3C and 3G, at the end of the 37-day directed differentiation, the parental lines, as expected, showed little to no expression of the full-length FIX transcript, whereas the gene-corrected differentiated lines (HB1: 4#9, 4#21, and 4#25; HB2: 3A+ and 3B+) showed robust expression of FIX. This was further confirmed by immunostaining for the FIX protein in these cells (Figures 3D and 3H). Although both parental and gene-corrected iPSCs look similar on day 0 in terms of their FIX expression, the gene-corrected lines exhibit strong expression of FIX protein at the end of the differentiation process (Figure 3D, top versus bottom panel). We see similar upregulation of FIX expression in the HB2 iPSC lines after gene correction, as both 3A+ and 3B+ acquire strong positivity for the FIX protein, unlike the parental clones 3 and 6 (Figure 3H). We can also detect FIX in the culture supernatant (Figures S9A and S9B). We also confirmed these findings by a western blot. Interestingly, the parental and gene-corrected iPSC-HLCs produce comparable amounts of hAlb in the culture supernatant, whereas the gene-corrected iPSC-HLCs produce significantly more FIX (2- to 6-fold) in the culture supernatant compared with the parental iPSC lines (Figure S10). Thus, based on multiple lines of evidence, we demonstrate complete functional restoration of FIX expression in multiple patient-derived HLCs through a CRISPR/Cas9-based genomic correction strategy.
Transplantation and In Vivo Functionality of the In-Vitro-Differentiated iPSC-HLCs
We next wanted to see whether these in-vitro-differentiated, patient-derived, gene-corrected iPSC-HLCs were viable and functional in our mouse model of hemophilia B. We therefore intrasplenically transplanted these iPSC-HLCs into multiple quadruple KO hemophilia B cohorts to examine their viability, functionality, and safety. We used cadaveric hHeps as a positive control in each experiment and PBS or medium as negative controls. The animals immediately underwent cyclical NTBC withdrawal and restoration to support the engraftment and expansion of these HLCs. They were also periodically bled in between cycles, and the serum was used to screen for the presence of hAlb (a surrogate marker for the efficiency of engraftment), FIX, and a one-stage FIX activity assay (an activated partial thromboplastin time [aPTT] clotting assay) was performed. Figures 4A and 4B show results from engraftments of two patient-derived, gene-corrected iPSC-HLCs along with one example of cadaveric hepatocytes. Both iPSC-HLC cell lines show increasing levels of engraftment (hAlb), FIX levels, FAH, and clotting activity, similar to that observed with cadaveric hepatocytes (Figure 4C). Similar results were observed with two cell lines from the second patient, using the human Cytokeratin 18 (CK18) marker instead of FAH (Figure S11). These data suggest that the transplanted iPSC-HLCs were viable and functional for nearly 10 months. It is worth noting that the expression of hAlb in the animals transplanted with cadaveric hHeps was much more robust and reproducible (Figure 4C), perhaps reflecting lower engraftment efficiencies or incomplete differentiation of iPSC cells to hepatocytes. Although multiple groups have achieved in vitro differentiation of embryonic stem cell (ESC)- and iPSC-derived cells to HLCs, there have been few reports of their long-term engraftment (>7–60 days), in vivo viability, and function.
Figure 4. In-Vitro-Differentiated iPSC-HLCs Show Long-Term Engraftment and Function in Our Mouse Model.

(A) We tracked engraftment of the iPSC-HLCs by an ELISA for hAlb in the sera of transplanted animals. As can be seen, the corrected iPSC line (HB2 EPC Cl3 A+) was present and producing Albumin in the recipient mice for 5–8 months. At clinical or experimental endpoints, liver tissue from the transplanted animal was also found to contain clusters of Fah- and FIX-positive cells, confirming the long-term presence, engraftment, viability, and function of iPSC-HLCs.
(B) We made similar observations in other transplanted animals, where hAlb was present in the recipient mice for 5–8 months. At clinical or experimental endpoints, liver tissue from the transplanted animal was also found to contain clusters of Fah- and FIX-positive cells, confirming the long-term presence, engraftment, viability, and function of iPSC-HLCs.
(C) Cadaveric hepatocytes (TRL HUM4100) were transplanted in the same cohort as a positive control for the transplantation procedure and immunohistochemical staining. Unlike iPSC-HLCs, cadaveric hepatocytes show a strong and sustained presence of hAlb in the mouse serum. They also show a larger alleviation of the clotting defect (from 10% to 50%), as measured by a one-stage FIX assay. At the experimental endpoints, clusters of Fah- and FIX-positive hHeps can be found in these recipient livers, as shown.
DISCUSSION
Due to its monogenic nature and a broad therapeutic index (FIX levels of 10%–150% are well tolerated), hemophilia B remains an ideal candidate for alternate therapies such as gene and cell therapy. After a series of successes in small and large animal models, gene therapy using AAV vectors has seen some recent success in clinical trials. At the same time, cellular immune responses against the virus have emerged as an obstacle, potentially resulting in loss of expression and other complications (Herzog, 2015; Sun et al., 2017). Transient immune suppression protocols have been developed to blunt these responses (Nienhuis et al., 2017). In addition, the size of the capsid and the viral vector limits the payload and, therefore, the applicability to other diseases. More recently, many groups have attempted permanent in vivo gene editing using zinc-finger nucleases, CRISPR/Cas9, and other DNA-editing enzymes (Guan et al., 2016; Huai et al., 2017; Nguyen and Anegon, 2016; Park et al., 2016). These studies continue to involve viral vectors and are plagued by concerns about immunogenicity, off-target genomic edits, lower efficiencies, and toxicity (Hinderer et al., 2018).
Another potential approach that may be curative is a whole-liver transplant because FIX is produced and secreted by the liver. However, organ transplantation is severely hampered by the shortage of donor livers and the need for constant immunosuppression. The potential for using isolated cells from either donor livers or from autologous stem cells can significantly mitigate these challenges. A cell therapy-based approach for hemophilia B could potentially provide a new therapeutic approach. There are three major cell sources for hepatocytes with their own respective advantages and disadvantages: heterologous cadaveric hepatocytes (immune reaction seen, limited availability), pluripotent stem cell (ESC and iPSC)-derived HLCs (patient-customizable with little immune reaction, unclear tumorigenic potential, scalable, and amenable to HDR-mediated genomic editing), and induced HLCs (iHeps) derived by direct reprogramming of fibroblasts to HLCs (lack of an expansion step can limit the cell numbers but also limit the tumorigenic risk at the same time, high genetic and epigenetic variability). The recent identification of epithelial cell adhesion molecule (EpCAM)+ bipotential stem cells from the adult liver opens up newer avenues for such cell therapy-based approaches because they provide a renewable source of hepatocytes from patients without the risk of reprogramming. Although these bipotential stem cell-derived hepatocytes have been shown to engraft in vivo, reaching the therapeutic benchmarks in terms of engraftment and repopulation efficiencies is still a bit away (Broutier et al., 2016; Huch et al., 2015).
In this study, we have demonstrated the utility of heterologous hepatocytes and explore the potential use of autologous iPSC-derived HLCs for cell therapy. For this study, we developed a new mouse model of hemophilia that is amenable to the engraftment and expansion of hHeps. We use the Fah/NTBC mode of selection to support the engraftment and expansion of the transplanted hepatocytes. Although this genetic reliance on the absence of Fah may have ordinarily limited the translatability of our approach, more recently,Nygaard et al. (2016) have adapted this selection principle for use in donors with a normal genetic background by using a small molecule called CEPHOBA (small-molecule inhibitor of fumarylacetoacetate hydrolase). Akin to a genetic defect in FAH, CEPHOBA allows for selective expansion of transplanted cells in the recipient. Although CEPHOBA is not Food and Drug Administration (FDA)-approved or in clinical use, the availability of small molecules like it allows us to envision a future where donor cells could be safely and selectively expanded in the recipient liver.
Although heterologous cell sources such as cadaveric hepatocytes are one alternative, use of autologous iPSC-derived HLCs as a renewable cell source would be ideal. Their ability to support HDR recombination, gene editing, and clonal proliferation allows in vitro screening and testing to avoid random integrations and off-target effects. Their autologous origin mitigates the risk of immune reaction and the need for constant immunosuppression. Many studies have shown the directed differentiation of ESCs and iPSCs into HLCs, and this has opened up the possibility of using iPSCs as an autologous and renewable source of cells. Although iPSCs have been differentiated into HLCs with remarkable in vitro functionality, their use in vivo has been limited (Basma et al., 2009; Baxter et al., 2015; Cayo et al., 2012; Duncan, 2016; Gieseck et al., 2014, 2015; Mallanna and Duncan, 2013; Touboul et al., 2010). Only a few studies have demonstrated long-term engraftment of iPSC-HLCs (Kim et al., 2011), and fewer still have used these cells for therapeutic purposes (Chen et al., 2015).
In this study, we examined the efficacy of generating iPSCs from hemophilia B patients, correcting the genetic defect in them by genomic editing and then directing these gene-corrected iPSCs into HLCs for use in cell therapy. In one case, we used CRISPR/Cas9-based point correction to replace the mutated base pair and restore it to the native sequence, whereas, in the other instance, we developed a more universal correction approach wherein a full-length FIX cDNA was knocked into exon 1 of the endogenous human FIX gene. This ensured expression of the full-length FIX protein under endogenous regulatory elements irrespective of where the patient’s original mutation was present.
After validation of the correction, we directed the differentiation of the parental and gene-corrected iPSCs into HLCs. We validated their hepatocyte-like phenotype by multiple assays, such as Albumin secretion, expression of hepatocyte-specific genes, downregulation of pluripotency-specific genes, RNA analysis, PAS staining, etc. Finally, we transplanted these iPSC-derived HLCs into our mouse model of hemophilia B, followed by serial sampling of the mouse plasma for hAlb expression, and examined the mouse liver at the clinical or experimental endpoints. In these transplanted liver sections, we were able to identify regions of viable and functional iPSC-HLCs by the presence of Fah (the mouse is FAH-deficient), FIX, and other human markers such as Albumin and/or hCK18. Some of the recipient animals also showed circulating hAlb in their serum; however, these levels were 5- to 20-fold lower than those obtained from cadaveric hHeps. We also found modest increases in the clotting efficiency of recipient mice (from less than 10% to ~25% of WT activity). Thus, even though iPSC-HLCs are present and functional in the recipient liver for up to a year, their therapeutic effect could be vastly improved. Interestingly, a study of 1,400 severe hemophilia B patients found that, with 15%–20% of FIX levels, joint bleeding rates approach zero, suggesting that even such small repopulation efficiencies by these iPSC-HLCs might be therapeutically effective. One other approach to further augment the potential of iPSC-HLCs would be to use the hyper-functional R338L (Padua variant) FIX, which results in 6- to 8-fold increase in FIX-specific activity (expressed as units activity per milligram protein) over the WT sequence (Crudele et al., 2015; Simioni et al., 2009).
Several issues may explain the limited therapeutic effect of iPSc-HLCs. The number of iPSC-HLCs transplanted is much lower because of the heterogeneity inherent to the differentiation process. Cell engraftment may be compromised by the enzymatic dissociation used to harvest HLCs; single-cell suspensions are reported to perform worse than cell aggregates or clumps (Gieseck et al., 2014). HLCs may not proliferate as well as the cadaveric hepatocytes in response to injury because of the lack of full maturation. The sudden change in the microenvironment from in vitro culture to in vivo may inhibit iPSC-HLC function and viability (Goldman and Gouon-Evans, 2016). The lower engraftment of iPSC-HLCs could also be a function of the number of cells transplanted (less than one million cells in most cases) or the mode of delivery. Intrasplenic transplantation of in-vitro-differentiated HLCs is also plagued by risks associated with thrombotic events, internal hemorrhages, and other ancillary causes.
We believe that improved and scalable differentiation and delivery techniques could help us target more iPSC-HLCs to the liver in addition to improving their overall viability and functionality. It is, however, important to remember that, unlike cadaveric hepatocytes, the large-scale reproducible production and transplantation of these iPSc hepatocytes remains challenging and expensive at present.
EXPERIMENTAL PROCEDURES
Generation and Maintenance of Rag2−/− IL2rγ−/− Fah−/− FIX−/−-Deficient Mice
The FIX-deficient hemophilic mice were previously generated as described in Wang et al. (1997). Positive ESC clones were injected into C57BL6 blastocysts as described, and the resulting chimeric males were bred to C57BL6 and 129Sv to establish an inbred line of mutant mice. Genotypes of mice were established by Southern blot hybridization using tail biopsy DNA. An inbred line of hemophilic mice was established through repeated crossing for more than 7 generations. The Rag2−/− IL2rγ−/− Fah−/− line was generated by crossing the Fah−/− mouse with the IL2rγ−/− (B6.129S4-Il2rgtm1Wjl/J) mouse from The Jackson Laboratory and the Rag2−/− mouse (RagN12) from Taconic Farms. The triple knockout mouse line was then crossed with the hemophilic line to generate the quadruple knockout strain that was hemophilic and permissive to engraftment and expansion of hHeps. Genotyping was done with the same primers and conditions as described before and according to the protocols provided by vendors. The quadruple knockout Rag2−/− IL2rγ−/− Fah−/− FIX−/− mice were kept on 2.5 mg/mL NTBC (100%) in the drinking water. Mice were kept in temperature- and humidity-controlled animal quarters with a 12-hr light-dark cycle. Purified human FIX (Benefix, Pfizer) diluted in PBS was injected intravenously (retro-orbitally) into mice in the event of any bleeding or hemophilic complications. All animal procedures were performed in accordance with protocols approved by the Salk Institute Animal Care and Use Committee. Both male and female mice were used for this work.
Mouse Plasma Collection
Blood samples were collected from the retro-orbital plexus into 0.1 vol of 3.2% sodium citrate. After two sequential centrifugation steps (4,500 × g and 14,500 × g), the plasma was stored at −70°C for all future analyses.
ELISA
The levels of hAlb protein in the mouse serum were measured by a sandwich ELISA using capture and detection antibodies from Bethyl Laboratories (A80-129A at 1:100 primary and A80-129P at 1:75,000) according to the manufacturer’s instructions. The ELISA was performed in duplicates for at least 2 or more biological replicates per experimental group. The unknown values were extrapolated based on a standard curve and multiplied by the initial dilution factor to calculate the absolute concentration. Levels of FIX protein in the mouse sera or in the culture supernatant were likewise measured by a sandwich ELISA using antibodies from Affinity Biologicals (GA-FIX-EA, matched antibody pair for hFIX ELISA). The unknown values were extrapolated based on a standard curve and multiplied by the initial dilution factor to calculate the absolute concentration.
One-Stage FIX Activity Test
Factor IX activity in mouse sera was determined by a one-stage FIX activity test assay as follows. Fifty microliters of factor IX-deficient human plasma (George King Biomedical, Overland, KS), 50 µL of a 1:5 (or 1:10) dilution of mouse test plasma in HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer (50 mM HEPES), and 50 µL of aPTT reagent (Pacific Hemostasis, Thermo Fisher Scientific) were incubated at 37°C in an ST4 coagulometer (Stago, NJ). After 3 min, clotting was initiated by addition of 50 µL of 33 mM CaCl2 (Pacific Hemostasis, Thermo Fisher Scientific) in HEPES buffer (Life Technologies). Factor IX activity of duplicate samples was determined from a log-log standard curve that was constructed from the clotting time results for dilution (1:5 to 1:1,280) of pooled normal mouse plasma.
Although this assay relies on the principle of aPTT, it is actually a modification of the original method developed by Langdell et al. (1953). The one-stage factor IX activity assay performs the aPTT test in the presence of standardized factor IX-deficient plasma so that the degree of correction of clotting of the factor IX-deficient plasma is directly related to the amount of factor IX activity that is supplemented by the addition of the animal plasma sample. We also see some FIX activity in our hemophilic animals when measured by the one-stage FIX activity assay, even though there is little to no FIX protein in circulation. This is a technical artifact because of the lower sensitivity of the one-stage FIX activity assay at lower ranges (<15%).
Western Blot from Mouse Serum Samples
Mouse plasma or culture supernatants were subjected twice to barium citrate adsorption (Bajaj and Birktoft, 1993). Four microliters of 1 M BaCl2 was added to 50 µL of mouse plasma or culture supernatant, incubated at room temperature for 5 min, and centrifuged at 3,800 × g for 10 min. The precipitated proteins were dissolved in 25 µL of citrate-saline buffer and precipitated again by BaCl2. The pellets were dissolved in 75 µL of citrate-saline buffer, and 10 µL samples were electrophoresed through an SDS 12% polyacrylamide gel. The gel was blotted on a polyvinylidene fluoride membrane (Immobilon-P, Millipore), followed by sequential incubations with solutions containing rabbit anti-human factor IX antibody or anti-hAlb antibody. Chemiluminescence reagent (PerkinElmer) was used as a substrate to detect antibody-bound protein bands.
Intrasplenic Transplantation of Cadaveric Hepatocytes
Many studies have shown that hepatocytes (approximately 20–40 µm in diameter) are transplanted into rodents via direct splenic puncture, whereby they enter the portal vein branches and are entrapped in the proximal hepatic sinusoids (Weber et al., 2009). Cryopreserved cadaveric hepatocytes were primarily sourced from Triangle Research Laboratories (now with Lonza) or from other vendors for comparison (Xenotech, Lonza, Celsis, etc.). Cells were thawed according to the manufacturer’s instructions and cultured in vitro for 2 days before isolating and transplanting them into adult Rag2−/− IL2rγ−/− Fah−/− FIX−/− mice as described previously (Bissig et al., 2007). Typically, one to three million cells were transplanted per animal, and the speed and extent of repopulation was largely dependent on this initial number. Although dose dependence was not studied, we did find that animals that received more hepatocytes did repopulate faster and to a greater extent. The percentage of repopulation also depends on other factors related to the surgery itself and the number of cycles of NTBC withdrawal. Briefly, mice were anesthetized with isoflurane and kept on a heating pad during the whole procedure. They were administered Benefix retro-orbitally before any invasive procedure at doses recommended by the manufacturer. A mid-abdominal or lateral incision was performed, and the spleen was prepared for injection. The lower pole of the spleen was injected with the three different cell types re-suspended in PBS. Closing of the abdominal muscle layer was performed with 4.0 silk sutures, and the skin was stapled or sutured. NTBC was immediately withdrawn in a stepwise gradation, with each step lasting 2 days before completely stopping the drug (25%, 12%, 6%, and 3% of 2.5 mg/L colony maintenance concentration). After 3 weeks of the first cycle, the mice were put back on 100% NTBC-containing water (2.5 mg/L) for 10 days and then returned to 0% NTBC as described in the daily steps. This withdrawal cycle was repeated multiple times over the course of a year or until the animal died. Animals were monitored during this period and were euthanized as they approached the clinical or experimental endpoints. All iPSC-HLC transplant experiments were done in groups of 3–4 and were repeated at least twice. All groups from one patient (parental and corrected) were differentiated together and transplanted on the same day in similar animals (age and health). We have depicted representative data from individual animals in each cohort. For the cadaveric hepatocyte transplants, we transplanted over 40 animals during the course of the entire study, and representative results are depicted. Typically, 4- to 8-week-old animals were used for the transplantation to account for the year-long repopulation times. Both male and female mice were used as recipients for this study, as determined by availability.
Blood Cell Preparation, Culturing, and Storage
Blood samples from two anonymized hemophilia B patients with a severe FIX deficiency (less than 1% activity) were obtained from University of North Carolina at Chapel Hill after due informed consent and approval by the local research ethics committee. The Salk institutional review board (IRB) further authorized the use of these de-identified patient samples. Up to 50 mL of patient blood was drawn into Vacutainers with sodium citrate and shipped at room temperature. The samples were then loaded on a Ficoll gradient and centrifuged to isolate the PBMCs. The buffy coat was transferred to a new tube, and then the volume was raised to 10 mL with PBS. The cells were resuspended in 5 mL red blood cell (RBC) lysis buffer for 5–10 min and then centrifuged to separate the cells from the lysed membranes. The cells were resuspended in PBS and washed again. Cells were counted and used for freezing, expansion, and DNA and RNA extraction. The resulting PBMCs were cultured under two growth conditions: EPC medium (L-EPC) to expand the adherent endothelial cells (Chang et al., 2013) and serum free medium/mononuclear cells (SFM/MNC) medium to culture the non-adherent progenitor cells (Ye et al., 2013). Isolated PBMCs were also frozen and used for DNA isolation to identify the mutations in the patient sample.
The PBMCs derived from the Ficoll gradient were cultured under two different conditions. One batch was resuspended in 2 mL of expansion medium (EM) (QBSF-60 stem cell medium containing 50 µg/mL ascorbic acid, 50 ng/mL c-kit ligand (SCF), 10 ng/mL IL-3, 2 U/mL erythropoietin (EPO), 40 ng/mL IGF-1, 1 µM dexamethasone, and 1% penicillin/streptomycin) and transferred to one well of a 12-well plate. After an initial decrease in cell number, there was robust proliferation. The cells were incubated in a 37°C, 5% CO2 incubator. The medium was changed every other day by centrifugation to remove the medium and replaced with fresh medium. The second batch of MNCs was used for late EPC isolation by culturing on matrigel-coated plates in endothelial growth medium-2MV (CC-3202, Lonza) (basal endothelial medium-2 [EBM], 20% human serum [HyClone fetal bovine serum (FBS)], vascular endothelial growth factor [VEGF], basic fibroblast growth factor [bFGF], insulin growth factor [IGF], gentamicin, ascorbic acid, and hydrocortisone). Late-outgrowth EPCs (late EPCs) formed approximately 18 days after culturing in endothelial growth medium (EGM)-2MV and exhibited a high proliferative capacity. They also had a cobblestone appearance and expanded with subsequent passaging. To passage, cells were washed with PBS and trypsinized with 0.05% trypsin-EDTA.
Integration-free Reprogramming of Non-adherent PBMCs and Adherent EPCs into iPSCs
After 14 days of cell expansion, both classes of PBMCs were harvested and transduced by Oct4, Sox2, Klf4, and c-Myc Sendai viral vectors (CytoTune-iPSC 2.0 Sendai Reprogramming Kit, Life Technologies) at an MOI of 10. The transduction was performed by centrifugation at 2,000 rpm for 30 min, in medium supplemented with cytokines and Y27632 (inhibitor of Rho-associated, coiled-coil containing protein kinase [ROCK-i]). The day after transduction (day 1), Sendai viruses were removed by centrifuging the cell suspension, and the cells were resuspended in fresh SFM supplemented with cytokines and ROCK-i (Y27632) for 2 days. Finally, the cells were seeded onto 10-cm dishes with 0.1% gelatin and inactivated mouse embryonic fibroblasts (MEFs). The medium was gradually switched from SFM/MNC or EPC medium to CDF-12 (DMEM/F12, knockout serum replacement [KOSR], β-mercaptoethanol [BME], bFGF, non-essential amino acids [NEAA], and Glutamax), which is more suitable for iPSC growth and proliferation. The medium was refreshed every other day. When the iPSC colonies emerged, they were manually picked and replated onto matrigel-coated plates for expansion in STEM-TeSR. Cells were fed every day, and clones were expanded for additional characterization.
iPSC Culture and Maintenance
iPSC lines were maintained on 6-well plates coated with matrigel with daily TeSR medium changes. At 70%–80% confluence, cells were passaged using dispase (37°C for 5–7 min), rinsed, and mechanically dissociated with a glass pipette. For differentiation experiments, the cell lines were harvested by accutase, counted, and then plated with ROCK (Y27632).
Characterization of iPSCs
IPSC lines were rinsed with 1× PBS, fixed with 4% paraformaldehyde, and stored in PBS for staining. Cells were permeabilized with PBST (PBS supplemented with 0.1% Triton X-100) for 10 min at room temperature. Non-specific antigens were then blocked by incubating the cells with PBST+BSA at room temperature for 1 hr. Cells were incubated overnight at 4°C in PBS-BSA containing 10% donkey serum and the following specific primary antibodies: 1:500 anti-human OCT4 (Cell Signaling Technologies), 1:500 anti-human NANOG (Cell Signaling Technologies), 1:500 anti-human SSEA4 (Cell Signaling Technologies),), 1:500 anti-TRA-1–60 (Cell Signaling Technologies), 1:500 anti-SOX2 (Cell Signaling Technologies), and 1:200 anti-Lin28 (R&D Systems). Following 3 washes with PBS, cells were incubated with appropriate Alexa Fluor conjugated secondary antibodies at 1:200 dilution. After 3 washes with PBS, the samples were incubated for 10 min with DAPI (1 µg/mL) in PBS, followed by a final wash in PBS. Fluorescence images were captured at 10× or 20× on a Zeiss LSM 710/780 confocal microscope and processed using ImageJ.
Teratoma Formation Assay
iPSCs were expanded to a 10-cm plate on MEFs, collected by scraping, and injected subcutaneously. Approximately 1 × 107 cells were injected in 100 µL DMEM containing 10% FBS above each shoulder into immunecompromised NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice (The Jackson Laboratory, Sacramento, CA). Mice were euthanized, and tissues were collected when tumor growths reached the approved experimental or clinical endpoints. Growths were dissected, fixed in 4% formalin for 48 hr, and then transferred to PBS. Tissues were then embedded in paraffin, sectioned, and stained with H&E for histological analysis.
Genomic DNA Extraction
Genomic DNA was isolated by either of the following two approaches. Cells were lysed in TAIL DNA isolation buffer (1% SDS [50ml of 10%], 0.1M NaCl [10ml of 5M], 0.1M EDTA [100ml of 0.5M], and 0.05M Tris [pH = 8.0] [25ml of 0.5M]) at 50°C for at least 1 hr. After incubation, DNA was extracted by isopropanol precipitation and resuspended in 100 µL Invitrogen Tris EDTA (TE) buffer. Alternatively, cells were lysed in the Episelect QuickExtract DNA isolation solution according to the manufacturer’s protocol.
Surveyor Assay
A surveyor assay in HEK293T cells was used to evaluate the cleavage efficiency of the shortlisted guides. Cell growth medium (DMEM containing 10% FBS and 1% Pen/Strep) was aspirated from each well, washed once with PBS, replaced with 2 mL of Opti-MEM (Gibco), and incubated at 37°C. 1 µg of pX330/pX450 vectors with Cas9 and the guides was lipofected into HEK293T cells using Lipofectamine 2000 transfection reagent (Life Technologies, Carlsbad, CA) following manufacturer’s instructions. 4–6 hr later, the medium was aspirated from the transfected cells and replaced with 2 mL growth medium. These cells were harvested after 72 hr using DNA extraction buffer (Episelect Technologies, San Diego, CA) according to the manufacturer’s instructions. The DNA was quantified by picogreen and analyzed for insertions or deletions (indels) created by Cas9/guide RNA (gRNA) using a Surveyor assay (Integrated DNA Technologies). Cells were transfected with the pX330/pX450 Cas9 vector with the shortlisted guides. Genomic DNA was isolated from the transfected cells after 36–48 hr. The genomic sequence around the target site was PCR-amplified using primers flanking the target locus. Each reaction contained the following: 12.5 µL 2× Phusion PCR Master Mix, 10 µL nuclease-free water, 1.5 µL forward and reverse primers (1:50 dilution), and 1 µL gDNA template (1:100 dilution). The sample was then amplified under optimal conditions. When the locus-specific PCR products were generated, PCR samples were melted and annealed using a thermocylcer program suggested by the manufacturer (Surveyor Mutation Detection Kit, Integrated DNA Technologies) to give either homoduplexes or mismatched heteroduplexes, the latter being sensitive to Surveyor nuclease. The re-annealed products were then treated with Surveyor nuclease, which detects and cleaves at the sites of indel mutations generated by on-target Cas9 activity. When treated with Surveyor nuclease, any mismatches caused by indel formation following endogenous NHEJ repair of genomic sites cleaved by CRISPR will be cut, yielding cleavage products of the expected sizes on an 8% Tris-borate-EDTA (TBE) gel (Life Technologies, Carlsbad, CA).
Nucleofection of iPSCs, Colony Isolation, and Screening for CRISPR/Cas9 Correction
Adherent iPSCs were dissociated using Tryple (Gibco), centrifuged, and resuspended in TeSR containing 10 mM ROCK-i (Y27632). Cells (1–2 × 106) were washed to remove accutase and resuspended in 100 µL Lonza nucleofection solution (VPH-5012, according to the manufacturer’s protocol). The following amounts of DNA were added: 1 µg pX330 Cas9+gRNA, 4 µg HDR template, and 0.5 µg GFP. The samples were then nucleofected using the B-016 protocol on the Amaxa nucleofector. After nucleofection, TeSR was added, and cells were transferred to a 12-well matrigel-coated plate. After 24–48 hr of incubation, the nucleofected iPSCs were harvested by accutase and sorted for GFP expression. The GFP-positive nucleofected cells were centrifuged at 1,500 rpm for 5 min and then plated onto 10-cm MEF plates at single-cell density for colony screening or harvested for genomic DNA analysis. The medium was changed every other day for these dishes, and after around 2 weeks, decent-sized iPSC colonies were visible. These colonies were manually picked and checked for correction of the point mutation by a PCR-based screen.
The iPSc clones were picked from the 10-cm dish into a 24-well plate coated with irradiated MEFs. When the isolated iPSC clones were grown to the appropriate size, each well was split into duplicate 24-well matrigel plates. Roughly two-thirds of each colony were added to a matrigel plate for genomic DNA isolation, whereas the other third was carried to another matrigel plate designated for clonal maintenance and expansion. Genomic DNA was isolated using Episelect DNA isolation buffer according to the manufacturer’s instructions. To screen genomic DNA samples, primers were designed, and the target locus was amplified by PCR to yield an integration-specific product that could be distinguished from the WT by restriction digestion (PstI/XhoI) or by size.
The primers and guides used for the correction strategies used in this study can be found in Table 1.
Table 1.
Primers and Guides Used for the Correction Strategies Used in This Study
| Primer Name | Sequence (5′to 3′) | |
|---|---|---|
| 1 | HB2 Ex 5 Guide A | GTAACATGTAACATTAAGAA |
| 2 | HB2 Ex 5 Guide B | GTAAAAATAGTGCTGATAACA |
| 3 | HB2 Ex 5 TV2 FP | TTCAAGAACTGGCAAGAACTG |
| 4 | HB2 Ex 5 TV2 RP | CGGAATCATTCTGACACCAA |
| 5 | HB1 Ex2/3 Guide A1 | CACCGGAGATATAATAGCGGTAAAC |
| 6 | HB1 Ex2/3 Guide A2 | AAACGTTTACCGCTATTATATCTCC |
| 7 | HB1 Ex2/3 Guide B1 | CACCGGAACCTCGAGAGAGAATGCA |
| 8 | HB1 Ex2/3 Guide B2 | AAACTGCATTCTCTCTCGAGGTTCC |
| 9 | HB1 Ex2/3 Guide C1 | CACCGCGGCCAAAGAGATATAATAG |
| 10 | HB1 Ex2/3 Guide C2 | AAACCTATTATATCTCTTTGGCCGC |
| 11 | HB1 Ex2/3 Guide D1 | CACCGAAATTCTGAATCGGCCAAAG |
| 12 | HB1 Ex2/3 Guide D2 | AAACCTTTGGCCGATTCAGAATTTC |
| 13 | hFIX Ex2–3 Surv_SR_FP2 | TTGGCTCCATGCCCTAAAGAG |
| 14 | hFIX Ex2–3 Surv_SR_RP2 | TGTGAAGCCCTAGGGAGGAT |
| 15 | HB1 Ex2/3 FP4 | GGCAACCATATTCTGAA |
| 16 | HB1 Ex2/3 RP4 | AATGCAAACTTTCATTAGGTTC |
| 17 | hFIX exon1 Guide A | GCAGCGCGTGAACATGATCA |
| 18 | hFIX Exon1 Guide B | TCTGCCTTTTAGGATATCTAC |
| 19 | hFIX Exon1 Guide C | TTTAGGATATCTACTCAGTGC |
| 20 | hFIX Ex1 surv_FP1 | ACTGATGAACTGTGCTGCCA |
| 21 | hFIX Ex1 surv_RP1 | TACTTACCAACCTGCGTGCT |
| 22 | hFIX Ex1 surv_FP2 | TGGGTCCCACTGATGAACTG |
| 23 | hFIX Ex1 surv_RP2 | ACCAGTACTTACCAACCTGCG |
| 24 | hFIX Ex1 surv_FP3 | GTCCCACTGATGAACTGTGC |
| 25 | hFIX Ex1 surv_RP3 | CTGCGTGCTGGCTGTTAGA |
| 26 | hFIX Ex1 scr_FP2 | TGACAAAGATACGGTGGGTCC |
| 27 | hFIX Ex1 scr_RP2 | TGCTGGCTGTTAGACTCTTCAATA |
| 28 | hFIX Ex1 scr_FP3 | ACTTGTCCCAAGAGGCCATT |
| 29 | hFIX Ex1 scr_RP3 | GTGCTGGCTGTTAGACTCTTCA |
| 30 | FIX uni corr Ex-ex junction FP1 | GTGCTGAATGTACAGTTTTTCTTG |
| 31 | FIX uni corr Ex-ex junction RP1 | AAGCTTGATGCTTAAGTGAGCT |
| 32 | FIX uni corr Ex-ex junction FP2 | GTGCTGAATGTACAGTTTTTCTTGATCATG |
| 33 | FIX uni corr Ex-ex junction RP2 | CAAAACAACCTGCCAAGGGAATTG |
Directed Differentiation of iPSCs to HLCs
iPSCs were maintained on matrigel and cultured in STEM-TeSR medium. For differentiation, cells were harvested with accutase, counted, and plated on matrigel in STEM-TeSR medium with daily medium changes. Cells were plated at 30%–40% confluency to start with. When 80% confluent, the cells were treated with STEM-TeSR containing 2% DMSO for 48 hr (seen to improve efficiencies for some lines). After 2 days in TeSR + DMSO, cells were switched to the differentiation medium. The base medium comprised RPMI, Anti/anti, polyvinyl alcohol (PVA; 1 g/L), Glutamax, NEAA, human transferrin, completely defined (CD) lipids, ITS (insulin transferrin selenium), and thioglycerol. Spent medium was collected from the differentiating cells every day to assess differentiation as a function of hAlb secretion. Differentiation began with base medium containing 100 ng/mL Activin A, heparin (10 µg/mL), insulin (4 µg/mL), and Wnt3a (50 ng/mL) for 3 days with daily medium change. This was followed by treating the cells with completely defined medium (CDM)-PVA, heparin, insulin, Activin A (100 ng/mL), and fetal calf serum (FCS, 0.2%) for 48 hr. The FCS concentration was increased to 2% for the next 24 hr, and then FGF10 was added to the same medium composition at 10 ng/mL for 24 hr. This was followed by switching the medium composition to base medium, heparin (10 µg/mL), insulin (4 µg/mL), FCS (2%), and FGF10 (25 ng/mL) for 4 days. After 4 days, cells were transferred to base medium with heparin, insulin, BMP4 (20 ng/mL), and FGF2 (10 ng/mL) for 3 days. At this point, the hepatic progenitors were switched to base medium containing hepatocyte base media (HBM; Lonza) supplemented with the HCM single-quots (Lonza), heparin (10 µg/mL), insulin (4 µg/mL), Oncostatin M (20 ng/mL), and hepatocyte growth factor (HGF; 10 ng/mL) for 5 days. After this time, the medium was changed every other day for 8 days with base medium supplemented with heparin (10 µg/mL), insulin (4 µg/mL), Oncostatin M (35–40 ng/mL), HGF (50 ng/mL), and dexamethasone (1 µM) for ~8 days. In the end, the cells were treated with base medium, heparin (10 µg/mL), insulin (4 µg/mL), FH1 (20 mM), FPH1 (20 mM), HGF (50 ng/mL), and dexamethasone (1 µM) for 5–7 days. Finally, for additional maturation, we supplement the above medium composition with 8-Bromo cyclic AMP (cAMP) for another 2 days.
Immunocytochemical Staining
Cells were fixed in 3.2% (v/v) paraformaldehyde for 10 min at room temperature or overnight at 4°C, washed three times with PBS, and permeabilized in 0.5% Triton X-100 for 5 min at room temperature. Cells were blocked for 1 hr with 2% (w/v) BSA (Sigma-Aldrich) and 5% (v/v) normal donkey serum (Jackson ImmunoResearch) in PBS and then incubated in primary antibody at 4°C overnight. Cells were washed three times in PBS and incubated for 1 hr with secondary antibodies. Stained cells were finally incubated in DAPI for 5 min before washing. Cultures were either imaged on plates or mounted on glass microscope slides in Fluomount-G. Images were taken at either 10× or 20× on a Zeiss LSM 710/780 confocal microscope and processed using ImageJ.
PAS Staining
PAS staining was performed using a periodic acid-Schiff kit (Sigma, 395B) according to the manufacturer’s instructions with minor adaptations. Briefly, cells were fixed in 4% formaldehyde for 1–2 min, washed with PBS, stained for 5 min with 1% periodic acid, and washed with distilled water prior to incubation with Schiff’s reagent for 15 min. After washing three times with water, cells were counterstained for 90 s with hematoxylin solution and washed again three times with water prior to microscopic examination and imaging.
Statistical Analysis
The data reported are the average of biological or technical replicates along with the SE in each case. For the transplanted animals, representative images for each of the classes are depicted. Statistical analyses, where applicable, were conducted with GraphPad Prism software version 4.0 (GraphPad).
Supplementary Material
Highlights.
Human hepatocytes, transplanted into a mouse model, secrete FIX at therapeutic levels
Gene-corrected patient iPSCs can be differentiated into hepatocytes
Transplantation of hepatocytes is viable and functional in a mouse model for a year
Feasibility of autologous and heterologous cell therapy for treatment of hemophilia B
Acknowledgments
The authors would like to acknowledge the help and support provided by all core facilities at the Salk Institute: Stem Cell Core, Functional Genomics Laboratory, ARD/SAF, and the Biophotonics Core. The authors are also grateful to the Histology Core at the Sanford Burnham Prebys Medical Discovery Institute for tissue processing and histology services. This work was supported by the Waitt Advanced Biophotonics Core Facility of the Salk Institute with funding from NIH-NCI CCSG (P30 014195), a NINDS Neuroscience Core Grant (NS072031), and the Waitt Foundation. I.M.V. is an American Cancer Society Professor of Molecular Biology and holds the Irwin and Joan Jacobs Chair in Exemplary Life Science. This work was supported in part by grants from the NIH Cancer Center (Core Grant P30 CA014195-38), Ipsen, the H.N. and Frances C. Berger Foundation, the Glenn Center for Aging Research, Leona M. and Harry B. Helmsley Charitable Trust grant 2017-PG-MED001, and the California Institute for Regenerative Medicine (CIRM-TR4-06809). S.R. was supported by the CIRM-TR4-06809 grant for most of this study. The authors would also like to acknowledge the assistance of Brenda I. Nielsen at UNC-Chapel Hill with facilitating informed consent and sample collection from patients with hemophilia B. Finally, this project would not have been possible without the willing participation of the patients and their families.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and eleven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.121.
AUTHOR CONTRIBUTIONS
I.M.V. was responsible for designing the study and writing and planning the manuscript. S.R. contributed to designing, planning, executing, and analyzing all in vitro and in vivo experiments in addition to writing the manuscript. N.T. contributed to the planning and executing of experiments with S.R. K.T.G. and D.W. were involved in the planning and execution of some of the correction and differentiation experiments along with S.R. T.M. and B.M.L. were involved in gene editing for one of the iPSC lines. P.E.M. was involved in collecting peripheral blood samples from hemophilia B patients for the purpose of this study and contributed to the final editing of the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Arruda VR, Samelson-Jones BJ. Gene therapy for immune tolerance induction in hemophilia with inhibitors. J. Thromb. Haemost. 2016;14:1121–1134. doi: 10.1111/jth.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj SP, Birktoft JJ. Human factor IX and factor IXa. Methods Enzymol. 1993;222:96–128. doi: 10.1016/0076-6879(93)22009-5. [DOI] [PubMed] [Google Scholar]
- Basma H, Soto-Gutiérrez A, Yannam GR, Liu L, Ito R, Yamamoto T, Ellis E, Carson SD, Sato S, Chen Y, et al. Differentiation and transplantation of human embryonic stem cell-derived hepatocytes. Gastroenterology. 2009;136:990–999. doi: 10.1053/j.gastro.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter M, Withey S, Harrison S, Segeritz CP, Zhang F, Atkinson-Dell R, Rowe C, Gerrard DT, Sison-Young R, Jenkins R, et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol. 2015;62:581–589. doi: 10.1016/j.jhep.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissig KD, Le TT, Woods NB, Verma IM. Repopulation of adult and neonatal mice with human hepatocytes: a chimeric animal model. Proc. Natl. Acad. Sci. USA. 2007;104:20507–20511. doi: 10.1073/pnas.0710528105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A, Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis Definitions in hemophilia: communication from the SSC of the ISTH. J. Thromb. Haemost. 2014;12:1935–1939. doi: 10.1111/jth.12672. [DOI] [PubMed] [Google Scholar]
- Broutier L, Andersson-Rolf A, Hindley CJ, Boj SF, Clevers H, Koo BK, Huch M. Culture and establishment of self-renewing human and mouse adult liver and pancreas 3D organoids and their genetic manipulation. Nat. Protoc. 2016;11:1724–1743. doi: 10.1038/nprot.2016.097. [DOI] [PubMed] [Google Scholar]
- Cayo MA, Cai J, DeLaForest A, Noto FK, Nagaoka M, Clark BS, Collery RF, Si-Tayeb K, Duncan SA. JD induced pluripotent stem cell-derived hepatocytes faithfully recapitulate the pathophysiology of familial hypercholesterolemia. Hepatology. 2012;56:2163–2171. doi: 10.1002/hep.25871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WY, Lavoie JR, Kwon SY, Chen Z, Manias JL, Behbahani J, Ling V, Kandel RA, Stewart DJ, Stanford WL. Feeder-independent derivation of induced-pluripotent stem cells from peripheral blood endothelial progenitor cells. Stem Cell Res. (Amst.) 2013;10:195–202. doi: 10.1016/j.scr.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li Y, Wang X, Zhang W, Sauer V, Chang CJ, Han B, Tchaikovskaya T, Avsar Y, Tafaleng E, et al. Amelioration of Hyperbilirubinemia in Gunn Rats after Transplantation of Human Induced Pluripotent Stem Cell-Derived Hepatocytes. Stem Cell Reports. 2015;5:22–30. doi: 10.1016/j.stemcr.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crudele JM, Finn JD, Siner JI, Martin NB, Niemeyer GP, Zhou S, Mingozzi F, Lothrop CD, Jr, Arruda VR. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood. 2015;125:1553–1561. doi: 10.1182/blood-2014-07-588194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delorme MA, Adams PC, Grant D, Ghent CN, Walker IR, Wall WJ. Orthotopic liver transplantation in a patient with combined hemophilia A and B. Am. J. Hematol. 1990;33:136–138. doi: 10.1002/ajh.2830330211. [DOI] [PubMed] [Google Scholar]
- Duncan SA. Liver Capsule: Multipotent stem cells and their lineage restriction to hepatocytes. Hepatology. 2016;64:1330. doi: 10.1002/hep.28727. [DOI] [PubMed] [Google Scholar]
- Gibas A, Dienstag JL, Schafer AI, Delmonico F, Bynum TE, Schooley R, Rubin RH, Cosimi AB. Cure of hemophilia A by orthotopic liver transplantation. Gastroenterology. 1988;95:192–194. doi: 10.1016/0016-5085(88)90310-1. [DOI] [PubMed] [Google Scholar]
- Gieseck RL, 3rd, Hannan NR, Bort R, Hanley NA, Drake RA, Cameron GW, Wynn TA, Vallier L. Maturation of induced pluripotent stem cell derived hepatocytes by 3D-culture. PLoS ONE. 2014;9:e86372. doi: 10.1371/journal.pone.0086372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieseck RL, 3rd, Vallier L, Hannan NR. Generation of Hepatocytes from Pluripotent Stem Cells for Drug Screening and Developmental Modeling. Methods Mol. Biol. 2015;1250:123–142. doi: 10.1007/978-1-4939-2074-7_9. [DOI] [PubMed] [Google Scholar]
- Goldman O, Gouon-Evans V. Human Pluripotent Stem Cells: Myths and Future Realities for Liver Cell Therapy. Cell Stem Cell. 2016;18:703–706. doi: 10.1016/j.stem.2016.05.019. [DOI] [PubMed] [Google Scholar]
- Goodeve AC. Hemophilia B: molecular pathogenesis and mutation analysis. J. Thromb. Haemost. 2015;13:1184–1195. doi: 10.1111/jth.12958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Ma Y, Li Q, Sun Z, Ma L, Wu L, Wang L, Zeng L, Shao Y, Chen Y, et al. CRISPR/Cas9-mediated somatic correction of a novel coagulator factor IX gene mutation ameliorates hemophilia in mouse. EMBO Mol. Med. 2016;8:477–488. doi: 10.15252/emmm.201506039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RW. Hemophilia Gene Therapy: Caught Between a Cure and an Immune Response. Mol. Ther. 2015;23:1411–1412. doi: 10.1038/mt.2015.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinderer C, Katz N, Buza EL, Dyer C, Goode T, Bell P, Richman LK, Wilson JM. Severe Toxicity in Nonhuman Primates and Piglets Following High-Dose Intravenous Administration of an Adeno-Associated Virus Vector Expressing Human SMN. Hum. Gene Ther. 2018 doi: 10.1089/hum.2018.015. Published online February 12, 2018. https://doi.org/10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed]
- Huai C, Jia C, Sun R, Xu P, Min T, Wang Q, Zheng C, Chen H, Lu D. CRISPR/Cas9-mediated somatic and germline gene correction to restore hemostasis in hemophilia B mice. Hum. Genet. 2017;136:875–883. doi: 10.1007/s00439-017-1801-z. [DOI] [PubMed] [Google Scholar]
- Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MM, Ellis E, van Wenum M, Fuchs SA, de Ligt J, et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell. 2015;160:299–312. doi: 10.1016/j.cell.2014.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs F, Snoeys J, Feng Y, Van Craeyveld E, Lievens J, Armentano D, Cheng SH, De Geest B. Direct comparison of hepatocyte-specific expression cassettes following adenoviral and nonviral hydrodynamic gene transfer. Gene Ther. 2008;15:594–603. doi: 10.1038/sj.gt.3303096. [DOI] [PubMed] [Google Scholar]
- Kim J, Nam HY, Kim TI, Kim PH, Ryu J, Yun CO, Kim SW. Active targeting of RGD-conjugated bioreducible polymer for delivery of oncolytic adenovirus expressing shRNA against IL-8 mRNA. Biomaterials. 2011;32:5158–5166. doi: 10.1016/j.biomaterials.2011.03.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight R, Stanley S, Wong M, Dolan G. Hemophilia therapy and blood-borne pathogen risk. Semin. Thromb. Hemost. 2006;32(Suppl 2):3–9. doi: 10.1055/s-2006-946908. [DOI] [PubMed] [Google Scholar]
- Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol. Rev. 2000;174:21–34. doi: 10.1034/j.1600-0528.2002.017408.x. [DOI] [PubMed] [Google Scholar]
- Langdell RD, Wagner RH, Brinkhous KM. Effect of anti-hemophilic factor on one-stage clotting tests: a presumptive test for hemophilia and a simple one-stage anti-hemophilic factor assay procedure. J. Lab. Clin. Med. 1953;41:637–647. [PubMed] [Google Scholar]
- Mallanna SK, Duncan SA. Differentiation of hepatocytes from pluripotent stem cells. Curr. Protoc. Stem Cell Biol. 2013;26 doi: 10.1002/9780470151808.sc01g04s26. Unit 1G.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merion RM, Delius RE, Campbell DA, Jr, Turcotte JG. Orthotopic liver transplantation totally corrects factor IX deficiency in hemophilia B. Surgery. 1988;104:929–931. [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ, Harrington C, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, Della Peruta M, Lheriteau E, Patel N, Raj D, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TH, Anegon I. Successful correction of hemophilia by CRISPR/Cas9 genome editing in vivo: delivery vector and immune responses are the key to success. EMBO Mol. Med. 2016;8:439–441. doi: 10.15252/emmm.201606325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienhuis AW, Nathwani AC, Davidoff AM. Gene Therapy for Hemophilia. Mol. Ther. 2017;25:1163–1167. doi: 10.1016/j.ymthe.2017.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nygaard S, Barzel A, Haft A, Major A, Finegold M, Kay MA, Grompe M. A universal system to select gene-modified hepatocytes in vivo. Sci. Transl. Med. 2016;8:342ra79. doi: 10.1126/scitranslmed.aad8166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Lee DR, Sung JJ, Kim DW. Genome-editing technologies for gene correction of hemophilia. Hum. Genet. 2016;135:977–981. doi: 10.1007/s00439-016-1699-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simioni P, Tormene D, Tognin G, Gavasso S, Bulato C, Iacobelli NP, Finn JD, Spiezia L, Radu C, Arruda VR. X-linked thrombophilia with a mutant factor IX (factor IX Padua) N. Engl. J. Med. 2009;361:1671–1675. doi: 10.1056/NEJMoa0904377. [DOI] [PubMed] [Google Scholar]
- Stonebraker JS, Bolton-Maggs PH, Michael Soucie J, Walker I, Brooker M. A study of variations in the reported haemophilia B prevalence around the world. Haemophilia. 2012;18:e91–e94. doi: 10.1111/j.1365-2516.2011.02588.x. [DOI] [PubMed] [Google Scholar]
- Sun J, Yuan Z, Abajas YL, Szollosi DE, Hu G, Hua B, Xiao X, Li C. A Retrospective Study of the Cytokine Profile Changes in Mice with FVIII Inhibitor Development After Adeno-Associated Virus-Mediated Gene Therapy in a Hemophilia A Mouse Model. Hum. Gene Ther. 2017 doi: 10.1089/hum.2017.094. Published online October 26, 2017. https://doi.org/10.1089/hum.2017.094. [DOI] [PubMed]
- Touboul T, Hannan NR, Corbineau S, Martinez A, Martinet C, Branchereau S, Mainot S, Strick-Marchand H, Pedersen R, Di Santo J, et al. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology. 2010;51:1754–1765. doi: 10.1002/hep.23506. [DOI] [PubMed] [Google Scholar]
- Wang L, Zoppè M, Hackeng TM, Griffin JH, Lee KF, Verma IM. A factor IX-deficient mouse model for hemophilia B gene therapy. Proc. Natl. Acad. Sci. USA. 1997;94:11563–11566. doi: 10.1073/pnas.94.21.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Groyer-Picard MT, Franco D, Dagher I. Hepatocyte transplantation in animal models. Liver Transpl. 2009;15:7–14. doi: 10.1002/lt.21670. [DOI] [PubMed] [Google Scholar]
- Ye L, Muench MO, Fusaki N, Beyer AI, Wang J, Qi Z, Yu J, Kan YW. Blood cell-derived induced pluripotent stem cells free of reprogramming factors generated by Sendai viral vectors. Stem Cells Transl. Med. 2013;2:558–566. doi: 10.5966/sctm.2013-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.