

SUMMARY
Cancer stem cells promote neoplastic growth, in part by deregulating asymmetric cell division and enhancing self-renewal. To uncover mechanisms and potential therapeutic targets in glioma stem cell (GSC) self-renewal, we performed a genetic suppressor screen for kinases to reverse the tumor phenotype of our Drosophila brain tumor model and identified dCdk5 as a critical regulator. CDK5, the human ortholog of dCdk5 (79% identity), is aberrantly activated in GBMs and tightly aligned with both chromosome 7 gains and stem cell markers affecting tumor-propagation. Our investigation revealed that pharmaceutical inhibition of CDK5 prevents GSC self-renewal in vitro and in xenografted tumors, at least partially by suppressing CREB1 activation independently of PKA/cAMP. Finally, our TCGA GBM data analysis revealed that CDK5, stem cell, and asymmetric cell division markers segregate within non-mesenchymal patient clusters, which may indicate preferential dependence on CDK5 signaling and sensitivity to its inhibition in this group.
In Brief
Glioblastoma is the most common and deadliest form of brain tumor and can withstand current therapies due to the resilience of glioma stem cells (GSCs). Mukherjee et al. examine Cdk5 and its role in promoting stemness in asymmetric division of brain tumor stem cells in Drosophila and mice.
INTRODUCTION
Cancer stem cells possess self-renewal properties, yet differ from normal stem cells by their genetic instability and deregulated asymmetric division, which together enhance self-renewal and clonal proliferation (Mukherjee and Brat, 2017; Zabala et al., 2016). Core regulators of asymmetric cell division have been well described in Drosophila and many have mammalian orthologs with similar functions (Mukherjee and Brat, 2017). Asymmetric cell division mechanisms have also been investigated in the context of neoplastic diseases (Mukherjee et al., 2015). In our lab, we have successfully translated the Drosophila brain tumor model to disrupted asymmetric cell division in human brain tumors in order to uncover mechanisms and potential therapeutic targets relevant to glioblastoma (GBM), World Health Organization (WHO) grade IV (Mukherjee et al., 2016).
GBMs are a deadly form of brain tumor that are highly heterogeneous and include tumor cell clones with divergent genetic alterations and differentiation programs. Glioma stem cells (GSCs) are a small, but biologically critical GBM subpopulation that control growth and differentiation dynamics (Liebelt et al., 2016; Osuka and Van Meir, 2017). GSCs, often identified by their expression of CD133, are resistant to ionizing radiation therapy (Bao et al., 2006) and to temozolomide chemotherapy, the latter at least partially due to high MGMT expression in hypoxic regions (Persano et al., 2012). The ability of GSCs to direct glioma growth and evade therapy depends on their cooption of specific canonical stem cell signaling pathways, including Notch, WNT, and Shh (Clement et al., 2007; Wang et al., 2010), by which they acquire programs of self-renewal, propagation, and therapeutic resistance (Sengupta et al., 2012). A rational strategy to treat GBM would be to therapeutically target pathways on which GSCs depend (Felsher, 2010).
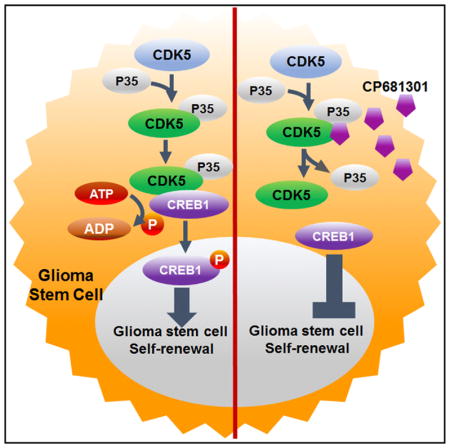
Cyclin-dependent kinase 5 (CDK5) is an unconventional Cdk that regulates developmental and adult neurogenesis, as well as cell survival in post-mitotic neurons (Lagace et al., 2008). In the brain, CDK5 normally remains inactive until it binds with its partners P35 and/or P39 (Shah and Lahiri, 2014). Aberrant CDK5 activity plays a critical role in the growth and propagation of multiple forms of cancers (Pozo et al., 2013; Yushan et al., 2015). CDK5 is highly expressed in GBM (Yushan et al., 2015), possibly due to its location on chromosome 7, which is one of the most frequent sites of copy number gains in primary (isocitrate dehydrogenase [IDH] wild-type) GBM (Ozawa et al., 2014). In this study, we present CDK5 as a promoter of self-renewal through its deregulation of asymmetric cell division in brain tumor stem cells, at least in part through its PKA/cyclic AMP-independent phosphorylation and activation of CREB1. We also show that antagonizing this CDK5-CREB1 pathway suppresses both self-renewal of GSCs and glioma growth.
RESULTS
dCdk5 Is Overexpressed in Drosophila Brain Tumor Stem Cells and Its Reduction Ameliorates Tumor Growth
In previous work, we established that Drosophila Brat and its human ortholog, TRIM3, regulate asymmetric cell division (Chen et al., 2014; Mukherjee et al., 2016). Reduced TRIM3 in GBMs is directly associated with glioma cell division and self-renewal. We also previously established a brat-RNAi model in Drosophila neuroblasts that generates brain tumors in adult flies (Mukherjee et al., 2016) that are composed almost entirely of tumor stem cells, are actively proliferating (Figure S1A), and have disrupted asymmetric cell division properties (Mukherjee et al., 2016). We next used the brat-RNAi system to generate eye tumors in Drosophila, which produce a visible and non-lethal phenotype. Genetic and phenotypic analysis of eye overgrowth phenotypes have been successfully used to delineate oncogenic signaling pathways such as RAS, Hippo, GSK3β, PTEN, and BMP2 (Gonzalez, 2013). To search for kinases that antagonize the tumor stem cell phenotype and that could represent potential therapeutic targets, we performed a genetic screen where we crossed RNAi lines for specific kinase genes with ey-GAL4 > brat-RNAi stock and determined the extent of reduction in the brat-RNAi eye tumor in the progeny. In the initial screen of 29 genes (Table S1), we identified Drosophila Cdk5 (dCdk5) (Figure 1A, v) and its activation partner, dP35 (Figure 1A, iv) as potential candidates, because their suppression by RNAi completely reversed the Drosophila brat-RNAi-mediated eye tumor phenotype (Figure 1A, ii). It is noteworthy that dCdk5 and dP35 knockdown in normal eye, in the absence of brat-RNAi, does not result in a phenotype (not shown). From these results, we hypothesized that dCdk5 may act as an oncogene that promotes CNS tumor growth. To investigate this further, we examined dCdk5 protein expression and distribution in brat-RNAi-mediated adult brain tumors, which are characterized by dominant population of stem cells that express the marker Mira (Miranda). We found that this brain tumor stem cell population strongly and specifically express activated phospho (Y15) dCdk5 (Lin et al., 2007) (Figure 1C, i and ii), while other cells did not. We next asked whether the suppression of dCdk5 or its partner, dP35, could reverse the brat-RNAi brain tumor phenotype that is caused by disrupted asymmetric cell division. Indeed, dp35-RNAi and dcdk5-RNAi both markedly reduced the proliferation of tumor stem cells (phosphorylated histone 3 [PH3], in white) (Figures 1C, iv and v, and S1B) and the number of cells expressing the stem cell marker Mira (in green) (Figures 1C, vii and viii, and S1C). The tumor volume itself was considerably reduced (Figure 1C, vii and viii), although the rescues were incomplete. These experiments established that dCdk5 and dP35 regulate cell proliferation and self-renewal properties of Drosophila brain tumor stem cells.
Figure 1. CDK5 Regulates Self-Renewal and Asymmetric Cell Division in Drosophila Neuroblasts.

(A) Drosophila eye screen. (i) Drosophila normal eye. (ii) brat-RNAi using eyeless-GAL4 drives overgrowth of the eye. Using this phenotype, we performed a suppressor screen for kinases and found that reducing dcdk5 (v) and its activating partner, dp35 (iv), can reverse the overgrowth successfully. Suppression of two other kinases, dS6k (iii) and dEip63E (vi), did not revert the overgrowth completely. Graphs of the suppressor screen experiments show statistical significance.
(B) Diagrams represent Drosophila P35 and Cdk5 are identical to human P35 (CDK5R1) and human CDK5 by 66% and 79%, respectively.
(C) brat-RNAi adult brain tumor shows high Mira expression (green) (i), indicating deregulated asymmetric division that generated these tumor stem cells. These cells also express high levels of active dCdk5 (white) (ii) compared with normal cells. RNAi suppression of dP35 and dCdk5 in brat-RNAi tumors reduced cell proliferation (PH3 staining, white) (iii, iv, and v) and the self-renewal marker (Mira, green) (vi, vii, and viii). Statistics are in Figure S1.
(D) We tested the potency of Cdk5 as an asymmetric cell division regulator by knocking down dCdk5 and by overexpressing human CDK5 specifically in neuroblasts. During asymmetric division, larval neuroblast mother cells (Mira positive, green) generate progenitors that are Mira negative and eventually differentiate (i, ii, and iii). Knock down of dCdk5 using RNAi specifically in neuroblasts reduced Mira expression without any visible change in daughter cells (red) (iv, v, and vi). However, ectopic expression of hCDK5 in neuroblasts disrupted the asymmetric division, generating more Mira-positive cells (vii, viii, and ix). Statistics are in Figure S1.
(E) CP681301, a specific CDK5 inhibitor, was fed to adult flies with brat-RNAi tumors. After 10 days of feeding, adult tumors lost expression of stem cell markers (Mira, green) (i and ii) and active phosphorylated dCdk5 expression (white) (iii and iv).
(F) Suppressing dCdk5 activity with CP681301 increased the median survival of the brat-RNAi tumor-carrying flies by 2.85-fold. p < 0.0001.
CDK5 Overexpression Deregulates Asymmetric Cell Division of Drosophila Neuroblasts
Drosophila neuroblasts normally divide asymmetrically, where after division one daughter cell keeping its self-renewal properties and expressing Mira and the other Mira-negative daughter cell losing the ability to self-renew (Figure 1D, i). Because suppression of dCdk5 had the capacity to suppress brat-RNAi tumor stem cell proliferation (Figure 1C, v), we hypothesized that the CDK5 signaling pathway might also regulate asymmetric cell division. To test this, we first suppressed dCdk5 using the RNAi system in neuroblasts with the inscuteable-GAL4 driver. We observed more than 50% reduction of the stem cell marker Mira (Figures 1D, iv, and S1G) and a high expression of neuronal cell marker ElaV in these cells. Drosophila neuroblasts normally do not express ElaV. However, dCdk5 suppression in these cells using two different RNAi lines (Bloomington Stock Center), showed neuronal differentiation (Figures S2A and S2B). We then investigated if increased CDK5 signaling deregulates the neuroblast self-renewal process by taking advantage of the human CDK5-overexpressing transgenic fly line (Zografos et al., 2016). The CDK5 protein sequence has 79% identity to dCdk5, and the human CDK5 activator protein P35 (CDK5R1) has 66% identity with dP35 (Figure 1B). Overexpressed CDK5 in fly neuroblasts greatly disrupted self-renewal. We saw a large accumulation of neuroblasts, as confirmed by the aberrant expression of Mira (Figure 1D, vii), which is typically lost following asymmetric division, when daughter cells generate differentiated neurons or glia. We also observed significant loss of ElaV, which most likely indicates that neuroblast proliferation is occurring at the expense of their differentiation (Figures S2A and S2B). Thus, our data suggest that elevated levels of CDK5 can deregulate the asymmetric division process, shifting toward more neuroblast generation.
We next tested whether a CDK5 inhibitor could suppress the self-renewal properties of Drosophila brat-RNAi brain tumor stem cells. We collected 0- to 2-day-old adult Drosophila harboring brat-RNAi tumors and fed them a highly specific CDK5 inhibitor (CP681301, 1 mM; Pfizer). We observed that CP681301 reduced active phospho-dCdk5 (Y15) (white) in tumor cells (Figures 1E, iii and iv, and S1E) after 10 days of feeding and reduced the self-renewal properties of stem cells as shown by the absence of Mira (green) (Figures 1E, i and ii, and S1D). CP681301 also increased the expression of neuronal marker ElaV in these cells, showing that in flies, dCdk5 inhibition causes differentiation (Figure S1F). Finally, we measured the lifespan of flies with tumors and found that CP681301 increased the median survival by 2.8-fold, although the tumors remained fatal (Figure 1F). Together, these data indicate that CDK5 acts as an oncogene in fly tumors by deregulating asymmetric division and promoting stemness, and that CDK5 inhibition can ameliorate the self-renewal capacity of tumor stem cells in flies. Our data also showed that Drosophila and human CDK5 proteins are highly conserved, even though the organisms are taxonomically distant.
CDK5 Is Overexpressed in GBMs and CDK5 Knockdown Reduces the Expression of Self-Renewal Genes in GSCs
The substantial homology between Drosophila and human CDK5, as well as the evidence that dCdk5 regulates self-renewal in Drosophila brain tumor stem cells and neuroblasts, led us to investigate CDK5 for parallel functions in patient-derived GBMs. Our analysis of the complete lower grade glioma (LGG) and GBM data from The Cancer Genomic Atlas (TCGA) revealed that CDK5 is frequently amplified in GBM (83%) and to a greater degree than in LGGs (15%) (Figure 2A). Moreover, CDK5 mRNA expression is significantly higher in GBMs than in LGGs (Figure S3A). LGGs and GBMs are now classified based on the mutation status of isocitrate dehydrogenase 1/2 (IDH1/2). The large majority of GBMs are wild-type for IDH, whereas most LGGs are IDH mutants. We found that CDK5 mRNA expression was significantly higher in only the GBMs (Figure 2B) compared to the IDH wild-type LGGs, but similar among IDH mutant GBMs and LGGs cases (Figure S3B). Some of these differences in CDK5 expression are likely due to frequent copy number gains of chromosome 7 in IDH wild-type GBMs, but not in IDH mutant GBMs, in agreement with previous findings that CDK5 is one of the few amplified genes on chromosome 7 that correlates with high mRNA expression (Ozawa et al., 2014).
Figure 2. High CDK5 mRNA and Protein Expression and Close Association with Stem Cell Markers in GBM.

(A) Most (82.8%) GBMs have high CDK5 gene copy number compared with 14.6% in LGGs.
(B) Comparing IDH1 wild-type LGGs and GBMs, we found that GBMs have high CDK5 mRNA expression.
(C) Immunohistological staining of LGG and GBM tumor tissues shows high CDK5 protein expression, correlating with mRNA data.
(D) We further investigated four patient-derived GBM neurosphere lines and found that they all express high levels of CDK5 and p35/25 proteins compared to the control NHNP cells. These neurosphere lines also express high levels of OLIG2 and SOX2, two important stem cell markers.
(E) Examination of TCGA RNA-seq RSEM data using similarity matrix analysis revealed that mRNA expressions of CDK5 and CDK5R1 (p35) positively correlate with stem cell and asymmetric cell division markers and negatively correlate with differentiation markers.
(F) To see if our data can reflect the TCGA data, we used FACS to isolate GSCs based on CD133 expression. We found that total CDK5 and activated phosphorylated CDK5 (pCDK5-Y15) expression is high in GSCs.
(G) To see whether CDK5 can regulate self-renewal markers in GSCs, we knocked down CDK5 using CDK5 siRNA and found that stem cell markers such as OLIG2 and SOX2 downregulated. **p < 0.01, ****p < 0.0001.
To determine if a high level of CDK5 mRNA corresponds to elevated CDK5 protein expression, we first performed immunohistochemistry on institutional GBM and LGG tissue samples and found that CDK5 was highly expressed in GBMs compared to LGGs (Figure 2C). We next examined CDK5 and hP35 (CDK5R1) protein expression in cultured, patient-derived GBM cells grown as neurospheres and found that they were both expressed at higher levels than normal human neural progenitor cells (NHNPs) (Lonza) (Figure 2D). GBM neurosphere lines also highly expressed the self-renewal proteins SOX2 and OLIG2 (Suvà et al., 2014; Trépant et al., 2015) (Figure 2D).
Based on our finding in Drosophila neuroblasts and brain tumor stem cells (Figures 1 and S1A), we hypothesized that CDK5 may directly regulate GSC properties promoting growth and maintenance of GBMs. To test this, we first established in a similarity matrix analysis that CDK5 and CDK5R1 expressions are correlated with the expression of self-renewal SOX2, OLIG2 (Suvà et al., 2014; Trépant et al., 2015), PROM1 (CD133) genes, and apically localized asymmetric cell division markers, NOTCH1, PARD6A, PROM1 (CD133), and GPSM2 (LGN) genes in GBM samples from the TCGA RNA sequencing (RNA-seq) and the U133 microarray data. We also showed that CDK5 and CDK5R1 are negatively correlated with the differentiation markers GFAP, GALC, APOE, and basally localized asymmetric cell division marker NUMB (Figures 2E and S3C). In particular, correlations between CDK5 and SOX2 (p < 0.0001) and OLIG2 (p < 0.0001) were extremely strong (Figure S3D). To determine if these TCGA results obtained from bulk tumor samples were also valid in a pure GSC population, we separated GSCs and non-GSCs from two patient-derived neurosphere lines (GBM121 [no EGFR amplification] and GBM39 [EGFRvIII]), based on CD133 and CDK5 expression. Although GSCs did not show a higher level of CDK5 protein than non-GSCs (Figures 2F, S3E, and S3F), they had greater expression of active phos-pho-CDK5 protein (Y15) (Figures 2F, S3E, and S3F) and self-renewal markers (CD133 and OLIG2) (Figure 2F). To address whether CDK5 can regulate self-renewal markers in GSCs, we knocked down CDK5 using CDK5 small interfering RNA (siRNA), which reduced both SOX2 and OLIG2 (Figure 2G). These data suggest that CDK5 could be a regulator of self-renewal properties of GBMs, much like dCdk5 in Drosophila neuroblasts.
CDK5 Inhibitor CP681301 Inhibits GSC Self-Renewal
Based on these results, we hypothesized that CDK5 is fundamental for the self-renewal properties of GSCs and tested two pharmacologic CDK5 inhibitors, including Roscovitine and the recently developed compound CP681301, for their effects on viability and stem cell properties. First we tested Roscovitine, a purine derivative well-known CDK5 inhibitor, on neurosphere lines and the MTT assay revealed that it is cytotoxic at 20 μM concentration (Figure S3G). Roscovitine also suppressed the expression of stem cell markers at earlier time points (Figure S3H). Several studies, however, have shown that Roscovitine can also inhibit CDK1, CDK2, CDK7, and CDK9 in addition to CDK5 (Yin et al., 2015). CP681301, on the other hand, has been developed as a CDK5 inhibitor with much greater specificity (Pozo et al., 2013). Activation of CDK5 depends on its binding with P35, a 35 kDa molecular weight protein. The CDK5-P35 complex is largely distributed in the perinuclear region and at the plasma membrane (Shah and Lahiri, 2014). Studies have shown that CDK5-P35 binding is unstable and that P35 can be cleaved by calpain into a smaller fragment called P25, which results in a more stable CDK5-P25 complex that localizes to both the cytosol and nucleus (Kusakawa et al., 2000). Both CDK5-P35 and CDK5-P25 show kinase activity (Shah and Lahiri, 2014). To determine if the CDK5 inhibitor CP681301 can suppress the kinase activity of both of these complexes, we tested the drug in a range of concentrations starting from 0.1 nM to 1 μM. Our results showed that CP681301 completely abolished activities of both CDK5-P35 and CDK5-P25 at a concentration of 1 μM (Figure 3A).
Figure 3. CDK5 Regulates Self-Renewal Properties in GSCs.

(A) To determine if CDK5 regulates self-renewal of GSCs, first we tested the potency of a highly specific CDK5 inhibitor called CP681301 from Pfizer. Using active CDK5-p35 and CDK5-p25 as enzymes, we established a dose-response curve for CP681301 in order to find the doses that successfully inhibit kinase catalytic activity of both protein complexes in vitro. 1 μM CP681301 was the optimal concentration that suppressed 100% of CDK5-p35 and CDK5-p25 catalytic activity.
(B) CP681301 (1 μM) for 96 hr is significantly less toxic to NHNPs (control) than GSCs. *, <0.05, ***, <0.001, ****, <0.0001.
(C) CP681301 treatment for 48 hr suppressed active CDK5 and reduced stem cell markers CD133, OLIG2, and SOX2 and cell proliferation marker KI67 in GSCs but did not change differentiation markers (GFAP).
(D and E) Using a limiting dilution assay (D) and visual demonstration (E), we established that CP681301 also reduces the self-renewal properties of GSCs.
(F) Immunocytochemistry showed that GSCs from two neurosphere lines have reduced levels of active CDK5 and stem cell markers SOX2 and OLIG2 (Figure S6).
Next, we tested the cytotoxic effect of CP681301 on GBM neurosphere cultures and NHNPs (normal human neuro-progenitors) in order to determine if there was differential toxicity among neoplastic and non-neoplastic cells. We found that 1 μM was variably cytotoxic to the tested GSC cultures but was not toxic to NHNPs at 96 hr (Figures 3B and S3I). We also found that treatment of GSCs with CP681301 for 48 hr moderately suppressed the expression of CDK5 total protein but had a more marked suppressive effect on the tyrosine-15 (Y15) phosphorylation status, which is associated with CDK5 kinase activation (Figure 3C) (Zukerberg et al., 2000). As mentioned, CP681301 is more specific to CDK5 than its closest relative CDK2, whose phosphorylation and activation is not affected in these cells upon 1 μM CP681301 treatment (Figure S4A). Interestingly, the inactivation of CDK5 by CP681301 was also associated with a reduction of the stem cell markers CD133, OLIG2, and SOX2 (Figure 3C) and the cell proliferation marker Ki67 (Figure 3C), but did not change GFAP and TUJ1 expression upon differentiation conditions (Figures 3C, S4B, and S4C). However, we have not completely ruled out that other lines of differentiation, such as oligodendrocytes, may occur.
To examine the effects of CDK5 inactivation by CP681301 on GSC self-renewal function, we tested the neurosphere formation using the extreme limiting dilution assay (ELDA) (http://bioinf.wehi.edu.au/software/elda/) (Hu and Smyth, 2009) and found that sphere formation rate is dramatically suppressed within 72 hr (Figure 3D). We performed the ELDA on GSCs after treating them with CDK5 siRNA for 72 hr. The results also show significant reduction in self-renewal under low CDK5 (Figure S5E). We also performed a visual primary sphere formation experiment in the absence and presence of CP681301 for 96 hr and found that even at early time points, the rate of sphere formation drops down from 77.87% to 52.31% in GBM121 GSCs and from 77.93% to 53.88% in GBM39 GSCs (Figure 3E). In these same cell lines with reduced sphere formation, we also showed that drug treatment was associated with reduced CDK5 phosphorylation, as well as OLIG2 and SOX2 expression (Figures 3F and S5A–S5C).
CP681301 Reduces Cell Proliferation in Xenograft Tumors but Not Normal Brain Tissue
After observing the effect of CP681301 on GBM121 and GBM39 GSCs in vitro, we generated xenograft orthotopic tumors in NSG mice. We orthotopically injected 1 × 105 GSCs from GBM121 and GBM39 into mouse brains and allowed them to grow for 2 months, at which time they measured from 0.3–0.4 mm (Figure 4A). We then removed the brains and established ex vivo or-ganotypic explant slice cultures (300-μm) to directly observe glioma growth dynamics and the expression of stem cell markers in the presence of 0, 10, or 50 μM CP681301. After 48 hr, CP681301 reduced the phosphorylation of CDK5 as well as the expression of the proliferation marker Ki67 (Figure 4B). Western blot data also showed reduced expression of the stem cell markers CD133, SOX2, and OLIG2 (Figures 4C and S5H–S5J). We also tested the contralateral brain hemispheres (without neoplastic cells) and found that CP681301 did not affect the viability of normal tissue or affect cell self-renewal markers or active CDK5 status (Figure S5G). On H&E and immunohistochemical stains, we did not see evidence of neurodegeneration, cell proliferation (Ki67) or increased expression of phospho-CDK5 and phos-pho-CREB1 (not shown). Together, these data indicate that CP681301 reduces CDK5 activation, stem cell marker expression, and cell proliferation in mouse xenografts ex vivo.
Figure 4. CDK5 Inhibitor Suppresses Self-Renewal and Proliferation of GBM Xenograft Tumors in Explant Cultures.

(A) Diagram shows the mice xenograft tumor implantation and explant culture flow chart. Two months after implantation, GBM121 and GBM39 both produce considerably large tumors on the right side of the brains.
(B) Tumor sections (300 μm) were cultured on Petri dishes for 48 hr with 0, 10, and 50 μM CP681301. Immunohistochemical analysis shows significant reduction of active CDK5 and Ki67 staining in the tumor in cultures sections treated with CP681301 relative to control.
(C) Western blots performed on protein lysates created from xenografted tumors show downregulation of stem cell markers and confirm suppression of CDK5 phosphorylation through ABL and cell proliferation (Ki67).
(D) Investigating putative downstream targets of CDK5, we found that phosphorylated (p)-RB (inactive RB), p-MYC (active MYC), and p-CREB1 are all suppressed by CP681301 treatment.
(E) Among these, the expression of CREB1 and CDK5 are strongly correlated in TCGA U133 GBM data.
(F) Additionally, TCGA RNA-seq RSEM data showed that CREB1 is positively correlated with iTPC transcription factors SOX2, OLIG2, SALL2, and POU3F2.
(G) Analysis of TCGA RNA-seq RSEM data also showed that CREB1 is strongly correlated with all four of these transcription factors, whereas MYC is only correlated with two of these.
(H) Immunocytological data also supports that CDK5 inhibition reduces CREB1 phosphorylation. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
CDK5 Activates the Self-Renewal Regulator CREB1 in GSCs
To determine the downstream pathways by which CDK5 regulates the self-renewal properties of GSCs, we examined the expression and activation of established self-renewal and proliferation-related oncogenic proteins, retinoblastoma (RB), c-MYC, and cAMP response element binding protein 1 (CREB1), in presence and absence of CP681301. We found that the phosphorylation of RB (Ser807/811) and CREB1 (Ser133) (Figures S6A and S6B) were reduced following CDK inhibition (Figure 4D). Activation of CREB occurs by Serine 133 phosphorylation (Mayr and Montminy, 2001) and our western blot and immunocytochemistry data (Figures 4H and S5D) indicates CDK5 activates CREB1 in this manner. To determine which of these CDK5 targets were most likely to regulate self-renewal properties in GBMs, we examined the correlation between RB, c-MYC, and CREB1 with four induced tumor-propagating cell (iTPC) transcription factors, SOX2, OLIG2, SALL2, and POU3F2 (Suvà et al., 2014), using RNA-seq data from TCGA. Among these, we found that CREB1 correlated most strongly with all four transcription factors, compared to c-MYC and RB (Figure 4G). Additionally, CDK5 also correlated with CREB1 in U133 microarray data (p < 0.001) (Figure 4E). Our similarity matrix analysis of TCGA RNA-seq data revealed that CREB1 is directly correlated with all iTPC transcription factors but is negatively correlated with differentiation markers (Figure 4F). We further examined the promoters of these iTPC transcription factors using Patch 1.0 transcription factor binding predictor software and found that the predicted promoter regions (1,000 bases upstream of the coding region) of all four factors found from Eukaryotic Promoter Database of Swiss Institute of Bioinformatics contain at least one full CRE motif for CREB1 binding, whereas only OLIG2, SOX2, and SALL2 promoters have c-MYC binding sites (Figure S6E). We also used JASPER transcription factor motif predictor and found that all these promoter regions have multiple CREB1 binding sites (Table S2). Finally, we verified our analysis using CREB Target Gene Database from Salk and using Human CRE prediction portal (http://natural.salk.edu/CREB/). We found that three out of four transcription factors (OLIG2, SOX2, and POU3F2) have cAMP response element (CRE) sites (Zhang et al., 2005). In addition to activating CREB, we also found that pharmacological suppression of CDK5 could downregulate the expression of potential CREB1 target genes, such as SOX2 and OLIG2 (Figure 4C).
We next tested CP681301 for its effects on CREB1 in ex vivo xenograft organotypic cultures by immunohistochemistry and western blotting and found that CREB1 phosphorylation was substantially suppressed after treatment (Figures 5A and 5B). Finally, in order to find the specific effect of CREB1 on GSCs self-renewal programs, we knocked down CREB1 using siRNA, which resulted in the reduction of self-renewal behavior measured by ELDA (Figure S5F) and expression of critical self-renewal transcription factors, including OLIG2, POU3F2, and SALL2, indicating that CREB1 indeed regulates stem cell programs in GSCs (Figure S6C). To determine how CDK5 regulates CREB1, we performed a co-immunoprecipitation (coIP) assay and found that CREB1 pulled down CDK5 (Figure 5C). This is a step forward on the existing knowledge, because previous studies have suggested that signal transduction intermediaries, such as protein kinase A (PKA), mediate activation of CREB by CDKs in neurons in dopaminergic neurotransmission (Zhong et al., 2014). To determine whether CDK5 can form complex with and can phosphorylate CREB1 in the absence of cAMP and PKA, we performed an in vitro kinase assay using only recombinant active CDK5-P35 and CDK5-P25 with recombinant CREB1 as a substrate. The results clearly show that both CDK5-P35 and CDK5-P25 phosphorylated CREB1 (Figure 5D). We used Tau, an established substrate of CDK5, as a control (Figure S6D). We also established that CP681301 (0.5 μM and 1 μM) was capable of suppressing CREB Ser133 phosphorylation by CDK5 (Figure 5D), similar to in situ GSC data.
Figure 5. CDK5 Directly Binds to and Phos-phorylates to Activate CREB1 in GSCs.

(A) CP681301 treatment of mice xenografted explant culture tumors induced a clear down-regulation of CREB1 phosphorylation.
(B) Western blot data using these tumor tissues also show downregulation of CREB1 phosphorylation.
(C) We then investigated the mechanism by which CDK5 regulates CREB1. We found that Cdk5 can directly bind with CREB1, and CP681301 reduces the binding of CDK5 to CREB1.
(D) An in vitro kinase assay showed that CDK5 can phosphorylate CREB1 at serine 133, and CP681301 can suppress this phosphorylation in a dose-dependent manner.
(E) Diagram showing our proposed model where CDK5 directly phosphorylates and activates CREB1 without the requirement of cAMP/PKA in GSCs.
CDK5 Expression Segregates with Non-mesenchymal GBMs
The finding that CDK5 may regulate self-renewal properties in GSCs by directly phosphorylating CREB independently of PKA/cAMP opens further avenues of inquiry, with potential clinical significance. To determine relevance to specific molecular subtypes of GBMs, we performed an unsupervised clustering analysis on data available from TCGA related to the self-renewal and asymmetric cell division markers, as well as CDK5, CDK5R1, and CREB1. Using most recent classification of GBM including neural, classic, and mesenchymal subtypes (Wang et al., 2018), our analysis of TCGA RNA-seq RSEM data from 129 patients revealed an interesting dichotomy, in which the majority of non-mesenchymal tumors (75.64%) were associated with high expression of CDK5, CDK5R1, CREB1, and self-renewal and asymmetric cell division markers, whereas the majority of the mesenchymal GBM subtypes (80.39%) showed high expression of differentiation markers (Figures 6A and 6B). These data suggest an enhanced stem cell signature in non-mesenchymal forms of GBM in which is self-renewal makers are expressed to a greater extent than in the mesenchymal subtype.
Figure 6. Expression Profile of CDK5 and Stem Cell Genes Segregate in Non-mesen-chymal GBMs.

(A) Using TCGA RNA-seq RSEM data, we examined whether the expression of CDK5, CDKR1, and CREB1, along with the self-renewal and asymmetric cell division marker genes, have a clinical significance. Unsupervised hierarchical clustering analysis indeed show that a majority of the non-mesenchymal GBMs (cluster 1) express high levels of these genes, whereas mesenchymal GBMs have high expression of differentiation markers (cluster 2).
(B) Our detailed analysis showed that 62.96% of non-mesenchymal tumors fall in cluster 1 (p < 0.001), whereas ~83.33% of mesenchymal tumors fall in cluster 2 (p < 0.001).
DISCUSSION
The roles of CDK5 in neurogenesis and in support of post-mitotic neuronal survival have been established for more than two decades, yet its potential influence on stem cell regulation is less clear (Hellmich et al., 1992; Smith-Trunova et al., 2015). In a previous study, when floxed cdk5 was excised by estrogen-induced nestin-driven Cre specifically in mice adult hippocampal subgranular neural stem cells, these cells failed to grow, suggesting that CDK5 has a fundamental role in regulating stem cell division and survival in the brain (Lagace et al., 2008). Considering the diverse targets of CDK5, such as RB, MYC, as well as Drp1 (Xie et al., 2015), CDK5 is likely to be one part of this complex self-renewal signaling network. Our current findings indicate that CDK5 is more deeply involved in regulating self-renewal properties by directly regulating CREB pathway and its downstream signaling network in GSCs.
Our investigation using Drosophila brain tumor stem cells and human GSCs establishes a direct role of CDK5 signaling in the regulation of self-renewal properties and asymmetric division. Asymmetric cell division is a central property of neural stem cells and the canonical pathways that control this delicate process in Drosophila neuroblasts have been well established (Egger et al., 2011; Knoblich, 2010). However, the impact of CDK5 on these pathways has not been explored. Our results showed that dCdk5 and human CDK5 could promote fly brain tumor stem cells and GSCs generated from deregulated asymmetric cell division, clearly indicating that CDK5 regulates self-renewing division in these populations.
In evaluating how CDK5 maintains the self-renewal properties that promote the proliferation and growth of GSCs, we found that CDK5 directly binds to and activates CREB1, a transcription factor with established significance in many cancer types, including breast cancer, leukemias and lymphomas, colorectal cancer, and glioma (Daniel et al., 2014), also reviewed in the Human Protein Atlas (Uhlén et al., 2005). Our analysis of TCGA data also shows that CREB1 correlates with the tumor-propagating cell transcription factors SOX2, OLIG2, SALL2, and POU3F2, each of which contains CREB1 binding sites in their promoters. Phosphorylation and activation of CREB1 has previously been shown to be dependent on cAMP (hence the name, cAMP response element binding protein) and PKA (Gonzalez and Montminy, 1989). cAMP levels regulate a variety of signaling pathways, including as PKA, p38, and MEK/ERK (Arthur and Cohen, 2000; Fujita et al., 2002; Grewal et al., 2000; Wiggin et al., 2002; Xing et al., 1996), which, in turn, control CREB1 activation. Our data indicate that CDK5, which has previously been shown to activate CREB1 through cAMP/PKA in dopamine neurons present in the ventral tegmental area (Zhong et al., 2014), can directly bind with and phosphorylate CREB1 in a PKA/cAMP-independent manner, at least in GSCs.
In gliomas, CREB regulates cell proliferation through (1) activating cyclin D1, cyclin B1, and PCNA, and residing downstream of ERK and AKT pathways (Sampurno et al., 2013) and (2) EGFR and PDGFR signaling (Mantamadiotis et al., 2012). Cell proliferation through CREB phosphorylation can also be induced by the absence of PTEN in a PI3K/AKT independent fassion (Gu et al., 2011). Our analysis of TCGA patient data shows that CREB is highly expressed in GBMs and positively correlated with CDK5 expression, supporting our finding that they are within the same pathway. Nuclear activated phospho-CREB seen by immunohistochemistry, a strong correlation between CREB and iTPC transcription factors and the suppression of stem cell markers with CREB downregulation support the notion that CREB regulates self-renewal properties in GSCs. CREB regulates proliferation of neural stem cells, both in the developing brain across taxa (Mantamadiotis et al., 2012) and in the neurogenic zones of the adult vertebrate brain (Mantamadiotis et al., 2012). Given the fact that CREB activation is essential in initiating stem cell proliferation, it is not surprising that GSCs coopt this signaling pathway for enhancing self-renewing division. Thus, suppression of CREB can pacify the growth and proliferation of these cells, corroborating previous findings (Mantamadiotis et al., 2012).
GBMs and LGGs have now been well characterized and molecularly classified (Brat et al., 2015; Ceccarelli et al., 2016). Our analysis of TCGA data confirms higher CDK5 expression in GBM than in LGG (using the combined IDH wild-type and mutant cases), which likely corresponds to frequent copy number gains on chromosome 7 in GBM (Ozawa et al., 2014). Both IDH wild-type LGG and GBM show frequent gains of chromosome 7, but because CDK5 mRNA expression is more highly expressed in GBMs, other factors critical to aggressive behavior, such as the development of hypoxia and necrosis, may cause further upregulation during disease progression. The associations of CDK5 with stem cell and asymmetric cell division markers in GBM samples support our mechanistic findings in Drosophila and mouse models, as does the interesting finding of strong negative correlations with differentiation markers. Finally, an unsupervised analysis of the GBM gene expression data showed that the CDK5 signaling pathway is highly enriched in non-mesenchymal GBMs, suggesting that these mechanisms are likely more active in specific subsets that could be sensitive to targeted therapies.
In our current study, we found that CDK5 plays a substantial role in the regulation of GSCs, adding to the findings of previous studies that have suggested that CDK5 is important for the growth of normal neural progenitors (Lagace et al., 2008). Using CDK5 knock down experiments and a specific CDK5 inhibitor CP681301, we established that GSCs are more sensitive to the suppression of this pathway than normal neural stem cells, supporting the conclusion that GSCs are more dependent on the activation of stem cell programs directed by CDK5 through CREB. This is an important finding, as targeting CDK5 signaling to treat GBMs could potentially eliminate CDK5-addicted GSCs and reduce the likelihood of recurrence.
EXPERIMENTAL PROCEDURES
All work with human cell lines as well as animal models was done at Emory University. The protocols to use human cells in laboratories were approved by Emory IRB and the use of animal models for the experiments was approved by Emory Institutional Animal Care and Use Committee (IACUC).
Drosophila Stocks and Genetic Cross
All Drosophila stocks were maintained at 25°C unless otherwise mentioned. Inscuteable-GAL4, UAS-hCDK5, UAS-bratRNAi, eyeless-GAL4, UAS-p35RNAi, UAS-cdk5RNAi, UAS-s6kaRNAi, UAS-Eip63ERNAi, and 26 other RNAi stocks (Table S1) were obtained from Bloomington Drosophila Stock Center.
eyeless-GAL4; UAS-brat-RNAi viable stock carried eye tumor. Flies from this stock were crossed with the RNAi lines tested. Eye tumor sizes were scored between −4 and +4, where 0 was considered as normal eye. Mann-Whitney U test was performed to find significance in the degree of rescue for each gene. Approximately 50 flies were checked per gene.
p35 and cdk5 RNAi stocks were used to knockdown these gene’s expressions in brat-RNAi brain tumor to see the rescue. At least 25 flies were tested.
UAS-Cdk5RNAi and UAS-hCDK5 stocks were used to test the effect of CDK5 downregulation and upregulation in Drosophila neuroblasts. 3rd instar Larvae were dissected and stained to check the changes in neuroblasts.
Drosophila Adult Brain Dissection and Staining
Adult Drosophila brains were dissected out in 1 × PBS by carefully removing the cuticle surrounding the brain. The brain was fixed with 4% paraformaldehyde in 1 × PBS for 90 min by end over end mixing. The fixed brains were washed with 0.5% Triton X-100 in 1 × PBS for 30 min to 1 hr. Brains were then incubated for 2 days at 4°C in primary antibody in 0.5% Triton X-100 in 1 × PBS with 10% BSA (antibody dilution buffer). After primary antibody incubation, brains were washed with 0.5% Triton X-100 in 1 × PBS 3 times, 15 min each. The secondary antibody was also diluted in antibody dilution buffer and incubated overnight at 4°C. Finally, the brains were washed 3 times and added to Vectashield Dapi (H-1200) (Vector Laboratories) mounting media and kept for 2 days in dark at 4°C. The brains were finally mounted using Vectashield Dapi (H-1200) mounting media. Primary antibodies are: Mira (Miranda) (1:200) (gift from Dr. Chengyu Lee) and phospho-Cdk5 (1:200) (sc-12918-R) (Santa Cruz). Pictures were taken using Olympus FV-1000 confocal microscope.
Drosophila Larval Brain Dissection and Staining
3rd instar larval brains were dissected out in 1 × PBS, fixed in 4% paraformaldehyde. Staining was performed following the standard protocol by Daul et al. (2010). Brains were finally mounted using Vectashield Dapi (H-1200) (Vector Laboratories) mounting media. The primary antibody was Mira (1:200). Pictures were taken using Olympus FV-1000 confocal microscope.
Drosophila Drug Feeding and Lifespan Study
Drosophila adult flies between 0–2 days old were fed with the CDK5 inhibitor CP681301 at 1 mM dilution in 100% ethanol spread on top of the food. Flies were kept at 18°C, and food was changed every other day. We keep the flies at 18°C to ensure that they get enough drug in their system before they perish due to the tumor. The control foods carry the vehicle, 100% ethanol.
TCGA Data Analysis
DNA copy number alteration (CNA), RNA seq, and U133 microarray TCGA data were obtained from cBioPortal and Ceccarelli et al. (2016). Graph pad Prism V6.0 was used to analyze the data and t test was performed to find out the significance of the data. IDH1 WT and IDH1 mut GBM (glioblastoma) and LGG (lower grade glioma) patients were selected manually.
Similarity Matrix Analysis
GBM RNA-seq and U133 microarray data from TCGA is used to generate similarity matrix. Spearman correlation distance analysis was applied using Morpheus online software from Broad Institute.
GBM Stem Cell Isolation
GBM39 (EGFR vIII-positive) and GBM121 (PDGFRA overexpression, no EGFR amplification) neurospheres were separated using Accutase, stained with PE-tagged CD133 antibody (Miltenyi Biotech) and CD133+ and CD133− cells were sorted through fluorescence-activated cell sorting (FACS) Aria 2.0 cell sorter.
Histology
Human GBM tissues and xenografted human tumor organotypic cultures were subjected to 10% formalin fixation and paraffin embedded sections were immune-stained. Mounted slides were observed using 20× magnification, and pictures were taken using Luminera camera. Primary antibodies used: CDK5 (1:200) (Santa Cruz), p-CDK5 (1:200) (Santa Cruz), p-CREB1 (1:100) (Cell Signaling Tech), and KI67 (1:200) (Abcam).
Western Blot
Standard western blot protocol was used to test different markers in GSCs and in co-immunoprecipitation. Proteins were isolated from GSCs and 10% SDS gels. Specific protein bands were visualized using chemiluminescent dye West Pico. Primary antibodies used were CDK5 (1:1,000), p-CDK5 (Y15) (1:1,000), OLIG2 (1:1,000) (Santa Cruz), CREB (1:1,000), p-CREB (1:1,000), SOX2 (1:1,000), (Cell Signaling Tech), KI67 (1:2,000) (Abcam), and Tubulin (1:10,000) (Developmental Studies Hybridoma Bank).
Co-immunoprecipitation
For co-immunoprecipitation experiments, CP681301 untreated and treated GSCs were collected and were fixed with 1% paraformaldehyde and lysed using NP-40. The paraformaldehyde fixation cross-linked the proteins for co-immunoprecipitation experiments. Co-immunoprecipitation was performed using protein G Dynabeads (Thermo-Fisher). Finally, eluted lysate was heated at 70°C for 30 min to reverse the cross-link and to release the proteins from their respective complexes. For the IP, we used CREB antibody and for detection, we used CREB, CDK5, and p35/25 antibodies.
In Vitro CP681301 Efficiency Test
Thermo Fisher Scientific tested the inhibitory efficiency of CP681301 on active CDK5/P25 and CDK5/P35 in vitro. The Z′-LITE Kit was used to perform the assay in a 384-well assay plate.
CDK5 Inhibitor Tests on GSCs
Two CDK5 inhibitors were tested on GBM121 and GBM39 GSCs. The traditional Roscovitine was used at 20-μM concentration and Pfizer drug CP681301 was used at 1-μM concentration and found to be effective. The stock solutions were made in DMSO. The working solutions were made diluting them in media. Cells were incubated for 48 hr in media with the inhibitors or with the DMSO only, and the results were obtained.
Extreme Limiting Dilution Assay
To perform extreme limiting dilution assay (ELDA), we seeded GSCs at 1, 20, 40, 60, 80, and 100 cells per well of 96-well plates. We used at least 24 wells for each of the limiting dilution of GSCs. After 24 hr of plating the cells, we added DMSO (vehicle) and CP681301 (CDK5 inhibitor). We counted the number of wells with spheres at the end of the experiment. We performed Chi-square statistical analysis using the following website: http://bioinf.wehi.edu.au/software/elda/. On the x axis, the number of cell dilutions are plotted. The y axis denotes “log fraction nonresponding.” The greater the number of single cells present at the end of the experiment, the less self-renewal is present, resulting in a more horizontal curve. When more cells make spheres as a result of self-renewing division, the curve is steeper. We tracked spheres up to 96 hr and took images of wells to demonstrate the rate sphere formation.
Fluorescence Immunocytochemistry
For immunocytochemistry, we collected the GSCs as single cell suspension, fixed them with 4% paraformaldehyde, and followed the protocol described by Mukherjee et al. (2016). Primary antibodies include: p-CDK5 (Y15) (1:100), OLIG2 (1:100) (Santa Cruz), p-CREB (1:100), and SOX2 (1:100) (Cell Signaling Tech).
Mice Xenograft Tumor Generation
GSCs from GBM121 and GBM39 were collected based on their CD133 marker expression using flow cytometry. Cells were then injected at 1 × 105 concentration at the right hemisphere of the neonatal NSG female mice. 10–12 mice were used to inject each of the GSCs.
Organotypic Explant Culture
Mice brains with xenografted tumors were harvested in 1 × sterile PBS. The brains were cut into about 300 μM sections using vibratome. The sliced tumor tissues and contralateral normal brains were added to porous membrane fed with media from the bottom. The CDK5 inhibitor CP681301 was added in all media at 0, 10 μM, and 50 μM concentration. The experiments were done for up to 48 hr. The tissues are then collected and were subjected to both histological staining and western blot analysis. 10–12 mice were tested for each type of GSCs.
In Vitro Kinase Assay
Recombinant active CDK5/P25 and CDK5/P35 proteins (Millipore) were used as enzymes. Active Tau protein (Anaspec: 55556-50) and active CREB1 (Life Tech: 10074-H10E-5) proteins were used as substrates. The in vitro kinase assay buffer, ATP (Cell Signaling Tech), and 0, 0.5 μM, and 1 μM of CP681301 were added and mixed well. The reaction was incubated at 30°C for 30 min. Reaction was stopped by adding sample buffer and heated to 70°C for 10 min. Samples were kept at −20°C until used in western blot. Phosphorylation antibodies were used to detect the protein band.
siRNA Experiments
CDK5 and CREB1 siRNAs were received from Origene Technologies. 3 siRNAs and one scrambled siRNA were transfected using Lipofectamine 3000 in GBM121 and GBM39 GSCs. The cells were incubated for 72 hr and the proteins were isolated and ran in western blot to visualize the effect of reduced CDK5 and CREB1 on stem cell markers.
CREB Binding Site Determination on Promoters
To determine CREB binding sites on the promoters of OLIG2, SOX2, SALL2, and POU3F2, we used Eukaryotic Promoter Database from Swiss Institute of Bioinformatics (https://epd.vital-it.ch/cgi-bin/get_doc?db=hgEpdNew&format=genome&entry=OLIG2_1). Using 500 bases upstream of the transcription start site, we determined the presence or absence of CREB binding sites using JASPER transcription factor motif database. We also used Patch 1.0 online software to find CREB binding sites. Finally, to ensure our claim, we used the CRE prediction tool available from Marc Montminy lab of Salk institute.
Image Analysis
Image analyses was performed using ImageJ program. We measured cell fluorescence with this program and obtained the corrected total cell fluorescence, where CTCF = integrated density – (area of selected cell × mean fluorescence of background readings). We quantified the western blot band intensities through densitometric analysis using ImageJ and normalized the band intensities with the loading control band, such as Tubulin.
Statistics
To determine the significance of genetic rescue in our fly genetic screen, we performed Mann-Whitney U test. For fluorescence image quantification and densitometry analysis we performed parametric t test. For extreme limiting dilution assay (ELDA), we performed Chi-square test.
Unsupervised Hierarchical Clustering Analysis
TCGA GBM samples with available RNA-seq gene expression data (Ceccarelli et al., 2016; Wang et al., 2018) were analyzed (n = 129). Normalized RSEM expression data were log2 transformed and patients were classified into GBM subtypes: classical (n = 40), G-CIMP (n = 8), mesenchymal (n = 50), and proneural (n = 28). Unsupervised hierarchical clustering was performed for the genes of interest (CDK5, CDK5R1, stem cell markers, and differentiation markers) using Euclidean distance and average clustering. Fisher’s exact test was performed to test the association of the Heatmap clusters with GBM subtypes. Heatmaps were generated using R 3.2.2 software.
Supplementary Material
Highlights.
Drosophila Cdk5 promotes stemness by altering asymmetric division of brain tumor stem cells
CDK5 regulates self-renewal by directly activating CREB1 independently of PKA/cAMP
CDK5 inhibitor CP681301 reduces self-renewal in mouse glioma xenografts
CDK5 and asymmetric stem cell division markers segregate in non-mesenchymal tumors
Acknowledgments
We thank Drs. Kenneth Moberg, Erwin Van Meir, Zixu Mao, Adam Marcus (Winship Cancer Institute at Emory University), Mario Suva (Massachusetts General Hospital, Harvard University), and Monica Venere (Cleveland Clinic) for their valuable guidance. We thank Dr. Cheng-Yu Lee (Univ. of Michigan) for the Mira antibody. Our special thanks go to Drs. James Bibb (University of Alabama) and Dr. Karin Pozo (UT Southwestern) for advice on the CDK5 inhibitor CP681301. We thank Pfizer for providing the CDK5 inhibitor. We especially thank Dr. Ernestine Mahar, Dr. Edmund Waller, Dr. Melissa Gilbert-Ross, Dr. Anna Kenney, Dr. Neil Anthony, Dr. Abhinav Dey, Dr. Bing Yu, Dr. Debanjan Bhattacharya, Dr. Satoru Osuka, Zhaobin Zhang, Diane Alexis, and Jennifer Sheldon for their assistance with flow cytometry, confocal imaging, fly stocks, and mouse colony maintenance. Finally, we thank the Emory Integrated Cellular Imaging Core, Emory Cancer Pathology and Tissue Core, Winship Bio-informatics and Statistics Core facility, Bloomington Drosophila Stock Center, and the Division for Animal Resources for their advice and contributions to this manuscript. This work was also supported by US Public Health Service NIH (R01 CA176659 and CA149107 to D.J.B. and K25CA181503 to J.K.), the Win-ship Cancer Institute NCI Cancer Center (P30CA138292), and the Georgia Research Alliance (to D.J.B.).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and two tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.04.016.
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conception and Design, S.M. and D.J.B.; Development of Methodology, S.M.; Acquisition of Data, S.M., C.T.-B., E.K., A.N., H.D., M.C., C.Z., B.D., M.R., S.S., J. Kowalski, J. Kong, and D.J.B.; Analysis and Interpretation of Data, S.M., R.R., and D.J.B.; Writing – Review of the Manuscript, S.M., C.T.B., E.K., A.N., M.C., C.Z., B.D., M.R., S.S., J. Kowalski, J. Kong, R.R., and D.J.B.; Administrative, Technical, or Material Support, C.T.-B., R.R., and D.J.B.
References
- Arthur JS, Cohen P. MSK1 is required for CREB phosphorylation in response to mitogens in mouse embryonic stem cells. FEBS Lett. 2000;482:44–48. doi: 10.1016/s0014-5793(00)02031-7. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, et al. Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. TCGA Research Network. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164:550–563. doi: 10.1016/j.cell.2015.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Kong J, Tucker-Burden C, Anand M, Rong Y, Rahman F, Moreno CS, Van Meir EG, Hadjipanayis CG, Brat DJ. Human Brat ortholog TRIM3 is a tumor suppressor that regulates asymmetric cell division in glioblastoma. Cancer Res. 2014;74:4536–4548. doi: 10.1158/0008-5472.CAN-13-3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel P, Filiz G, Brown DV, Hollande F, Gonzales M, D’Abaco G, Papalexis N, Phillips WA, Malaterre J, Ramsay RG, Mantamadiotis T. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis. 2014;3:e108. doi: 10.1038/oncsis.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daul AL, Komori H, Lee CY. Immunofluorescent staining of Drosophila larval brain tissue. Cold Spring Harb Protoc. 2010;5 doi: 10.1101/pdb.prot5460. https://doi.org/10.1101/pdb.prot5460. [DOI] [PubMed] [Google Scholar]
- Egger B, Gold KS, Brand AH. Regulating the balance between symmetric and asymmetric stem cell division in the developing brain. Fly (Austin) 2011;5:237–241. doi: 10.4161/fly.5.3.15640. [DOI] [PubMed] [Google Scholar]
- Felsher DW. MYC inactivation elicits oncogene addiction through both tumor cell-intrinsic and host-dependent mechanisms. Genes Cancer. 2010;1:597–604. doi: 10.1177/1947601910377798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Meguro T, Fukuyama R, Nakamuta H, Koida M. New signaling pathway for parathyroid hormone and cyclic AMP action on extracellular-regulated kinase and cell proliferation in bone cells. Checkpoint of modulation by cyclic AMP. J Biol Chem. 2002;277:22191–22200. doi: 10.1074/jbc.M110364200. [DOI] [PubMed] [Google Scholar]
- Gonzalez C. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat Rev Cancer. 2013;13:172–183. doi: 10.1038/nrc3461. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Grewal SS, Fass DM, Yao H, Ellig CL, Goodman RH, Stork PJ. Calcium and cAMP signals differentially regulate cAMP-responsive element-binding protein function via a Rap1-extracellular signal-regulated kinase pathway. J Biol Chem. 2000;275:34433–34441. doi: 10.1074/jbc.M004728200. [DOI] [PubMed] [Google Scholar]
- Gu T, Zhang Z, Wang J, Guo J, Shen WH, Yin Y. CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 2011;71:2821–2825. doi: 10.1158/0008-5472.CAN-10-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich MR, Pant HC, Wada E, Battey JF. Neuronal cdc2-like kinase: a cdc2-related protein kinase with predominantly neuronal expression. Proc Natl Acad Sci USA. 1992;89:10867–10871. doi: 10.1073/pnas.89.22.10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Knoblich JA. Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol. 2010;11:849–860. doi: 10.1038/nrm3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- Lagace DC, Benavides DR, Kansy JW, Mapelli M, Greengard P, Bibb JA, Eisch AJ. Cdk5 is essential for adult hippocampal neurogenesis. Proc Natl Acad Sci USA. 2008;105:18567–18571. doi: 10.1073/pnas.0810137105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebelt BD, Shingu T, Zhou X, Ren J, Shin SA, Hu J. Glioma stem cells: signaling, microenvironment, and therapy. Stem Cells Int. 2016;2016:7849890. doi: 10.1155/2016/7849890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Lin TY, Juang JL. Abl deregulates Cdk5 kinase activity and subcellular localization in Drosophila neurodegeneration. Cell Death Differ. 2007;14:607–615. doi: 10.1038/sj.cdd.4402033. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Papalexis N, Dworkin S. CREB signalling in neural stem/progenitor cells: recent developments and the implications for brain tumour biology. BioEssays. 2012;34:293–300. doi: 10.1002/bies.201100133. [DOI] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Brat DJ. Asymmetric Cell Division in Development, Differentiation and Cancer. Springer; 2017. Molecular programs underlying asymmetric stem cell division and their disruption in malignancy; pp. 401–421. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Kong J, Brat DJ. Cancer stem cell division: when the rules of asymmetry are broken. Stem Cells Dev. 2015;24:405–416. doi: 10.1089/scd.2014.0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Tucker-Burden C, Zhang C, Moberg K, Read R, Hadjipanayis C, Brat DJ. Drosophila Brat and human ortholog TRIM3 maintain stem cell equilibrium and suppress brain tumorigenesis by attenuating Notch nuclear transport. Cancer Res. 2016;76:2443–2452. doi: 10.1158/0008-5472.CAN-15-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. J Clin Invest. 2017;127:415–426. doi: 10.1172/JCI89587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa T, Riester M, Cheng YK, Huse JT, Squatrito M, Helmy K, Charles N, Michor F, Holland EC. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26:288–300. doi: 10.1016/j.ccr.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persano L, Pistollato F, Rampazzo E, Della Puppa A, Abbadi S, Frasson C, Volpin F, Indraccolo S, Scienza R, Basso G. BMP2 sensitizes glioblastoma stem-like cells to Temozolomide by affecting HIF-1α stability and MGMT expression. Cell Death Dis. 2012;3:e412. doi: 10.1038/cddis.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K, Castro-Rivera E, Tan C, Plattner F, Schwach G, Siegl V, Meyer D, Guo A, Gundara J, Mettlach G, et al. The role of Cdk5 in neuro-endocrine thyroid cancer. Cancer Cell. 2013;24:499–511. doi: 10.1016/j.ccr.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampurno S, Bijenhof A, Cheasley D, Xu H, Robine S, Hilton D, Alexander WS, Pereira L, Mantamadiotis T, Malaterre J, Ramsay RG. The Myb-p300-CREB axis modulates intestine homeostasis, radiosensitivity and tumorigenesis. Cell Death Dis. 2013;4:e605. doi: 10.1038/cddis.2013.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Marrinan J, Frishman C, Sampath P. Impact of temozolomide on immune response during malignant glioma chemotherapy. Clin Dev Immunol. 2012;2012:831090. doi: 10.1155/2012/831090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Lahiri DK. Cdk5 activity in the brain - multiple paths of regulation. J Cell Sci. 2014;127:2391–2400. doi: 10.1242/jcs.147553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith-Trunova S, Prithviraj R, Spurrier J, Kuzina I, Gu Q, Giniger E. Cdk5 regulates developmental remodeling of mushroom body neurons in Drosophila. Dev Dyn. 2015;244:1550–1563. doi: 10.1002/dvdy.24350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suvà ML, Rheinbay E, Gillespie SM, Patel AP, Wakimoto H, Rabkin SD, Riggi N, Chi AS, Cahill DP, Nahed BV, et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell. 2014;157:580–594. doi: 10.1016/j.cell.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trépant AL, Bouchart C, Rorive S, Sauvage S, Decaestecker C, Demetter P, Salmon I. Identification of OLIG2 as the most specific glioblastoma stem cell marker starting from comparative analysis of data from similar DNA chip microarray platforms. Tumour Biol. 2015;36:1943–1953. doi: 10.1007/s13277-014-2800-5. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Björling E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson AC, Angelidou P, Asplund A, Asplund C, et al. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics. 2005;4:1920–1932. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- Wang J, Wakeman TP, Lathia JD, Hjelmeland AB, Wang XF, White RR, Rich JN, Sullenger BA. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28:17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Hu B, Hu X, Kim H, Squatrito M, Scarpace L, deCarvalho AC, Lyu S, Li P, Li Y, et al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. Cancer Cell. 2018;33:152. doi: 10.1016/j.ccell.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin GR, Soloaga A, Foster JM, Murray-Tait V, Cohen P, Arthur JS. MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol Cell Biol. 2002;22:2871–2881. doi: 10.1128/MCB.22.8.2871-2881.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Wu Q, Horbinski CM, Flavahan WA, Yang K, Zhou W, Dombrowski SM, Huang Z, Fang X, Shi Y, et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat Neurosci. 2015;18:501–510. doi: 10.1038/nn.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- Yin X, Qi Y, Ren M, Wang S, Jiang H, Feng H, Cui S. Roscovitine treatment caused impairment of fertilizing ability in mice. Toxicol Lett. 2015;237:200–209. doi: 10.1016/j.toxlet.2015.06.014. [DOI] [PubMed] [Google Scholar]
- Yushan R, Wenjie C, Suning H, Yiwu D, Tengfei Z, Madushi WM, Feifei L, Changwen Z, Xin W, Roodrajeetsing G, et al. Insights into the clinical value of cyclin-dependent kinase 5 in glioma: a retrospective study. World J Surg Oncol. 2015;13:223. doi: 10.1186/s12957-015-0629-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabala M, Lobo N, Qian D, van Weele L, Heiser D, Clarke M, Liu H, Lathia J. Overview: Cancer Stem Cell Self-Renewal. Elsevier Cambridge; 2016. [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong P, Liu X, Zhang Z, Hu Y, Liu SJ, Lezama-Ruiz M, Joksimovic M, Liu QS. Cyclin-dependent kinase 5 in the ventral tegmental area regulates depression-related behaviors. J Neurosci. 2014;34:6352–6366. doi: 10.1523/JNEUROSCI.3673-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zografos L, Tang J, Hesse F, Wanker EE, Li KW, Smit AB, Davies RW, Armstrong JD. Functional characterisation of human synaptic genes expressed in the Drosophila brain. Biol Open. 2016;5:662–667. doi: 10.1242/bio.016261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM, Gertler FB, Vidal M, Van Etten RA, Tsai LH. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–646. doi: 10.1016/s0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.