

SUMMARY
Dendritic filopodia select synaptic partner axons by interviewing the cell surface of potential targets, but how filopodia decipher the complex pattern of adhesive and repulsive molecular cues to find appropriate contacts is unknown. Here we demonstrate in cortical neurons that a single cue is sufficient for dendritic filopodia to reject or select specific axonal contacts for elaboration as synaptic sites. Super-resolution and live-cell imaging reveals that EphB2 is located in the tips of filopodia and at nascent synaptic sites. Surprisingly, a genetically encoded indicator of EphB kinase activity, unbiased classification, and a photoactivatable EphB2 reveal that simple differences in the kinetics of EphB kinase signaling at the tips of filopodia mediate the choice between retraction and synaptogenesis. This may enable individual filopodia to choose targets based on differences in the activation rate of a single tyrosine kinase, greatly simplifying the process of partner selection and suggesting a general principle.
eTOC Blurb
Mao et al. provide a mechanism for local control of synaptic specificity. A transfectable indicator of EphB activity reveals that the decision of filopodial to make synapses or to seek new targets is guided by kinetically distinct signals.
INTRODUCTION
The pattern of synaptic connections underlies proper brain function. Yet the physical proximity of a dendrite and an axon is insufficient to predict the presence of a synaptic connection, indicating that neurons possess specific mechanisms to enable the selection of the appropriate synaptic partners (Kasthuri et al., 2015). Establishment of the appropriate pattern of synaptic contacts requires dendritic filopodia to select amongst a host of potential target axons and decide whether to retract and seek new axonal contacts or to generate a stable synaptic connection. Movement of dendritic filopodia allows for sampling of many different cellular interactions that drive engagement of adhesive and repulsive cues present on potential targets. However, the signals that enable the decision to select a small subset of these dendritic-axonal contacts as targets to be stabilized and form synapses remain unclear.
At sites of contact between axons and filopodia, if a synapse is not generated, or the target is incorrect, the filopodium retracts or moves away from the contact. The choice of whether a particular filopodium is stabilized or repulsed to seek out a new possible contact occurs rapidly over a few minutes, suggesting that local signaling at the site of contact determines whether the contact will be stabilized or lost. An important class of factors thought to be related to target selection is the transcellular synaptic organizing proteins. Transsynaptic organizing proteins function as pairs of interacting molecules, with one member in the presumptive presynaptic neuron and a second in the postsynaptic neuron (Dalva et al., 2007; Shen and Scheiffele, 2010). Yet whether these molecules are localized to filopodia as they generate new synaptic sites is not known.
To discriminate among the multiple axons in close contact with each dendritic protrusion, a filopodium could use a complex combination of distinct repulsive and attractive cues. Alternatively, in principle a single molecule could generate sufficient variability in signaling to specify the fate of a given filopodium. For example, distinct calcium dynamics downstream of the NMDA receptor have been shown to be sufficient to govern whether a synapse is strengthened or depressed (Feldman, 2012; Higley and Sabatini, 2012). Of the potential transsynaptic organizing proteins that might control target selection, the EphBs and ephrin-Bs are particularly attractive. The EphB family of receptor tyrosine kinases (RTKs) function early in development when filopodia are abundant and regulate filopodial motility (Kania and Klein, 2016; Lai and Ip, 2009). Loss of function mutations to EphBs result in defective filopodial movement, while reconstitution experiments indicate that the EphB-dependent filopodial motility relies on the EphB-ephrin-B interaction (Kayser et al., 2008). Not only are EphBs required for normal filopodial movement, but EphBs can generate adhesive transcellular interactions that might stabilize filopodia by binding axonal ephrin-Bs (Kania and Klein, 2016; Lai and Ip, 2009). EphB binding to axonal ephrin-B is required for presynaptic specialization formation (McClelland et al., 2009), while postsynaptic EphBs are required for normal excitatory synapse development in vitro and in vivo (Henkemeyer et al., 2003; Kayser et al., 2006; Kayser et al., 2008). In the mature synapse, EphBs are found within the core of the postsynaptic complex (Perez de Arce et al., 2015), suggesting the possibility that they are an early factor important for synapse initiation. However, it remains unknown whether EphBs are found within filopodia as filopodia interact with molecular cues to select axons for nascent synaptic contacts.
Here we describe the dynamic intracellular signals that direct the behavior of individual filopodia to initiate synaptogenesis or to retract and seek new contacts between cortical neurons. We find that EphB2 regulates the actions of filopodia through a combination of precise subcellular localization and induction of specific intracellular signals within filopodia. EphB2 is localized to the tips of moving and stable dendritic filopodia. In stable synaptic filopodia, EphB2 is found adjacent to its ligand ephrin-B1, presynaptic marker vesicular glutamate transporter 1 (VGLUT1), and synaptic release sites. Using a dual-color ratiometric vector-encoded indicator GPhosEphB and an unbiased classification approach, we demonstrate that the choice of individual filopodia to retract or make stable axo-dendritic contacts is governed by specific differences in EphB signaling: retraction is initiated by a rapid increase in EphB activity occurring within minutes, while slower activation results in formation of stable contacts. The impact of EphB kinase activation on filopodial retraction is validated in HEK cells and cultured cortical neurons using a genetically encoded photoactivatable EphB2 protein. These studies identify a mechanism that enables individual filopodia to locally decide their own fates, thereby simplifying the selection of appropriate synaptic targets.
RESULTS
EphB2 is found at synaptic sites in dendritic filopodia
Ephrin-B1 and EphB2 are required for synapse formation and are found at mature synaptic sites (Kayser et al., 2006; McClelland et al., 2009; Perez de Arce et al., 2015). To determine where EphB and ephrin-Bs function as axo-dendritic contacts are initiated, we investigated the localization of EphB2 and ephrin-B1 during a phase of rapid EphB-dependent synapse formation when filopodia are abundant (days in vitro [DIV]7-10) (Kayser et al., 2008; McClelland et al., 2009). mNeptune transfected cortical neurons were immunostained for endogenous EphB2, ephrin-B1, and the presynaptic marker VGLUT1 (Kayser et al., 2006; McClelland et al., 2009; Zhou et al., 2012). Using four-color imaging, with endogenous EphB2 and ephrin-B1 imaged by super-resolution Stimulated Emission Depletion (STED) microscopy and VGLUT1 and mNeptune imaged with confocal microscopy (Figure 1A, (Hruska et al., 2015)). Consistent with previous work (Kayser et al., 2008), many contacts were found along the shaft of the dendrite as indicated by colocalization of EphB2, ephrin-B1 and VGLUT1 (Figure 1A). In addition, many dendritic filopodia were EphB2+ and colocalized with ephrin-B1 and VGLUT1 puncta, with ephrin-B1 more frequently and closely associated with VGLUT1 (Figures S1A–S1F).
Figure 1. EphB receptors are located on the surface of filopodia and colocalize with ephrin-B1, VGLUT1.

(A) Combined two-color super-resolution STED and confocal imaging demonstrate that EphB2 in filopodia (green, STED) colocalizes with ephrin-B1 (red, STED) and VGLUT1 (purple, confocal). Neurons were transfected with mNeptune (gray, confocal). Dashed lines show the morphology of transfected neurons. Arrows indicate an example of colocalization of EphB2, ephrin-B1 and VGLUT1 in filopodia. Scale bar = 1 μm. (B) The proportion of EphB2+ and EphB2− filopodia (n = 256). (C) Quantification of EphB2, ephrin-B1 and VGLUT1 colocalization in EphB2+ filopodia (n = 189). (D) Surface staining of EphB2 (green) in DIV7-10 neurons transfected with tdTomato. Arrows indicate EphB2+ filopodia. Dashed lines show the morphology of transfected neurons. Bottom figures show that acid treatment removes surface staining of EphB2. Scale bar = 2 μm. See also Figure S1.
To determine whether the transsynaptic organizers EphB2 and ephrin-B1 were found at potential synaptic sites at the tips of filopodia, we quantified the proportion of EphB2+ filopodia apposed to ephrin-B1 and VGLUT1 puncta. Four-color STED/confocal imaging revealed that most filopodia contained EphB2 (~74%, 189/256, Figures 1A and 1B; Hruska et al., 2015). Of the filopodia containing EphB2 puncta, ~ 43% of EphB2+ filopodia (81/189) colocalized with or were adjacent to both ephrin-B1 and VGLUT1 puncta (Figures 1A and 1C). Consistent with the well described presynaptic localization of ephrin-B1 and the selective function of ephrin-B1 in recruiting presynaptic specializations (Figures S1A–S1E) (McClelland et al., 2010; McClelland et al., 2009), VGLUT1 puncta were rarely found at filopodia containing EphB2 but lacking ephrin-B1 (<10%, Figure 1C). Moreover, live-cell staining for EphB2 revealed that EphB2 is enriched on the cell surface in the dendritic shaft and within the majority of dendritic filopodia (72%, 237/328, Figures 1D, S1G–S1H, Hanamura et al., 2017; Perez de Arce et al., 2015). Thus, EphB2 and ephrin-B1 are localized appropriately at sites of contact between filopodia and axons to control the initial steps of synapse development, suggesting that EphBs function on the surface of dendritic filopodia and may interact in trans with presynaptic ephrin-B1.
EphB in stable filopodia colocalizes with synaptic release sites
To determine whether stable filopodia containing EphB2 might form functional synapses, we conducted live-cell imaging of synaptic release sites using FM dye. Neuronal morphology was visualized by transfecting neurons with mTurquoise2 (Goedhart et al., 2012), and the localization of EphB2 in living cells was determined by co-expressing EphB2-YFP (Kayser et al., 2008). In DIV7-10 neurons stable filopodia were defined as those that moved less than 1 μm/frame during the 30 minute imaging period (Kayser et al., 2008) and presynaptic release sites were visualized with spontaneously loaded FM 4–64 (See Methods for detail, Figure 2A) (Wilhelm et al., 2010). Stable filopodia were more likely to colocalize with synaptic sites if EphB2-YFP was present (69 ± 6 %, n = 9) than if EphB2-YFP was absent (49 ± 6 %, n = 9, p = 0.024, t-test, Figures 2B–2D). These findings suggest that EphB2 in filopodia may drive the initiation of functional synapse formation. Together with our STED imaging results, these findings suggest that the localization of EphB2 to tips of filopodia may determine which axo-dendritic contacts are stabilized to become synaptic sites.
Figure 2. EphB receptors cluster at the tip of moving and stable filopodia, and colocalize with synaptic release sites.

(A) Experimental procedure for labeling of presynaptic release sites as in ((Wilhelm et al., 2010), See Methods for details). (B) Representative images show filopodia stable for 30 minutes in neurons transfected with mT2 (Arrows). (C) The same dendrite as in B overlaid with images of EphB2-YFP (yellow) and synaptic release sites (red, labeled with FM4-64). Arrows indicate EphB2-YFP+ filopodial tips colocalized with FM4-64. Dashed lines show the morphology of transfected neurons. Scale bar = 5 μm. (D) Quantification of colocalization (EphB2+: 69 ± 6%, n = 9; EphB2-: 49 ± 6%, n = 9, p = 0.024, t-test). * p < 0.05. Error bars indicate SEM (E) Representative images from 30-minute time-lapse movies of DIV7-10 neurons transfected with EphB2-YFP. Two types of filopodia were identified (Stable and Moving filopodia). Arrows indicate EphB2+ tips across time window. Scale bar = 2 μm. See also Movie S1.
EphB clusters in both moving and stable dendritic filopodia
While stable EphB2-containing filopodia are often sites of synaptic transmission, moving filopodia explore potential axonal contacts (Kayser et al., 2008). Therefore, we asked whether EphBs might mediate these distinct cellular behaviors by localizing differently in moving and stable filopodia. As expected from Figure 2C, live-cell imaging of neurons transfected with EphB2-YFP revealed that clusters of EphB2-YFP were often found in the tips of stable filopodia (65%, 156/240, Figure 2E, Movie S1). However, similar numbers of clusters of EphB2 were seen in moving filopodia (78%, 83/106, Figure 2E, Movie S1), suggesting that differences in the localization of EphB2 cannot explain the distinct functions of EphBs in moving and stable filopodia, and supporting the previous findings that EphBs are required for both filopodial motility and synaptogenesis (Kayser et al., 2006; Kayser et al., 2008).
Design and validation of genetically-encoded fluorescent reporters for tyrosine kinase activity
Since clusters of EphB2 are localized to both moving and stable filopodia, we hypothesized that specific differences in the kinetics or magnitude of EphB2 kinase signaling might determine whether a filopodium is repulsed or stabilized upon contact with an axon. To test this hypothesis, we designed a genetically-encoded tyrosine phosphorylation indicator called Green Phosphorylation (GPhos) to visualize the subcellular activity of tyrosine kinases (TKs). The indicator is composed of a single, circularly-permutated EGFP core flanked on the N-terminus by two consensus phosphorylation sites for specific tyrosine kinases, and on the C-terminus by a relatively promiscuous SH2 domain that binds to phosphorylated tyrosine residues (Figure 3A) (Nakai et al., 2001; Pawson, 2004; Songyang et al., 1993). To validate our design and enable straightforward testing, we first made a reporter for the intracellular TK Fyn using a circularly-permutated EGFP fused with a peptide containing the two consensus sites for tyrosine phosphorylation (YEEIVGEFKIYEEI, Figure S2, (Songyang et al., 1993)), which undergo phosphorylation following Fyn activation, and the Fyn SH2 domain. SH2 domain binding to the phosphorylated tyrosine increased fluorescence intensity of the EGFP core (Figure S2). To generate a quantitative ratiometric tool, we fused a second fluorescent protein monomeric red fluorescent protein (mRFP, Figures S2B and S2C) (Campbell et al., 2002) that has a similar bleaching rate to EGFP, with a rigid five-turn alpha-helical linker to the indicator core (Figures 3A, S2B and S2D).
Figure 3. The GPhos indicators selectively report activity of EphB or EphA tyrosine kinases.

(A) Design of single- and dual-color GPhos indicators. GPhosEphB and GPhosEphA indicator differ only in the phospho-peptide region (shown in blue). (B) GPhosEphB was immunoprecipitated from HEK293T cells transfected with GPhosEphB indicator and EphB2 or EphA4 receptor and probed for phosphorylation (PY99). Ephrin-B2 treatment resulted in phosphorylation of GPhosEphB only in cells transfected with EphB2. Lower western blots show expression controls. (C) Ratiometric pseudocolor images of single optical sections collected every three minutes from cells transfected with EphB2 and either GPhosEphB (top) or GPhosEphA (bottom). Cells were treated with activated ephrin-B2 as indicated by the orange bar. The pseudocolored lookup table (16 colors) indicates the ratiometric GPhos signal. (D) Quantification of the effects of ephrin-B2 treatment on GPhos signal in HEK293T cells transfected with EphB2 (GPhosEphB: n = 6; GPhosEphA: n = 4, p < 0.0001, two-way ANOVA). * p < 0.05, ** p < 0.01, *** p < 0.001. Western blot shows phosphorylation state of the immunoprecipitated FLAG-EphB2 receptor at 5, 15, and 30 minutes after activation with ephrin-B2. (E) As in C, but ratiometric images of single optical sections were collected when HEK293T cells were transfected with EphA4 and either GPhosEphB or GPhosEphA. GPhosEphA signal increased with ephrin-A1 treatment. (F) Quantification of the effects of ephrin-A treatment (GPhosEphB: n = 10; GPhosEphA: n = 10, p < 0.0001, two-way ANOVA). ** p < 0.01, *** p < 0.001,**** p < 0.0001. Western blot shows phosphorylation state of the immunoprecipitated FLAG-EphA4 receptor at 5, 15, and 30 minutes after activation with ephrin-A1. (G) Ratiometric images of TKI mouse neurons transfected with GPhosEphB indicator and treated with activated ephrin-B2. (H) Quantification of the effects of ephrin-B2 treatment on GPhos signal in transfected neurons (n = 11). (I) Ratiometric images of TKI neurons transfected with GPhosEphB indicator and blocked with 1-NA-PP1 before treatment with activated ephrin-B2. (J) Quantification of the effects of 1-NA-PP1 on ephrin-B treatment (n = 8). The pseudocolor lookup table (fire) indicates the ratiometric GPhos signal in neurons. Scale bars = 5. Error bars indicate SEM in Figures D, F, H, J. See also Figures S2, S3 and Movies S2–S5.
GPhos indicators are phosphorylatable (Figure S3A) and report both increases and decreases in tyrosine kinase activity (Figure S3B). Selective indicators for Ephs were created using peptides containing autophosphorylation sites of the juxtamembrane domains of EphB2 or EphA4 (GPhosEphB: YIDPFTYEDP; GPhosEphA: YVDPFTYEDP; Figure 3A). HEK293T cells transfected with each indicator and either EphB2 or EphA4, and treated with activated soluble ephrin-B2 or ephrin-A1 show selective phosphorylation of GPhosEphB in only EphB2 transfected cells (Figure 3B). Live-cell imaging of EphB2 transfected cells stimulated with ephrin-B revealed an increase of GPhos signal in single optical sections within only GPhosEphB transfected cells, but not within cells transfected with GPhosEphA (GPhosEphB: n = 6; GPhosEphA: n = 4, p < 0.0001, two-way ANOVA, Figures 3C and 3D, Movie S2). GPhosEphB also effectively reported rapid changes in EphB2 activation and the activity of EphB1 and EphB3 (Figures S2E–S2K). Similarly, ephrin-A stimulation of cells transfected with EphA4 increased the GPhos signal in only the GPhosEphA transfected cells (GPhosEphB: n = 10; GPhosEphA: n = 10, p < 0.0001, two-way ANOVA, Figures 3E and 3F, Movie S3). The increase in ratiometric indicator signal was similar to the time course of receptor activation visualized by western blot analysis (Figures 3D and 3F). Importantly, the majority of the GPhos signal was found near the cell membrane (Figures 3C and 3E), suggesting that the indicators can report spatially restricted signals.
To determine whether the GPhosEphB indicator effectively reports EphB activity in neurons (which often express both EphBs and EphAs), we confirmed that transfection of the GPhos indicator does not affect synapse density (Figures S3C and S3D), and accurately reflects changes in EphB kinase activity (Figure S3E). We then tested the specificity of the GPhosEphB indicator for EphB activity in a triple kinase knock-in (TKI) mouse model that allows kinase activity of EphBs but not EphAs to be selectively inhibited with 1-NA-PP1 (Figure S3F) (Soskis et al., 2012). Treatment of GPhosEphB transfected neurons cultured from TKI mice with soluble ephrin-B resulted in a robust increase in GPhosEphB signal (Figures 3G and 3H, Movie S4). However, 1-NA-PP1 application completely blocked the effects of ephrin-B treatment (Figures 3I and 3J, Movie S5). Similarly, transfection of rat cortical neurons with dominant-negative EphB receptor blocked the ability of ephrin-B to induce GPhosEphB signal (Figures S3G and S3H). Together these findings indicate that GPhosEphB can selectively and effectively report EphB kinase activity in neurons.
Restricted spatial and temporal patterns of GPhos indicator after focal activation of ephrin-B1
To determine the spatial extent of EphB signaling in neurons, we examined the ability of axonal ephrin-B1 to induce dendritic EphB signaling. mTurquoise2-tagged ephrin-B1 (mT2-eB1) was transfected into neurons. As expected from data of Figures 1 and S1, and our previous work (McClelland et al., 2009), mT2-eB1 localized into puncta on the surface of axons (Figure S4A). To visualize contacts between ephrin-B1 expressing axons (mT2-eB1 transfected, cyan) and EphB signaling in dendrites (GPhosEphB transfected, fire) neuronal cultures were first transfected with mT2-eB1 at DIV3-5, and then with GPhosEphB at DIV5-7 (See Methods for detail; Figures 4A, 4D and 4G). Neurons were imaged once every 0.25–1 minute for 15–30 minutes at DIV7-10. At sites of contact, the GPhosEphB signal induced by mT2-eB1 puncta was restricted to a small region near the apparent point of contact (Figure 4A). The average area of GPhosEphB signal (0.41 ± 0.09 μm2, n = 28) was significantly smaller than the average area of mT2-eB1 puncta (0.67 ± 0.06 μm2, n = 28, p = 0.02, paired t-test). To test whether the GPhosEphB signal was due to EphB2-ephrin-B1 mediated signaling, neurons containing ephrin-B1 puncta that maintained contact with dendrites during the 15–30 minute imaging period were fixed and stained for EphB2 (Figure 4B, see Methods for experimental detail). Retrospective analysis indicates that 80% of sites with persistent mT2-eB1 puncta had both EphB2 and GPhosEphB signals (12/15, n = 6; Figures 4A–4C). Together these findings indicate that EphB kinase activation occurs in a focal area when activated by ephrin-B1.
Figure 4. Focal activation of EphB in dendrites by axonal ephrin-B1.

(A) An example of persistent contact between axonal mT2-eB1 puncta (cyan) and GPhosEphB (fire) transfected dendrite. Images of RFP channel at the last frame of 15 min movie were shown (pseudocolored, green). The ROI shows the contact between axonal mT2-eB1 puncta and GPhosEphB transfected dendrite (2 min and 15 min). Arrows indicate the colocalization of eB1 puncta (cyan) and persistent GPhos signal (fire). (B) The RFP image (pseudocolored, green) after fixation is shown. Confocal image of EphB2 (red) colocalizes with eB1 puncta (cyan). (C) Quantification of the colocalization of mT2-eB1, EphB2 and GPhos. (D) As in A, an example of persistent contact between axonal mT2-eB1 and GPhosEphB transfected dendrite. Dashed lines show the morphology of axons. Arrows indicate sites of contact. Arrowheads indicate control ROIs used in E. (E) Quantification of GPhos signal in D. (F) Quantification of summary data of GPhos signals in control and mT2-eB1 contacting sites. Data are shown as the average GPhosEphB signal when the mT2-eB1 puncta are in contact with the axon or in control ROIs (arrowheads in D). (Ctrl: 1.00 ± 0.01, n = 19; eB1: 1.23 ± 0.04, n = 19, p ≤0.001, paired t-test). *** p = 0.001. (G) Examples of transient contact between moving axonal mT2-eB1 puncta and GPhosEphB transfected dendrite. Arrows and dashed lines as in D. (H) Quantification of GPhos signal in G. (I) As in F, quantification of GPhos signal in control and mT2-eB1 contacting sites from pooled data (Ctrl: 1.00 ± 0.01, n = 9; eB1: 1.24 ± 0.04, n = 9, p ≤ 0.001, paired t-test). *** p = 0.001. Control ROIs were selected at sites in dendritic shaft without axonal contacts and GPhosEphB signals. Scale bars = 2 μm. Error bars indicate SEM in Figures F and I. See also Figures S4 and Movies S6–S7.
While many mT2-eB1 puncta remained stationary during the imaging period, some mT2-eB1 puncta move in a saltatory fashion within axons of transfected neurons (Movies S6 and S7). At sites of contact, GPhosEphB signals found within dendrites had different patterns of GPhosEphB activity depending on the behavior of the mT2-eB1 puncta (Figure S4B). At dendrites contacting a stable mT2-eB1puncta (Figure 4D), GPhosEphB signals were persistent and significantly elevated (Ctrl: 1.00 ± 0.01, n = 19; eB1: 1.23 ± 0.04, n = 19, p ≤ 0.001, paired t-test, Figures 4D–4F, Movie S6). In contrast, at sites of contact between dendrites and moving mT2-eB1puncta, the GPhosEphB signal was significantly elevated only when the puncta were in apparent contact with the dendrite; the signal returned to the baseline after the mT2-eB1 puncta moved along the axon in a saltatory manner (Ctrl: 1.00 ± 0.01, n = 9; eB1: 1.24 ± 0.04, n = 9, p ≤ 0.001, paired t-test, Figures 4G–4I, Movie S7). While the duration of these GPhos signals differed, the magnitude of the signals at persistent or transient mT2-eB1 puncta did not (Persistent: 1.23 ± 0.04 vs. Transient: 1.24 ± 0.04, p = 0.885, t-test). Thus, GPhosEphB can report both sustained and transient signals, and axonal ephrin-B1 induces a spatially restricted EphB activation at sites of axo-dendritic contact.
Dynamics of EphB signaling are related to filopodial motility and stabilization
Clusters of EphB2 are found in both moving and stable filopodia. To test whether differences in the spatial or temporal dynamics of EphB signaling may enable filopodia to decide whether to connect to or retract from a contact with an axon, we conducted live-cell imaging of neurons transfected with GPhosEphB and visualized contacts between dendritic filopodia and axons. Axons were visualized by transfecting a different set of neurons with mTurquoise2 (mT2) using the protocol described in the Methods (cyan, Figures 5A and 5D). Images of dendrites contacted by axons were collected 30 to 60 minutes once every minute.
Figure 5. Distinctive patterns of EphB activity in Retracting and Connecting filopodia.

(A) An example of retracting filopodia. Axons are labeled with mTuquoise2 and indicated by dashed lines. Ratiometric images (pseudocolored) show transient high GPhosEphB signal (middle panel). Arrowhead indicates focal GPhosEphB signal (ROI). (B) Quantification of GPhosEphB signal in A. (C) GPhosEphB signal in the Retracting filopodia group. Gray lines indicate GPhos signal in individual filopodia and the green line represents mean (n = 9). Time 0 indicates the time filopodia contact a labeled axon (vertical dashed line). The bar indicates the fraction of filopodia in contact with the axons (Black = 100%, White = 0%). Control mean shaft GPhosEphB is indicated by the black line. (D) An example of Connecting filopodia as in A. (E) Quantification of GPhosEphB signal in D. (F) As in C, but the mean GPhosEphB signal in the Connecting filopodia is shown as a red line (n = 12). (G) Illustration of Retracting & Connecting filopodia behavior and EphB signaling. The slope was calculated as the difference between the baseline and the peak value divided by the time from the baseline to the peak. (H) Comparison of the kinetics of average GPhosEphB signal. Retracting group was best fit with a peak model. The Connecting group was best fit with an exponential model. (I) The slope of GPhosEphB signal in Retracting filopodia was significantly sharper than that in Connecting filopodia (Retracting: 0.31 ± 0.04, n = 9; Connecting: 0.17 ± 0.04, n = 12, p = 0.021, Mann Whitney U test) * p < 0.05. Scale bars = 2 μm. Error bars indicate SEM in Figure I. See also Figure S5 and Movies S8–S9.
We observed two general types of filopodia: filopodia that moved during the imaging period (moving filopodia) and filopodia that remained in stable contact with an axon during the imaging period (connected filopodia). Similar to what we saw with mT2-eB1, EphBs were focally activated within both moving and connected filopodia, and this activity appeared as discrete puncta of GPhosEphB signals (Figures 5A and 5D). The area of the GPhosEphB signal in filopodia was not significantly different from the area of the signal induced by the axonal mT2-eB1 (mT2-eB1/GPhos puncta: 0.41 ± 0.09 μm2, n = 28; endogenous GPhos puncta: 0.27 ± 0.02 μm2, n = 53, p = 0.063, t-test). These findings are consistent with our previous results that EphBs cluster in moving and stable filopodia, and they support the notion that endogenous axonal ephrin-B1 (Figures 1A and S1A–S1F) (McClelland et al., 2009) likely induce EphB activation in dendritic filopodia.
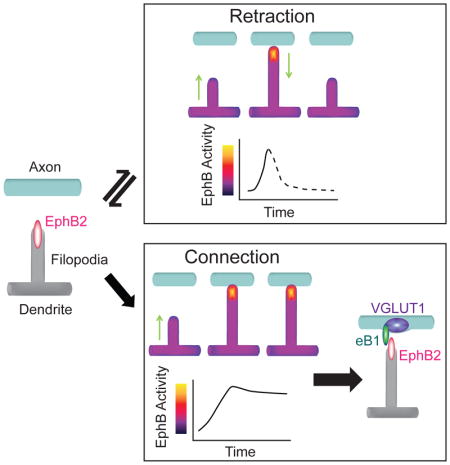
There were two types of behavior in moving dendritic filopodia: a filopodium contacted an axon and retracted, resulting in a transient axo-dendritic contact (Retracting filopodia), or a filopodium extended to contact and remained in contact with the axon to generate a stable axo-dendritic contact (Connecting filopodia). Both Retracting and Connecting filopodia showed increased levels of EphB kinase activation relative to the dendritic shaft. Remarkably, distinct patterns of EphB kinase activity reliably predicted whether a filopodium retracted from an axon or be stabilized and remained in contact with an axon. The differences in EphB2 kinase behavior were not reflected in the average area of activation in Retracting or Connecting filopodia (Retracting: 0.26 ± 0.04 μm2, n = 9; Connecting: 0.22 ± 0.04 μm2, n = 12, p = 0.483, t-test). Instead, the behavior appeared to be linked to the rate of activation of the GPhosEphB signal.
To begin to determine whether magnitude or rate of EphB activation might drive differences in filopodial behavior, we examined the kinetics of the GPhos signal in Retracting and Connecting filopodia. After the initial contact with axons, the average GPhosEphB signal in Retracting filopodia increased rapidly to reach a maximum of ~140% of baseline dendritic levels before retraction (Figures 5A–5C, see Methods for detail of analysis, n = 9; Movie S8). In contrast, the average GPhosEphB signal in Connecting filopodia reached the maximum after ~3 minutes (136%, n = 12, Figures 5D–5F, Movie S9) and remained elevated after initial contact. Despite the functional difference in the behavior of these filopodia, there was no significant difference between the maximum increase in the EphB signal between Retracting and Connecting filopodia (Retracting: 136 ± 7 %, n = 9; Connecting: 136 ± 10 %, n = 12; p = 0.145, Mann-Whitney U). In contrast, the time from the baseline was significantly different between Retracting and Connecting filopodia (Time to peak: Retracting: 1.4 ± 0.2 min, n = 9; Connecting: 6.8 ± 1.7 min, n = 12, p = 0.017, Mann-Whitney U). These findings suggest that the kinetics of EphB kinase activity within individual filopodia may drive specific filopodial behaviors.
To test whether differences in the kinetics of EphB kinase activity determine how filopodia behave, we next calculated the rate of change in EphB kinase activity between Retracting and Connecting filopodia (Figures 5G and 5H). Although the maximum signal increases in Retracting and Connecting filopodia are not significantly different, the rate of increase (slope) in GPhosEphB activity in the Retracting filopodia was significantly faster than that in the Connecting filopodia (Retracting: 0.31 ± 0.04, n = 9; Connecting: 0.17 ± 0.04, n = 12, p = 0.021, Mann-Whitney U, Figure 5I). These findings suggest that the rate of the rising phase of EphB activity may determine whether filopodia initiate synapse formation or retract and seek new axonal contacts. Consistent with this possibility, key features related to kinase activity (including signal slope, amplitude, and variance) were sufficient to decode whether a filopodium remained in contact with an axon or retracted at a rate significantly above chance (98% classification efficiency with held-out validation; Range 92.5–100%; Figure S5). These data support a model where rapid (≤ 1 min) activation of EphBs induces filopodial retraction, whereas sustained or slow and more variable activation of EphBs induces filopodial stabilization.
EphB signaling controls filopodial movement
To investigate how EphB signaling controls filopodial behavior, we first examined how bath application of activated soluble ephrin-B impacted filopodial movement (Dalva et al., 2000). Consistent with the model that sustained EphB2 kinase activation results in filopodial stabilization, the bath application of ephrin-B increased overall GPhos signal slowly (Figures S7E–S7G) and decreased filopodial movement (Figures 6A–6H).
Figure 6. The dynamics of EphB activity determine filopodial behavior.

Slow activation of EphB2 decreases filopodial movement. (A) Dendritic filopodia before and after application of control (FC). (B) Colored lines indicate the distance moved by each filopodium in (A). (C) As in A, but images after application of activated ephrin-B2. (D) As in B. (E) Quantification of GPhosEphB signal in control (Before: 1.3 ± 0.04; After: 1.4 ± 0.09, n = 4, p = 0.358, paired t-test). (F) Quantification of distance moved in control (Before: 0.6 ± 0.06; After: 0.5 ± 0.09, n = 4, p = 0.091, paired t-test). (G) Quantification of GPhosEphB signal in ephrin-B2 treated group (Before: 1.1 ± 0.04; After: 1.5 ± 0.06, n = 4, p ≤ 0.001, paired t-test). ***p ≤ 0.001. (H) Quantification of distance moved in ephrin-B2 group (Before: 0.6 ± 0.04; After: 0.4 ± 0.05, n = 4; p = 0.013, paired t-test). *p < 0.05. (I) Upper: Illustration of the focal application of activated ephrin-B. Lower: Illustration of the EphB signaling induced by exogenous ephrin-B (cyan). (J) An example of Retracting filopodia after ephrin-B treatment (cyan). Arrowheads indicate GPhosEphB signal. (K) An example of Connecting filopodia after ephrin-B treatment as in J. (L) Graph of GPhosEphB in Retracting (green) and Connecting (red) filopodia. Thin lines represent individual filopodial GPhosEphB signals. Means are thick lines. (M) Graph depicting the mean, variance and slope of individual filopodia that have made contact with presynaptic elements (green = Retracting, red = Connecting). Classifying filopodia using a trained linear kernel support vector machine (using held-out data) reveals that Retracting and Connecting filopodia can be correctly identified 91% the time (classification accuracy range across restarts = 83 – 100%). Scale bars = 2 μm. Error bars indicate SEM in Figure E–H. See also Figure S6 and Movies S10–S11.
The differences in EphB signaling in Retracting and Connecting filopodia suggest two possible models for how EphB controls the behavior of filopodia: 1. The decision might be controlled by the magnitude of the signal; or 2. The decision might be controlled by the rate of increase in the signal. To determine whether specific types of EphB signaling might cause certain filopodial behaviors, endogenous EphB receptor signaling was activated in GPhosEphB transfected neurons by focal application of fluorescently-labeled activated soluble ephrin-B (647-eB), and live-cell imaging was conducted (Figure 6I).
If a difference in magnitude of the EphB signaling is responsible for filopodial retraction or stabilization, we expect stronger activation of EphB signaling might induce filopodial retraction, whereas weaker activation of EphB signaling might induce filopodial stabilization. However, the differences in filopodial behavior were not likely to be driven by the magnitude of EphB signaling because the maximum increase in amplitude of GPhosEphB signal at 647-eB puncta was not significantly different between groups (Retracting: 138 ± 5%, n = 33; Connecting: 136 ± 7%, n = 20, p = 0.515, Mann Whitney U. Figures 6 and S6A–S6E). The average size of 647-eB puncta (0.28 ± 0.03 μm2) in Retracting filopodia was not significantly different from the average size of 647-eB puncta in Connecting filopodia (0.27 ± 0.03 μm2, p = 0.726, t-test), There was also no correlation between the size of 647-eB puncta and the GPhosEphB signal in either Retracting or Connecting group (Figure S6G). These findings suggest that ephrin-B concentration may not be related to cluster size. Regardless, while increases in EphB kinase activity are associated with changes in the motility of dendritic filopodia, the magnitude of EphB signaling alone cannot generate distinct filopodial behavior.
If the rate of EphB activation drives filopodial behavior, rapid increases in EphB activity should result in retraction while a slow increase in EphB activity should result in formation of a stable contact (Figure 5H). Consistent with our models, fast-rising EphB kinase activity caused filopodia to retract (Figure 6J, Movie S10), while slowly rising EphB kinase activity resulted in stable contacts between filopodia and 647-eB puncta (Time to the peak/Retracting: 1.4 ± 0.1 min, n = 31; Time to the peak/Connecting: 5.6 ± 1.1 min, n = 20, p ≤ 0.001, Mann-Whitney U, Figures 6J–6K, S6A–S6E, Movies S10 and S11). Moreover, the slopes of GPhosEphB signal in induced Retracting vs. Connecting filopodia were significantly different (Retracting: 0.29 ± 0.03, n = 31; Connecting: 0.2 ± 0.04, n = 20, p = 0.033, Mann-Whitney U, Figure S6F). Importantly, both the slopes and the time to peak in the ephrin-B-induced Retracting and Connecting groups were not significantly different from those found in neurons responding to endogenous ephrin-B cues (p > 0.1 for all comparisons, Mann-Whitney U, Figures 5 and 6). These data support the hypothesis that EphB activity within microdomains of filopodia determines filopodial behavior.
To test whether the EphB signaling governing filopodial retraction vs. connection resulting from the exogenous addition of ephrin-B was mediated by similar mechanisms as in the endogenous events, we asked whether the same unbiased classification criteria developed for classification of the endogenous EphB signals could be effective at classifying exogenously driven ephrin-B filopodial behavior. We found that a classifier trained on the endogenously activated GPhosEphB data was also effective at classifying exogenously activated GPhosEphB signals in filopodia (Figures 6L–6M, 98% endogenous vs. 91% exogenous classification efficiency with held-out validation; Exogenous eB Range 83.3–100%). These data strongly support the hypothesis that EphB activity within microdomains of filopodia determines filopodial behavior.
To test directly whether rapid activation of EphB2 is sufficient to induce filopodial retraction, we engineered a photoactivatable form of EphB2 by fusing the cryptochrome 2 (CRY2) domain of Arabidopsis thaliana to full-length FLAG-tagged EphB2 (fEphB2, Figure 7A). We generated several different constructs tagged with fluorescent proteins (mCherry or iRFP670) and fused with either a full-length CRY2 (EphB2-CRY2, (Kennedy et al., 2010)), an oligomerization promoting variant of CRY2 (EphB2-Oligo, (Taslimi et al., 2014)), or a truncated human codon-optimized version of CRY2 (EphB2-CRY2hm, (Chang et al., 2014)). Stimulation with blue light (440nm or 470nm) resulted in EphB2 kinase activation (Figures 7B, S7C and S7D). Kinase-dead versions (KD) of fEphB2-CRY2hm-iRFP670 (fEphB2-CRY2) showed no response to light stimulation or ephrin-B treatment (Figure 7C). fEphB2-CRY2 localized to the cell surface in neurons (Figures S7A and S7B). Consistent with published reports on CRY2 activation kinetics, photostimulation results in a rapid (15~30 sec) phosphorylation of fEphB2-CRY2 with similar activation rates regardless of photoactivation strength (Figures S7C–S7G, (Kim et al., 2014)). In contrast, ephrin-B treatment results in a slower activation of EphB kinase in neurons indicated by western blot (Figures S7C–S7G). Importantly, the kinetics of the rising phase of photoactivation of EphB2 is similar to the rate of EphB2 kinase activation found before filopodial retraction (Figures 5C and S7C–S7G).
Figure 7. Fast activation of EphB2 induces filopodial retraction.

(A) Design of photoactivatable EphB2. Xfp: fluorescent protein (mCherry or iRFP670). (B) Western blots of HEK293T cells transfected with EphB2-CRY2 variants and treated with activated ephrin-B2 (30 mins) or blue light (440nm, 1 min) (CRY2-cherry: fEphB2-CRY2-mCherry; Oligo-cherry: fEphB2-CRY2-Oligo-mCherry; CRY2hm-cherry: fEphB2-CRY2hm-mCherry; CRY2-iRFP670: fEphB2-CRY2-iRFP670) (C) Western blots of HEK293T cells transfected with wild-type (WT) or kinase-dead (KD) versions of fEphB2-CRY2hm-iRFP670 (fEphB2-CRY2) and treated with ephrin-B2 or blue light. (D) HEK293T cells transfected with WT or KD versions of fEphB2-CRY2 and imaged every 10 seconds. Photostimulation was conducted by a single scan with the 470 nm laser. Arrows indicate filopodia retracting after photostimulation. (E) Quantification of fEphB2-CRY2 transfected filopodia remaining after photostimulation (n = 12, * p = 0.013, ** p = 0.002, *** p ≤ 0.001, paired t-test). (F) Quantification of fEphB2-KD-CRY2 transfected filopodia remaining after photostimulation (n = 13, p > 0.05, paired t-test). (G) Neuron transfected with WT or KD versions of fEphB2-CRY2 and imaged every 10 seconds. Arrows as in D. Dashed lines show the border of photostimulated region. (H) Quantification of filopodia retraction after photostimulation (PHS) (p = 0.025, ANOVA; WT-PHS+: n = 21, vs. WT-PHS-: n = 11, p = 0.019; WT-PHS+: n = 21, vs. KD-PHS: n = 14, p = 0.036, post hoc: Fisher’s LSD test). (I) Quantification of the distance filopodia moved after photostimulation (WT-PHS+: n = 50 vs. WT-PHS-: n = 25, p = 0.025, t-test). Scale bars = 2 μm. Error bars indicate SEM in Figures E, F, H, I. See also Figure S7 and Movies S12–S13.
To determine the impact of rapid EphB2 activation, we first asked if EphB2 might mediate filopodial behavior in non-neuronal cells that contain dynamic filopodia. HEK293T cells were transfected with either fEphB2-CRY2 (WT) or fEphB2-KD-CRY2 (KD) and protected from room light exposure. Transfected cells were imaged every 10 seconds for 3–6 minutes before and after photoactivation of fEphB2-CRY2 with 470nm light. Photostimulation resulted in a significant loss of filopodia within 10 seconds after EphB activation in WT, but not in KD transfected cells (Figures 7D–7F, Movie S12). Photostimulation of EphB2 results in ~30% loss of filopodia by 90 seconds after stimulation (Remaining filopodia: 68 ± 4%, vs. 100%, n = 12, p < 0.001, paired t-test, Figure 7E). These findings suggest that rapid activation of EphB2 can drive filopodial retraction in HEK293T cells.
To determine whether rapid activation of EphB2 might cause retraction of filopodia in neurons, cortical neurons were transfected with fEphB2-CRY2 (WT) or fEphB2-KD-CRY2 (KD) and tdTomato and imaged every 10 seconds for 3–6 minutes at DIV9-10. Regions of interest in transfected neurons were stimulated with a single scan of 470nm light simultaneously with imaging (Region of interest: noise region outlined by the dashed line at time = 0, Figure 7G, Movie S13). There was no effect of photostimulation on filopodial movement or number in neurons transfected with KD (Figures 7G–7I, Movie S13). However, photostimulation of neurons expressing WT drove significant increase in the percentage of retracting filopodia (40 ± 8%, n = 21, in WT with stimulation vs. Ctrl: 12 ± 9%, n = 11, p = 0.019, Fisher’s LSD post hoc, Figures 7G–7H). Moreover, photostimulation of WT resulted in a significant decrease in the length of filopodia (p = 0.025, t-test, Figure 7I). These findings indicate that rapid activation of EphB2 drives the retraction of filopodia in HEK293T cells and neurons and suggest that differences in the kinetics of EphB signals within filopodia control the decision to retract from or stabilize at a contact.
EphB signaling in stable filopodia is linked to synaptic differentiation
We next asked whether filopodia making sustained contacts with axons had elevated EphB kinase activity. EphB kinase activity in Connected filopodia was persistently and significantly elevated above baseline (average signal in filopodia: 118 ± 3%, n = 22; average baseline signal in shaft: 100 ± 1%, n = 16, p ≤ 0.001, t-test, Figures 8A–8C, Movie S14). The level of GPhosEphB activation within Connected filopodia was similar to the levels found in Connecting filopodia 20 minutes after initial contact (Connected filopodia: 118 ± 3%, n = 22; Connecting filopodia: 126 ± 6%, n = 12, p = 0.179, t-test). These findings suggest that sustained EphB kinase signaling may be associated with long-lasting axo-dendritic contacts. Consistent with this hypothesis, elevated EphB kinase signaling was also found at sites of contact between a dendritic shaft and axon (Figures S8A–S8C), where many synapses form in immature neurons (Kayser et al., 2006; Kayser et al., 2008).
Figure 8. EphB signaling is elevated in stable filopodia and colocalized with VGLUT1 puncta.

(A) An example of Connected filopodia (axonal contact maintained for >30min) contacting an mT2 labeled axon. Dashed lines show the morphology of axons. Arrowheads indicate GPhosEphB signal. (B) Quantification of GPhosEphB signal in A. (C) Quantification of GPhosEphB signal in the pooled data set (ROI: n = 22, indicated by arrowheads; Ctrl: n = 16, p ≤ 0.001, K–S Test). (D) Arrowheads indicate a stable filopodium. Images of RFP channel at the last frame of 30-minute movie and after fixation. (E) Images of GPhosEphB signal in the filopodium in D. (F) The same filopodium as in D and E shown overlaid with VGLUT1 staining (white). Dashed lines show the morphology of the transfected neuron. Arrowheads indicate GPhosEphB signal colocalized with VGLUT1. (G) As in D, arrows point to a stable filopodium. (H) Images of GPhosEphB signal in the filopodium in G. (I) As in F, the filopodium was shown overlaid with staining of VGLUT1. (J) Quantification of GPhosEphB signal in the VGLUT1+ filopodium from D–F. (K) Quantification of GPhosEphB signal in the VGLUT1− filopodium from G–I. (L) Average GPhos signal in VGLUT1+ filopodia was significantly higher than that in VGLUT1− filopodia (VGLUT1+: 1.30 ± 0.05, n = 36; VGLUT−: 1.10 ± 0.04, n = 20, p = 0.005, t-test). Scale bars = 2 μm. ** p < 0.01. Error bars indicate SEM in Figures C and L. See also Figure S8 and Movies S14–S15.
Since EphB2 puncta are found in stable filopodia with functional release sites (Figure 2), we next sought to determine whether stable filopodia with elevated EphB activity are more likely to colocalize with presynaptic markers than stable filopodia with low levels of EphB activity. Retrospective analysis of GPhosEphB transfected neurons stained with the presynaptic marker VGLUT1 after live imaging (see Methods for experimental detail), indicates that EphB signals are significantly higher in stable filopodia that colocalized with VGLUT1 puncta than in stable filopodia that did not colocalize with VGLUT1 puncta (VGLUT1-: 110 ± 5 %, n = 20; VGLUT1+: 130 ± 4 %, n = 36, p = 0.005, t-test, Figures 8D–8L, Movie S15). These findings suggest that stable elevated levels of EphB kinase activity are found in filopodia at synaptic sites. Occasionally we were able to observe moving filopodia that became stable during our imaging period (Figures S8D–S8G). Interestingly, these filopodia tended to be stabilized at sites that also colocalize with VGLUT1 puncta after reconstruction (Figure S8F). These findings are consistent with the model that sustained EphB activity results in Connected filopodia and suggest that sustained EphB activity within stable filopodia may drive the decision of a filopodium to initiate synaptogenesis.
DISCUSSION
In this study, we find that EphB2 RTKs are positioned and signal to enable selection of appropriate synaptic contacts between neurons. EphB receptor tyrosine kinases are important for both synapse formation and the movement of dendritic filopodia (Kayser et al., 2008) and are clustered on the surface near the tips of filopodia. In stable filopodia EphB2 colocalizes with ephrin-B1, VGLUT1, and synaptic release sites, indicating that EphBs are likely to initiate the formation of nascent synaptic contacts. Interestingly both moving and stable filopodia contain clusters of EphB receptors, suggesting that localization of EphB proteins cannot explain the difference in filopodial behaviors. Visualization of the dynamics of EphB signaling in living neurons reveals distinct kinetics in filopodia that behave differently. Fast-rising EphB signaling is correlated with filopodial retraction whereas slow-rising EphB signaling is found in filopodia that make stable contacts. Both retraction and stabilization require a certain similar threshold of activation. Stable filopodia with slightly elevated levels of EphB activity appear to be nascent synaptic sites. These data suggest that the dynamics of EphB kinase activity are linked to the behavior of filopodia and the decision to generate a synaptic contact. Thus, the process of partner selection by individual filopodia could be reduced to the integration of signaling by a single tyrosine kinase, EphB2, illustrating how local differences in signal kinetics can provide a model that may simplify the complex process of target selection and synaptogenesis.
EphB receptor signaling and filopodial movement
It has long been appreciated that Eph-ephrin binding can induce repulsion and attraction (Flanagan and Vanderhaeghen, 1998). Unlike other cell adhesion molecules such as Netrins, which drive repulsion or attraction through binding of different receptors (Hong, 1999; Hopker et al., 1999), EphB-ephrin-B binding induces repulsion or attraction via the same receptor (McLaughlin et al., 2003; Zimmer et al., 2003). Previous work suggests that EphB functions are associated with differences in the amplitude of the EphB signals. For instance, in the tectum, EphB expressing interstitial branches of retinal ganglion cells can be repelled by high concentrations and attracted by low concentrations of ephrin-B1 (McLaughlin et al., 2003). It remains unclear, however, how EphB activation plays different roles in repulsion and attraction. Our data indicate that EphB signaling can occur much faster than previously known and that the rate of change in EphB signal rather than the absolute magnitude, appears to enable the cell to discriminate differences, which allow for the initiation of distinct cellular behaviors.
During contact-mediated synaptogenesis, EphB signaling mediates both filopodial retraction and synapse formation (Kayser et al., 2008). Our data suggest that distinct EphB signaling in moving dendritic filopodia controls the decision of filopodia to retract or stay connected to an axonal partner. EphB kinase activation reached similar levels in both retracting and connecting filopodia, suggesting that the decision of a filopodia to retract or stay in contact with an axon is not solely dependent on the level of EphB activation. Instead, filopodial retraction appears to be driven by rapid increases in EphB signals, while stabilization of contacts is generated by slow-rising signals. Consistent with this model, slow EphB activation by bath application of ephrin-B reduces filopodia motility, while fast EphB activation driven by photostimulation induces retraction. Photoactivation of EphB2 occurs over ten to fifteen seconds, suggesting that to fully resolve the kinetics of EphB signaling in filopodia, EphB kinase activity need to be imaged at a faster rate. These experiments may reveal additional kinetic components to decision making in filopodia.
Whether a filopodium retracts or stabilizes appears to be determined by kinetically distinct EphB signals. This model is supported by the findings that simple differences in the rate of EphB kinase activation are sufficient to drive distinct modes of filopodial behavior. Indeed, our data suggest that correct connections between dendrites and the appropriate axons are selected by the presence of two types of EphB-specific signaling. This model suggests that in cases where EphB signaling is blocked, inappropriate EphB-ephrin-B interactions would fail to be rejected and incorrect connections that are normally lost would be maintained; however under these circumstances, synapse formation principally mediated by the EphB-ephrin-B interactions, would be intact. These data are consistent with recent findings that downregulating EphB kinase activation while maintaining normal EphB-ephrin-B interactions has little impact on synapse number (Soskis et al., 2012). In these animals, we predict that EphB kinase blockade would result in defective target selection and generate defects in the specificity of synaptic connections. It will be important to test whether this is the case.
How are differences in the rate of these signals controlled? For calcium signaling, chelation of calcium ions controls both the duration and spatial extent of the signal (Augustine et al., 2003; Higley and Sabatini, 2012). Phosphatase activity may provide a similar constraint on intracellular EphB kinase signaling. While work has focused on the function of a number of phosphatases in presynaptic differentiation (Takahashi and Craig, 2013), evidence for phosphatase activity (Pten, PP1) has also been linked to the regulation of filopodial motility and synapse formation (Fuentes et al., 2012; Luikart et al., 2008; Terry-Lorenzo et al., 2005). However, it is not clear whether these phosphatases might regulate EphB kinase function in filopodia. While phosphatases are an attractive candidate for the regulation of EphB signaling, it is also possible that regulation of EphB activity is accomplished through another mechanism, such as receptor cleavage, endocytosis, or cis-binding to ephrin ligands (Kania and Klein, 2016). Significant effort will be needed to determine the mechanism responsible for regulation of kinetics of EphB activity in filopodia.
EphB receptor and the initiation of synapse formation
During development, dendritic filopodia are involved in establishing axo-dendritic contacts, where nascent synapses can form within 30 minutes of initial contact (Friedman et al., 2000; Jin and Garner, 2008). Consistent with previous findings in EphB triple knockout animals and using shRNA knockdown (Henkemeyer et al., 2003; Kayser et al., 2006; Kayser et al., 2008), our data indicate that EphBs are positioned and signal within neurons to control these key early steps in synaptogenesis and suggest that EphB signaling is linked to the choice of individual filopodia to differentiate into synapses.
To initiate new synaptic contacts, transsynaptic synaptogenic factors need to be positioned at the sites of contact between dendrites and axons. EphB2 appears to regulate both the stabilization of contacts and filopodial movement, providing a cue that may control the process of synaptic selection. Previous work indicates that EphB2 is required for ~ 40% of excitatory contacts made in cortex and functions postsynaptically by binding to its presynaptic ligand ephrin-B1 (Kayser et al., 2008; McClelland et al., 2009). Our super-resolution imaging indicates that as synapses are forming, EphB2, ephrin-B1, and VGLUT1 colocalize in ~30% of filopodia. In addition, live-cell imaging experiments indicate that stable filopodia that contain EphB2 colocalize with functional synaptic release sites at a similar ratio and filopodia with elevated EphB kinase activity were significantly more likely to be found at synaptic sites. In filopodia that extend and make a stable contact with an axon, EphB signaling increases slowly and becomes sustained. This may enable recruitment of VGLUT1+ synaptic vesicles (Figure S8). Thus, EphBs localized in dendritic filopodia appear to actively signal to generate functional synaptic contacts. Then through the well-established direct extracellular interactions between EphBs and the NMDAR and the intracellular interactions with adaptor proteins that bind to AMPARs, such as GRIP (Dalva et al., 2007; Kayser et al., 2006; Sheffler-Collins and Dalva, 2012), EphB could then recruit additional synaptic proteins leading to the maturation of synapses.
Development of genetically encoded TK indicators
Despite the importance of TKs for cell signaling and adaptive plasticity, there is little known regarding the dynamics of TK activity within living cells. GPhos indicators report EphB and EphA activity selectively, suggesting that the tool can selectively allow visualization of the dynamic TK signaling, enabling live-cell TK biochemistry. For instance, the activation kinetics of Eph kinases have long been thought to be slow (> 15 min), however, the GPhosEphB indicator demonstrates that local changes in kinase activity occur rapidly (≤ 1 min). It will be important to determine the local kinetics of other TKs using GPhos based reporters.
FRET-based genetically encoded indicators of kinase activity have been developed for monitoring ERK, Src, PKA, PKB, and PKC activities (Oldach and Zhang, 2014). Our indicators adapt the core of circularly-permutated EGFP similar to GCaMP (Chen et al., 2013; Looger and Griesbeck, 2012; Nakai et al., 2001), flanked with the phosphorylation sites from specific kinases of interest. With GPhos, changes in kinase activity are visualized as increases in fluorescence intensity, making imaging of our probe relatively straightforward. Considering cell morphology, focus drift, background noise, and variance in transfection, we introduced a second fluorescent protein and fused it via a rigid alpha-helical linker into the indicator core. The GPhos dual-color system enables the normalization of changes in fluorescent intensity (GPhos) to stable baseline fluorescence (mRFP). This approach effectively attenuates variability and could be applied to other indicators, including those used for calcium.
Signaling microdomains of tyrosine kinases
Little has been understood about the impact of the timing and spatial localization of kinase activity in the control of specific cellular behaviors. Our data provide an example of how differences in signaling kinetics of a kinase can result in distinct cellular outcomes. Eph signaling can drive a host of different cellular events from actin depolymerization, resulting in repulsion and growth cone collapse, to synapse formation and control of NMDAR synaptic localization (Kania and Klein, 2016; Sheffler-Collins and Dalva, 2012). Yet in each of these pathways, there appears to be little crosstalk. How can a single molecule function selectively in multiple downstream pathways within the postsynaptic complex? For EphB signaling in filopodia, it appears that specificity of signaling may be encoded in part by restricting the signal to a small domain or subregion of the cell. Thus, rather than generating large-scale, cell-wide changes, EphB signaling generates specificity by being both spatially and temporally restricted. We propose that distinct cellular behaviors driven by a single signaling protein can be controlled by differences in the pattern, localization, and timing of kinase signaling. Thus similar to calcium (Augustine et al., 2003; Higley and Sabatini, 2012), microdomains of kinase activity within cells may be essential to generate specificity of tyrosine kinase signaling.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact Matthew B Dalva (matthew.dalva@jefferson.edu).
Experimental Model and Subject Details
Animals
All animal studies were performed according to the Institutional Animal Care and Use Committee guidelines at Thomas Jefferson University. Triple kinase knock-in (TKI) mice with point-mutations to the kinase domains of EphB1, EphB2, and EphB3 were maintained on a 12 hr light/dark cycle and accommodated in breeding pairs (Soskis et al., 2012). E17-18 time pregnant Long Evans rats and wild-type CD-1 mice were purchased from Charles River Laboratories.
Rat cortical neuronal culture
Unless indicated specifically, cultured cortical neurons were used throughout the paper. Cortical neuron cultures were generated from E17-18 Long Evans rats as previously described (Dalva et al., 2000; Kayser et al., 2006). Embryos were harvested at E17-18 and brains were isolated in ice-cold HEPES buffered (20 mM) Hank’s balanced salt solution (HBSS). Meninges were removed using fine forceps. The striatum and hippocampi were separated and discarded and cortices were collected. Cortices were incubated with 10 u/ml papain (Worthington Biochemical Corporation) in HBSS for 2–5 minutes at 37°C. After three washes in HBSS with 0.01 g/ml trypsin inhibitor (Sigma), the corticies were gently triturated with fire-polished glass Pasteur pipette 10–15 times to obtain a homogenous cell suspension. Bubbling was avoided and the pipette was maintained within the cell suspension during the trituration. Dissociated neurons were plated on poly-D-lysine and laminin (both from BD Biosciences) coated glass coverslips (12mm, 150–200K/coverslip), 35mm glass dishes (250K/dish), 6-well plates (2M/well), or 10 cm dishes (8M/dish). Neurons were cultured in Neurobasal medium (Invitrogen), supplemented with B-27 supplement (Invitrogen), glutamine (Sigma), and penicillin/streptomycin (Sigma), and maintained in a humidified incubator with 5% CO2 at 37°C (Kayser et al., 2006).
Mouse cortical neuronal culture
Mouse neurons were dissociated from brains of newborn pups (postnatal day 1) with knock-in mutations of EphB1, EphB2, and EphB3 (Soskis et al., 2012). The mouse neuron dissociation protocol was adapted from (Hilgenberg and Smith, 2007; Kayser et al., 2008). Dissection of mouse brains was similar to that for rat brains. To improve neuronal survival, a dissecting solution was used as described (Hilgenberg and Smith, 2007). After brain dissection and incubation with papain, mouse cortices were washed with 2 HI (high inhibitor solution, 10 mg/ml trypsin inhibitor, 10 mg/ml BSA, 50 μM APV) and 3 LI (low inhibitor solution, 1 mg/ml trypsin inhibitor, 1 mg/ml, BSA, 50 μM APV). Cortices were triturated 4–5 times through a fire-polished Pasteur pipette. The cell suspension was spun at 200 rpm for 30 seconds at 4°C. The supernatant was transferred into a new tube and spun at 800 rpm for 5 minutes at 4°C. The pellet was resuspended in Neurobasal medium with the supplements as described above. Neurons were plated on 35mm glass dishes (500K/dish), 6-well plates (2M/well), or 10 cm dishes (8M/dish).
HEK 293T cell culture
HEK293T cells were grown in Dulbecco’s Modified Eagle Medium (Invitrogen), 10% fetal bovine serum (Hyclone, Atlanta Biologicals), penicillin/streptomycin, and glutamine.
Method Details
Generation of the GPhos indicator core
The indicator (GPhos) consists of a single circularly-permutated GFP core adapted from GCaMP (Nakai et al., 2001), two N-terminal consensus phosphorylation sites, and a C-terminal relatively promiscuous SH2 domain (Figure 3A) (Songyang et al., 1993). Each fragment was fused by overhang restriction enzyme sites (Figure 3A). With the available updated versions of GCaMP, we have continually upgraded our GPhos core with GCaMP6 (Chen et al., 2013). In the following description, uppercase indicates sequence of the targeted protein and lower case indicates bases added for cloning purposes. For GPhosEphB and GPhosEphA, the selected sites were from the juxtamembrane tyrosine phosphorylation domain of EphB2 (AA573-582) and EphA4 (AA596-605). The amino acid sequence used for the GPhosEphB indicator was gsYIDPFTYEDPag, encoded by the forward primer: 5′-gatctggatccTATATAGACCCTTTCACCTATGAAGATCCTgccggcc -3′. For the GPhosEphA indicator, the amino acid sequence used was gsYVDPFTYEDPag, encoded by the forward primer: 5′-gatctggatccTATGTGGATCCCTTTACATACGAAGACCCCgccggcc -3′. For the GPhosFyn indicator, the amino acid sequence used was gsYEEIVGEFKIYEEImgle, encoded by the primer sequence 5′-ggatccTACGAGGAGATCGTTGGTGAAATCTACGAAGAGATCatgggcctcgag -3′. Generation of the GPhos constructs was accomplished by annealing the appropriate forward and reverse primers to yield DNA fragments with overhangs for insertion into BglII and XhoI sites.
In addition, a unique BamHI was added to each primer to facilitate future cloning and enable rapid validation of appropriate insert integration. After phosphorylation, DNA fragments were cloned into the BglII and XhoI sites using standard techniques (Figure 3A). Notably this approach enabled us to easily change the phosphorylation sites on the indicator. The second element of the GPhos indicator is the domain that selectively binds the phosphorylated tyrosines. For the initial indicators, we used the Fyn SH2 domain. We first generated primers to amplify a region from AA131 to AA255 of the mouse Fyn kinase. This region contains the SH2 domain, flanked by 17 N-terminal amino acids and 12 C-terminal amino acids. N-terminal primers contained an MluI site while C-terminal primers had a NotI site to enable cloning into the vector. Again, to facilitate future cloning and enable rapid validation of insert integration, we added an AgeI site before the NotI site in the C-terminal primer.
Generation of the dual color indicator
To generate the rigid linker connecting the second fluorescent protein to the GPhos indicator, we used a DNA fragment encoding 5-alpha helical turns. The amino acid sequence used to generate the rigid linker was LAEAAAKEAAAKEAAAKEAAAKEAAAKAAA (Arai et al., 2004). Forward and reverse primers were designed with 5′ BglII and 3′ BamHI overhangs, annealed together, then cloned into the GPhos vector via the BglII and BamHI sites. A unique SalI site was added at the 5′ end after the BglII site to enable cloning of the fluorescent molecule. The primers to amplify full-length mRFP contained a 5′ BglII and 3′ SalI site. After PCR, the fragment was ligated to the N-terminus of the GPhos core via BglII and SalI sites in front of the 5-alpha helical linker. After introducing the second fluorescent molecule and modification, we failed to detect any FRET (not shown), suggesting that dual-color ratiometric indicators are resistant to FRET.
Generation of the photoactivatable EphB2
To generate photoactivatable EphB2, FLAG-tagged EphB2 (fEphB2) (Dalva et al., 2000) was cloned into the pDONR 221 vector (Gateway Cloning, Invitrogen) to make the entry clone pENTR fEphB2. Then three restriction enzyme sites (MfeI, BamHI, NdeI) were introduced before the EphB PDZ binding domain by site-directed mutagenesis. The location of the restriction enzyme sites was chosen based on the location of YFP in our EphB2-YFP construct (Kayser et al., 2006). CRY2 (Arabidopsis cryptochrome 2)-mCherry was cloned by PCR with primers flanking the CRY2-mCherry sequence with MfeI and NdeI sites. The PCR fragment was digested, cut, and ligated into pENTR-fEphB2 with a ligation kit (Roche Applied Science) (Figure 7A). Then the pENTR-fEphB2-CRY2-mCherry was cloned into the destination vector by Gateway recombination to generate the expression clone pFUG-fEphB2-CRY2-mCherry (fEphB2-CRY2). Consistent with a previous finding that CRY2 homo-oligomerizes after blue light exposure (Chang et al., 2014; Duan et al., 2017), we also successfully induced EphB2 phosphorylation of fEphB2-CRY2 with blue light. Laser lines of 440, 470 and 530nm were tested. As expected only the 440nm and 470nm lasers were able to induce phosphorylation of EphB2 fused with various CRY2 domains. We first developed a number of photoactivatable EphB2 constructs based on different available versions of CRY2 and validated each of them (Chang et al., 2014; Taslimi et al., 2014). After testing fEphB2-CRY2 (Kennedy et al., 2010), fEphB2-CRY2-Oligo (Taslimi et al., 2014) and fEphB2-CRY2hm (Chang et al., 2014), we found that, while all three were effectively photoactivated (Figure 7B), fEphB2-CRY2hm generated the most consistent results. To make fEphB2-CRY2hm compatible with transfection of tdTomato in neurons, we replaced mCherry in fEphB2-CRY2hm with iRFP670 (Shcherbakova and Verkhusha, 2013), which can be excited with 633nm laser light. fEphB2-KD-CRY2hm was generated from FLAG-tagged EphB2-KD (Dalva et al., 2000) using the same approach as described above for fEphB2. Then, fEphB2-CRY2hm-iRFP670 and fEphB2-KD-CRY2hm-iRFP670 were used for photostimulation in HEK cells and cultured neurons.
Expression Constructs
EphB2-YFP, FLAG-EphB2, FLAG-EphA4, FLAG-EphB1, EphB3, EphB2DN, FynDN, FynCA were generated and used previously (Dalva et al., 2000; Kayser et al., 2006; Takasu et al., 2002). mTurquoise2-ephrinB1 was generated by inserting mTurquoise2 (a gift from Dr. Joachim Goedhart (Goedhart et al., 2012) after the HA tag of HA-ephrin-B1(McClelland et al., 2009) using restriction enzyme sites and a ligation kit (Roche Applied Science). tdTomato and mRFP were gifts from Dr. Roger Tsien (Campbell et al., 2002). mNeptune (a gift from Dr. Michael Lin) (Zhou et al., 2012) and the GCaMP core domain (Chen et al., 2013) were obtained from Addgene. Using Gateway recombination (Invitrogen), DNA fragments were cloned into a pFUG vector containing a human ubiquitin promoter (hUb) (Hruska et al., 2015), a pENTR vector containing a Synapsin promoter (a gift from Dr. Peter Scheiffele), or a vector containing the pCAG promoter (a gift from Dr. Artur Kania). Both the pENTR vector and pCAG vector were converted to be Gateway compatible. A fragment of the ccdB gene (inhibiting E. coli growth) for negative selection, flanked by 5′ attP5 and 3′ attP4 sites for specific recombination, was cloned downstream of the promoters.
HEK cell transfection
HEK293T cells were transfected using the calcium phosphate precipitation method (Xia et al., 1996). Briefly, The pH of HeBS (274 mM NaCl, 10 mM KCl, 1.4 mM Na2HPO4.7H2O, 15 mM D-glucose, 42 mM HEPES) was adjusted by NaOH to yield pH from 7.03, 7.05, 7.07, 7.09, 7.11, to 7.14. HeBS was filter-sterilized, aliquoted and stored at −20°C. Each pH was tested to determine the one providing the best transfection efficiency. To prepare transfection mixture, 100 μl of HeBS with the most effective pH was added in an Eppendorf tube. In a different tube, 10 μl 2.5M CaCl2 was diluted to 90 μl HEPES-buffered ddH2O (2.5 mM HEPES). Then DNA was added into to CaCl2-HEPES tube, mixed by pipetting 6–8 times and added dropwise to the HeBS tube to initiate precipitation. The mixture was bubbled 6–8 times and added on HEK cells (one well in 6-well plate) dropwise. Transfected cells were imaged or lysed 16–24 hours after transfection.
Transfection of neuronal culture
Neurons were transfected using Lipofectamine 2000 (Invitrogen), as described previously (Kayser et al., 2006; McClelland et al., 2009). Transfection mixture was prepared (for two coverslips or one 35mm glass-bottom dish) as following: 2 μl Lipofectamine 2000 was added in 100 ul neurobasal medium (plain) in a polystyrene tube (USA scientific). DNA was added to 100 μl neurobasal medium in an Eppendorf tube. After 10 minutes, DNA mixture was slowly added to Lipofectamine mixture dropwise. The total 200 μl mixture was bubbled 3–5 times and incubated at room temperature for another 10 minutes. Before adding the mixture on neuronal culture, conditioned media of neuronal culture was replaced with warm plain neurobasal medium (300 ul for one well in 24-well plate and 800 ul for one 35mm glass-bottom dish) and kept in water bath at 37°C. The neuronal culture was incubated with the transfection mixture for 2 hours. Then transfection media was replaced with filter-sterilized warm conditioned media and the neuronal culture was returned to the incubator. For single transfection, neurons were transfected on DIV3-7 or DIV0 and imaged DIV7-10. For sequential transfections, neurons were first transfected with mTurquoise2 or mT2-ephrin-B1 on DIV3-5, then maintained in an incubator at 37°C. Two days later at DIV5-7, the same neurons were transfected with the second construct, GPhosEphB. These two transfections normally resulted in different sets of neurons. Each set of neurons was transfected with only one construct. Live-cell imaging was conducted on DIV7-10. GPhosEphB signal in dendrites and dendritic filopodia was imaged at the sites in contact with axons expressing of mT2 or mT2-ephrin-B1. In the rare cases that neurons were cotransfected with both constructs (mTurquoise2 and GPhosEphB, or mT2-ephrin-B1 and GPhosEphB), they were excluded from imaging.
FM dye labeling
To examine cultured rat cortical neurons (DIV7-10), conditioned media was replaced with ACSF (140 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 20 mM glucose, and 10 mM HEPES, pH 7.2) and neurons were incubated with FM4-64 (Molecular Probes, 10 μM) for 15 minutes. After three 10 minute rinses with ACSF, neurons were imaged for 30 minutes using a Leica SP5 Confocal microscope. The first frame was used to represent the loaded image. During the 30 minutes imaging time, FM4-64 was spontaneously unloaded. A final wash was given and a Z-stack image was taken to represent the background image (Figure 2A) (Wilhelm et al., 2010). The loaded image was aligned and subtracted with the background image in ImageJ (NIH). The subtracted image was thresholded to show FM4-64 puncta and merged with the EphB2-YFP channel of the loaded image to show the colocalization of EphB2-YFP and FM4-64 puncta. The morphology and movement of dendritic filopodia were determined by cell-filling mTurquoise2. The EphB localization was determined by EphB2-YFP clusters.
Antibodies for immunostaining
The following primary antibodies were used for immunostaining: goat anti-EphB2 (1:200–1:500, R&D), mouse anti-EphB2 (1:200–1:500, Invitrogen), mouse anti-HA (1:1000, BioLegend), mouse anti-FLAG (1:2000,Sigma), rabbit anti-actin (1:1000, Sigma), goat anti-ephrin-B1 (1:200, R&D systems), guinea pig anti-VGLUT1 (1:5000, Millipore). Secondary antibodies Atto 425, Dylight 488 or Cy2, Dylight 647 or Cy5, and Cy3 from Jackson ImmunoResearch, Rockland or Abcam were used from 1:200 to 1:500.
Immunostaining and live-cell surface staining
Cultured rat neurons were washed once with ACSF. Then neurons were fixed with 4% paraformaldehyde and 2% sucrose for eight minutes. Neurons were washed three times with PBS and blocked with 1% ovalbumin and 0.2% cold water fish gelatin for one hour with 0.01% Saponin (Sigma). Next neurons were incubated with primary antibodies for 1–2 hours. After three washes with PBS, neurons were incubated with secondary antibodies for 45 minutes. Finally, neurons were washed three times with PBS and mounted with Mowiol (Valnes and Brandtzaeg, 1985).
For live-cell staining, live cultured rat neurons were incubated with primary antibodies at 37°C, 5% CO2 for eight minutes (Hanamura et al., 2017; Lander et al., 1998; Perez de Arce et al., 2015). After one brief wash with warm ACSF, neurons were fixed with 4% paraformaldehyde and 2% sucrose for 8 minutes. Neurons were washed three times with PBS and blocked with 1% ovalbumin and 0.2% cold-water fish gelatin for one hour without detergent (to prevent permeabilization). Neurons were then incubated with secondary antibody for 45 minutes. Finally, neurons were washed three times with PBS and mounted with Mowiol. Some neurons were blocked again with blocking buffer containing Saponin (to permeabilize membranes) and then stained with the same primary antibody but a different secondary antibody to visualize the total expression of the protein. In acid-stripping experiments, after incubation with primary antibody, neurons were placed on ice and washed once with ice-cold ACSF and then incubated with 0.2M acetic acid/0.5M NaCl for 10 minutes (Carroll et al., 1999). Cells were washed with ice-cold PBS three times before fixation. Then the neurons were fixed, blocked, incubated with the secondary antibody, washed and mounted as described above.
Treatment of the cells
To block EphB activity, TKI mouse neurons were treated with 1-NA-PP1 (Cayman Chemical). 1-NA-PP1 was first dissolved in DMSO and then diluted in ACSF to a final concentration of 1 μM. The same amount of DMSO without 1-NA-PP1 was diluted in ACSF and added to neurons to test whether DMSO would affect imaging. Considering the minimal amount of DMSO (0.25 ul in 1 ml ACSF), we did not find any change in GPhos signal (data not shown). TPA or PP2 (Sigma) was diluted in ACSF to a final concentration of 100 nM (TPA), 3 μM (PP2). Mouse ephrin-B2-FC, ephrin-B1-FC, ephrin-A1-FC or FC (R&D Systems) was clustered with anti-human IgG FC (Jackson ImmunoResearch) and then diluted in ACSF or neurobasal to a final concentration of 250 ng/ml. For local application, glass pipettes were pulled by Flaming/Brown micropipette puller (tip opening ~ 3–5 μm) and filled with clustered ephrin-B-FC.
Micromanipulator (Narishige) controlled the fine positioning of the glass pipette near the imaged neuron (20–50 μm). Ephrin-B-FC was given by gravity.
When HEK293T cells and neurons were prepared for photostimulation, after transfection, cells were protected from all light until imaged and care was taken to search for transfected cells using wavelengths that would not drive photoactivation (>530nm). For initial validation of EphB2 photoactivatable constructs, dishes of transfected HEK293T cells were exposed to 440 nm laser light for 1 minute. The effects of light intensity on the kinetics of EphB phosphorylation were tested using 470nm (3mW, DC2100, Thorlabs) and 440 nm (25 mW, PSU-III-FDA, CNI laser) blue light because CRY2 can be activated effectively with these wavelengths (Kennedy et al., 2010). Finally, HEK cells were illuminated with 470 nm light for 5s and then lysed at 0.25, 0.5, 1, 2, 15 minutes or 440 nm light for 15s and then lysed at 0.25, 0.5, 1, 2, 5, 15 minutes. Each experiment was repeated > 3 times.
Synaptosome fractionation preparation
Synaptosome fractionation was conducted as previously described (Gurd et al., 1974; Hanamura et al., 2017; Hruska et al., 2015). Briefly, P21 CD-1 mouse brain was homogenized on ice in HEPES–buffered sucrose (0.32 M sucrose, 4mM HEPES, pH7.4), containing fresh protease inhibitor and PMSF. The homogenized sample was centrifuged at 1000g for 15 minutes at 4°C, and the supernatant (S1) was centrifuged at 10,000g at 4°C to yield the crude synaptosomal pellet (P2). P2 was re-suspended in the HEPES-buffered sucrose, and centrifuged again to yield the washed crude synaptosomal fraction (P2′). P2′ was lysed again by hypoosmotic shock using ice cold H2O plus protease inhibitors. P2′ was centrifuged at 25,000g to yield a supernatant (S3) and a pellet (P3). P3 was re-suspended and spun through a sucrose gradient (0.8M, 1.0M and 1.2M sucrose). Synaptic Plasma membranes (SPM) was recovered from the layer between 1.0 and 1.2M sucrose. SPM was re-suspended, added Triton X-100 to 0.5% and centrifuged at 32,000g to yield the PSD-1T pellet. The PSD-1T pellet was re-suspended, added TritionX-100 to 0.5% and centrifuged at 200,000g to yield the PSD-2T pellet. In a separate step, the PSD-1T was incubated with ice-cold 3% sarcosyl and centrifuged at 200,000g to yield PSD-S pellet. The protein concentration of all fractions was measured using DC™ protein Assay (Bio-Rad). An equal amount of protein was denatured with boiled Laemelli sample buffer and loaded in each lane for Western blotting. Primary antibodies used in this experiments are mouse anti-PSD95 (K28/43, 1:5000, Neuromab), rabbit anti-NR1 (GluN1) (1:500, Millipore), Rabbit anti-VAMP 1/2/3 (1:2500, Synaptic Systems), mouse anti-synaptophysin (1:5000, Synaptic Systems) and goat anti-ephrinB1(1:200, R&D systems).
Immunoprecipitation and Western Blotting
Cells were washed once with ice-cold PBS and lysed in RIPA buffer (20mM Tris-Cl pH 7.5, 140 mM NaCl, 2 mM EDTA) containing fresh 10 mM NaF (Sigma), 1% NP40 (Thermo Scientific), 0.5% C24H39NaO4 (sodium deoxycholate), 1mM Na3VO4, 1 mM PMSF and proteinase inhibitor cocktail (all from Sigma) (Dalva et al., 2000). Lysates were centrifuged at 14,000 rpm for 25 minutes at 4°C. The supernatant was incubated with primary antibody (rabbit anti-GFP, Invitrogen; goat anti-EphB2, R&D systems; mouse or rabbit anti-FLAG, Sigma) for an hour. Then pre-incubation protein G agarose beads (blocked with 1% BSA in RIPA buffer) were added (Invitrogen) and rotated at 4°C for an hour. The beads were then washed three times and denatured with boiled Laemelli sample buffer. The immunoprecipitated proteins and lysates were separated on an SDS-PAGE gel (6–15% gel depending on the molecular weight of the protein of interest) and transferred to PVDF membrane (Millipore). Immunoblots were blocked and probed for phosphorylated tyrosines using mouse monoclonal anti-p-Tyr (PY99, 1:100, Santa Cruz), or rabbit anti-p-EphB (p*B2, 1:1000, (Dalva et al., 2000)). The loading controls were probed for rabbit anti-GFP (1:2500, Invitrogen), goat anti-EphB2 (1:500, R&D Systems), mouse anti-EphB2 (1:500, Invitrogen), or mouse or rabbit anti-FLAG (1:1000, Sigma). Horseradish peroxidase-conjugated secondary antibodies (anti-mouse and anti-rabbit, Jackson ImmunoResearch) were used at 1:10,000. HRP-conjugated anti-goat secondary antibody (R&D Systems) was used at 1:2000. Western Lightning® Plus-ECL (PerkinElmer, Waltham, MA) was used for detection of HRP activity.
Image acquisition
Images were collected using a Leica SP2, Leica SP5, Leica SP8, or a Leica Confocal microscope equipped with a Yokagowa spinning disk CSU 10. Confocal images were collected as Z-stacks at 0.3 to 0.5 μm intervals. Sequential scanning was adapted to avoid bleed-through artifacts. Super-resolution STED images were collected at a single focal plane using a Leica TCS SP5. Images were processed using Leica Application Suite Advanced Fluorescence Software (LAS AF software) and ImageJ (NIH) (Hruska et al., 2015; Kayser et al., 2008). For live-cell imaging, neurons or HEK cells were removed from the incubator and placed in ACSF. Before stimulation or application of reagents, cells were imaged for 10 to 30 minutes to obtain baseline measurements. Images were acquired every 0.1 to 3 minutes as either single focal planes or Z-stacks. For neurons, only cells with pyramidal shape were chosen to be imaged. Both fluorescent light and laser light were kept at low levels to avoid photobleach. Stable or Connected filopodia were defined as those that moved less than 1 μm/frame during the 30 minute imaging period (Kayser et al., 2008). For photostimulation, one scan with the 470nm laser line was applied to the whole field during live-cell imaging of HEK cells or to an ROI during live-cell imaging of neurons. The output of each scan = 10 uW. Images were collected every 10 seconds in a single focal plane for HEK cell imaging and in a 3–4 frame Z-stack for neuronal imaging.
Registration of live-cell imaging and images after fixation
First, cultured DIV7-10 neurons were imaged for 30 to 60 minutes. The location of each imaged neuron was recorded using a computer-controlled stage. The live-cell imaging of GPhosEphB was processed and analyzed to generate ratiometric images. After the last time point of live-cell imaging, neurons were fixed with 4% paraformaldehyde and 2% sucrose immediately to capture the final morphology of the imaged dendritic filopodia and stained with primary antibody (EphB2 or VGLUT1) as stated above. The location of imaged neurons was saved to be reloaded for reimaging after fixation. Permanent markers in different colors were used at the four corners of the glass dish for additional reference of reimaging. After immunostaining, each live-imaged neuron was found using the recorded location and was reimaged to visualize EphB2 or VGLUT1 puncta. Importantly, the mRFP component of the GPhosEphB indicator could be visualized after fixation to show neuronal morphology (Figures 4A, 4B, 8D and 8G). Finally, the images from post-fixation and live-cell imaging were registered and the level of GPhosEphB signal was determined at the site of contact or in the stable filopodia. To prevent experimenter bias, the level of EphB activity acquired from live-cell imaging was calculated prior to examine immunostaining images.
Modeling and analysis
The ability to discriminate Retracting and Connecting filopodia was tested by assessing the cross-validated classification accuracy of different observations by three classifiers implemented in Matlab. Two linear classifiers (a linear Support Vector Machine and linear discriminant) and a non-linear classifier (logistic regression) were employed. The classifiers were trained and tested on three features of each observation: the slope of the signal from t = −1 to the peak; the mean during contact; and the variance of the signal during contact. Similar results were obtained by using only pairs of the three features. A 5-fold cross-validation scheme was used. Such procedure was iterated 500 times in order to obtain a distribution of classification accuracies for statistical testing.
Quantification and Statistical Analysis
Image analysis
Image analysis was conducted off-line using ImageJ with both custom and standard macros. For super-resolution STED data analysis, raw STED images were deconvoluted by using SP5 LAS AF using 80 nm microscope resolution (determined using 40 nm yellow/green (505/515) beads, cat #: F8795, Thermo Fisher Scientific). The images then subjected to background subtraction (mean intensity of all pixels in the images) followed by a Gaussian blur (2 pixel size for STED, 0.7 pixel size for Confocal). The morphology of dendritic filopodia was defined by mNeptune. Only elongated protrusions without enlarged heads were defined as filopodia. The puncta of EphB2, ephrin-B1, and VGLUT1 were thresholded as described previously (Hruska et al., 2015; Kayser et al., 2008; McClelland et al., 2009). To determine the relationship between EphB2, ephrin-B1, and VGLUT1, the proportion of filopodia containing these puncta was determined manually. The colocalization of VGLUT1 with EphB2 or ephrin-B1 was determined using a previously published method (McClelland et al., 2009). Briefly, binary masks of each label were generated and the location of each puncta was cataloged. Using the catalog of masks of each punctum, the colocalization and percent overlap of each colocalized punctum was determined in a field using ImageJ. The distance between puncta was measured by drawing a line between the center of mass of each punctum. The centers were defined by using an ImageJ macro (https://imagej.nih.gov/ij/macros/DrawEllipse.txt).
For live-cell imaging, any time series with focal plane drifting or photobleaching was discarded. Images were first smoothed using a standard Gaussian blur (radius = 0.5–2 pixels). To obtain a ratiometric image, we used the ratio-plus macro (see: http://rsbweb.nih.gov/ij/plugins/ratio-plus.html) to calculate the ratio of GFP/RFP. The background was measured as the average signal of an approximately 50 × 50 pixel region with no transfected cells. Infinity values were set to zero using the custom ScaleRatio macro. The resulting images were quantified by measuring average ratio values using standard ImageJ tools. Ratio change was calculated as F/F = (F – F0)/F0 (Yuste and Konnerth, 2005). The average signal within imaged neurons before ephrin application was considered as F0.
For analyzing focal activation of GPhosEphB, we first noticed that GPhos signal showed a spatially restricted rather than diffuse pattern. The average size of the GPhos signal was calculated by thresholding the ratiometric image with 20% above average signal in the shaft where little dynamics of GPhos signals were detected. Then, we drew an ROI in the dendritic shaft as the baseline and an ROI at axo-dendritic contacts or filopodia tips where GPhos signals are found. The sizes of ROIs were limited by the width of filopodia and set in each image at 0.13 to 0.2 μm2 based on the average size of endogenously activated GPhos signal (~ 0.27 μm2). For analysis of GPhos signal in filopodia, the average intensity of an ROI defining the area of Gphos activity was measured in the tip of the filopodia (0.13 to 0.2 μm2) in the ratiometric images. The raw ratiometric value was normalized to the control level, which is defined as the average intensity of a control region with the same size in the dendritic shaft during same imaging period. The time 0 is defined as filopodia are in contact with the axons. The average of GPhos signal before -1 min was set as a baseline. The rate of change (slope) of GPhos signal in filopodia was defined as the difference between the peak signal and the baseline divided by the time from -1 to the peak signal. For rare cases in which the slope was negative, infinity, or zero (n = 2), the data were excluded. For calculating the size of ephrin-B puncta, either overexpressed mT2-eB1 or exogenous 647-eB puncta were thresholded and measured as described previously (Hruska et al., 2015; Kayser et al., 2008; McClelland et al., 2009).
For analysis of filopodia movement before and after ephrin-B treatment, 15 minutes movie imaged before and after treatment were chosen. The maximal activation of EphB2 was found at 15–30 minute after ephrin-B addition (Figure 3); therefore images to be analyzed were collected 15–30 minutes after ephrin-B treatment. The ImageJ plug-in Manual tracking (https://imagej.nih.gov/ij/plugins/track/track.html) was used to measure the average distance filopodia moved during 15 minutes. GPhos signal was calculated as described above.
For quantification of filopodia numbers and distance moved before and after photostimulation, 10 frames (every 10 second) were chosen before and after photostimulation. The frame at photostimulation was set at time 0. The first frame before photostimulation was set at time -10s. The number of filopodia in HEK cells were counted manually in each frame and normalized to the number of filopodia in HEK cell at frame of -10s. For neuron experiments, the distance of filopodia in each frame was measured. Distance moved forwards was indicated as + μm and distance moved backwards was indicated as – μm. Because of the limits of microscopy resolution, only distance moved >1 μm was considered effective. Finally, the percentage of retracting filopodia in neurons before and after photostimulation was calculated.
Statistical analysis
All experiments were conducted a minimum of three times. Sample size (n) is indicated in the figure legends or in the text corresponding to the number of experiments was performed. Statistical analysis was conducted using GraphPad Prism (GraphPad Software, San Diego, CA) or Sigmaplot (Systat Software, San Jose, CA). Means were given with standard errors of the mean (± SEM) throughout. T-test was used for two-group comparison. Paired t-test was used for matched two-group comparison. Mann-Whitney U test was used for non-parametric two-group comparison. ANOVA was used for multiple group comparison. Two-way ANOVA was used for comparison with two independent variables. Kolmogorov-Smirnov test (K-S test) was used to test whether two sets of data have the same distribution. Extra-sum-of-squares F test was used to compare the goodness-of-fit of fitted curves.
Supplementary Material
Movie of data shown in Figure 2E. EphB2-YFP is shown as black on a white background. Dark dots in the filopodia tips represent clusters of EphB2. Images were collected every minute. Scale bar = 2 μm
Movie of data shown in Figure 3C. HEK293T cells were transfected with EphB2 and either GPhosEphB (top) or GPhosEphA (bottom). The white circle indicates the addition of activated ephrin. Images were collected every three minutes. Scale bars = 5 μm
Movie of data shown in Figure 3E. HEK293T cells were transfected with EphA4 and either GPhosEphB (top) or GPhosEphA (bottom). The white circle indicates the addition of activated ephrin. Images were collected every three minutes. Scale bars = 5 μm
Movie of data shown in Figure 3G. An example of a mouse EphB TKI neuron transfected with GPhosEphB. The white circle indicates the addition of activated ephrin-B2. Images were collected every minute. Scale bar = 5 μm
Movie of data shown in Figure 3H. An example of a mouse EphB TKI neuron transfected with GPhosEphB. The white square indicates the addition of 1-NA-PP1. The white circle represents the addition of activated ephrin-B2. Images were collected every minute. Scale bar = 5 μm
Movie of data shown in Figure 4D. An example of a neuron transfected with GPhosEphB contacting stable axonal mTurquoise2-ephrinB1 puncta. EphB2 signal is persistent. Images were collected every 15 seconds. Scale bar = 2 μm
Movie of data shown in Figure 4G. An example of a neuron transfected with GPhosEphB contacting moving mTurquoise2-ephrinB1 puncta. EphB2 signal is transient. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 5A. An example of a dendritic filopodium retracting from an axon (transfected with mTurquoise2). GPhosEphB showed an elevated signal before filopodia retraction. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 5D. An example of a dendritic filopodium that is stabilized after contacting an axon (transfected with mTurquoise2). GPhosEphB showed persistent signal. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 6J. An example of a dendritic filopodium retracting from a clustered ephrin-B punctum (Cyan). The fast EphB signal induced retraction. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 6K. An example of a dendritic filopodium that is stabilized when contacting a clustered ephrin-B punctum (Cyan). The slow EphB signal induced stabilization. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 7D. Examples of HEK293T cells transfected with fEphB2-CRY2 or fEphB2-KD-CRY2. Images were collected once every 10 seconds. Photoactivation is indicated by the white circle in the upper right of the movies. Scale bar = 2 μm
Movie of data shown in Figure 7G. Examples of neurons transfected with fEphB2-CRY2 or fEphB2-KD-CRY2 and tdTomato. Images were collected once every 10 seconds. Photoactivation is indicated by the white circle in the upper right and increased brightness in the movie in the region of interest (ROI). Scale bar = 2 μm
Movie of data shown in Figure 8A. An example of a dendritic filopodium that is stably contacting an axon (transfected with mTurquoise2). GPhosEphB showed persistent signal. Images were collected every minute. Scale bar = 2 μm
Movie of data shown in Figure 8E and 8H. Left: an example of a stable filopodium that has a high GPhos signal. Right: an example of a stable filopodium that has a low GPhos signal. Images were collected every two minutes. Scale bar = 2 μm
Highlights.
EphBs colocalize with ephrinB1 and VGLUT1 and release sites in stable filopodia
GPhos tyrosine kinase (TK) indicators enable in situ biochemistry of Eph and TKs
Moving and stable filopodia contain EphBs but their EphB TK signaling is distinct.
Differences in the kinetics of EphB signaling guide selection/rejection of contacts
Acknowledgments
We thank members of the Dalva laboratory and Dr. Le Ma (Jefferson) for helpful discussions and Dr. Michael Greenberg (Harvard Medical School) for providing the EphB TKI mice. SRD is supported by grants RO11DC011558, RO11DC016222, and U01NS094191 from the National Institutes of Health. MBD is supported by RO1 grants MH086425 and DA022727 and a grant from the Vickie and Jack Farber Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
YTM wrote the paper, refined the indicators, designed and conducted experiments, JXZ developed indicators and conducted and designed experiments, KH conducted experiments, SDB and GI conducted modeling experiments, MBD wrote the paper, designed experiments, and conceived the design of indicators.
DECLARATION OF INTERESTS
The authors declare no competing financial interests. A US Patent (9012617) has been issued to MBD and JXZ for GPhos and use of TK indicators.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arai R, Wriggers W, Nishikawa Y, Nagamune T, Fujisawa T. Conformations of variably linked chimeric proteins evaluated by synchrotron X-ray small-angle scattering. Proteins. 2004;57:829–838. doi: 10.1002/prot.20244. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci U S A. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll RC, Beattie EC, Xia H, Luscher C, Altschuler Y, Nicoll RA, Malenka RC, von Zastrow M. Dynamin-dependent endocytosis of ionotropic glutamate receptors. Proc Natl Acad Sci U S A. 1999;96:14112–14117. doi: 10.1073/pnas.96.24.14112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KY, Woo D, Jung H, Lee S, Kim S, Won J, Kyung T, Park H, Kim N, Yang HW, et al. Light-inducible receptor tyrosine kinases that regulate neurotrophin signalling. Nature communications. 2014;5:4057. doi: 10.1038/ncomms5057. [DOI] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, McClelland AC, Kayser MS. Cell adhesion molecules: signalling functions at the synapse. Nature reviews Neuroscience. 2007;8:206–220. doi: 10.1038/nrn2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- Duan L, Hope J, Ong Q, Lou HY, Kim N, McCarthy C, Acero V, Lin MZ, Cui B. Understanding CRY2 interactions for optical control of intracellular signaling. Nature communications. 2017;8:547. doi: 10.1038/s41467-017-00648-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. The spike-timing dependence of plasticity. Neuron. 2012;75:556–571. doi: 10.1016/j.neuron.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JG, Vanderhaeghen P. The ephrins and Eph receptors in neural development. Annu Rev Neurosci. 1998;21:309–345. doi: 10.1146/annurev.neuro.21.1.309. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Fuentes F, Zimmer D, Atienza M, Schottenfeld J, Penkala I, Bale T, Bence KK, Arregui CO. Protein tyrosine phosphatase PTP1B is involved in hippocampal synapse formation and learning. PLoS One. 2012;7:e41536. doi: 10.1371/journal.pone.0041536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J, von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, van Weeren L, Gadella TW, Jr, Royant A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93% Nature communications. 2012;3:751. doi: 10.1038/ncomms1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurd JW, Jones LR, Mahler HR, Moore WJ. Isolation and partial characterization of rat brain synaptic plasma membranes. Journal of neurochemistry. 1974;22:281–290. doi: 10.1111/j.1471-4159.1974.tb11591.x. [DOI] [PubMed] [Google Scholar]
- Hanamura K, Washburn HR, Sheffler-Collins SI, Xia NL, Henderson N, Tillu DV, Hassler S, Spellman DS, Zhang G, Neubert TA, et al. Extracellular phosphorylation of a receptor tyrosine kinase controls synaptic localization of NMDA receptors and regulates pathological pain. PLoS biology. 2017;15:e2002457. doi: 10.1371/journal.pbio.2002457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkemeyer M, Itkis OS, Ngo M, Hickmott PW, Ethell IM. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J Cell Biol. 2003;163:1313–1326. doi: 10.1083/jcb.200306033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Calcium signaling in dendritic spines. Cold Spring Harbor perspectives in biology. 2012;4:a005686. doi: 10.1101/cshperspect.a005686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgenberg LG, Smith MA. Preparation of dissociated mouse cortical neuron cultures. Journal of visualized experiments : JoVE. 2007:562. doi: 10.3791/562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K. A ligand-gated association between cytoplasmic domains of UNC5 and DCC family receptors converts netrin-induced growth cone attraction to repulsion. Cell. 1999;97:927–941. doi: 10.1016/s0092-8674(00)80804-1. [DOI] [PubMed] [Google Scholar]
- Hopker VH, Shewan D, Tessier-Lavigne M, Poo M, Holt C. Growth-cone attraction to netrin-1 is converted to repulsion by laminin-1. Nature. 1999;401:69–73. doi: 10.1038/43441. [DOI] [PubMed] [Google Scholar]
- Hruska M, Henderson NT, Xia NL, Le Marchand SJ, Dalva MB. Anchoring and synaptic stability of PSD-95 is driven by ephrin-B3. Nat Neurosci. 2015;18:1594–1605. doi: 10.1038/nn.4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Garner CC. Molecular mechanisms of presynaptic differentiation. Annu Rev Cell Dev Biol. 2008;24:237–262. doi: 10.1146/annurev.cellbio.23.090506.123417. [DOI] [PubMed] [Google Scholar]
- Kania A, Klein R. Mechanisms of ephrin-Eph signalling in development, physiology and disease. Nat Rev Mol Cell Biol. 2016 doi: 10.1038/nrm.2015.16. [DOI] [PubMed] [Google Scholar]
- Kasthuri N, Hayworth KJ, Berger DR, Schalek RL, Conchello JA, Knowles-Barley S, Lee D, Vazquez-Reina A, Kaynig V, Jones TR, et al. Saturated Reconstruction of a Volume of Neocortex. Cell. 2015;162:648–661. doi: 10.1016/j.cell.2015.06.054. [DOI] [PubMed] [Google Scholar]
- Kayser MS, McClelland AC, Hughes EG, Dalva MB. Intracellular and trans-synaptic regulation of glutamatergic synaptogenesis by EphB receptors. J Neurosci. 2006;26:12152–12164. doi: 10.1523/JNEUROSCI.3072-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser MS, Nolt MJ, Dalva MB. EphB receptors couple dendritic filopodia motility to synapse formation. Neuron. 2008;59:56–69. doi: 10.1016/j.neuron.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nature methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Kim JM, Lee M, Kim CY, Chang KY, Heo WD. Spatiotemporal control of fibroblast growth factor receptor signals by blue light. Chem Biol. 2014;21:903–912. doi: 10.1016/j.chembiol.2014.05.013. [DOI] [PubMed] [Google Scholar]
- Lai KO, Ip NY. Synapse development and plasticity: roles of ephrin/Eph receptor signaling. Current opinion in neurobiology. 2009;19:275–283. doi: 10.1016/j.conb.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Lander C, Zhang H, Hockfield S. Neurons produce a neuronal cell surface-associated chondroitin sulfate proteoglycan. J Neurosci. 1998;18:174–183. doi: 10.1523/JNEUROSCI.18-01-00174.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looger LL, Griesbeck O. Genetically encoded neural activity indicators. Current opinion in neurobiology. 2012;22:18–23. doi: 10.1016/j.conb.2011.10.024. [DOI] [PubMed] [Google Scholar]
- Luikart BW, Zhang W, Wayman GA, Kwon CH, Westbrook GL, Parada LF. Neurotrophin-dependent dendritic filopodial motility: a convergence on PI3K signaling. J Neurosci. 2008;28:7006–7012. doi: 10.1523/JNEUROSCI.0195-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland AC, Hruska M, Coenen AJ, Henkemeyer M, Dalva MB. Trans-synaptic EphB2-ephrin-B3 interaction regulates excitatory synapse density by inhibition of postsynaptic MAPK signaling. Proc Natl Acad Sci U S A. 2010;107:8830–8835. doi: 10.1073/pnas.0910644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland AC, Sheffler-Collins SI, Kayser MS, Dalva MB. Ephrin-B1 and ephrin-B2 mediate EphB-dependent presynaptic development via syntenin-1. Proc Natl Acad Sci U S A. 2009;106:20487–20492. doi: 10.1073/pnas.0811862106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin T, Hindges R, Yates PA, O’Leary DD. Bifunctional action of ephrin-B1 as a repellent and attractant to control bidirectional branch extension in dorsal-ventral retinotopic mapping. Development. 2003;130:2407–2418. doi: 10.1242/dev.00467. [DOI] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, Imoto K. A high signal-to-noise Ca(2+) probe composed of a single green fluorescent protein. Nat Biotechnol. 2001;19:137–141. doi: 10.1038/84397. [DOI] [PubMed] [Google Scholar]
- Oldach L, Zhang J. Genetically encoded fluorescent biosensors for live-cell visualization of protein phosphorylation. Chem Biol. 2014;21:186–197. doi: 10.1016/j.chembiol.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. Specificity in Signal Transduction: From Phosphotyrosine-SH2 Domain Interactions to Complex Cellular Systems. Cell. 2004;116:191–203. doi: 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- Perez de Arce K, Schrod N, Metzbower SW, Allgeyer E, Kong GK, Tang AH, Krupp AJ, Stein V, Liu X, Bewersdorf J, et al. Topographic Mapping of the Synaptic Cleft into Adhesive Nanodomains. Neuron. 2015;88:1165–1172. doi: 10.1016/j.neuron.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova DM, Verkhusha VV. Near-infrared fluorescent proteins for multicolor in vivo imaging. Nature methods. 2013;10:751–754. doi: 10.1038/nmeth.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffler-Collins SI, Dalva MB. EphBs: an integral link between synaptic function and synaptopathies. Trends in neurosciences. 2012;35:293–304. doi: 10.1016/j.tins.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Scheiffele P. Genetics and cell biology of building specific synaptic connectivity. Annu Rev Neurosci. 2010;33:473–507. doi: 10.1146/annurev.neuro.051508.135302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, et al. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- Soskis MJ, Ho HY, Bloodgood BL, Robichaux MA, Malik AN, Ataman B, Rubin AA, Zieg J, Zhang C, Shokat KM, et al. A chemical genetic approach reveals distinct EphB signaling mechanisms during brain development. Nat Neurosci. 2012;15:1645–1654. doi: 10.1038/nn.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Craig AM. Protein tyrosine phosphatases PTPdelta, PTPsigma, and LAR: presynaptic hubs for synapse organization. Trends in neurosciences. 2013;36:522–534. doi: 10.1016/j.tins.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasu MA, Dalva MB, Zigmond RE, Greenberg ME. Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science. 2002;295:491–495. doi: 10.1126/science.1065983. [DOI] [PubMed] [Google Scholar]
- Taslimi A, Vrana JD, Chen D, Borinskaya S, Mayer BJ, Kennedy MJ, Tucker CL. An optimized optogenetic clustering tool for probing protein interaction and function. Nature communications. 2014;5:4925. doi: 10.1038/ncomms5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry-Lorenzo RT, Roadcap DW, Otsuka T, Blanpied TA, Zamorano PL, Garner CC, Shenolikar S, Ehlers MD. Neurabin/protein phosphatase-1 complex regulates dendritic spine morphogenesis and maturation. Mol Biol Cell. 2005;16:2349–2362. doi: 10.1091/mbc.E04-12-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valnes K, Brandtzaeg P. Retardation of immunofluorescence fading during microscopy. The journal of histochemistry and cytochemistry : official journal of the Histochemistry Society. 1985;33:755–761. doi: 10.1177/33.8.3926864. [DOI] [PubMed] [Google Scholar]
- Wilhelm BG, Groemer TW, Rizzoli SO. The same synaptic vesicles drive active and spontaneous release. Nat Neurosci. 2010;13:1454–1456. doi: 10.1038/nn.2690. [DOI] [PubMed] [Google Scholar]
- Yuste R, Konnerth A. Imaging in neuroscience and development : a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 2005. [Google Scholar]
- Zhou XX, Chung HK, Lam AJ, Lin MZ. Optical control of protein activity by fluorescent protein domains. Science. 2012;338:810–814. doi: 10.1126/science.1226854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer M, Palmer A, Köhler J, Klein Rd. EphB-ephrinB bi-directional endocytosis terminates adhesion allowing contact mediated repulsion. Nature cell biology. 2003;5:869–878. doi: 10.1038/ncb1045. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie of data shown in Figure 2E. EphB2-YFP is shown as black on a white background. Dark dots in the filopodia tips represent clusters of EphB2. Images were collected every minute. Scale bar = 2 μm
Movie of data shown in Figure 3C. HEK293T cells were transfected with EphB2 and either GPhosEphB (top) or GPhosEphA (bottom). The white circle indicates the addition of activated ephrin. Images were collected every three minutes. Scale bars = 5 μm
Movie of data shown in Figure 3E. HEK293T cells were transfected with EphA4 and either GPhosEphB (top) or GPhosEphA (bottom). The white circle indicates the addition of activated ephrin. Images were collected every three minutes. Scale bars = 5 μm
Movie of data shown in Figure 3G. An example of a mouse EphB TKI neuron transfected with GPhosEphB. The white circle indicates the addition of activated ephrin-B2. Images were collected every minute. Scale bar = 5 μm
Movie of data shown in Figure 3H. An example of a mouse EphB TKI neuron transfected with GPhosEphB. The white square indicates the addition of 1-NA-PP1. The white circle represents the addition of activated ephrin-B2. Images were collected every minute. Scale bar = 5 μm
Movie of data shown in Figure 4D. An example of a neuron transfected with GPhosEphB contacting stable axonal mTurquoise2-ephrinB1 puncta. EphB2 signal is persistent. Images were collected every 15 seconds. Scale bar = 2 μm
Movie of data shown in Figure 4G. An example of a neuron transfected with GPhosEphB contacting moving mTurquoise2-ephrinB1 puncta. EphB2 signal is transient. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 5A. An example of a dendritic filopodium retracting from an axon (transfected with mTurquoise2). GPhosEphB showed an elevated signal before filopodia retraction. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 5D. An example of a dendritic filopodium that is stabilized after contacting an axon (transfected with mTurquoise2). GPhosEphB showed persistent signal. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 6J. An example of a dendritic filopodium retracting from a clustered ephrin-B punctum (Cyan). The fast EphB signal induced retraction. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 6K. An example of a dendritic filopodium that is stabilized when contacting a clustered ephrin-B punctum (Cyan). The slow EphB signal induced stabilization. Images were collected once every minute. Scale bar = 2 μm
Movie of data shown in Figure 7D. Examples of HEK293T cells transfected with fEphB2-CRY2 or fEphB2-KD-CRY2. Images were collected once every 10 seconds. Photoactivation is indicated by the white circle in the upper right of the movies. Scale bar = 2 μm
Movie of data shown in Figure 7G. Examples of neurons transfected with fEphB2-CRY2 or fEphB2-KD-CRY2 and tdTomato. Images were collected once every 10 seconds. Photoactivation is indicated by the white circle in the upper right and increased brightness in the movie in the region of interest (ROI). Scale bar = 2 μm
Movie of data shown in Figure 8A. An example of a dendritic filopodium that is stably contacting an axon (transfected with mTurquoise2). GPhosEphB showed persistent signal. Images were collected every minute. Scale bar = 2 μm
Movie of data shown in Figure 8E and 8H. Left: an example of a stable filopodium that has a high GPhos signal. Right: an example of a stable filopodium that has a low GPhos signal. Images were collected every two minutes. Scale bar = 2 μm