

Summary
Background
Chronic rhinosinusitis (CRS) is a heterogeneous chronic inflammatory disease generally divided based on the presence or absence of nasal polyps (NPs). One of the features of NPs is excessive fibrin deposition, which is associated with down-regulation of tissue plasminogen activator (t-PA) in NPs. As t-PA is expressed in epithelial cells, and epithelium is readily accessible to topical therapies, identifying compounds that can mediate the induction of t-PA would be a potential new strategy for the treatment of NPs.
Objective
The objective of this study was to determine whether short-chain fatty acids (SCFAs) can induce t-PA in airway epithelial cells via their known receptors GPR41 and GPR43.
Methods
We performed immunohistochemistry (IHC) to determine whether receptors for SCFAs, known as G protein-coupled receptor 41/free fatty acid receptor 3 (GPR41/FFAR3) and GPR43/FFAR2, are expressed in nasal tissue. Primary normal human bronchial epithelial (NHBE) cells were stimulated with different concentrations of SCFAs to test induction of t-PA, which was analysed by expression of mRNA and protein. Mediation of responses by SCFA receptors was evaluated by specific receptor gene silencing with siRNA.
Results
Immunohistochemistry study revealed that airway epithelial cells expressed GPR41 and GPR43. Acetic acid, propionic acid, butyric acid and valeric acid significantly induced t-PA expression from two- to tenfolds. The strongest inducer of t-PA from NHBE cells was propionic acid; cells stimulated with propionic acid released t-PA into the supernatant in its active form. Gene silencing of GPR41 and GPR43 revealed that induction of t-PA by SCFAs was dependent upon both GPR41 and GPR43.
Conclusions and Clinical Relevance
Short-chain fatty acids were shown to induce airway epithelial cell expression of t-PA via GPR41 and GPR43. Topical delivery of potent compounds that activate these receptors may have value by reducing fibrin deposition and shrinking nasal polyp growth.
Keywords: chronic rhinosinusitis, fibrin, GPR41, GPR43, t-PA
1. Introduction
Chronic rhinosinusitis (CRS) is a disease that is defined as inflammation of the nose and paranasal sinuses of at least 12 weeks of duration. CRS is one of the most common chronic diseases in the United States, which affects more than 10 million Americans and greatly impairs quality of life in affected patients.1-4 CRS is a heterogeneous disease and is frequently divided into 2 groups based on the presence or absence of nasal polyps (NPs): CRS with NPs (CRSwNP) and CRS without NPs (CRSsNP). Inflammatory patterns in these 2 diseases are different and also show heterogeneity.5,6 Pathologically, CRSwNP tissue often displays intense eosinophilic infiltration and a Th2-biased cytokine profile, especially in western countries.7 It is well known that severe asthma shows high comorbidity with CRSwNP.8 Furthermore, patients with CRSwNP often require repeated surgical intervention, despite the use of oral and/or topical corticosteroids. New medical approaches that can effectively shrink or eliminate NPs are clearly needed.
NPs usually present around the middle nasal meatus or paranasal sinuses as oedematous masses. Histologically, NPs consist of intense oedematous stroma and the formation of pseudocyst filled with plasma proteins accompanied by infiltration of inflammatory cells.9 Although plasma exudation from capillaries occurs as a result of profound inflammation, the precise mechanisms of retention of extravascular plasma proteins in NP submucosa have not been fully elucidated. A previous report from our laboratory demonstrated excessive fibrin deposition, and low levels of d-dimer, a fibrin degradation product, in NPs, suggesting that fibrinolysis is impaired in NPs.10 Profound fibrin deposition may mediate the retention of exuded plasma proteins and the formation of intense oedema in NP tissue. Activation of the coagulation cascade and deposition of fibrin are consequences of inflammation for host defence and may confine infections and support repair processes.11 However, dysregulation of the coagulation cascade is involved in the aetiology of many diseases in which excessive fibrin deposition occurs, including rheumatoid arthritis, adult respiratory distress syndrome and severe asthma.12-14 Ordinarily, fibrin degradation is mediated by plasmin, which is generated through cleavage of plasminogen by urokinase plasminogen activator (u-PA) and/or tissue plasminogen activator (t-PA). Our previous study demonstrated profound down-regulation of t-PA expression in NPs, but not u-PA.10 The data showed that t-PA was most prominently observed in epithelial and glandular cells in normal uncinate tissue (UT), but expression was lost in NP epithelium. Tissue plasminogen activator converts plasminogen to plasmin, which is then responsible for the degradation of cross-linked of fibrin. Loss of t-PA in NP tissue thus may contribute to the accumulation of cross-linked fibrin in NP tissue. As therapies for the management of polyp growth in patients with CRSwNP are at present limited, a new strategy to reduce NP through the induction of t-PA is worthy of exploration. Published studies of t-PA expression by endothelial cells show that cytokines,15,16 hormones 17 and drugs 18 can induce t-PA from endothelial cells or fibroblasts; regulation of t-PA expression in airway epithelial cells has not been studied extensively.
Short-chain fatty acids (SCFAs) are carboxylic acids with 1 to 6 carbon atoms. SCFAs are produced by microbiota in the distal small intestine and the colon by the anaerobic fermentation of non-digestive dietary residues.19 Acetic acid, propionic acid and butyric acid are important species of SCFAs and are present at millimolar levels in the gut.20 SCFAs produced in the gut lumen are readily absorbed by intestinal epithelial cells, circulate in the body and can be used as an energy source in peripheral tissues.21 Many recent reports have discovered diverse functions of SCFAs in inflammation, carcinogenesis, metabolism and allergic diseases.22–28 SCFAs are now recognized to be key molecules that regulate beneficial effects on human health.29 They can perform their functions via intermediary metabolism or via specific SCFA receptors, namely G protein-coupled receptor 41/free fatty acid receptor 3 (GPR41/FFAR3) and GPR43/FFAR2, which are 7-transmembrane-spanning G protein-coupled receptors. These receptors are expressed on the surface of a variety of cells including immune cells, adipocytes and gut epithelial cells.30,31 The activation of SCFA receptors can have various effects, depending on the disease state and the expressing cell type.32 Although butyric acid has been shown to induce t-PA in endothelial cells,33 there have not been any reports on SCFA regulation of t-PA in airway epithelial cells.
Based on the foregoing introduction, we believe that induction of t-PA expression in nasal or sinus epithelial cells would have a beneficial effect in blocking or reducing nasal polyp formation. The goal of these studies was therefore to test the hypothesis that SCFAs will induce t-PA expression by airway epithelial cells from the lungs or nose. The main finding that we report here is that SCFAs induce t-PA in human airway epithelial cells by a mechanism that is dependent on the known SCFA receptors GPR41 and GPR43.
2. Materials and Methods
2.1. Patient recruitment and clinical sample collection
Patients with CRS were recruited from the Allergy-Immunology and Otolaryngology Clinics of the Northwestern Medical Group (NMG) and the Northwestern Sinus Center at NMG. All subjects gave informed consent, and the protocol and consent forms governing procedures for study have been approved by the Institutional Review Board of Northwestern University Feinberg School of Medicine. All subjects met the criteria for CRS as defined by the American Academy of Otolaryngology-Head and Neck Surgery Chronic Rhinosinusitis Task Force.34,35 Sinonasal and NP tissues were obtained from routine functional endoscopic sinus surgery in patients with CRS. Details of the subjects'racteristics are included in Table 1. Further details are provided in the Supporting Information.
Table 1. Characteristics of the subjects.
| Control (n = 46) | CRSwNP (n = 52) | |
|---|---|---|
| Gender, male/female | 18/28 | 30/22 |
| Ethnicity, White/non-White | 32/14 | 43/9 |
| Age, y | 44.4 + 13.0 | 46.4 + 13.5 |
| Comorbidity, yes/no/unknown | ||
| Asthma | 2/44/0 | 31/21/0 |
| Atopy, yes/no/unknown | 8/35/3 | 30/14/8 |
| Steroid use, yes/no/unknown | 6/38/2 | 27/23/2 |
2.2. Elisa
Concentrations of t-PA in tissue homogenate and the supernatants of cultured cells were determined with a commercially available enzyme-linked immunosorbent assay (ELISA) kit according to the manufacturer's protocol (Assaypro, St. Charles, MO). The absorbance was measured using a Bio-Rad Spectrophotometer Model 680 Microplate Reader (Bio-Rad, Hercules, CA) with associated software applied to the sandwich enzyme immunoassay technique.
2.3. t-PA activity assay
The activity of t-PA was determined with a commercially available Human tPA Chromogenic Activity Assay Kit according to the manufacturer's protocol (Assaypro). The absorbance was measured using the Bio-Rad Spectrophotometer Model 680 Microplate Reader (Bio-Rad).
2.4. Cell viability assay
One hundred μL of NHBE cells (1 × 106 cells/well) was seeded in a collagen-coated 96-well tissue culture plates. Cells were cultured in BEGM without hydrocortisone for at least 24 hours before stimulation. Cells were exposed to concentrations of SCFA used in the activation studies, and viability was assayed as described below. A solution of 1% Triton X-100 (Sigma-Aldrich) was used as a positive control. Before stimulation, the medium was replaced, and NHBE cells were incubated with SCFAs. After 24 hours, 20 μL of MTS reagent (Promega, Madison, WI, USA) was added. The absorbance was measured after 2 hours at 490 nm using the Spectra max Gemini EM (Molecular Device, Sunnyvale, CA).
2.5. pH measurement
The pH of medium containing SCFAs was measured using Accumet Basic (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's protocol. The mean pH value was assessed using triplicate measurements for each sample.
2.6. Transepithelial electrical resistancemeasurement
NHBE cells (0.5 × 106 per plate) were plated in Corning 3460 Tran-swells (Corning, NY), and submerged cells were stimulated with 10 mmol/L of propionic acid for 48 hours. The transepithelial electrical resistance (TEER) was measured using an Epithelial Electronical Resistance Meter (Kanto Reagents, Tokyo, Japan) according to the manufacturer's protocol.
2.7. Immunohistochemistry, cell culture and treatments, real-time PCR, and Western blot analysis
Details can be found in the Supporting Information section in the online repository.
2.8. Statistical analysis
All data are presented as the mean ± SEM, unless otherwise mentioned. The data from culture experiments were analysed using the paired Student's t test. Differences between the groups were analysed with the Kruskal-Wallis ANOVA with Dunnett post hoc testing and a Mann-Whitney U test. In all cases, P < .05 was considered to be statistically significant. All statistical analyses were performed using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA) software.
3. Results
3.1. Epithelial cells expressed both GPR41 and GPR43 in sinonasal tissue
To assess the expression of GPR41 and GPR43 in sinonasal tissues and in NP tissues, we performed IHC analysis of surgical specimens from uncinate tissue (UT) of control and NPs from CRSwNP subjects (Figure 1). Expression of both GPR41 and GPR43 was observed in sinonasal epithelial cells as well as resident and infiltrating cells within the lamina propria.
Figure 1.
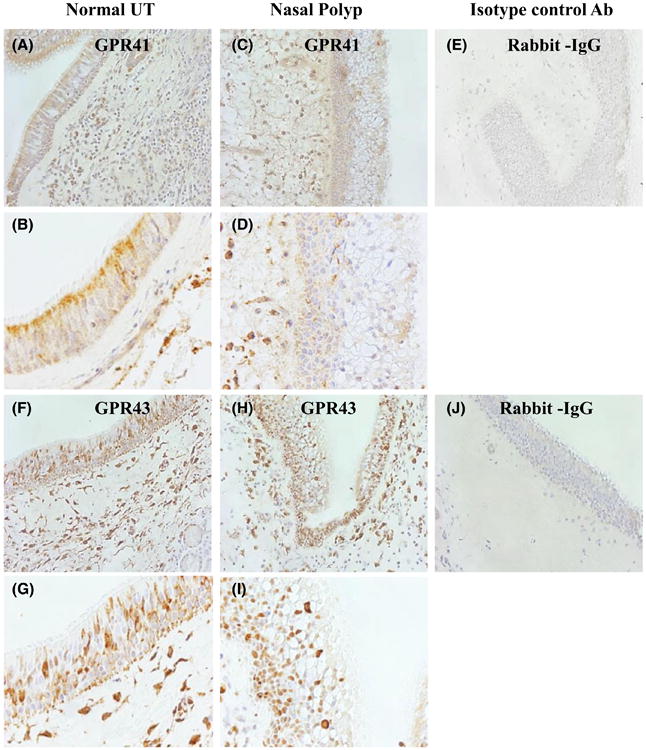
Immunohistochemical (IHC) detection of GPR41 (A-E) and GPR43 (F-J) in human sinonasal tissue. Representative immunostaining for GPR41 in UT from control subject (A, B) and in NP tissue (C, D), and staining for GPR43 in UT from a control subject (F, G) and in NP tissue (H, I). Representative isotype control antibody staining in NP tissue is shown (E, J). Magnification ×200 (A, C, E, F, H, J) and ×400 (B, D, G, I)
3.2. GPR41 and GPR43 mRNA expressions were up-regulated in nasal polyp tissue
Next, we wanted to know the expression levels of both GPR41 and GPR43 in sinonasal tissue. The mRNA from whole tissue extracts was analysed to compare the expression levels of both GPR41 and GPR43. As shown in Figure 2A,B, the expression of both GPR41 and GPR43 mRNA was ten- to 100-fold higher in NPs compared to control UT samples (GPR41; P < .001, GPR 43: P < .0001). As IHC analysis showed that both GPR41 and GPR43 were expressed in epithelial cells, we evaluated expression of both genes in fresh nasal cells collected by scraping. As with whole tissue extracts, the expression of both GPR41 and GPR43 was significantly higher (two to tenfold) in nasal scraping-derived epithelial cells from NPs compared to normal UT epithelial scrapings (GPR41; P < .05, GPR 43: P < .001, Figure 2C,D).
Figure 2.

Total RNA was extracted from nasal UT and NP tissues (A, B) and nasal epithelial cell scrapings from UT and NP tissues (C, D). Expression of GPR41 and GPR43 mRNA was analysed by RT-PCR. The expression of mRNA was normalized to the housekeeping gene β-Glucuronidase (GUSB).*P < .05, ***P < .001, ****P < .0001. (●; control UT, ■; NP tissue)
3.3. SCFAs induced t-PA from NHBE cells
We stimulated cultured submerged NHBE cells with SCFAs in vitro. As shown in Figure 3A, propionic acid, butyric acid and valeric acid strongly induced t-PA expression in a concentration-dependent manner, whereas acetic acid only weakly induced t-PA and hexanoic acid did not induce t-PA. The maximal concentrations of SCFAs to significantly induce t-PA in NHBE cells were 10 mmol/L for acetic acid (P < .05), 1 mmol/L for propionic acid (P < .05), 0.5 mmol/L for butyric acid (P < .05) and 1 mmol/L for valeric acid (P < .05); none of these concentrations induced harmful effects on the viability of NHBE cells (Figure S1). We measured t-PA protein levels in the supernatants from SCFA stimulated NHBE cells by ELISA. A 48-hour stimulation with SCFAs induced t-PA protein release from NHBE cells into the supernatant (Figure 3B). The levels of t-PA released into the supernatant by stimulation with propionic acid (P < .001), butyric acid (P < .05) and valeric acid (P < .05) were significantly higher (three- to fivefold) than levels in supernatants of non-stimulated NHBE cells. In the presence of SCFA in the medium, there were no significant pH changes (Figure S2).
Figure 3.

Submerged normal human bronchial epithelial (NHBE) cells were stimulated with indicated concentrations of SCFAs for 24 h. A, Cell lysates were harvested for RNA extraction to analyse t-PA mRNA expression by RT-PCR (n = 4-8); B, Submerged NHBE cells were stimulated with 10 mmol/L acetic acid, 10 mmol/L propionic acid, 1 mmol/L butyric acid, 10 mmol/L valeric acid and 10 mmol/L hexanoic acid. Supernatants were collected after 48 h of stimulation to analyse t-PA protein by ELISA (n = 6); C, Submerged NHBE cells were stimulated with 10 mmol/L propionic acid for 48 h. Then t-PA activity in the supernatants was measured by chromogenic activity kit (n = 5-6); D, NHBE cells were harvested from transwells after stimulation with 10 mmol/L of propionic acid for 48 h. Whole cell lysates and apical and basolateral medium were collected. Levels of t-PA in the cell lysates and medium were measured by ELISA (n = 8). Data shown are mean ± SEM of 4 independent experiments. * P < .05, **P < .01, ***P < .001, ****P < .0001 when compared to non-stimulated cells
3.4. Epithelial-derived t-PA was in the active form
To assess whether released t-PA was in the active form, we measured t-PA activity with a commercially available t-PA activity kit. As propionic acid was the strongest inducer of t-PA in NHBE cells, we used propionic acid in further experiments. We found that a significant amount of t-PA released from non-stimulated NHBE cells was in the active form. Supernatants from propionic acid stimulated NHBE cells showed significantly higher t-PA activity compared to non-stimulated NHBE cells and the increase in activity was proportionally similar to the increased level of t-PA mRNA (approx. threefold) (Figure 3C). These results indicate that at least a proportion of the t-PA released into the supernatant by both unstimulated and stimulated epithelial cells is in the active form.
3.5. SCFA-induced t-PA is secreted by NHBE cells onto both apical and basolateral surfaces
Most of the fibrin deposition that has been described in NP is found in the lamina propria within NPs.10 To test whether t-PA induced by SCFAs is released at the site of cross-linked fibrin, we tested whether the release was basolateral or apical. To clarify this point, NHBE cells were cultured in collagen-coated transwells. Cell lysates, basolateral media and apical supernatants were all collected after 48 hours of stimulation with 10 mmol/L propionic acid, and t-PA was measured by ELISA. Propionic acid significantly enhanced t-PA protein levels found in cell lysates as well as in both apical and basolateral media (Figure 3D). The results demonstrate that 78.5% of the total amount of t-PA produced by propionic acid stimulated NHBE was released and the remainder was retained in the cells. Although 66.8% of the released t-PA was found in the apical supernatants, a substantial amount (33.2%) was released to the basolateral side. To clarify whether the administration of propionate affects TEER, which might affect the passage of t-PA between the apical and basolateral layers, we also measured TEER. We found that the administration of propionate did not have any significant effect on TEER (Figure S3). These results indicate that induction of t-PA by SCFAs in vivo might be expected to increase the amounts of t-PA found within the tissue both above and below the basement membrane.
3.6. Induction of t-PA expression by SCFAs in NHBE cells is dependent on GPR41 and GPR43
GPR41 has been shown to signal through the PTX-sensitive Gαi/o family, whereas GPR43 signals through both Gαi/o and Gαq protein families which are PTX-insensitive.30,31 To clarify whether t-PA induction by SCFA was mediated by GPR41 or GPR43, NHBE cells were pretreated with PTX prior to stimulation by propionic acid. Figure 4A shows that PTX produced a dose-dependent inhibition of propionic acid-induced t-PA expression in NHBE cells, partially implicating GPR41. The minimum concentration of PTX to significantly attenuate propionic acid-induced t-PA expression was 1 ng/mL (P < .001); however, higher concentrations of PTX did not fully suppress t-PA expression, implying that some t-PA induction by propionic acid might be mediated by a PTX-sensitive (e.g Gαi/o family) signalling pathway. We next tested whether some of the propionic acid t-PA induction utilized a PTX-insensitive Gαq protein. YM-254890, an experimental compound that was isolated from the culture broth of Chromobacterium sp., is a selective Gαq/11 inhibitor that blocks the exchange of GDP for GTP during Gαq/11 activation.36,37 NHBE cells were pretreated with YM-254890 prior to stimulation with propionic acid. Pretreatment with 10 nmol/L of YM-254890 attenuated t-PA expression induced by propionic acid in NHBE cells (P < .05, Figure 4B), without affecting t-PA mRNA expression or cell viability (Figures S4 and S5). When NHBE cells were pretreated with both PTX (1 ng/mL) and YM-254890 (1000 nmol/L), the combined effect revealed greater inhibition of t-PA induction by propionate than either compound alone (Figure 4C).
Figure 4.

Pertussis toxin and YM-254890attenuate propionic acid-mediated t-PA induction in NHBE cells. Submerged NHBE cells were pretreated with PTX or PBS (A), YM-254890 or DMSO (B), PTX (1 ng/mL) and YM-254890 (1000 nmol/L) (C) (n = 5) prior to stimulation with 10 mmol/L of propionic acid for 24 h. Cell lysates were harvested for RNA extraction to analyse t-PA mRNA expression by RT-PCR. Data shown are mean ± SEM of independent experiments. *P < .05, **P < .01, ***P < .001
Although experiments with PTX and YM-254890 implicate GPR41 and GPR43, they provide only circumstantial support for the involvement of these 2 SCFA receptors. To confirm the involvement of GPR41 and GPR43 in propionic acid-mediated induction of t-PA, a small interfering RNA (siRNA) knock-down approach was employed. Concentrations of 40 nmol/L of one siRNA targeting GPR41 and another targeting GPR43 significantly suppressed expression of GPR41 and GPR43 proteins in NHBE cells as detected by Western blot analysis after transfection (P < .05, Figure 5A,B). Although the genes were not completely suppressed, the reduction of either gene alone was sufficient to partially inhibit t-PA induction by propionic acid (Figure 5C,D). The combination of siRNA against both receptors leads to nearly complete suppression of t-PA expression, as shown in Figure 5E, supporting an involvement of both GPR41 and GPR43 in SCFA-mediated induction of t-PA in NHBE cells.
Figure. 5.
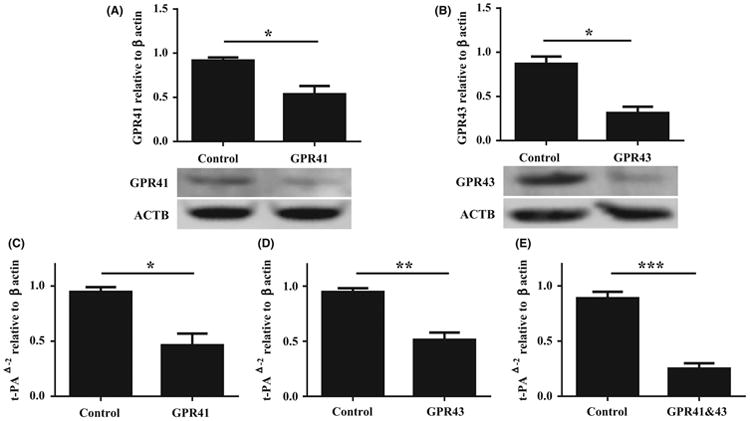
Small interfering RNA (siRNA) attenuate propionic acid-mediated t-PA induction in NHBE cells. (A, B) Submerged NHBE cells were transfected with 40 nmol/L of control siRNA and siRNA targeting human GPR41 (A) or GPR43 (B). Whole cell lysates were electrophoresed and transferred onto a PVDF membrane. Representative Western blot with anti-human GPR41 or anti-human GPR43 antibody is shown (bottom). Semiquantitative densitometry of 4 independent experiments. (C, D, E) Submerged NHBE cells were transfected with 40 nmol/L of control siRNA or siRNA targeting human GPR41 (C), GPR43 (D), or GPR41 and GPR43 (E) as indicated. Forty-eight hours after transfection, cells were stimulated with 10mM of propionic acid for 24 h (n = 6). Data shown are mean ± SEM of independent experiments. *P < .05, ** P < .01, ***P < .001
3.7. SCFAs induced t-PA from PNECs
Induction of t-PA by SCFA or synthetic agonists for their receptors, given therapeutically topically in the nose, could theoretically shrink nasal polyp tissues in which fibrin deposition plays a role in polyp growth. After demonstrating that SCFAs induced t-PA from NHBE cells, we next tested whether SCFAs can induce t-PA in primary nasal epithelial cells (PNECs) from nasal polyp. PNECs from nasal polyp were stimulated with varied concentrations of SCFAs. As shown in Figure 6, propionic acid stimulation of PNECs from nasal polyp enhanced the expression of t-PA. The approximate efficacy of SCFA as inducers of t-PA in PNECs was propionic acid acid > butyric acid = valeric acid. The minimum concentrations that significantly enhanced t-PA expression were 1 mmol/L of propionic acid (P < .05), 0.5 mmol/L of butyric acid (P < .05) and 10 mmol/L of valeric acid (P < .05), respectively.
Figure 6.

SCFAs induced t-PA expression from human primary nasal epithelial cells (PNECs). Submerged PNECs were stimulated with different concentrations of SCFAs for 24 h. Cell lysates were harvested for RNA extraction to analyse t-PA mRNA expression by RT-PCR (n = 4). Data shown are mean ± SEM of 4 independent experiments. *P < .05 when compared to non-stimulated cells
4. Discussion
Mucosal epithelium presents a first line of defence against many pathogens, allergens and environmental exposures. Epithelial cells have many functions that maintain homeostasis such as mucociliary action, maintenance of airway surface liquid, production of mucus and antimicrobial peptides, ion transport and maintenance of a structural barrier.38 Fibrin is produced during the process of epithelial repair,39 and epithelial-derived t-PA can take part in the repair process via LRP-1(Low density lipoprotein receptor-related protein 1).40 Dysregulation of epithelium and morphological changes in NPs have been shown.3,41 Prolonged inflammatory states also disrupt the balance of the coagulation and fibrinolysis cascades, leading to excessive fibrin deposition in the tissue. Takabayashi et al showed that excessive fibrin deposition is seen in NPs and that t-PA is down-regulated in NPs, further demonstrating that IL-4 and IL-13 inhibit t-PA induction in airway epithelial cells.10 They also reported that FXIIIA, a transglutaminase that cross-links fibrin in the final stage of the coagulation cascade, is elevated in NPs.42 Activated FXIII crosslinks fibrin between γ-glutamyl and ε-lysyl residues on adjacent fibrin chains in polymerized fibrin to yield the mature clot, and also crosslinks α2-plasmin inhibitor with fibrin. The cross-linking of fibrin reinforces its stiffness and rigidity that allows retention of plasma proteins. Notably, Takabayashi et al showed that M2 macrophages were major FXIII-A-expressing cells in NPs. Their findings align well with the view that the Th2-polarized inflammatory state facilitates fibrin deposition by decreasing fibrinolysis and enhancing the coagulation cascade in NPs. Together, these findings suggest that it is feasible to diminish NPs formation by inducing t-PA, causing removal of fibrin and decreased disease development and symptoms.
Induction of the t-PA gene and t-PA activity is regulated by several mechanisms.43 The regulation of t-PA gene transcription has thus far been investigated in a variety of cell types. Retinoic acid, butyric acid and statins are known to increase t-PA gene expression in endothelial cells, but the precise mechanism is not well understood.18,43,44 In the present study, we demonstrate that SCFAs can induce t-PA in NHBE cells and PNECs without any harmful effects on cell viability. Among several SCFAs, propionic acid tended to be the most effective agonist inducing t-PA in both NHBE cells and PNECs. Although the SCFAs induced t-PA at concentrations in the mmol/L level, these levels are similar to levels found in vivo and are in the same range as reports in other cell types.33 This level of potency may restrict the activation mechanism of both GPR41 and GPR43 by SCFAs to specific locations in the body. We showed that a significant amount of t-PA released from propionic acid stimulated NHBE cells was in the active form, and t-PA from transwell cultured NHBE cells was released both apically and basolaterally, indicating that some t-PA released from airway epithelial cells should reach the submucosal layer and effect fibrin degradation in the tissue.
SCFAs are generated in part by bacterial fermentation of dietary fibres by gut microbiota. SCFAs are produced at concentrations of ∼100 mmol/L in the gut lumen, and the major components are acetic acid (C2), propionic acid (C3) and butyric acid (C4).45 Previous studies have highlighted the variety of functions of SCFAs in inflammation, carcinogenesis, metabolism and allergy.22–28 There is interest in SCFA receptors as attractive pharmaceutical targets having a broad range of potential therapeutic applications.46 Thus, it is important to clarify the signalling partners associated with SCFA beneficial effects. We showed that the receptors, GPR41 and GPR43, are present in epithelial cells of both normal uncinate tissue and in NPs. This is the first report that these GPRs exist in the upper airway epithelium. Furthermore, the expression of both GPR41 and GPR43 was significantly higher in nasal scraping-derived epithelial cells from NPs compared to normal UT epithelial scrapings (Figure 2C,D). These results support the rationale of using topical administration of SCFAs to patients with NPs to induce t-PA and potentially decrease the size of the NPs.
GPCRs constitute the largest class of cell surface receptors, consisting of Gα, Gβ and Gγ subunits. GPCR is classified into 4 families according to their α subunit: Gi, Gs, G12/13 and Gq.47 The Gq family activates phospholipase and consists of 4 members: Gq, G11, G14 and G15/16 and their respective α subunits are thus Gαq, Gα11 and Gα14.48 On the other hand, the Gα15/16, and Gs and Gi families regulate adenylyl cyclase activity.43 PTX inhibits the α subunit of Gi proteins by locking the α subunits in a GDP-bound inactive state. Although GPR41 and GPR43 display 43% amino acid identity, they have different affinities for SCFAs.49 The rank order of potency for GPR41 is propionic acid ∼ butyric acid ∼ valeric acid > acetic acid, whereas the corresponding rank for GPR43 is acetic acid ∼ propionic acid > butyric acid > valeric acid.30,31 These potency rank orders for both GPRs are consistent with our results that propionic acid was, for the most part, the strongest inducer of t-PA in NHBE, and the response was inhibited by both Gαi/o and Gαq/11 inhibitors. We pretreated NHBE cells with both PTX and YM-254890 to test the combined inhibitory effect on t-PA induction. The result showed greater inhibition than either drug alone but revealed some residual t-PA that was not suppressed (Figure 4C), indicating either that the drugs did not completely block their targets or that other mechanisms might be present. Other potential mechanisms by which SCFAs could affect the cells might be via effects mediated by a transporter or pursuant to direct passage of the SCFAs across the cell membrane. Once inside the cell, SCFAs might affect signal transduction or transcriptional events, as reported elsewhere.26,45 It has been reported that SCFAs can act as histone deacetylase inhibitors (HDACi),26 and HDACi can also enhance t-PA induction in endothelial cells. We therefore tested whether trichostatin A, a broad HDACi, induces t-PA in NHBE cells. The results showed that trichostatin A increased t-PA expression (Figure S6). We also observed that t-PA induction by SCFAs was at least partially receptor dependent based on reduced response following siRNA gene knock-down of each receptor and an even greater reduction when both receptors were knocked down. We used primary nasal epithelial cells as well as NHBE cells, and found similar responses, suggesting that SCFAs can theoretically activate t-PA expression and release in the nose and sinuses of patients with CRS. Furthermore, we attempted to measure levels of SCFAs in nasal lavage fluids by gas chromatography; however, we could not detect SCFAs (data not shown). This could possibly reflect the large dilution of nasal lining fluid during lavage with a large volume of saline, could reflect partitioning of SCFAs in tissue vs the airway lumen or could reflect lower levels of SCFAs in nasal tissues than in the circulation. Nonetheless, based on the present findings, we suggest that it may be worthwhile to test whether intranasal administration of SCFAs, or other GPR41 and GPR43 agonists, can induce t-PA release and decrease nasal polyp size in vivo. Synthetic allosteric agonists for GPR41 and GPR43 have been reported, and agonists with much higher potency or pharmacodynamic attributes amenable to topical nasal use would be particularly promising in this regard.32
In NPs, infiltration of eosinophils accompanies other immune cells such as mast cells, basophils, dendritic cells and neutrophils.7,50–52 Mast cells in NPs have distinct phenotypes by tissue location, and are thought to contribute to the pathogenesis of NPs.50,53 These cells release cytokines and chemokines that orchestrate and accelerate inflammatory response in NPs. Importantly, these inflammatory cells also express GPR41 and/or GPR43.30,31 We performed immunofluorescence double staining of ECP (eosinophil cationic protein) for eosinophils and either GPR41 or GPR43. We found that eosinophils in NPs express GPR43 and weakly express GPR41 (Figure S7). SCFAs are known to modulate and control inflammatory responses via these receptors. Importantly, Trompette et al showed that high fibre diet or propionic acid administration in mice impaired Th2 polarization via GPR41 by suppressing expression of IL-4, IL-5, IL-13 and IgE, as well as reducing eosinophil infiltration in the lung.24 Another group also reported that high fibre diet or SCFA administration modified the development of allergic airway disease. This report provided evidence for a role of diet and acetic acid in the development of airway disease in humans.28 As Th2 cytokines are elevated in NPs and IL-13 can down-regulate t-PA, SCFAs might also indirectly be able to decrease the size of NPs by dampening type 2 inflammation in addition to inducing release of t-PA. We found that expression of both GPR41 and GPR43 mRNA was elevated in NPs in surgical samples (Figure 2A,B), which might reflect the infiltration of GPR41 or GPR43 expressing inflammatory cells in NPs, as well as increased expression by epithelial cells. Although further studies are required, we speculate that the administration of SCFAs might produce beneficial effects for treatment of NPs by more than one mechanism.
In summary, we showed that SCFAs, especially propionic acid, butyric acid and valeric acid, induced the expression and release of t-PA by airway epithelial cells, and that released t-PA was in the active form. We also found that the SCFA receptors GPR41 and GPR43 are expressed by nasal epithelial cells and can mediate the induction of t-PA following stimulation with SCFAs. Our results indicate that allosteric agonists for GPR41 and GPR43, including SCFAs, might be worthy of consideration as a new strategy for treatment of NPs.
Supplementary Material
Acknowledgments
Funding: information Foundation for the National Institutes of Health, Grant/Award Number: R37HL068546, U19AI106683
Footnotes
Conflict of Interest: The authors declare no conflict of interests.
Supporting Information: Additional Supporting Information may be found online in the supporting information tab for this article
References
- 1.Bhattacharyya N. Incremental health care utilization and expenditures for chronic rhinosinusitis in the United States. Ann Otol Rhinol Laryngol. 2011;120:423–427. doi: 10.1177/000348941112000701. [DOI] [PubMed] [Google Scholar]
- 2.Meltzer EO, Hamilos DL, Hadley JA, et al. Rhinosinusitis: establishing definitions for clinical research and patient care. J Allergy Clin Immunol. 2004;114(Suppl. 6):155–212. doi: 10.1016/j.jaci.2004.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kern RC, Conley DB, Walsh W, et al. Perspectives on the etiology of chronic rhinosinusitis: an immune barrier hypothesis. Am J Rhinol. 2008;22:549–559. doi: 10.2500/ajr.2008.22.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schleimer RP, Kato A, Peters A, et al. Epithelium, inflammation, and immunity in the upper airways of humans: studies in chronic rhinosinusitis. Proc Am Thorac Soc. 2009;6:288–294. doi: 10.1513/pats.200808-088RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomassen P, Vandeplas G, Van Zele T, et al. Inflammatory endotypes of chronic rhinosinusitis based on cluster analysis of biomarkers. J Allergy Clin Immunol. 2016;137:1449–1456. doi: 10.1016/j.jaci.2015.12.1324. [DOI] [PubMed] [Google Scholar]
- 6.Tan BK, Klingler AI, Poposki JA, et al. Heterogeneous inflammatory patterns in chronic rhinosinusitis without nasal polyps in Chicago, Illinois. J Allergy Clin Immunol. 2016;14:30956–3. doi: 10.1016/j.jaci.2016.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Zele T, Claeys S, Gevaert P, et al. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy. 2006;61:1280–1289. doi: 10.1111/j.1398-9995.2006.01225.x. [DOI] [PubMed] [Google Scholar]
- 8.Lin DC, Chandra RK, Tan BK, et al. Association between severity of asthma and degree of chronic rhinosinusitis. Am J Rhinol Allergy. 2011;25:205–208. doi: 10.2500/ajra.2011.25.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachert C, Gevaert P, Holtappels G, Cuvelier C, van Cauwenberge P. Nasal polyposis: from cytokines to growth. Am J Rhinol. 2000;14:279–290. doi: 10.2500/105065800781329573. [DOI] [PubMed] [Google Scholar]
- 10.Takabayashi T, Kato A, Peters AT, et al. Excessive fibrin deposition in nasal polyps caused by fibrinolytic impairment through reduction of tissue plasminogen activator expression. Am J Respir Crit Care Med. 2013;187:49–57. doi: 10.1164/rccm.201207-1292OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jennewein C, Tran N, Paulus P, Ellinghaus P, Eble JA, Zacharowski K. Novel aspects of fibrin(ogen) fragments during inflammation. Mol Med. 2011;17:568–573. doi: 10.2119/molmed.2010.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gabazza EC, Osamu T, Yamakami T, et al. Correlation between clotting and collagen metabolism markers in rheumatoid arthritis. Thromb Haemost. 1994;71:199–202. [PubMed] [Google Scholar]
- 13.Idell S. Adult respiratory distress syndrome: do selective anticoagulants help? Am J Respir Med. 2002;1:383–391. doi: 10.1007/BF03257165. [DOI] [PubMed] [Google Scholar]
- 14.Wagers SS, Norton RJ, Rinaldi LM, Bates JH, Sobel BE, Irvin CG. Extravascular fibrin, plasminogen activator, plasminogen activator inhibitors, and airway hyperresponsiveness. J Clin Invest. 2004;114:104–111. doi: 10.1172/JCI19569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medcalf RL, Kruithof EK, Schleuning WD. Plasminogen activator inhibitor 1 and 2 are tumor necrosis factor/cachectin-responsive genes. J Exp Med. 1988;168:751–759. doi: 10.1084/jem.168.2.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang YC, Yang SF, Huang FM, Tai KW, Hsieh YS. Induction of tissue plasminogen activator gene expression by proinflammatory cytokines in human pulp and gingival fibroblasts. J Endod. 2003;29:114–117. doi: 10.1097/00004770-200302000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Lansink M, Kooistra T. Stimulation of tissue-type plasminogen activator expression by retinoic acid in human endothelial cells requires retinoic acid receptor beta 2 induction. Blood. 1996;88:531–541. [PubMed] [Google Scholar]
- 18.Dunoyer-Geindre S, Fish RJ, Kruithof EK. Regulation of the endothelial plasminogen activator system by fluvastatin. Role of Rho family proteins, actin polymerisation and p38 MAP kinase. Thromb Haemost. 2011;105:461–472. doi: 10.1160/TH10-07-0444. [DOI] [PubMed] [Google Scholar]
- 19.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergman EN. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol Rev. 1990;70:567–590. doi: 10.1152/physrev.1990.70.2.567. [DOI] [PubMed] [Google Scholar]
- 21.Cherbut C. Motor effects of short-chain fatty acids and lactate in the gastrointestinal tract. Proc Nutr Soc. 2003;62:95–99. doi: 10.1079/PNS2002213. [DOI] [PubMed] [Google Scholar]
- 22.Tang Y, Chen Y, Jiang H, Robbins GT, Nie D. G-protein-coupled receptor for short-chain fatty acids suppresses colon cancer. Int J Cancer. 2011;128:847–856. doi: 10.1002/ijc.25638. [DOI] [PubMed] [Google Scholar]
- 23.Sleeth ML, Thompson EL, Ford HE, Zac-Varghese SE, Frost G. Free fatty acid receptor 2 and nutrient sensing: a proposed role for fibre, fermentable carbohydrates and short-chain fatty acids in appetite regulation. Nutr Res Rev. 2010;23:135–145. doi: 10.1017/S0954422410000089. [DOI] [PubMed] [Google Scholar]
- 24.Trompette A, Gollwitzer ES, Yadava K, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20:159–166. doi: 10.1038/nm.3444. [DOI] [PubMed] [Google Scholar]
- 25.Smith PM, Howitt MR, Panikov N, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fernando MR, Saxena A, Reyes JL, McKay DM. Butyrate enhances antibacterial effects while suppressing other features of alternative activation in IL-4-induced macrophages. Am J Physiol Gastrointest Liver Physiol. 2016;310:24. doi: 10.1152/ajpgi.00440.2015. [DOI] [PubMed] [Google Scholar]
- 27.Maslowski KM, Vieira AT, Ng A, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461:1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thorburn AN, McKenzie CI, Shen S, et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nat Commun. 2015;6:7320. doi: 10.1038/ncomms8320. [DOI] [PubMed] [Google Scholar]
- 29.Sivaprakasam S, Prasad PD, Singh N. Benefits of short-chain fatty acids and their receptors in inflammation and carcinogenesis. Pharmacol Ther. 2016;23:30049–3. doi: 10.1016/j.pharmthera.2016.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Poul E, Loison C, Struyf S, et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J Biol Chem. 2003;278:25481–25489. doi: 10.1074/jbc.M301403200. [DOI] [PubMed] [Google Scholar]
- 31.Brown AJ, Goldsworthy SM, Barnes AA, et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J Biol Chem. 2003;278:11312–11319. doi: 10.1074/jbc.M211609200. [DOI] [PubMed] [Google Scholar]
- 32.Ulven T. Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front Endocrinol. 2012;3:111. doi: 10.3389/fendo.2012.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunoyer-Geindre S, Kruithof EK. Epigenetic control of tissue-type plasminogen activator synthesis in human endothelial cells. Cardiovasc Res. 2011;90:457–463. doi: 10.1093/cvr/cvr028. [DOI] [PubMed] [Google Scholar]
- 34.Meltzer EO, Hamilos DL, Hadley JA, et al. Rhinosinusitis: establishing definitions for clinical research and patient care. Otolaryngol Head Neck Surg. 2004;131(Suppl. 6):S1–S62. doi: 10.1016/j.otohns.2004.09.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pearlman AN, Conley DB. Review of current guidelines related to the diagnosis and treatment of rhinosinusitis. Curr Opin Otolaryngol Head Neck Surg. 2008;16:226–230. doi: 10.1097/MOO.0b013e3282fdcc9a. [DOI] [PubMed] [Google Scholar]
- 36.Taniguchi M, Nagai K, Arao N, et al. YM-254890, a novel platelet aggregation inhibitor produced by Chromobacterium sp. QS3666. J Antibiot. 2003;56:358–363. doi: 10.7164/antibiotics.56.358. [DOI] [PubMed] [Google Scholar]
- 37.Takasaki J, Saito T, Taniguchi M, et al. A novel Galphaq/11-selective inhibitor. J Biol Chem. 2004;279:47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- 38.Podolsky DK. Mucosal immunity and inflammation. V. Innate mechanisms of mucosal defense and repair: the best offense is a good defense. Am J Physiol. 1999;1:G495–G499. doi: 10.1152/ajpgi.1999.277.3.G495. [DOI] [PubMed] [Google Scholar]
- 39.Perrio MJ, Ewen D, Trevethick MA, Salmon GP, Shute JK. Fibrin formation by wounded bronchial epithelial cell layers in vitro is essential for normal epithelial repair and independent of plasma proteins. Clin Exp Allergy. 2007;37:1688–1700. doi: 10.1111/j.1365-2222.2007.02829.x. [DOI] [PubMed] [Google Scholar]
- 40.Lin S, Racz J, Tai MF, et al. A role for low density lipoprotein receptor-related protein 1 in the cellular uptake of tissue plasminogen activator in the lungs. Pharm Res. 2016;33:72–82. doi: 10.1007/s11095-015-1763-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saitoh T, Kusunoki T, Yao T, et al. Relationship between epithelial damage or basement membrane thickness and eosinophilic infiltration in nasal polyps with chronic rhinosinusitis. Rhinology. 2009;47:275–279. doi: 10.4193/Rhin08.109. [DOI] [PubMed] [Google Scholar]
- 42.Takabayashi T, Kato A, Peters AT, et al. Increased expression of factor XIII-A in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2013;132:584–592. doi: 10.1016/j.jaci.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kruithof EK, Dunoyer-Geindre S. Human tissue-type plasminogen activator. Thromb Haemost. 2014;112:243–254. doi: 10.1160/TH13-06-0517. [DOI] [PubMed] [Google Scholar]
- 44.Huber D, Cramer EM, Kaufmann JE, et al. Tissue-type plasminogen activator (t-PA) is stored in Weibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood. 2002;99:3637–3645. doi: 10.1182/blood.v99.10.3637. [DOI] [PubMed] [Google Scholar]
- 45.Ganapathy V, Thangaraju M, Prasad PD, Martin PM, Singh N. Transporters and receptors for short-chain fatty acids as the molecular link between colonic bacteria and the host. Curr Opin Pharmacol. 2013;13:869–874. doi: 10.1016/j.coph.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 46.Bolognini D, Tobin AB, Milligan G, Moss CE. The pharmacology and function of receptors for short-chain fatty acids. Mol Pharmacol. 2016;89:388–398. doi: 10.1124/mol.115.102301. [DOI] [PubMed] [Google Scholar]
- 47.Haraguchi M, Border WA, Huang Y, Noble NA. t-PA promotes glomerular plasmin generation and matrix degradation in experimental glomerulonephritis. Kidney Int. 2001;59:2146–2155. doi: 10.1046/j.1523-1755.2001.00729.x. [DOI] [PubMed] [Google Scholar]
- 48.Del Rosso M, Fibbi G, Pucci M, Margheri F, Serrati S. The plasminogen activation system in inflammation. Front Biosci. 2008;13:4667–4686. doi: 10.2741/3032. [DOI] [PubMed] [Google Scholar]
- 49.Stoddart LA, Smith NJ, Milligan G. International Union of Pharmacology. LXXI. Free fatty acid receptors FFA1, -2, and -3: pharmacology and pathophysiological functions. Pharmacol Rev. 2008;60:405–417. doi: 10.1124/pr.108.00802. [DOI] [PubMed] [Google Scholar]
- 50.Takabayashi T, Kato A, Peters AT, et al. Glandular mast cells with distinct phenotype are highly elevated in chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2012;130:410–420. doi: 10.1016/j.jaci.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mahdavinia M, Carter RG, Ocampo CJ, et al. Basophils are elevated in nasal polyps of patients with chronic rhinosinusitis without aspirin sensitivity. J Allergy Clin Immunol. 2014;133:1759–1763. doi: 10.1016/j.jaci.2013.12.1092. https://doi.org/10.1016/j.jaci.2013.12.1092. Epub 2014 Mar 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poposki JA, Peterson S, Welch K, et al. Elevated presence of myeloid dendritic cells in nasal polyps of patients with chronic rhinosinusitis. Clin Exp Allergy. 2015;45:384–393. doi: 10.1111/cea.12471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gevaert P, Calus L, Van Zele T, et al. Omalizumab is effective in allergic and nonallergic patients with nasal polyps and asthma. J Allergy Clin Immunol. 2013;131:110–116. doi: 10.1016/j.jaci.2012.07.047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.