

Abstract
Background
Many genome- and epigenome-wide association studies (GWAS and EWAS) and studies of promoter methylation of candidate genes for inflammatory bowel disease (IBD) have demonstrated significant associations between genetic and epigenetic changes and IBD. Independent GWA studies have identified genetic variants in the BACH2, IL6ST, LAMB1, IKZF1, and MGAT3 loci to be associated with both IBD and immunoglobulin G (IgG) glycosylation.
Methods
Using bisulfite pyrosequencing, we analyzed CpG methylation in promoter regions of these five genes from peripheral blood of several hundred IBD patients and healthy controls (HCs) from two independent cohorts, respectively.
Results
We found significant differences in the methylation levels in the MGAT3 and BACH2 genes between both Crohn’s disease and ulcerative colitis when compared to HC. The same pattern of methylation changes was identified for both genes in CD19+ B cells isolated from the whole blood of a subset of the IBD patients. A correlation analysis was performed between the MGAT3 and BACH2 promoter methylation and individual IgG glycans, measured in the same individuals of the two large cohorts. MGAT3 promoter methylation correlated significantly with galactosylation, sialylation, and bisecting GlcNAc on IgG of the same patients, suggesting that activity of the GnT-III enzyme, encoded by this gene, might be altered in IBD. The correlations between the BACH2 promoter methylation and IgG glycans were less obvious, since BACH2 is not a glycosyltransferase and therefore may affect IgG glycosylation only indirectly.
Conclusions
Our results suggest that epigenetic deregulation of key glycosylation genes might lead to an increase in pro-inflammatory properties of IgG in IBD through a decrease in galactosylation and sialylation and an increase of bisecting GlcNAc on digalactosylated glycan structures. Finally, we showed that CpG methylation in the promoter of the MGAT3 gene is altered in CD3+ T cells isolated from inflamed mucosa of patients with ulcerative colitis from a third smaller cohort, for which biopsies were available, suggesting a functional role of this glyco-gene in IBD pathogenesis.
Electronic supplementary material
The online version of this article (10.1186/s13148-018-0507-y) contains supplementary material, which is available to authorized users.
Background
Inflammatory bowel disease (IBD) is a chronic intestinal inflammatory condition classified in two major forms—Crohn’s disease (CD) and ulcerative colitis (UC)—which exhibit etiologically and clinically distinct features. Nowadays, IBD affects 2.5–3 million people in Europe and causes considerable morbidity [1]. Despite numerous clinical, genetic, and other experimental studies, our understanding of IBD development and progression remains incomplete.
It is generally accepted that IBD represents an aberrant immune response to gut microbiota in genetically susceptible individuals [2]. Genome-wide association studies (GWAS) have identified over 200 genetic susceptibility loci, the majority of which were associated with both forms of IBD in genome-wide meta-analysis [3–7]. However, common genetic variants account only for 8.2 and 13.1% heritability of UC and CD, respectively [7]. Interaction of an individual’s gut microbiome, immune system, genetic background, and environmental factors, such as smoking, diet, drugs, and physical activity [2, 8–10], makes IBD a complex etiopathogenic entity. The challenge is therefore to identify additional factors involved in the development and progression of this disease, especially given its rapidly increasing incidence. It is probable that epigenetics play a key role in the interactions between environmental, microbial, and genetic factors that participate in IBD development and progression. These include DNA methylation and histone modifications, as well as some other epigenetic mechanisms [11–13]; for a review, see [14, 15].
DNA methylation remains the most studied epigenetic modification, readily assayed in a large number of individuals/samples. Hypermethylation of gene promoters is generally associated with gene silencing, while promoter hypomethylation is associated with gene activation [16]. Environmentally changed DNA methylation pattern may contribute to the development of many complex diseases by mediating the interplay between external and internal factors and the gene expression [17–21]. There are also data to suggest that the aforementioned environmental modifiers of IBD can also affect DNA methylation [17–19, 22]. Epigenetic component of IBD has been addressed in many studies, mostly by whole genome methylation analysis performed on peripheral blood mononuclear cells (PBMCs) or mucosal tissue, revealing regions differentially methylated between the disease and healthy state, as well as between CD and UC [11–13, 23–26].
The majority of eukaryotic proteins are modified by addition of complex oligosaccharides (glycans) through the process of glycosylation. Therefore, glycans are an integral part of nearly all membrane and secreted proteins, including components of the immune system [27]. Aberrant protein glycosylation is implicated in virtually every human complex disease, including inflammation [28–31]. Previous studies have suggested that N-glycosylation of secreted and membrane proteins might be regulated epigenetically and that aberrant glycosylation profiles in disease can arise through aberrant epigenetics [32–38]. A comprehensive review about the role of protein glycosylation in IBD has been given recently [39]. N-glycosylation of serum-circulating proteins (such as the acute phase proteins; immunoglobulin G, IgG; and immunoglobulin A, IgA) or whole plasma N-glycome (i.e., N-glycans present on all plasma proteins) has been the focus of IBD biomarker discovery [36, 40–43]. In addition, our partners from IBD consortium and others established that altered glycosylation of IgG, which is a key effector of the humoral immune system, has a role in balancing inflammation at the systemic level [42–46].
GWA studies indicated associations of IBD with several loci involved in protein glycosylation [47, 48]. More recently, the first GWAS of IgG glycosylation identified 16 loci specifically associated with changes in IgG glycosylation [49]. Interestingly, five of these loci showed pleiotropy with IBD: MGAT3, a glyco-gene encoding for a glycosyltransferase, GnT-III; LAMB1, a member of transmembrane glycoprotein family of extracellular matrix; the IL6ST, a signal transducer shared by many cytokines; IKZF1; and BACH2, transcription factors involved in B cell differentiation, activation, and maturation. Only the MGAT3 is a classical glyco-gene with a known function in IgG glycosylation, while the exact functional roles for other four GWAS hits in IgG glycosylation or IBD remain unknown.
In this study, we investigated promoter methylation differences in these five genes, associated with both IBD and IgG glycosylation, in peripheral whole blood of several hundred IBD patients from two independent cohorts. We also correlated promoter methylation data with IgG glycosylation data analyzed previously for the same IBD patients by our partners from the IBD consortium [43, 46, 50]. Peripheral blood was used for DNA methylation analysis and serum or plasma was used for glycan analysis, since one of our goals was the search for potential IBD biomarkers. As peripheral whole blood is a heterogeneous cell mixture with specific methylation pattern for each of the cell types [51], we also analyzed promoter methylation of our candidate genes in CD19+ B cells and CD3+ T cells isolated from peripheral blood mononuclear cells (PBMCs). B cells were of our particular interest since these cells produce IgG on their membrane and are precursors of plasma cells which secrete IgG. We have further explored if aberrant promoter methylation recorded in peripheral whole blood of IBD patients can be a proxy for epigenetic events occurring in the inflamed mucosa. To address this question, we analyzed DNA methylation from PBMCs, CD3+ T cells isolated from PBMCs, and CD3+ T cells isolated from inflamed colonic mucosa of UC patients from the third smaller cohort, for which biopsies were available.
Methods
Patient selection and ethics
Patients were recruited prospectively from Edinburgh, UK, and Florence, Italy, as a part of the IBD-BIOM project. The recruitment of patients from Edinburgh has been described elsewhere [13, 43]. Briefly, we recruited IBD patients prospectively as close as possible to the date of diagnosis from gastroenterology outpatient and endoscopy appointments between 2012 and 2015. We recruited symptomatic controls from gastroenterology clinics during the same period. In these individuals, we had excluded IBD and other organic bowel pathology following biochemical and/or endoscopic investigations. We recruited a further healthy volunteer cohort with no gastrointestinal symptoms. IBD patients were stratified by disease type (ulcerative colitis, UC, and Crohn’s disease, CD). Detailed genetic, phenotypic, and other data regarding IBD cases are given in Additional file 1: Tables S1 and S2. Florence cohort was collected through the network of the Italian Group for IBD (IG-IBD) since the beginning of 2001 and first described in 2005 [1] following an internal validation of phenotyping. Subsequently, longitudinal update has been performed on a yearly basis.
Ethical approvals were obtained from Tayside Committee on Medical Ethics B, and all patients and controls provided written, informed consent (LREC 06/S1101/16, LREC 2000/4/192).
Florence recruitment details
IBD patients were prospectively recruited as close as possible to the date of diagnosis from gastroenterology outpatient and endoscopy appointments between years 2012 and 2015 in different tertiary referral centers in San Giovanni Rotondo, Rome, Rozzano (Milan), Padua, and Florence, Italy. Symptomatic controls were recruited in the same centers (gastroenterology clinics) during the same period. In these individuals, IBD and other organic bowel pathology were excluded by biochemical and/or endoscopic investigations. IBD patients were stratified by disease type (ulcerative colitis, UC, and Crohn’s disease, CD). Samples were obtained with the same methodology (see further) and centrally collected at San Giovanni Rotondo, Italy.
Sample collection
We collected whole blood at the time of patient recruitment into 9-ml serum Z-clot activator tubes (Greiner), allowed them to clot at 4 °C for 60 min, and then centrifuged at 2500×g for 15 min. The serum was aliquoted off and stored at − 80 °C until further analysis.
A subset of patients and controls recruited in Edinburgh (Additional file 1: Table S3) underwent immunomagnetic cell separation to obtain CD19+ B cells. The methods have previously been detailed elsewhere [13]. Venepuncture using 9-ml K3 EDTA vacuette (Greiner) tubes was performed to obtain between of 18 and 36 ml of EDTA-buffered blood. An initial Ficoll (Ficoll-Paque, GE Healthcare, Bucks, UK) density gradient centrifugation was performed to obtain peripheral blood mononuclear cells. Cells labelled with antibody-coated microbeads (human CD8+ and CD19+ microbeads, 20 μl per 1 × 107 cells) were immunomagnetic separated using the autoMACs Pro cell separator (Miltenyi, Germany). CD19+ separations were performed following an initial CD8+ depletion step. Nucleic acids were extracted using AllPrep (Qiagen, Hilden, Germany) according to the manufacturer’s guidance and stored at − 80 °C.
Colonic biopsies from controls and UC patients with inactive and active form of disease were mechanically dissociated to prepare single-cell suspensions using Hanks’ balanced salt solution modified medium, without calcium chloride and magnesium sulfate (HBSS) (Sigma), with penicillin/streptomycin and gentamicin. PBMCs were obtained by density gradient centrifugation using Lymphoprep. CD3+ T cells (from biopsies and blood) were magnetically sorted by using the EasySep™ Human T Cell Enrichment Kit (STEMCELL) following the manufacturer’s instructions. Following cell isolation, DNA extraction was performed using the Invisorb Spin Tissue Mini Kit (Stratec Molecular) following the manufacturer’s instructions.
DNA methylation analysis
We analyzed promoter methylation of the candidate genes in the DNA from whole blood, as well as from the separated CD19+ B cells. In addition, for the MGAT3, which is a glycosyltransferase with direct and known function in IgG glycosylation [52], we analyzed promoter methylation—in DNA from PBMCs, CD3+ T cells isolated from PBMCs, and CD3+ T cells isolated from the colonic mucosa of healthy controls and UC patients (classified according to active and inactive form of the disease) of the third independent smaller subcohort collected by the Gastroenterology Department of Centro Hospitalar do Porto-Hospital de Santo António, Portugal (Additional file 1: Table S4). All specimens were subjected to histological examination and classification. All participants gave informed consent about all clinical procedures, and research protocols were approved by the ethics committee of CHP/HSA, Portugal (233/12(179-DEFI/177-CES).
For DNA methylation analysis, 500 ng of DNA from whole blood was bisulfite converted using EZ-96 DNA Methylation Gold kit (Zymo Research, Freiburg, Germany), and 100 ng of DNA from CD19+ B cells, PBMCs, and T cells was converted using EZ DNA Methylation Gold kit (Zymo Research, Freiburg, Germany) according to the manufacturer’s protocol. Two to six pyrosequencing assays were developed for promoter regions of each of the five candidate genes (BACH2, MGAT3, IL6ST, IKZF1, and LAMB1). The selection of analyzed CpG sites was random for assays 2–5 of the MGAT3 gene. CpG sites within the MGAT3 assay 1 were selected based on the GEO (Gene Expression Omnibus) database where methylation data were obtained using Illumina HumanMethylation450 BeadChip v1.1 technology. For the BACH2 gene, assays were selected based on location of differentially methylated CpGs in different cell lines tested by ENCODE project, using Illumina HumanMethylation450 BeadChip v1.1 technology (a newer version, the Infinium MethylationEPIC 850K was not available at the time). We used traditional bisulfite-based protocols which cannot discriminate between 5-methylcytosine (5-mC) and 5-hydroxymethylcytosine (5-hmC) as oxidative bisulfite (oxBS-450K) method can [53]. However, recent studies have shown that global DNA hydroxymethylation is very low in blood cells [54, 55]. Furthermore, hydroxymethylation is significantly depleted from promotors and CpG islands, while enriched in the gene bodies [53, 56].
Based on the estimated statistical power, we did initial screening on 60 patients for each pyrosequencing assay, after which we excluded those genes (pyrosequencing assays) that did not show any statistically significant differences between IBD patients and healthy controls. Pyrosequencing assays for LAMB1, IL6ST, and IKZF1 are shown in Additional file 2: Figure S1. We continued to analyze promoter methylation only in the BACH2 and MGAT3 genes. Specific regions were amplified using PyroMark PCR kit (Qiagen, Hilden, Germany). The cycling conditions for the BACH2 gene were as follows: initial polymerase activation step for 15 min at 95 °C followed by 50 cycles of 30 s denaturation at 95 °C, primer annealing for 30 s at primer-specific temperatures (Additional file 1: Table S5), and 30 s at 72 °C, with final extension at 72 °C for 10 min. The cycling protocol used for amplification of the MGAT3 gene fragments was described previously [35], with the annealing temperature adjusted to 55 °C for the fragment 1 performed on DNA from CD19+ B cells. For quantitative measurement of DNA methylation level at specific CpG sites, PCR-amplified bisulfite-converted DNA was sequenced using the PyroMark Q24 Advanced pyrosequencing system (Qiagen) according to the manufacturer’s recommendations. Sequences of PCR and pyrosequencing primers for the BACH2 and the MGAT3 genes are listed in Additional file 1: Table S5. EpiTect PCR Control DNA Set (methylated and unmethylated bisulfite-converted human DNA, Qiagen) was used as a control for PCR and pyrosequencing reactions.
Statistical analysis
The nonparametric Mann-Whitney U test was used to compare the methylation status of CpG sites encompassed by the pyrosequencing assays in the MGAT3 and BACH2 genes between the two independent groups: HC compared to each of CD or UC. Significance threshold was set at p < 0.05 with additional Bonferroni correction for multiple testing. Given that age was our primary concern as a potential confounder, we visualized the age in the three groups (CD, UC, and HC) for the samples included in each analysis as violin plots (Additional file 3: Figure S2) and assured there was no significant difference between the age groups (p > 0.05) using the Mann-Whitney U test. This was done to assure the validity and strengthen the rationale for the selection of statistical methods.
For the data of the MGAT3 promoter methylation from PBMCs, CD3+ T cells isolated from blood, and CD3+ T cells isolated from inflamed colonic mucosa (the Porto cohort), the Mann-Whitney U test was applied with Bonferroni correction accounting for 15 CpG sites.
Glycan analysis
Glycans present on IgG were analyzed from serum of over 1000 IBD (UC and CD) patients and healthy controls in the Edinburgh cohort using ultra performance liquid chromatography (UPLC) [43, 50]. In the Florence cohort, plasma samples of 3500 IBD patients and healthy controls was used for analysis of IgG glycopeptides by liquid chromatography coupled to mass spectrometry (LC-MS) [46]. The data for IgG glycosylation analysis were used in this work for correlation analysis with promoter methylation data of MGAT3 and BACH2 genes, with matching samples from the very same patients and healthy controls.
Isolation of IgG from blood plasma
IgG has been isolated from blood plasma by affinity chromatography using CIM Protein G 96-well plate (BIA Separations, Ajdovščina, Slovenia) and vacuum manifold (Pall Corporation, Port Washington, NY, USA) as previously described [57, 58]. In short, plasma samples (50–90 μl) were diluted with 1 × PBS, pH 7.4 in the ratio 1:7. All samples were filtered through 0.45 and 0.2-μm AcroPrep GHP filter plates (Pall Corporation) using vacuum manifold and immediately applied to preconditioned Protein G plate. After washing of the Protein G plate, IgG was eluted with 0.1 mol L−1 formic acid and immediately neutralized with ammonium bicarbonate to pH 7.0. Protein G plate was regenerated and stored at 4 °C.
IgG glycosylation analysis using ultra-performance liquid chromatography
N-glycans from isolated IgG in the Edinburgh cohort were released with PNGase F after drying 300 μl of each IgG elution fraction, labeled with 2-aminobenzamide and excess of regents removed by clean-up using hydrophilic interaction liquid chromatography solid phase extraction (HILIC-SPE). Fluorescently labeled and purified N-glycans were separated by HILIC-UPLC using Acquity UPLC instrument (Waters, Milford, MA, USA) as previously described [43]. Samples were separated into 24 peaks [57], and the amount of N-glycans in each chromatographic peak was expressed as a percentage of total integrated area (% area).
IgG glycosylation analysis using liquid chromatography coupled to mass spectrometry
In the Florence cohort, Fc-specific IgG glycopeptides were analyzed after IgG purification, overnight trypsin digestion at 37 °C, and reverse-phase purification on Chromabond C18 beads using vacuum manifold as described [46, 59]. Samples were analyzed using nanoliquid chromatography coupled to mass spectrometry (nanoLC-MS), on a nanoACQUITY UPLC system (Waters, Milford Massachusetts, USA) coupled to quadrupole-TOF-MS (Compact; Bruker Daltonics, Bremen, Germany) equipped with a sheath-flow ESI sprayer (capillary electrophoresis ESI-MS sprayer; Agilent Technologies, Santa Clara, USA) as previously described [46]. The nanoACQUITY UPLC system and the Bruker Compact Q-TOF-MS were operated under HyStar software version 3.2.
Data was processed as described previously [46, 60]. This resulted in the extraction of 16 IgG1, 16 IgG2/3, and 11 IgG4 glycoforms. The tryptic Fc-glycopeptides for IgG2 and IgG3 subclasses have identical peptide moieties in the Caucasian population and are therefore not distinguishable with this methodology. Annotation of the spectra was done based on accurate mass according to the relevant literature [40, 57].
Correlation analysis
Methylation data for the BACH2 (assay 2) and the MGAT3 (assays 1 and 2) genes (obtained for the two large cohorts) were filtered according to the peak quality by rejecting peaks marked as “failed” by the pyrosequencing software. Average methylation across all assayed CpG sites was calculated for each pyrosequencing assay in each cohort. Methylation results for individual patients were matched with their corresponding glycan profiles. Sizes of datasets and patient classes obtained after including complete records (i.e., both methylation data and glycan profiles present) are shown in Additional file 1: Table S6. Individual glycan structures were represented as relative abundances and batch-corrected. Percentage of structures with bisecting N-acetylglucosamine was calculated for each cohort as a derived trait at this point. Glycan structures identified by each method were translated to Oxford notation, and only the 13 structures present in both the Edinburgh and Florence datasets were considered for correlation. We used IgG1 data from the Florence cohort, as this isoform was the most abundant. Three additional derived traits were calculated: ratios of FA2B to FA2, FA2BG1 to FA2G1, and FA2BG2 to FA2G2.
Pearson correlation between CpG methylation data and 17 glycan features (13 structures and 4 derived traits) was calculated. Significance threshold was set at p < 0.05 with additional Bonferroni correction for 17-fold multiple testing.
Methylation of assayed CpG sites in promoters of the BACH2 and MGAT3 genes was correlated with measured glycan structures. Pearson correlation coefficient along with the associated p value was calculated between average CpG methylation (for all genes/assays) and each measured IgG glycan structure. Calculation was done on pairwise complete observations. Only correlations with the p value below 0.01 were considered further. Next, correlation coefficients for all CpG assays were calculated, which was used to rank glycan structures according to regulation by the assayed region. Glycan structures with the strongest correlation (either positive or negative) to CpG methylation were then used to explain regulatory effects. All calculations and data visualizations were done in R language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, Austria). Visualization of correlations was done using the R package “corrplot.”
Results
Promoter methylation of the candidate genes in whole blood and B cells of IBD patients
In order to assess the level of methylation in CpG islands of the five candidate genes (BACH2, MGAT3, IKZF1, LAMB1, and IL6ST), associated with both IBD and IgG glycosylation by GWAS, we developed several pyrosequencing assays for each of the genes (Fig. 1 and Additional file 2: Figure S1). We performed initial screening of the pyrosequencing assays on 60 patients. Overall cytosine methylation levels were very low for LAMB1 (average value per group < 8%), for IL6ST (< 3.5%), and for IKZF1 (< 4%) in the assayed portion of their promoters; therefore, we could not identify differential methylation. We then excluded these genes from further analysis.
Fig. 1.

Positions of the BACH2 and MGAT3 genes in the human genome and relative positions of the fragments analyzed for methylation level (pyrosequencing assays) within these genes. For each pyrosequencing assay (A1–A5), the region amplified by PCR is shown. Positions of the genes on the chromosomes are shown using chromosome models (red vertical lines). Coordinates are relative to the hg19 human genome assembly. The genes are displayed in the direction corresponding to their reading frames. Annotations (CpG islands and pyrosequencing assays) are to scale. TSS transcription start site
MGAT3 and BACH2 promoter methylation was analyzed in several hundred IBD patients and healthy controls from two independent cohorts (Additional file 1: Tables S1 and S2). In these genes, we analyzed methylation level at 47 CpGs covered by five pyrosequencing assays in the BACH2 gene: 21 CpG sites were in the promoter region, 1 CpG site was in first exon, and 25 CpG sites were located in the first intron of the gene. A total of 32 CpG sites, covered by five pyrosequencing assays, was analyzed for MGAT3: 18 CpG sites were located in the promoter region and 14 CpG sites in the first intron (Fig. 1). Most of those CpG sites were located within CpG islands of the both genes. We found differential CpG methylation between IBD patients and HC within the assay A2, located at 213–368 bp upstream (relative to the gene orientation) of the TSS in the BACH2 promoter and within the assays A1 and A2, located in the CpG island 1 of the MGAT3 gene. The same pattern of differential methylation at these CpG sites was observed in whole blood of patients and HC from two large independent cohorts (Fig. 2). CpG methylation level was generally low (up to 20%) in the assayed portion of the BACH2 promoter; however, significant differences between HC and CD methylation level were recorded at CpG sites 4, 5, 6, and 8 (Fig. 2a). For the assayed portion of the MGAT3 promoter, general methylation level was high, with all CpG sites showing a reproducibly significant difference between HC and both CD and UC. CpG sites 2, 13, and 15 showed significant differences only for CD but not for UC. Direction of change was different for the two genes—differentially methylated CpG sites within the BACH2 promoter were hypomethylated, while those for the MGAT3 gene were hypermethylated in disease compared to healthy individuals.
Fig. 2.

Box plot of CpG methylation in peripheral whole blood for the BACH2 and MGAT3 genes in the Edinburgh and Florence cohorts and in B cells from a subset of patients from Edinburgh cohort. Groups were compared using the Mann-Whitney U test with significance threshold of p = 0.05, corrected for multiple testing using the Bonferroni method. a Methylation levels were generally low in the assayed portion of the BACH2 gene promoter, with significant differences between HC and CD methylation at CpG sites 4, 5, 6, and 8 (replicated in both cohorts). For the MGAT3 gene, general methylation level was high, with all CpG sites showing a reproducibly significant difference between HC and both CD and UC, except for CpG sites 2, 13, and 15 for which reproducible significant differences were found only between HC and CD. b In B cells, isolated from PBMCs of a subset of the patients from the Edinburg cohort, differential methylation was found at the CpG position 5 of the BACH2 gene (assay 2) between HC and CD, while for the MGAT3 gene, differentially methylated were CpG sites 1–5, 12, and 13 between HC and CD. CD Crohn’s disease, UC ulcerative colitis, HC healthy controls
These results were confirmed on CD19+ B cells isolated from peripheral whole blood of the independent, smaller patient sample from the Edinburgh cohort (67 samples). The CpG sites 1–5 and 12–13 in the MGAT3 promoter were differentially methylated between CD and HC. Only CpG site 5 within the assay A2 of the BACH2 gene showed change in the methylation level between HC and CD in CD19+ B cells (Fig. 2b). There were no differences in the methylation level of the same CpG sites within assayed fragments of the BACH2 and MGAT3 genes between UC and HC.
It is worth noting that the same pattern of CpG methylation differences was observed in PBMCs of the IBD patients and HC from both large independent cohorts, and most of the CpG sites within the assayed portion of the MGAT3 promoter were also differentially methylated in CD19+ B cells from the subset of IBD patients from the Edinburgh cohort (Fig. 2a, b).
Promoter methylation of the MGAT3 gene in CD3+ T cells from PBMCs and inflamed colonic mucosa of UC patients
We included in our investigation biopsy samples of UC patients from an independent cohort from the Gastroenterology Department of Centro Hospitalar do Porto-Hospital de Santo António, Portugal. Given the technical challenges in obtaining DNA and RNA from a small number of purified cells from inflamed colonic mucosa, a subset of patients with active and inactive phase of UC was selected for methylation analysis from three sources: (1) PBMCs, (2) CD3+ T cells isolated from PBMCs, and (3) CD3+ T cells isolated from colonic mucosa (see also Additional file 1: Table S4).
Inter-individual variation of MGAT3 methylation level measured from PBMCs and from CD3+ T cells isolated from PBMCs was quite large—it varied from 47 to 94% and from 26 to 90%, respectively. Therefore, we could not find any difference in CpG methylation level between UC patients and HC in assayed fragments of the MGAT3 promoter, neither in PBMCs nor in CD3+ T cells isolated from PBMCs. However, we recorded a total of 7 (out of 15) differentially methylated CpG sites in CD3+ T cells isolated from colonic mucosa of UC patients with active disease compared with HC (Fig. 3). Overall, the methylation level of CpGs within assayed fragments of the MGAT3 promoter was high in CD3+ T cells from healthy colonic mucosa (between 77 and 98%). When compared to inflamed mucosa of UC patients with active phase of the disease, the same CpG sites were hypomethylated, with the highest difference at the CpG position 10 (13.24%; p = 5.08 × 10−5; Fig. 3). In inactive UC, no significant differences could be found after Bonferroni correction for multiple testing.
Fig. 3.
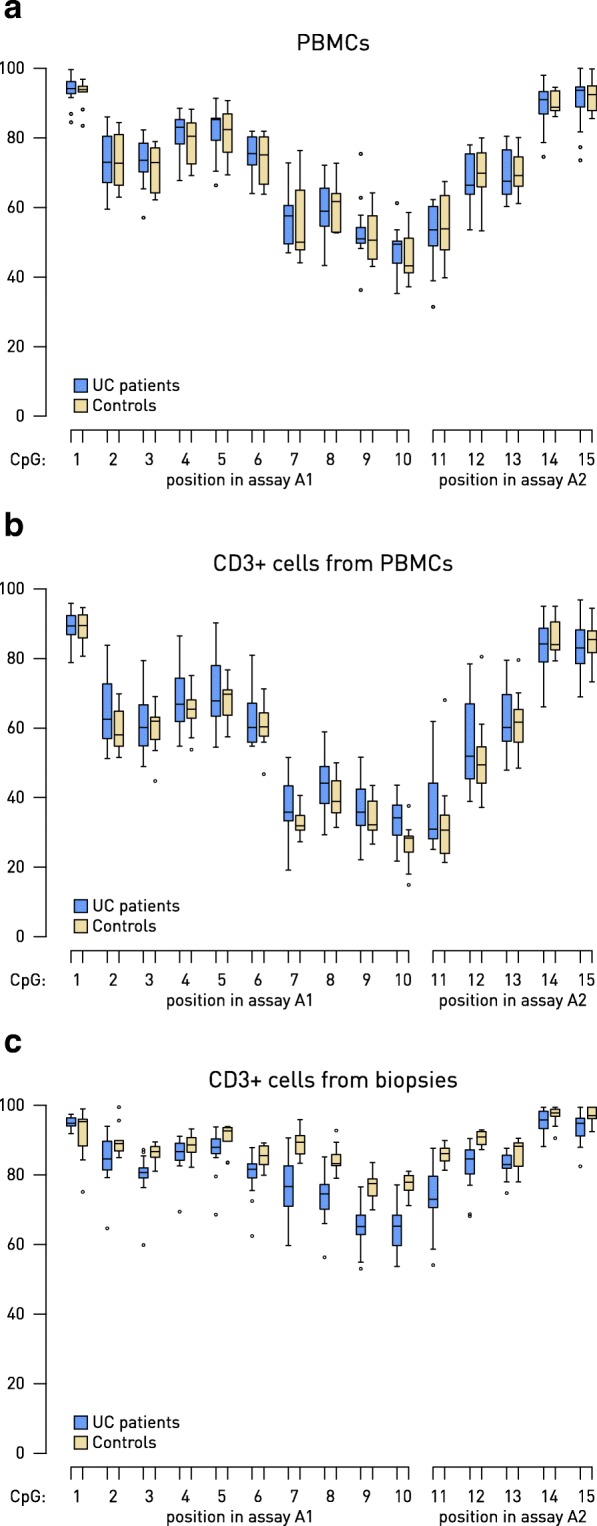
Box plot of CpG methylation level in the MGAT3 gene promoter (assays A1 and A2) analyzed from PBMCs (a), CD3+ T cells isolated from PBMCs (b), and CD3+ T cells isolated from inflamed colonic mucosa (c) from the independent cohort of Porto. Changes between UC patients with active disease and HC were statistically significant only in CD3+ T cells isolated from inflamed colonic mucosa at CpG positions 3 and 7–12 (p < 0.05 after Bonferroni correction for 15 hypotheses). PBMC peripheral blood mononuclear cells, UC ulcerative colitis, HC healthy controls
It is worth noting that the methylation pattern in CD3+ T cells isolated from inflamed colonic mucosa differed from the methylation patterns in PBMCs and for CD3+ T cells isolated from PBMCs. The latter two were very similar and had much lower methylation levels than that measured for CD3+ T cells from inflamed colonic mucosa (Fig. 3). Also, MGAT3 methylation level was increased in UC compared with HC when measured from PBMCs or CD3+ T cells isolated from PBMCs (hypermethylation), while it was decreased (hypomethylation) when measured from CD3+ T cells isolated from inflamed colonic mucosa in comparison with healthy mucosa.
Correlation between the MGAT3 and BACH2 promoter methylation and IgG glycosylation
There were statistically significant correlations that replicated across assays and cohorts between the MGAT3 promoter methylation and glycan structures FA2, FA2G2, FA2BG2, and FA2G2S1, as well as the derived trait of the ratio of FA2B to FA2 (Fig. 4a). All correlations except with FA2 were negative. No reproducible significant correlations could be found between BACH2 promoter methylation and the glycan structures (Fig. 4a).
Fig. 4.

Correlations between CpG methylation in the BACH2 and MGAT3 gene promoters and glycan structures measured from the same individuals of the Edinburgh and Florence cohorts, mapped to the glycan biosynthesis pathways. a Correlation coefficients between average CpG methylation in the assayed gene promoter fragments and glycan structure percentages are shown as blue (positive) or red (negative correlation) circles with their size and shade proportional to the correlation coefficient. Correlations without statistical significance (p > 0.05 after Bonferroni correction for multiple testing) are crossed. Columns represent 13 individual glycan structures and four derived traits (beige box). EDI Edinburgh cohort, FLO Florence cohort, Bisecting, percentage of all structures with bisecting N-acetylglucosamine, B/FA2 ratio of FA2B to FA2 structures, B/FA2G1 ratio of FA2BG1 to FA2G1 structures, B/FA2G2 ratio of FA2BG2 to FA2G2 structures. b Glycan biosynthesis pathways with the glycan structures, labels, and the enzymes mapped to correlation results for the MGAT3 gene. Light blue rectangles indicate positive, while light red rectangles indicate negative correlation between the glycan structures or traits and CpG methylation levels. Only correlations replicated across assays and/or cohorts are shown. The red rectangle around the MGAT3 enzyme reflects the negative correlation between CpG methylation and the derived trait B/FA2, which effectively measures enzyme activity at this step. MGAT3 N-acetilglucosaminyltransferase III (GnT-III), FUT8 fucosyltransferase 8, GalT1 galactosyltranserase 1, ST6GalT1 Beta-galactoside alpha-2,6-sialyltransferase 1
In order to infer a mechanistic pathway of the observed correlations, we mapped them to the glycan biosynthesis pathways (Fig. 4b). The ratio of bisecting glycans to FA2 was taken as an indicator of MGAT3 (GnT-III) activity. This interpretation allowed us to infer lower GnT-III enzymatic activity when the promoter of the MGAT3 gene was methylated. Increase in MGAT3 promoter methylation correlated with a decrease in certain galactosylated and sialylated structures (Fig. 4b). In addition to the decreased levels of bisecting GlcNAc on non-galactosylated glycans (B/FA2), the most significant effect of the MGAT3 promoter methylation on IgG glycome composition was a decrease of IgG galactosylation.
Discussion
Results from this study strongly indicate that the MGAT3 and BACH2 genes play an important role in IBD pathogenesis and suggest a possible disease pathway mediated by the pro-inflammatory properties of IgG antibodies acquired by alterations in Fc glycosylation. Our recent study, performed on a large cohort of over 1000 IBD patients, reported a significant difference in IgG glycome composition in both UC and CD compared to healthy controls [43, 46]. We found a decrease in quantity of galacosylated glycans in both CD and UC, as well as a decrease in sialylated glycans and an increase of bisecting GlcNAc on digalactosylated glycan structures on IgG in CD. Indeed, alternative N-glycosylation of an IgG molecule influences its function—pro-inflammatory and anti-inflammatory activity depends on the glycans added on the Cy2 domain of its Fc region [29]. These glycans are of a biantennary complex type with or without bisecting GlcNAc, core fucose, galactose, and sialic acid residues [61]. Recently, this was confirmed in a large multi-centric study of IgG glycome in IBD [46]. Therefore, glycan changes observed on IgG in peripheral blood of UC and CD patients are obviously associated with increased inflammatory potential of IgG, suggesting functional relevance of IgG glycosylation for IBD.
Here, we propose a possible mechanism underlying the aberrant IgG glycosylation pattern observed in IBD [43, 46]. Out of five candidate genes analyzed in this work, the MGAT3, a glycosyltransferase which participates in synthesis of IgG glycans, and the BACH2, a transcription factor and a master regulator of a network of genes relevant for B cell integrity [62, 63], showed differential methylation in peripheral blood of both CD and UC patients when compared to healthy individuals. Even though we identified changes in methylation level for both UC and CD compared to HC, the differences were more pronounced for CD. This is concordant with other studies that explored either whole genome methylation or promoter methylation of candidate genes in IBD [11]. The extent of the change in IgG glycome composition was also consistently higher in CD than UC compared to HC [43, 46].
The protein encoded by the MGAT3 gene (N-acetylglucosaminyltransferase III, GnT-III) is responsible for significant functional alteration of glycans on the Fc region of an IgG antibody. The GnT-III adds N-acetylglucosamine (GlcNAc) on β1,4-linked mannose in the three-mannose core of N-glycans, producing bisecting GlcNAc structures. In the same CD patients, who showed changed MGAT3 promoter methylation level in peripheral blood cells, a significant increase in the percentage of bisecting GlcNAc on glycans of circulating IgG antibodies was recorded, too. The association of the MGAT3 with both IgG N-glycosylation [49] and Crohn’s disease [4, 5] suggests that N-glycans with bisecting GlcNAc could be involved in CD pathogenesis through functional effect on IgG antibody.
Correlations between BACH2 and MGAT3 promoter methylation and glycan structures have given further insight into the changes of IgG glycosylation pattern mediated by those two genes (Fig. 4b). The MGAT3 promoter methylation probably led to decreased GnT-III enzymatic activity, as revealed by negative correlation between methylation and total bisecting glycans to FA2 ratio. Namely, GnT-III adds a bisecting GlcNAc to FA2. A further proof is the positive correlation between the MGAT3 promoter methylation and FA2, since it is not surprising that substrate accumulates when enzyme activity is decreased. More complex effects were observed on galactosylation and sialylation. The negative correlation between MGAT3 methylation and galactosylation of both, glycans with (FA2BG2) and without bisecting GlcNAc (FA2G2), suggests that the effect of increased galactosylation is not caused only by steric effects of bisecting GlcNAc, but also through some indirect effects of MGAT3 expression on galactosyltransferase activity. It seems as though galactosylation and sialylation are co-regulated with the addition of a bisecting GlcNAc catalyzed by GnT-III. Furthermore, Dekkers and co-workers recently reported that transfection of cells with MGAT3 causes an increase of IgG galactosylation [64].
Much weaker correlation was observed between BACH2 methylation and IgG glycosylation. This was expected since BACH2 is not a glycosyltransferase and thus is not directly involved in glycan biosynthetic pathways. However, weak positive correlations with A2G2, FA2G2, and FA2G2S1 structures, which involve galactosylation and sialylation, were observed, as well as weak negative correlation with fucosylated bianntenary structure FA2. This is interesting because GWA studies associated BACH2 with IgG galactosylation [49] as well as with various immune and inflammatory diseases including IBD [4, 5, 65–67] in which IgG acquires pro-inflammatory properties through decrease in galactosylation, sialylation, and fucosylation [43, 46]. Since BACH2 is orchestrating a gene regulatory network in B cells [62], we believe that some glyco-genes are also regulated by this transcription factor. Indeed, our in silico analysis identified several glyco-genes, mostly galactosyltransferases (including B4GALT1 and B4GALT2), to possess putative AP-1 and NFE2 binding sites for BACH2 transcription factor [63], suggesting that these galactosyltransferases could be controlled by BACH2 (Additional file 1: Tables S7 and S8). Our present efforts are focused on functional studies with hope to reveal a more complete view of the BACH2 role in IgG glycosylation.
Since DNA methylation pattern is tissue-specific, our goal was to ascertain if CpG methylation from blood could be a proxy for CpG methylation of the same candidate gene in the tissue where the inflammation is taking place. In fact, IBD is an immune-mediated disorder in which T cells are actively implicated in development of gut-mucosa inflammation [68]. Previous evidence has suggested that N-glycosylation of intestinal T cells is associated with UC pathogenesis and disease severity [50, 69]. Therefore, we analyzed MGAT3 promoter methylation in CD3+ T cells isolated from PBMCs and from intestinal mucosa of UC patients with active and inactive form of the disease and compared with MGAT3 promoter methylation from PBMCs of the same patients from the Porto cohort. We found 7 out of 15 differentially methylated CpG sites in CD3+ T cells isolated from colonic mucosa of UC patients with active form of the disease compared to CD3+ T cells from mucosa of healthy individuals. On the other hand, there were no differences in MGAT3 promoter methylation between patients with either active or inactive form of UC and healthy controls neither in PBMCs nor in CD3+ T cells isolated from PBMCs. This could be due to high dispersion in the methylation level when measured from PBMCs (47–94%) and CD3+ T cells isolated from PBMCs (26–90%), probably due to small sample size and dispersion in age. Namely, cell composition changes across age in whole blood, and it can explain dispersion of CpG methylation level observed in our sample [70]. Considering much smaller dispersion in values of methylation level (65–97%) measured from CD3+ T cells from lamina propria, the differences could be used as a signature for inflammation.
Conclusions
Taken together, our results suggest that the aberrant methylation observed in the MGAT3 gene in CD3+ T cells from intestinal mucosa of UC patients, B cells from peripheral blood, and the whole peripheral blood in UC and CD patients is a possible mechanism underlying inflammation due to a change in the immune system—either through the change of glycans on Fc region of IgGs or by modulating the glycosylation profile of glycoproteins on intestinal T cells. Others [24] have shown that some of their candidate genes changed promoter methylation level in whole biopsies, while some of the genes showed changes only in some cell types of the heterogeneous cell population from the epithelial and non-epithelial cells, pointing out the importance of cell separation from mucosal biopsies. Interestingly, one of the genes that showed differential methylation in the non-epithelial fraction, representing immune and stromal cells, was FUT7, the fucosyltransferase involved in sialyl Lewis X synthesis, a ligand in selectin-mediated adhesion of leukocytes to activated endothelium. Furthermore, Dias and collaborators proposed a molecular mechanism in IBD involving another glyco-gene, the MGAT5 (GnT-V), responsible for branching of N-glycans. They showed decreased expression of branched N-glycans on T cell receptor (TCR) of lamina propria associated with disease severity in patients with active UC [50]. Dysregulation of N-glycan branching on TCR contributes to a decreased threshold of T cell activation leading to a hyper-immune response which is a feature of UC patients. Taken together, our results and those of others suggest an important role of aberrant protein glycosylation (partly through epigenetic mechanisms) in IBD through dysregulation of the immune system. Also, in IBD diagnosis and treatment, it is important to find a non-invasive, specific, and clinically useful biomarkers in order to identify high-risk patients. Using MGAT3 hypermethylation together with the glycan traits as markers from peripheral blood of IBD patients seems promising in the disease identification.
Additional files
Supplementary Tables 1-8. Demographics of IBD patients and healthy controls (1-4), PCR primers (5), number of samples per analysis (6) and in silico analysis of transcription factor binding sites in gene promoters (7, 8). (DOCX 70 kb)
Figure S1. Position of the pyrosequencing assays for the genes LAMB1, IL6ST, and IKZF1 in the genome relative to CpG islands, annotated promoters, and exons. (PDF 523 kb)
Figure S2. Violin plots showing the age distribution in IBD patients (CD, UC) and healthy controls (HC). The groups were well matched by age, which was shown by Mann-Whitney U test: no significant differences between groups were found at the level p = 0.05. (PDF 704 kb)
Acknowledgements
The authors would like to thank Stephanie Scott for her organizational and administrational contribution. The study has been funded by the EU FP7 grant European Commission IBD-BIOM (contract # 305479), EU FP7 Regional Potential Grant INTEGRA-Life (contract # 315997), European Structural and Investment Funds grant for the Croatian National Centre of Research Excellence in Personalized Healthcare (contract # KK.01.1.1.01.0010), and Croatian Science Foundation grant EpiGlycoIgG (contract # 3361). Financial support from Portugal (PI: SSP): FEDER—Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020—Operacional Programme for Competitiveness and Internationalisation (POCI), Portugal 2020, and by Portuguese funds through FCT—Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Inovação in the framework of the project (POCI-01/0145-FEDER-016601; PTDC/DTP-PIC/0560/2014) was received. SSP also acknowledges the European Crohn’s and Colitis Organization (ECCO) and the “Broad Medical Research program at Crohn’s and Colitis Foundation of America-CCFA” for funding. SSP acknowledges the Portuguese Group of Study on IBD (GEDII) for funding. A.M.D. [PD/BD/105982/2014] also acknowledges FCT for funding.
IBD-BIOM consortium: Daniel Kolarich (Department of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, Potsdam, Germany), Manfred Wuhrer (Center for Proteomics and Metabolomics, Leiden University Medical Center, Leiden, The Netherlands; Division of BioAnalytical Chemistry, VU University Amsterdam, Amsterdam, the Netherlands), Dermot P. B. McGovern (F. Widjaja Family Foundation Inflammatory Bowel and Immunobiology Research Institute, Cedars-Sinai Medical Center, Los Angeles), Iain K. Pemberton (IP Research Consulting SAS, Paris, France), Daniel IR Spencer (Ludger Ltd., Culham Science Centre, Oxford, UK, Daryl L. Fernandes (Ludger Ltd., Culham Science Centre, Oxford, UK), Rahul Kalla, Kate O’Leary, Alex T Adams, Hazel Drummond, Elaine Nimmo, Ray Boyapati, David C Wilson (Centre for Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK), Ray Doran (Ludger Ltd., Culham Science Centre, Oxford, UK), Igor Rudan (all, Centre for Population Health Sciences, University of Edinburgh, Edinburgh, UK), Paolo Lionetti (Paediatric Gastroenterology Unit, AOU Meyer, Viale Pieraccini, Florence, Italy), Natalia Manetti (Department of Medical and Surgical Sciences, Division of Gastroenterology, University Hospital Careggi, Florence, Italy), Fabrizio Bossa (Department of Medical Sciences, Division of Gastroenterology, IRCCS-CSS Hospital, Viale Cappuccini, Rotondo, Italy), Paola Cantoro, Anna Kohn (Division of Gastroenterology, S. Camillo Hospital, Rome, Italy), Giancarlo Sturniolo (Gastrointestinal Unit, University of Padua, Padua, Italy), Silvio Danese (IBD Unit, Humanitas Research Institute, Rozzano, Milan, Italy), Mariek Pierik (Maastricht University Medical Centre (MUMC), Maastricht, the Netherlands), and David C. Wilson (Centre for Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK).
Funding
This independent research was generously supported by the following grants: EU FP7 research grant IBD-BIOM (contract # 305479) to JS, VA, GL, and VZ; EU FP7 Regional Potential Grant INTEGRA-Life (contract # 315997) to GL and VZ; European Structural and Investment Funds grant for the Croatian National Centre of Research Excellence in Personalized Healthcare (contract # KK.01.1.1.01.0010) to GL and VZ; Croatian Science Foundation grant EpiGlycoIgG (contract # 3361) to VZ; FEDER COMPETE 2020 POCI, Portugal 2020, and Portuguese funds through FCT (contracts # POCI-01/0145-FEDER-016601 and PTDC/DTP-PIC/0560/2014) to SP; and FTC (contract # PD/BD/105982/2014) to AMD.
Availability of data and materials
All original data can be obtained from the authors upon request.
Abbreviations
- CD
Crohn’s disease
- EWAS
Epigenome-wide association study
- GlcNAc
N-acetylglucosamine
- GWAS
Genome-wide association study
- HC
Healthy control
- IBD
Inflammatory bowel disease
- IgG
Immunoglobulin G
- LC-MS
Liquid chromatography coupled to mass spectrometry
- PBMCs
Peripheral blood mononuclear cells
- UC
Ulcerative colitis
Authors’ contributions
Study design was conceived by VZ, AV, GL, and SP. Sample provision was provided by JS, NTV, NAK, ERN, VA, RD’I, and SP. Blood and biopsy processing was conducted by AMD and AL. DNA methylation analyses were carried out by MK, DM, PD, IS, and IB. Glycan analysis was carried out by IT, JŠ, MŠ, and GR. Statistical and correlation analyses were carried out by AV, MK, and DM. Drafting of the manuscript was carried out by VZ, AV, MK, DM, and IT-A. All authors were involved in critical review, editing, revision, and approval of the final manuscript.
Ethics approval and consent to participate
For the Edinburgh and Florence cohorts, ethical approvals were obtained from Tayside Committee on Medical Ethics B, and all patients and controls provided written, informed consent (LREC 06/S1101/16, LREC 2000/4/192). For the patients from Portugal, all clinical procedures and research protocols were approved by the ethics committee of CHP/HSA, Portugal (233/12(179-DEFI/177-CES); all participants gave their informed consent.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Marija Klasić and Dora Markulin contributed equally to this work.
Electronic supplementary material
The online version of this article (10.1186/s13148-018-0507-y) contains supplementary material, which is available to authorized users.
Contributor Information
Vlatka Zoldoš, Email: vzoldos@biol.pmf.hr.
IBD consortium:
Daniel Kolarich, Manfred Wuhrer, Dermot P. B. McGovern, Iain K. Pemberton, Daniel I. R. Spencer, Daryl L. Fernandes, Rahul Kalla, Kate O’Leary, Alex T. Adams, Hazel Drummond, Elaine Nimmo, Ray Boyapati, David C. Wilson, Ray Doran, Igor Rudan, Paolo Lionetti, Natalia Manetti, Fabrizio Bossa, Paola Cantoro, Anna Kohn, Giancarlo Sturniolo, Silvio Danese, Marieke Pierik, and David C. Wilson
References
- 1.Burisch J, Pedersen N, Čuković-Čavka S, Brinar M, Kaimakliotis I, Duricova D, Shonová O, Vind I, Avnstrøm S, Thorsgaard N, et al. East–West gradient in the incidence of inflammatory bowel disease in Europe: the ECCO-EpiCom inception cohort. Gut. 2013;63:588–597. doi: 10.1136/gutjnl-2013-304636. [DOI] [PubMed] [Google Scholar]
- 2.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 3.Anderson CA, Boucher G, Lees CW, Franke A, D'Amato M, Taylor KD, Lee JC, Goyette P, Imielinski M, Latiano A, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–252. doi: 10.1038/ng.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franke A, Balschun T, Sina C, Ellinghaus D, Häsler R, Mayr G, Albrecht M, Wittig M, Buchert E, Nikolaus S, et al. Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL) Nat Genet. 2010;42:292–294. doi: 10.1038/ng.553. [DOI] [PubMed] [Google Scholar]
- 5.Franke A, McGovern DP, Barrett JC, Wang K, Radford-Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47:979–986. doi: 10.1038/ng.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bonaz BL, Bernstein CN. Brain-gut interactions in inflammatory bowel disease. Gastroenterology. 2013;144:36–49. doi: 10.1053/j.gastro.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Pituch-Zdanowska A, Banaszkiewicz A, Albrecht P. The role of dietary fibre in inflammatory bowel disease. Gastroenterol Rev. 2015;3:135–141. doi: 10.5114/pg.2015.52753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubin DT, Hanauer SB. Smoking and inflammatory bowel disease. Eur J Gastroenterol Hepatol. 2000;12:855–862. doi: 10.1097/00042737-200012080-00004. [DOI] [PubMed] [Google Scholar]
- 11.McDermott E, Ryan EJ, Tosetto M, Gibson D, Burrage J, Keegan D, Byrne K, Crowe E, Sexton G, Malone K, et al. DNA methylation profiling in inflammatory bowel disease provides new insights into disease pathogenesis. J Crohn’s Colitis. 2015;10:77–86. doi: 10.1093/ecco-jcc/jjv176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nimmo ER, Prendergast JG, Aldhous MC, Kennedy NA, Henderson P, Drummond HE, Ramsahoye BH, Wilson DC, Semple CA, Satsangi J. Genome-wide methylation profiling in Crohnʼs disease identifies altered epigenetic regulation of key host defense mechanisms including the Th17 pathway. Inflamm Bowel Dis. 2012;18:889–899. doi: 10.1002/ibd.21912. [DOI] [PubMed] [Google Scholar]
- 13.Ventham NT, Kennedy NA, Adams AT, Kalla R, Heath S, O'Leary KR, Drummond H, consortium IB, consortium IC, Wilson DC, et al: Integrative epigenome-wide analysis demonstrates that DNA methylation may mediate genetic risk in inflammatory bowel disease. Nat Commun 2016, 7:13507. [DOI] [PMC free article] [PubMed]
- 14.Low D. DNA methylation in inflammatory bowel disease and beyond. World J Gastroenterol. 2013;19:5238. doi: 10.3748/wjg.v19.i32.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ventham NT, Kennedy NA, Nimmo ER, Satsangi J. Beyond gene discovery in inflammatory bowel disease: the emerging role of epigenetics. Gastroenterology. 2013;145:293–308. doi: 10.1053/j.gastro.2013.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 17.Ambatipudi S, Cuenin C, Hernandez-Vargas H, Ghantous A, Le Calvez-Kelm F, Kaaks R, Barrdahl M, Boeing H, Aleksandrova K, Trichopoulou A, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8:599–618. doi: 10.2217/epi-2016-0001. [DOI] [PubMed] [Google Scholar]
- 18.Barrès R, Yan J, Egan B, Treebak Jonas T, Rasmussen M, Fritz T, Caidahl K, Krook A, O’Gorman Donal J, Zierath Juleen R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15:405–411. doi: 10.1016/j.cmet.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 19.Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, Blumberg RS. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012;336:489–493. doi: 10.1126/science.1219328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Symonds ME, Sebert SP, Budge H. The impact of diet during early life and its contribution to later disease: critical checkpoints in development and their long-term consequences for metabolic health. Proc Nutr Soc. 2009;68:416. doi: 10.1017/S0029665109990152. [DOI] [PubMed] [Google Scholar]
- 21.Vaissiere T, Hung RJ, Zaridze D, Moukeria A, Cuenin C, Fasolo V, Ferro G, Paliwal A, Hainaut P, Brennan P, et al. Quantitative analysis of DNA methylation profiles in lung cancer identifies aberrant DNA methylation of specific genes and its association with gender and cancer risk factors. Cancer Res. 2009;69:243–252. doi: 10.1158/0008-5472.CAN-08-2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milagro FI, Mansego ML, De Miguel C, Martinez JA. Dietary factors, epigenetic modifications and obesity outcomes: progresses and perspectives. Mol Asp Med. 2013;34:782–812. doi: 10.1016/j.mam.2012.06.010. [DOI] [PubMed] [Google Scholar]
- 23.Adams AT, Kennedy NA, Hansen R, Ventham NT, OʼLeary KR, Drummond HE, Noble CL, El-Omar E, Russell RK, Wilson DC, et al. Two-stage genome-wide methylation profiling in childhood-onset Crohnʼs disease implicates epigenetic alterations at the VMP1/MIR21 and HLA loci. Inflamm Bowel Dis. 2014;20:1784–1793. doi: 10.1097/MIB.0000000000000179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cooke J, Zhang H, Greger L, Silva A-L, Massey D, Dawson C, Metz A, Ibrahim A, Parkes M. Mucosal genome-wide methylation changes in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:2128–2137. doi: 10.1002/ibd.22942. [DOI] [PubMed] [Google Scholar]
- 25.Harris RA, Nagy-Szakal D, Mir SAV, Frank E, Szigeti, Kaplan JL, Bronsky J, Opekun A, Ferry GD, Winter H, Kellermayer R. DNA methylation-associated colonic mucosal immune and defense responses in treatment-naïve pediatric ulcerative colitis. Epigenetics. 2014;9:1131–1137. doi: 10.4161/epi.29446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karatzas PS, Gazouli M, Safioleas M, Mantzaris GJ. DNA methylation changes in inflammatory bowel disease. Ann Gastroenterol. 2014;27:125–132. [PMC free article] [PubMed] [Google Scholar]
- 27.Hart GW, Copeland RJ. Glycomics hits the big time. Cell. 2010;143:672–676. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Böhm S, Kao D, Nimmerjahn F. Sweet and sour: the role of glycosylation for the anti-inflammatory activity of immunoglobulin G. In: Fc Receptors. Basel: Springer International Publishing; 2014. p. 393–417. 393–417. [DOI] [PubMed]
- 29.Kaneko Y. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science. 2006;313:670–673. doi: 10.1126/science.1129594. [DOI] [PubMed] [Google Scholar]
- 30.National Research Council (U.S.). Committee on Assessing the Importance and Impact of Glycomics and Glycosciences., National Research Council (U.S.). Board on Chemical Sciences and Technology., National Research Council (U.S.). Board on Life Sciences.: Transforming glycoscience: a roadmap for the future. Washington, D.C.: National Academies Press; 2012. [PubMed]
- 31.Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol: Mech Dis. 2015;10:473–510. doi: 10.1146/annurev-pathol-012414-040438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horvat T, Deželjin M, Redžić I, Barišić D, Herak Bosnar M, Lauc G, Zoldoš V. Reversibility of membrane N-Glycome of HeLa cells upon treatment with epigenetic inhibitors. PLoS One. 2013;8:e54672. doi: 10.1371/journal.pone.0054672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horvat T, Mužinić A, Barišić D, Bosnar MH, Zoldoš V. Epigenetic modulation of the HeLa cell membrane N-glycome. Biochim Biophys Acta Gen Subj. 2012;1820:1412–1419. doi: 10.1016/j.bbagen.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 34.Horvat T, Zoldoš V, Lauc G. Evolutional and clinical implications of the epigenetic regulation of protein glycosylation. Clin Epigenetics. 2011;2:425–432. doi: 10.1007/s13148-011-0039-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klasić M, Krištić J, Korać P, Horvat T, Markulin D, Vojta A, Reiding KR, Wuhrer M, Lauc G, Zoldoš V. DNA hypomethylation upregulates expression of the MGAT3 gene in HepG2 cells and leads to changes in N-glycosylation of secreted glycoproteins. Sci Rep. 2016;6:24363. [DOI] [PMC free article] [PubMed]
- 36.Saldova R, Dempsey E, Pérez-Garay M, Mariño K, Watson JA, Blanco-Fernández A, Struwe WB, Harvey DJ, Madden SF, Peracaula R, et al. 5-AZA-2′-deoxycytidine induced demethylation influences N-glycosylation of secreted glycoproteins in ovarian cancer. Epigenetics. 2011;6:1362–1372. doi: 10.4161/epi.6.11.17977. [DOI] [PubMed] [Google Scholar]
- 37.Vojta A, Samaržija I, Bočkor L, Zoldoš V. Glyco-genes change expression in cancer through aberrant methylation. Biochim Biophys Acta Gen Subj. 2016;1860:1776–1785. doi: 10.1016/j.bbagen.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Zoldoš V, Horvat T, Novokmet M, Cuenin C, Mužinić A, Pučić M, Huffman JE, Gornik O, Polašek O, Campbell H, et al. Epigenetic silencing of HNF1A associates with changes in the composition of the human plasma N-glycome. Epigenetics. 2012;7:164–172. doi: 10.4161/epi.7.2.18918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Theodoratou E, Campbell H, Ventham NT, Kolarich D, Pučić-Baković M, Zoldoš V, Fernandes D, Pemberton IK, Rudan I, Kennedy NA, et al. The role of glycosylation in IBD. Nat Rev Gastroenterol Hepatol. 2014;11(10):588–600. [DOI] [PubMed]
- 40.Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- 41.Miyahara K, Nouso K, Saito S, Hiraoka S, Harada K, Takahashi S, Morimoto Y, Kobayashi S, Ikeda F, Miyake Y, et al. Serum glycan markers for evaluation of disease activity and prediction of clinical course in patients with ulcerative colitis. PLoS One. 2013;8:e74861. doi: 10.1371/journal.pone.0074861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinzaki S, Iijima H, Nakagawa T, Egawa S, Nakajima S, Ishii S, Irie T, Kakiuchi Y, Nishida T, Yasumaru M, et al. IgG oligosaccharide alterations are a novel diagnostic marker for disease activity and the clinical course of inflammatory bowel disease. Am J Gastroenterol. 2008;103:1173–1181. doi: 10.1111/j.1572-0241.2007.01699.x. [DOI] [PubMed] [Google Scholar]
- 43.Trbojevic Akmacic I, Ventham NT, Theodoratou E, Vuckovic F, Kennedy NA, Kristic J, Nimmo ER, Kalla R, Drummond H, Stambuk J, et al. Inflammatory bowel disease associates with proinflammatory potential of the immunoglobulin G glycome. Inflamm Bowel Dis. 2015;21:1237–1247. doi: 10.1097/MIB.0000000000000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plomp R, Ruhaak LR, Uh HW, Reiding KR, Selman M, Houwing-Duistermaat JJ, Slagboom PE, Beekman M, Wuhrer M. Subclass-specific IgG glycosylation is associated with markers of inflammation and metabolic health. Sci Rep. 2017;7:12325. doi: 10.1038/s41598-017-12495-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013;13:176–189. doi: 10.1038/nri3401. [DOI] [PubMed] [Google Scholar]
- 46.Simurina M, de Haan N, Vuckovic F, Kennedy NA, Stambuk J, Falck D, Trbojevic-Akmacic I, Clerc F, Razdorov G, Khon A, et al: Glycosylation of immunoglobulin G associates with clinical features of inflammatory bowel diseases. Gastroenterology. 2018;154(5):1320–1333.e10. [DOI] [PMC free article] [PubMed]
- 47.Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, Wesley E, Parnell K, Zhang H, Drummond H, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009;41:1330–1334. doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McGovern DPB, Jones MR, Taylor KD, Marciante K, Yan X, Dubinsky M, Ippoliti A, Vasiliauskas E, Berel D, Derkowski C, et al. Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum Mol Genet. 2010;19:3468–3476. doi: 10.1093/hmg/ddq248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lauc G, Huffman JE, Pucic M, Zgaga L, Adamczyk B, Muzinic A, Novokmet M, Polasek O, Gornik O, Kristic J, et al. Loci associated with N-glycosylation of human immunoglobulin G show pleiotropy with autoimmune diseases and haematological cancers. PLoS Genet. 2013;9:e1003225. doi: 10.1371/journal.pgen.1003225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dias AM, Dourado J, Lago P, Cabral J, Marcos-Pinto R, Salgueiro P, Almeida CR, Carvalho S, Fonseca S, Lima M, et al. Dysregulation of T cell receptor N-glycosylation: a molecular mechanism involved in ulcerative colitis. Hum Mol Genet. 2014;23:2416–2427. doi: 10.1093/hmg/ddt632. [DOI] [PubMed] [Google Scholar]
- 51.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlen SE, Greco D, Soderhall C, Scheynius A, Kere J. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krištić J, Zoldoš V, Lauc G. Complex genetics of protein N-glycosylation. In: Endo T, Seeberger PH, Hart GW, Wong C-H, Taniguchi N, editors. Glycoscience: biology and medicine. Tokyo: Springer Japan; 2014. pp. 1–7. [Google Scholar]
- 53.Stewart SK, Morris TJ, Guilhamon P, Bulstrode H, Bachman M, Balasubramanian S, Beck S. oxBS-450K: a method for analysing hydroxymethylation using 450K BeadChips. Methods. 2015;72:9–15. doi: 10.1016/j.ymeth.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kroeze LI, Aslanyan MG, van Rooij A, Koorenhof-Scheele TN, Massop M, Carell T, Boezeman JB, Marie JP, Halkes CJ, de Witte T, et al. Characterization of acute myeloid leukemia based on levels of global hydroxymethylation. Blood. 2014;124:1110–1118. doi: 10.1182/blood-2013-08-518514. [DOI] [PubMed] [Google Scholar]
- 55.Sanchez-Guerra M, Zheng Y, Osorio-Yanez C, Zhong J, Chervona Y, Wang S, Chang D, McCracken JP, Diaz A, Bertazzi PA, et al. Effects of particulate matter exposure on blood 5-hydroxymethylation: results from the Beijing truck driver air pollution study. Epigenetics. 2015;10:633–642. doi: 10.1080/15592294.2015.1050174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368–1380. doi: 10.1016/j.cell.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pucic M, Knezevic A, Vidic J, Adamczyk B, Novokmet M, Polasek O, Gornik O, Supraha-Goreta S, Wormald MR, Redzic I, et al. High throughput isolation and glycosylation analysis of IgG-variability and heritability of the IgG glycome in three isolated human populations. Mol Cell Proteomics. 2011;M111(010090):10. doi: 10.1074/mcp.M111.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trbojevic-Akmacic I, Ugrina I, Lauc G. Comparative analysis and validation of different steps in glycomics studies. Methods Enzymol. 2017;586:37–55. doi: 10.1016/bs.mie.2016.09.027. [DOI] [PubMed] [Google Scholar]
- 59.Selman MH, Derks RJ, Bondt A, Palmblad M, Schoenmaker B, Koeleman CA, van de Geijn FE, Dolhain RJ, Deelder AM, Wuhrer M. Fc specific IgG glycosylation profiling by robust nano-reverse phase HPLC-MS using a sheath-flow ESI sprayer interface. J Proteome. 2012;75:1318–1329. doi: 10.1016/j.jprot.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 60.Jansen BC, Falck D, de Haan N, Hipgrave Ederveen AL, Razdorov G, Lauc G, Wuhrer M. LaCyTools: a targeted liquid chromatography-mass spectrometry data processing package for relative quantitation of glycopeptides. J Proteome Res. 2016;15:2198–2210. doi: 10.1021/acs.jproteome.6b00171. [DOI] [PubMed] [Google Scholar]
- 61.Fujii S, Nishiura T, Nishikawa A, Miura R, Taniguchi N. Structural heterogeneity of sugar chains in immunoglobulin G. Conformation of immunoglobulin G molecule and substrate specificities of glycosyltransferases. J Biol Chem. 1990;265:6009–6018. [PubMed] [Google Scholar]
- 62.Igarashi K, Ochiai K, Itoh-Nakadai A, Muto A. Orchestration of plasma cell differentiation by Bach2 and its gene regulatory network. Immunol Rev. 2014;261:116–125. doi: 10.1111/imr.12201. [DOI] [PubMed] [Google Scholar]
- 63.Kuwahara M, Suzuki J, Tofukuji S, Yamada T, Kanoh M, Matsumoto A, Maruyama S, Kometani K, Kurosaki T, Ohara O, et al. The Menin–Bach2 axis is critical for regulating CD4 T-cell senescence and cytokine homeostasis. Nat Commun. 2014;5:3555. [DOI] [PMC free article] [PubMed]
- 64.Dekkers G, Plomp R, Koeleman CA, Visser R, von Horsten HH, Sandig V, Rispens T, Wuhrer M, Vidarsson G. Multi-level glyco-engineering techniques to generate IgG with defined Fc-glycans. Sci Rep. 2016;6:36964. doi: 10.1038/srep36964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chu X, Pan C-M, Zhao S-X, Liang J, Gao G-Q, Zhang X-M, Yuan G-Y, Li C-G, Xue L-Q, Shen M, et al. A genome-wide association study identifies two new risk loci for Graves’ disease. Nat Genet. 2011;43:897–901. doi: 10.1038/ng.898. [DOI] [PubMed] [Google Scholar]
- 66.Dubois PCA, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, Zhernakova A, Heap GAR, Ádány R, Aromaa A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sawcer S, Hellenthal G, Pirinen M, Spencer CC, Patsopoulos NA, Moutsianas L, Dilthey A, Su Z, Freeman C, Hunt SE, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Macdonald TT, Monteleone G. Immunity, inflammation, and allergy in the gut. Science. 2005;307:1920–1925. doi: 10.1126/science.1106442. [DOI] [PubMed] [Google Scholar]
- 69.Vossenkamper A, Hundsrucker C, Page K, van Maurik A, Sanders TJ, Stagg AJ, Das L, MacDonald TT. A CD3-specific antibody reduces cytokine production and alters phosphoprotein profiles in intestinal tissues from patients with inflammatory bowel disease. Gastroenterology. 2014;147:172–183. doi: 10.1053/j.gastro.2014.03.049. [DOI] [PubMed] [Google Scholar]
- 70.Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31. doi: 10.1186/gb-2014-15-2-r31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables 1-8. Demographics of IBD patients and healthy controls (1-4), PCR primers (5), number of samples per analysis (6) and in silico analysis of transcription factor binding sites in gene promoters (7, 8). (DOCX 70 kb)
Figure S1. Position of the pyrosequencing assays for the genes LAMB1, IL6ST, and IKZF1 in the genome relative to CpG islands, annotated promoters, and exons. (PDF 523 kb)
Figure S2. Violin plots showing the age distribution in IBD patients (CD, UC) and healthy controls (HC). The groups were well matched by age, which was shown by Mann-Whitney U test: no significant differences between groups were found at the level p = 0.05. (PDF 704 kb)
Data Availability Statement
All original data can be obtained from the authors upon request.