

Abstract
Background
Small studies have implicated plasminogen activator inhibitor-1 (PAI-1) as a predictor of cardiovascular events; however, these findings have been inconsistent.
We sought out to examine the potential role of PAI-1 as a marker for major adverse cardiovascular events (MACE).
Methods
We systematically reviewed all indexed studies examining the association between PAI-1 and MACE (defined as death, myocardial infarction, or cerebrovascular accident) or restenosis. EMBASE, Web of Science, Medline, and the Cochrane Library were searched through October 2016 to identify relevant studies, supplemented by letters to authors and review of citations. Studies reporting the results of PAI-1 antigen and/or activity levels in association with MACE in human subjects were included.
Results
Of 5961 articles screened, we identified 38 articles published between 1991 to 2016 that reported PAI-1 levels in 11,557 patients. In studies that examined PAI-1 antigen and activity levels, 15.1% and 29.6% of patients experienced MACE, respectively. Patients with MACE had higher PAI-1 antigen levels with a mean difference of 6.11 ng/mL (95% CI, 3.27-8.96). This finding was similar among patients with and without known coronary artery disease. Comparatively, studies that stratified by PAI-1 activity levels were not associated with MACE. In contrast, studies of coronary restenosis suggest PAI-1 antigen and activity levels are negatively associated with MACE.
Conclusions
Elevated plasma PAI-1 antigen levels are associated with MACE. Definitive studies are needed to ascertain if PAI-1 acts simply as a marker of risk or if it is indeed a bona fide therapeutic target.
Electronic supplementary material
The online version of this article (10.1186/s12959-018-0166-4) contains supplementary material, which is available to authorized users.
Keywords: Plasminogen activator inhibitor-1, Biomarkers, Mortality, Myocardial infarction, Meta-analysis
Background
Obstructive coronary artery disease (CAD) is the leading cause of mortality in the western world. The cornerstone of therapy for CAD remains revascularization and secondary medical therapy to modify risk factors. The fibrinolytic system has implications for both approaches to disease management. First, percutaneous coronary intervention (PCI) with implantation of a coronary stent remains the predominant method of coronary revascularization [1]. However, complications such as in-stent restenosis and stent thrombosis following PCI limit its efficacy. Thus, in the peri-revascularization period, preventing thrombotic events is paramount until the vessel’s endothelial lining and function are restored. Second, long term therapy with antiplatelet and/or oral anticoagulation is an integral part of secondary preventive medical therapy. Thus, dysregulation of the fibrinolytic pathways may increase the risk of complications from revascularization therapy and diminish the efficacy of long term medical therapy to reduce the risk of recurrent events.
The fibrinolytic system is activated by the conversion of plasminogen to plasmin by serine proteases such as tissue or urokinase-type plasminogen activator (t-PA and u-PA, respectively). In contrast, fibrinolysis is inhibited by plasminogen activator inhibitor-1 (PAI-1), which is a member of the serine protease inhibitor (serpin) family. Ultimately, thrombosis risk is influenced by the balance between PAI-1 and t-PA. Thus, an increase in the PAI-1 levels in plasma can induce a hypercoagulable state [2]. PAI-1 is released by vascular endothelial cells, hepatocytes, adipocytes, cardiomyocytes, fibroblasts, and platelets [3, 4]. In healthy humans, plasma levels of PAI-1 exceed t-PA by a ratio of over 4:1 with most of PAI-1 being cleared by the liver [5]. In pathologic conditions, PAI-1 production can be upregulated by pro-inflammatory factors such as TNFα, TGFβ, and insulin [6]. Elevated plasma PAI-1 levels have been associated with impaired fibrinolytic activity in stroke and coronary artery disease [7]. Moreover, PAI-1 antigen and activity levels are elevated in patients with type 2 diabetes [8], hyperinsulinemia [9], and those with insulin resistance [10, 11]. Yet, a definitive assessment of the impact of elevated PAI-1 as a biomarker or therapeutic target has yet to be evaluated. Accordingly, we performed a systematic review and meta-analysis of PAI-1 antigen and activity levels and their relationship with major adverse cardiovascular events (MACE) in humans.
Methods
Literature search strategy
Literature searches were guided by a medical librarian with expertise in systematic reviews (S.V.) using a combination of key terms and index headings related to PAI-1, coronary disease (informed by the Cochrane review search strategy for coronary heart disease in exercise-based cardiac rehabilitation [12]), and the Cochrane Highly Sensitive search strategy to eliminate articles on animal studies in Medline. The search was additionally peer-reviewed by a second medical librarian (R.S.). Once finalized, the search strategy was then translated to other bibliographic databases (see Additional file 1 for the full Medline search). The final search was conducted on October 2016 in Medline (Ovid) (In-Process & Other Non-Indexed Citations and Ovid MEDLINE(R) 1946-), Embase (Ovid) (Embase Classic + Embase 1947-), Cochrane Library (Ovid) (from inception), and Web of Science (Thomson Reuters) (all indexes, from inception). Search results were exported to EndNote X7 (Thomson Reuters, New York, USA) and duplicates eliminated using the program’s duplicate identification feature and manual inspection. A review protocol was produced but not registered in a database.
Titles and abstracts were screened by two independent reviewers (R.J. and P.M.) using Covidence (Melbourne, Australia). Full articles were retrieved in cases of missing abstracts. Corresponding authors were contacted for additional information when necessary.
Inclusion and exclusion criteria and quality assessment
Studies were included if they met the following criteria: (1) PAI-1 antigen or activity levels were reported; (2) the population studied comprised individuals aged 18 years or older; (3) components of MACE (death, myocardial infarction, and cerebrovascular events including stroke and transient ischemic attacks) or restenosis were reported; (4) articles were published in English. Exclusion criteria included: (1) PAI-1 polymorphism studies examining the association between 4G/5G and adverse events; (2) animal or in vitro studies; and (3) studies reporting hazard ratios only. Full text data extraction was conducted by two independent evaluators (R.J. and P.M.). Each reviewer independently extracted patient population characteristics, group sizes, PAI-1 antigen and activity levels, follow-up duration, and MACE and restenosis. All discrepancies were resolved by consensus prior to locking the database for analysis (Tables 1 and 2). Included observational studies were evaluated for quality and risk of bias using the Newcastle-Ottawa Quality Assessment Scale [13] by two independent evaluators (R.J. and P.M.) with disagreements resolved by consensus. Visual funnel plot inspection was used to screen for publication bias.
Table 1.
Studies reporting PAI-1 antigen levels (ng/mL) and major adverse cardiovascular events and restenosis
| Event | No Event | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Year | Study Design | Follow-up (months) | Population of interest | N | PAI-1 (IU/mL) | Range | Death | MI | Restenosis | CVA | N | PAI-1 (IU/mL) | Range |
| Sane et al. [50] | 1991 | Cohort | 24 | Fibrinolytics | 24 | 57 | 58 | 24 | n/a | n/a | n/a | 315 | 54 | 53 |
| Cortellaro et al. [51] | 1993 | Case-Control | 24 | Subgroups Combined | 58 | 11.3 | 0.7 | 13 | 17 | n/a | 20 | 87 | 7.3 | 0 5 |
| Brannstrom et al. [52] | 1995 | Cohort | 46 | Anticoagulant | 38 | 21.9 | 15.1 | 38 | n/a | n/a | n/a | 167 | 16.7 | 11.8 |
| Juhan-Vague et al. [53] | 1996 | Cohort | 24 | MI | 106 | 18.2 | 8 2 | 40 | 66 | n/a | n/a | 2700 | 14.8 | 8.9 |
| Nordt et al. [54] | 1998 | Cohort | 12 | Fibrinolytics | 5 | 28.8 | 14.3 | n/a | n/a | 5 | n/a | 26 | 27.1 | 15.4 |
| Alaigh et al. [55] | 1998 | Cohort | 6 | Elective PCI | 28 | 20.75 | 11.06 | n/a | n/a | 28 | n/a | 45 | 24.5 | 13.85 |
| Moss et al. [56] | 1999 | Cohort | 26 | MI | 81 | 25 | 18 | 25 | 56 | n/a | n/a | 964 | 29 | 28 |
| Redondo et al. [57] | 2001 | Cohort | 24 | MI | 37 | 40.725 | 22.28 | 2 | 5 | 30 | n/a | 157 | 42.65 | 21.02 |
| Fornitz et al. [58] | 2001 | Cohort | 6 | Elective PCI | 7 | 82.6 | 26.6 | n/a | n/a | 7 | n/a | 12 | 72.2 | 27 |
| Bogaty et al. [59] | 2001 | Case-Control | 48 | MI | 23 | 23.83 | 19.04 | n/a | 8 | n/a | n/a | 77 | 18.9 | 16.24 |
| Ganti et al. [60] | 2002 | Cohort | n/a | MI | 4 | 80.68 | 16.38 | 4 | n/a | n/a | n/a | 38 | 61 | 21.95 |
| Lip et al. [61] | 2002 | Cohort | 12 | Stroke | 27 | 56.5 | 30.7 | 27 | n/a | n/a | n/a | 59 | 45.9 | 23.3 |
| Inoue et al. [62] | 2003 | Cohort | 6 | MI | 24 | 28 | 4 | n/a | n/a | 24 | n/a | 42 | 29 | 4 |
| Christ et al. [20] | 2005 | Cohort | 6 | Elective PCI | 25 | 14.8 | 0.7 | n/a | n/a | 25 | n/a | 55 | 16.8 | 2.1 |
| Robinson et al. [63] | 2007 | Cohort | 5 to 51 | Coronary Heart Disease | 19 | 36.3 | 17.9 | 2 | 2 | n/a | 2 | 79 | 45.4 | 25.6 |
| Katsaros et al. [24] | 2008 | Cohort | 6 to 8 | Elective PCI | 12 | 11.69 | 8.05 | n/a | n/a | 12 | n/a | 61 | 22.78 | 18.76 |
| Thogersen et al. [64] | 2009 | Case-Control | n/a | Healthy | 50 | 38.27 | 16.79 | n/a | 50 | n/a | n/a | 56 | 29 | 15.75 |
| Akkus et al. [65] | 2009 | Cohort | 12 | Cardiogenic Shock | 33 | 116.5 | 97.26 | 33 | n/a | n/a | n/a | 27 | 71.33 | 54.79 |
| Pineda et al. [66] | 2010 | Cohort | 36 | MI | 25 | 65.13 | 53.3 | 4 | 21 | n/a | n/a | 117 | 70.1 | 48.34 |
| Wennberg et al. [67] | 2012 | Case-Control | 168 | Healthy | 469 | 57.28 | 25.83 | n/a | 469 | n/a | n/a | 895 | 51.23 | 25.11 |
| Yano et al. [68] | 2013 | Cohort | 20 | Smokers | 66 | 65.73 | 64.57 | n/a | 11 | n/a | 55 | 744 | 42.97 | 36.54 |
| Yano et al. [69] | 2014 | Cohort | 30 | Hypertension | 42 | 31.67 | 16.12 | 4 | 13 | n/a | 16 | 548 | 28.33 | 14.87 |
| Knudsen et al. [70] | 2014 | Case-Control | 12 | HIV | 54 | 111 | 8.5 | 3 | 51 | n/a | n/a | 54 | 92 | 7 |
| Golukhova et al. [71] | 2015 | Cohort | 28 | Elective PCI | 23 | 72.75 | 29.86 | 2 | 9 | 11 | n/a | 71 | 49.75 | 23.16 |
CVA cerebrovascular accident
Table 2.
Studies reporting PAI-1 activity levels (IU/mL) and major adverse cardiovascular events and restenosis
| Event | No Event | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reference | Year | Study Design | Follow-up (months) | Population of interest | N | PAI-1 Activity (U/mL) | Range | Death | MI | Restenosis | N | PAI-1 Activity (U/mL) | Range |
| Sane et al. [50] | 1991 | Cohort | 24 | Fibrinolytics | 28 | 17 | 16 | n/a | n/a | 28 | 328 | 19 | 21 |
| Shah et al. [72] | 1992 | Cohort | 9 | Elective PCI | 28 | 8 | 7.1 | n/a | n/a | 28 | 40 | 12 | 8 |
| Gray et al. [73] | 1993 | Cohort | 0.1 | MI | 13 | 20.6 | 11 | n/a | 13 | n/a | 85 | 20.1 | 7.9 |
| Malmberg et al. [74] | 1994 | Case-Control | 90 | MI | 53 | 23.75 | 13.34 | 20 | 33 | n/a | 55 | 18 | 10.97 |
| Brack et al. [75] | 1994 | Cohort | 4 | Elective PCI | 16 | 4.63 | 4.71 | n/a | n/a | 16 | 30 | 5.77 | 5.06 |
| Nordt et al. [54] | 1998 | Cohort | 12 | Fibrinolytics | 5 | 8.7 | 8.3 | n/a | 5 | n/a | 26 | 9.9 | 8.2 |
| Jansson et al. [76] | 1998 | Cohort | 120 | MI | 54 | 9.1 | 5.1 | 54 | n/a | n/a | 69 | 10.6 | 7.1 |
| Wiman et al. [77] | 2000 | Case-Control | 3 | MI | 61 | 22.1 | 17.5 | n/a | 61 | n/a | 95 | 18.2 | 16.5 |
| Wiman et al. [77] | 2000 | Case-Control | 3 | Ml | 25 | 15.4 | 13.6 | n/a | 25 | n/a | 38 | 17.8 | 12.4 |
| Prisco et al. [78] | 2001 | Cohort | 18 | MI | 18 | 11.27 | 7.64 | n/a | n/a | 18 | 36 | 15.8 | 27.49 |
| Prisco et al. [78] | 2001 | Cohort | 18 | Elective PCI | 6 | 8.33 | 8.1 | n/a | n/a | 6 | 42 | 7.57 | 9.52 |
| Sargento et al. [79] | 2003 | Cohort | 12 | MI | 7 | 6.34 | 1.56 | 5 | 2 | n/a | 80 | 4.47 | 1.84 |
| Marcucci et al. [19] | 2006 | Case-Control | 22 2 | MI | 109 | 22 | 9.09 | 54 | 55 | n/a | 411 | 24.25 | 10.63 |
| Schoebel et al. [80] | 2008 | Cohort | 2 | MI | 18 | 3.7 | 1.8 | n/a | n/a | 18 | 42 | 5.3 | 3.2 |
Wiman et al. [77] is presented twice as data for men and women were reported separately
Statistical analysis
The primary clinical endpoint for this study was MACE – a composite of death, myocardial infarction, or cerebrovascular events. The secondary endpoint included components of the primary as well as coronary restenosis in patients undergoing coronary revascularization. Mean circulating PAI-1 antigen (ng/mL) and activity (IU/mL) levels and their associated standard deviations were used for analyses. Fourteen studies reported median and interquartile ranges, which were converted to approximated means and standard deviations using the method described by Wan et al. [14].
All analyses were performed using Review Manager (RevMan) 5.3 (Cochrane Collection, Copenhagen, Denmark). PAI-1 antigen and activity levels were compared between patients with or without the outcomes of interest either as absolute values or dichotomized as high vs. low. Random effects models stratified by study design and study quality were used to generate pooled mean differences with 95% confidence intervals. Post-hoc meta regression was performed to account for timing of blood draw and acute phase reactions in studies of patients presenting with acute myocardial infarction or stroke.
Results
Included studies
Study selection
After excluding duplicate articles, 5961 titles and abstracts were screened, of which 340 underwent full review and 38 were ultimately included (Fig. 1, Tables 1 and 2). Study populations were heterogeneous, including patients presenting with stable angina, acute coronary syndrome, and non-cardiac diseases. Study sample sizes ranged from 19 to 2806. Most studies were of moderate quality (see Additional file 1: Tables S1 and S2 for details of study quality assessments). Funnel plots are shown in Additional file 1: Figures S1 and S2. In pooled analyses of all studies reporting PAI-1 antigen levels (n = 8999), 1362 events were reported, including 234 deaths, 795 myocardial infarctions, 101 cerebrovascular events, and 142 restenoses (Table 1). In all studies that examined PAI-1 activity levels (n = 1490), 441 events were reported, including 133 deaths, 194 myocardial infarctions, and 114 cases of restenosis (Table 2).
Fig. 1.
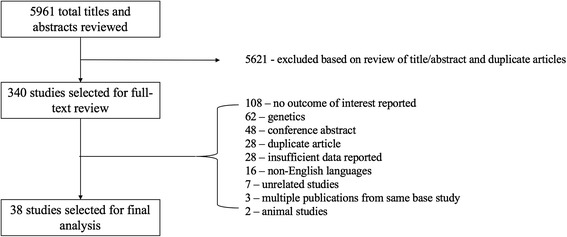
Flow diagram of the included PAI-1 studies for meta-analysis
PAI-1 and clinical outcomes
Major adverse cardiovascular events
PAI-1 antigen levels were higher in those with MACE with a mean difference of 6.11 ng/mL (95% CI, 3.27-8.96, P < 0.001) – a difference that was present irrespective of study design (Fig. 2). When restricted to high-quality studies, PAI-1 antigen levels in patients with MACE were 5.22 ng/mL (95% CI, 2.97-7.54, P < 0.001; Additional file 1: Figure S3). Among seven studies reporting morning blood draws between 7:00 and 10:00 am, PAI-1 antigen levels were higher in those with MACE with a mean difference of 4.61 ng/mL (95% CI, 1.49-7.74, P = 0.004; Additional file 1: Figure S4). Meta-regression analysis of timing of the blood draw and acute phase studies was not predictive of the heterogeneity in our selected studies nor did it contribute to a greater understanding of the impact of PAI-1 in its association with MACE (Additional file 1: Table S3).
Fig. 2.
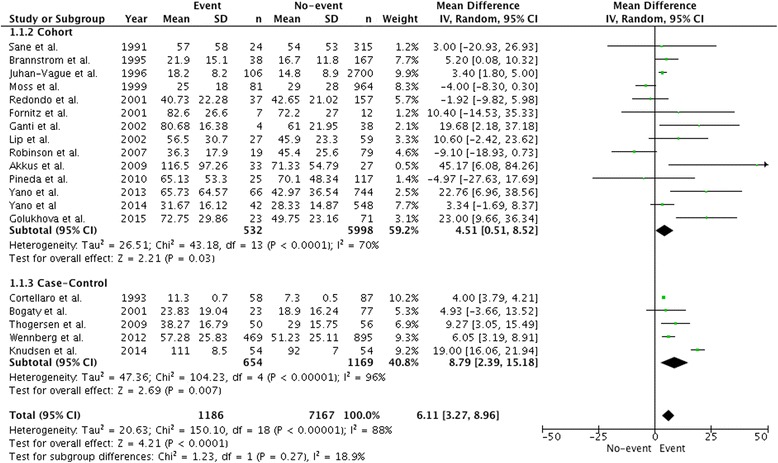
Comparison of mean PAI-1 antigen levels (ng/mL) in patients with major adverse cardiac events and control patients. Data is expressed as a mean difference and analyzed using a random effects model
In contrast to PAI-1 plasma antigen levels, there was no significant difference in PAI-1 activity levels between those with vs. without MACE (mean difference 0.59 IU/mL (95% CI, − 1.63-2.80, P = 0.60; Fig. 3). No association between PAI-1 activity levels and MACE was observed when the analysis was restricted to three high-quality studies with a mean difference of 1.14 IU/mL (95% CI, − 3.37-5.65, P = 0.62; Additional file 1: Figure S5).
Fig. 3.
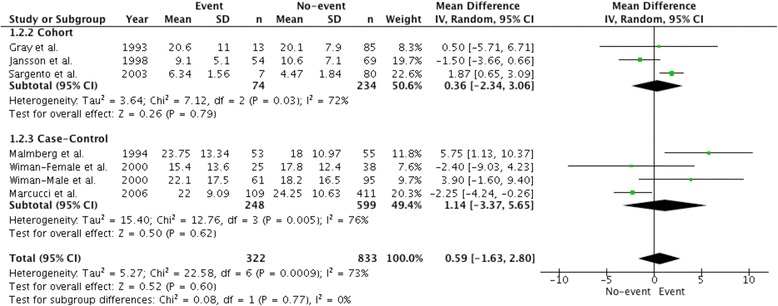
Comparison of mean PAI-1 activity levels (IU/mL) in patients with major adverse cardiac events and control patients. Data is expressed as a mean difference and analyzed using a random effects model
Major adverse cardiovascular events: Pre-existing versus no known coronary artery disease
Overall, among patients without prior coronary artery disease, 12.2% had an event. PAI-1 antigen levels in those without previously known coronary artery disease were on average 6.44 ng/mL higher in those with MACE relative to those without (95% CI, 2.64-10.25, P < 0.001; Fig. 4a). In studies that included patients with known CAD, 19.3% had MACE. PAI-1 antigen levels in those with known CAD were on average 5.49 ng/mL higher in those with MACE than those without (95% CI, 0.36-10.63, P = 0.04; Fig. 4b). No difference in PAI-1 activity levels between event and control groups was observed in the five studies reporting PAI-1 activity levels and MACE (Additional file 1: Figure S6).
Fig. 4.
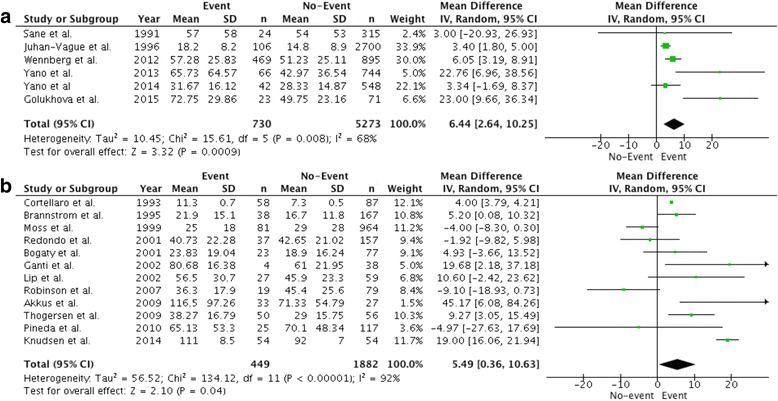
Comparison of mean PAI-1 antigen levels (ng/mL) in patients with primary and secondary major adverse cardiac events and control patients. Data is expressed as a mean difference and analyzed using a random effects model. a PAI-1 levels (ng/mL) in patients with primary major adverse cardiac events. b PAI-1 levels (ng/mL) in patients with secondary major adverse cardiac events
Death
In the five studies that reported mortality data, 126 deaths were observed (17.2% of patients). PAI-1 antigen levels were higher among patients who died (mean difference: 10.34 ng/mL (95% CI, 1.90-18.79, P = 0.02; Additional file 1: Figure S7).
Restenosis
In the six studies that examined restenosis following percutaneous coronary intervention (with and without coronary stent implantation), 101 events were observed (29.5% of patients). PAI-1 antigen levels were lower in those with restenosis with a mean difference of − 2.43 ng/mL (95% CI, − 4.48-(− 0.37), P = 0.02; Fig. 5a). An additional six studies provided restenosis rates and PAI-1 activity levels. These studies reported restenosis in 119 patients (17.9%). PAI-1 activity was lower in those with restenosis with a mean difference of − 1.73 IU/mL (95% CI: -2.80-(− 0.67), P = 0.001; Fig. 5b).
Fig. 5.
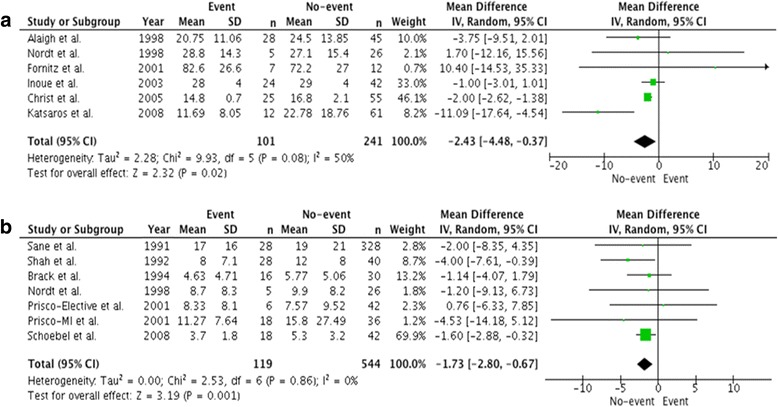
Comparison of mean PAI-1 antigen and activity levels in patients with restenosis and control patients. Data is expressed as a mean difference and analyzed using a random effects model. a Comparison of mean PAI-1 antigen levels (ng/mL) in patients with restenosis. b Comparison of mean PAI-1 activity levels (IU/mL) in patients with restenosis
High versus low PAI-1 levels
Three studies stratified their data by high versus low PAI-1 antigen levels. These studies reported a MACE rate of 54.4% [15–17]. High PAI-1 antigen levels were associated with a 58% greater risk of MACE compared to low PAI-1 antigen levels (RR 1.58, 95% CI: 1.42-1.76, P < 0.0001; Fig. 6).
Fig. 6.
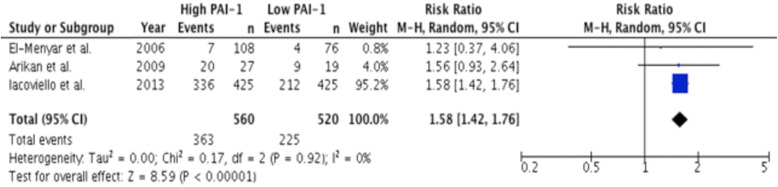
Comparison of risk of major adverse cardiac events in patients stratified by PAI-1 antigen levels (ng/mL). Data is expressed as a risk ratio and analyzed using a random effects model
Discussion
Incident and recurrent cardiovascular events remain important adverse outcomes despite major advances in revascularization and medical therapy. PAI-1 has been associated with MACE, but whether it is solely a marker for these events or a mediator with the potential of representing a unique therapeutic target is uncertain. Our analysis set out to evaluate the current state of evidence linking PAI-1 antigen and activity levels with these outcomes. Our study suggests that elevated PAI-1 antigen levels are associated with major adverse cardiac events in both primary and secondary event populations. In addition, elevated PAI-1 antigen levels were associated with all-cause mortality. While the populations studied were heterogeneous, the robustness of the association suggests that PAI-1 warrants further study as a marker and potential mediator of adverse cardiovascular events.
Our findings build upon the growing evidence that PAI-1 is a biomarker for MACE in patients with CAD. Tofler et al. identified elevated PAI-1 antigen levels to be predictive of cardiovascular disease [18]. In addition, previous studies of elevated PAI-1 antigen and activity levels predicted acute coronary syndrome after coronary stenting [15, 19–22]. Furthermore, Song et al. recently identified a causal relationship between elevated PAI-1 levels and incident CAD [23]. Our study expands on these findings by identifying elevated PAI-1 antigen levels as being associated with MACE in both primary (incident) and secondary event populations thereby suggesting a broader relevance of PAI-1 antigen levels. In addition, we demonstrate the potential applicability of PAI-1 antigen levels in predicting restenosis, consistent with a previous report by Katsaros et al. [24], which identified that patients with the lowest PAI-1 antigen tertile had a 9.5-fold increased risk of in-stent restenosis in patients managed with modern drug-eluting stents. However, our study, as with those mentioned above, are unable to ascertain if PAI-1 is a mediator or simply a marker of these events. Further study is needed to establish this important distinction.
The association of PAI-1 activity with MACE did not meet our pre-specified thresholds for significance. Although this finding suggests that PAI-1 antigen levels may be more robust as a biomarker, PAI-1 activity is a functional measure of the entire PAI-1 content in the plasma. The measurement of PAI-1 antigen captures the entire PAI-1 content in the sample in the form of free and active PAI-1 (which we refer to as PAI-1 activity), PAI-1 complexed to t-PA or u-PA, and latent PAI-1. Although PAI-1 antigen and activity levels are correlated, antigen levels will not necessarily reflect PAI-1 activity levels [25]. Indeed, at time of acute trauma such as plaque rupture, t-PA will complex with PAI-1 at a 1:1 ratio reducing detectable PAI-1 activity but not PAI-1 antigen levels. In addition, PAI-1 activity is influenced by experimental techniques during sample isolation such as freeze-thaw or sonication [26], low temperature, low pH, and high salt concentrations [27]. Factors which influence PAI-1 activity levels which impacted the significance of our findings include the method of PAI-1 extraction and isolation [26–30], time of blood draw [31], intra- and inter-assay variability in PAI-1 activity and antigen levels [32], and baseline risk factors which influences PAI-1 levels such as smoking [33, 34], high-fat diet [35], and maximal exercise [36]. Finally, in addition to important biological differences, manifest differences in the quality and power of studies examining antigen and activity levels existed which may explain the divergent results.
In-stent restenosis (ISR) is a result of neointimal formation or intimal thickening that narrows the vascular lumen following PCI [37]. The detailed molecular mechanism behind the pathophysiology of ISR has been reviewed elsewhere [38]. Briefly, studies have revealed that the initial recruitment of inflammatory cells is subsequently followed by smooth muscle cells (SMC) and myofibroblasts recruitment, which creates the extracellular matrix that narrows the vascular lumen [38]. SMCs achieve their peak proliferation at 48-96 h post-injury in the media and intima and return to their baseline following re-endothelialization of that artery within 8 weeks [39]. Conflicting evidence exists in the literature in the role of PAI-1 in cell migration. PAI-1 binding to low-density lipoprotein receptor-related protein 1 (LRP1) in SMCs promotes cell migration [40]. However, PAI-1 complexed to vitronectin has been demonstrated to inhibit cell migration and adhesion [41]. Thus, biological plausibility exists to link PAI-1 and restenosis following coronary intervention.
Clinically, low PAI-1 antigen and activity levels have been found to be associated with increased restenosis in our study; however, several limitations exist in these studies. First, these selected studies range from 1991 to 2008, during which time the intervention of choice evolved from balloon angioplasty to bare-metal stents to drug-eluting stents, which reduced the rate of ISR observed today [1]. Second, anti-proliferative agents that coat drug-eluting stents such as paclitaxel promote PAI-1 transcription and translation, impacting PAI-1 levels at the site of injury [42]. Third, PAI-1 activity cannot detect PAI-1 complexed to LRP1 found on smooth muscle cells and endothelial cells as they are no longer in circulation. Finally, since ISR occurs months following intervention, it remains unclear if baseline levels alone would be as predictive as repeated measurements. Repeat measurements of PAI-1 levels in these patients which would provide a comprehensive assessment of temporal PAI-1 levels from baseline to follow-up angiography or re-intervention. Nonetheless, despite these limitations we were able to link basal PAI-1 levels and restenosis. Future studies looking at modern revascularization techniques and temporal patterns of PAI-1 are warranted.
The value of PAI-1 as a biomarker has been questioned. First, PAI-1 expression is influenced by multiple pro-inflammatory conditions and is associated with various cardiovascular risk factors [6, 43]. For example, metabolic syndrome, obesity [44] and hyperinsulinemia [9]/insulin resistance [10] have all been linked with increased PAI-1 levels. In adjusted analysis, the predictive ability of elevated PAI-1 levels has not been independent of other cardiovascular risk factors and its additive benefit in risk prediction models has been lacking [45]. For example, Yarmolinsky et al. [3] reported that patients with diabetes had a significantly higher level of plasma PAI-1, which was associated with MACE. However, diabetics are at increased risk of both index and recurrent events. Accordingly, further studies are needed in more homogenous populations to ascertain the performance of PAI-1 in each individual cohort.
Our study is not without limitations. Relevant data could not be obtained from certain studies and patient level data were not available. In addition, the broad inclusion criteria resulted in a heterogeneous study population with differing PAI-1 measurement techniques. Variations in assays and standardizations as well as natural variations in PAI-1 levels may have influenced our results. For instance, considerable PAI-1 diurnal changes have been observed in previous studies [46, 47]. Most selected studies were of modest sample size and of low or moderate quality with only seven studies deemed to be of high quality [48]. The small number of studies may have limited the detection of small study effects or publication bias in funnel plots [49]. Finally, the large variation in study dates (1991 to 2016) spans a broad range of pharmacologic and revascularization practices, particularly coronary stent development, the introduction of dual antiplatelet therapy, and broadening indications for oral anticoagulation therapy. Accordingly, these findings may not be applicable in patients with specific risk profiles or those on contemporary medical therapy. Finally, while our study is provocative in the association demonstrated interventional studies are needed to link PAI-1 levels mechanistically to MACE.
Conclusion
PAI-1 plasma levels are promising markers for MACE; however, high quality studies in well-defined populations are still needed to robustly evaluate the performance of PAI-1 as a clinical biomarker. Whether PAI-1 is a bona fide therapeutic target remains to be established.
Additional file
Association between plasminogen activator inhibitor-1 and cardiovascular events: a systematic review and meta-analysis. (DOCX 4410 kb)
Acknowledgements
We thank Risa Shorr (R.S.), MLIS (The Ottawa Hospital, General Campus, Ottawa, Ontario, Canada) for peer-review of the MEDLINE literature search strategy.
Funding
R.J. was funded by the Vanier CIHR Canada Graduate Scholarship for his graduate studies.
Availability of data and materials
All data used for the systematic review and meta-analysis is present in the main manuscript in Tables 1 and 2.
Abbreviations
- CAD
Coronary artery disease
- MACE
Major adverse cardiovascular events
- PAI-1
Plasminogen activator inhibitor-1
- PCI
Percutaneous coronary intervention
- t-PA
Tissue-type plasminogen activator
- u-PA
Urokinase-type plasminogen activator
Authors’ contributions
R.J. and B.H. participated in the design, data extraction and analysis, and drafted the manuscript. P.M. participated in data extraction and analysis, and drafted the manuscript. F.D.R. helped design and drafted the manuscript. T.S., P.S., and Y.J. participated in data extraction. S.V. helped design the search strategy utilized for the systematic review. M.A.F. and A.L. was involved in critical revision of the manuscript. Finally, all authors read and approved the final draft of the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12959-018-0166-4) contains supplementary material, which is available to authorized users.
References
- 1.Simard T, Hibbert B, Ramirez FD, Froeschl M, Chen YX, O'Brien ER. The evolution of coronary stents: a brief review. Can J Cardiol. 2014;30:35–45. doi: 10.1016/j.cjca.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 2.Lucore CL, Sobel BE. Interactions of tissue-type plasminogen activator with plasma inhibitors and their pharmacologic implications. Circulation. 1988;77:660–669. doi: 10.1161/01.CIR.77.3.660. [DOI] [PubMed] [Google Scholar]
- 3.Yarmolinsky J, Bordin Barbieri N, Weinmann T, Ziegelmann PK, Duncan BB, Ines Schmidt M. Plasminogen activator inhibitor-1 and type 2 diabetes: a systematic review and meta-analysis of observational studies. Sci Rep. 2016;6:17714. doi: 10.1038/srep17714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. 2012;227:493–507. doi: 10.1002/jcp.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Taeye B, Smith LH, Vaughan DE. Plasminogen activator inhibitor-1: a common denominator in obesity, diabetes and cardiovascular disease. Curr Opin Pharmacol. 2005;5:149–154. doi: 10.1016/j.coph.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Srikanthan K, Feyh A, Visweshwar H, Shapiro JI, Sodhi K. Systematic review of metabolic syndrome biomarkers: a panel for early detection, management, and risk stratification in the west Virginian population. Int J Med Sci. 2016;13:25–38. doi: 10.7150/ijms.13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thogersen AM, Jansson JH, Boman K, Nilsson TK, Weinehall L, Huhtasaari F, Hallmans G. High plasminogen activator inhibitor and tissue plasminogen activator levels in plasma precede a first acute myocardial infarction in both men and women - evidence for the fibrinolytic system as an independent primary risk factor. Circulation. 1998;98:2241–2247. doi: 10.1161/01.CIR.98.21.2241. [DOI] [PubMed] [Google Scholar]
- 8.Meigs JB, O'Donnell CJ, Tofler GH, Benjamin EJ, Fox CS, Lipinska I, Nathan DM, Sullivan LM, D'Agostino RB, Wilson PW. Hemostatic markers of endothelial dysfunction and risk of incident type 2 diabetes: the Framingham offspring study. Diabetes. 2006;55:530–537. doi: 10.2337/diabetes.55.02.06.db05-1041. [DOI] [PubMed] [Google Scholar]
- 9.Trost S, Pratley R, Sobel B. Impaired fibrinolysis and risk for cardiovascular disease in the metabolic syndrome and type 2 diabetes. Curr Diab Rep. 2006;6:47–54. doi: 10.1007/s11892-006-0052-5. [DOI] [PubMed] [Google Scholar]
- 10.Sobel BE, Taatjes DJ, Schneider DJ. Intramural plasminogen activator inhibitor type-1 and coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1979–1989. doi: 10.1161/01.ATV.0000091250.53231.4D. [DOI] [PubMed] [Google Scholar]
- 11.Brazionis L, Rowley K, Jenkins A, Itsiopoulos C, O'Dea K. Plasminogen activator inhibitor-1 activity in type 2 diabetes - a different relationship with coronary heart disease and diabetic retinopathy. Arterioscler Thromb Vasc Biol. 2008;28:786–791. doi: 10.1161/ATVBAHA.107.160168. [DOI] [PubMed] [Google Scholar]
- 12.Anderson L, Oldridge N, Thompson DR, Zwisler AD, Rees K, Martin N, Taylor RS. Exercise-based cardiac rehabilitation for coronary heart disease: Cochrane systematic review and Meta-analysis. J Am Coll Cardiol. 2016;67:1–12. doi: 10.1016/j.jacc.2015.10.044. [DOI] [PubMed] [Google Scholar]
- 13.G Wells, B Shea, D O'Connell, J Peterson, V Welch, M Losos, P Tugwell. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses, 2010. http://www.ohri.ca/programs/clinical_epidemiology/oxford.asp. Accessed 4 Jan 2017.
- 14.Wan X, Wang W, Liu J, Tong T. Estimating the sample mean and standard deviation from the sample size, median, range and/or interquartile range. BMC Med Res Methodol. 2014;14:135. doi: 10.1186/1471-2288-14-135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iacoviello L, Agnoli C, De Curtis A, di Castelnuovo A, Giurdanella MC, Krogh V, Mattiello A, Matullo G, Sacerdote C, Tumino R, et al. Type 1 plasminogen activator inhibitor as a common risk factor for cancer and ischaemic vascular disease: the EPICOR study. BMJ Open. 2013;3:e003725. doi: 10.1136/bmjopen-2013-003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Menyar AA, Altamimi OM, Gomaa MM, Dabdoob W, Abbas AA, Abdel Rahman MO, Bener A, Albinali HA. Clinical and biochemical predictors affect the choice and the short-term outcomes of different thrombolytic agents in acute myocardial infarction. Coronary artery disease. 2006;17:431–7. doi: 10.1097/00019501-200608000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Arikan H, Koc M, Tuglular S, Ozener C, Akoglu E. Elevated Plasma Levels of PAI-1 Predict Cardiovascular Events and Cardiovascular Mortality in Prevalent Peritoneal Dialysis Patients. Ren Fail. 2009;31:438–45. doi: 10.1080/08860220902963772. [DOI] [PubMed] [Google Scholar]
- 18.Tofler GH, Massaro J, O'Donnell CJ, Wilson PW, Vasan RS, Sutherland PA, Meigs JB, Levy D, D'Agostino RB., Sr Plasminogen activator inhibitor and the risk of cardiovascular disease: the Framingham heart study. Thromb Res. 2016;140:30–35. doi: 10.1016/j.thromres.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcucci R, Brogi D, Sofi F, Giglioli C, Valente S, Liotta AA, Lenti M, Gori AM, Prisco D, Abbate R, Gensini GF. PAI-1 and homocysteine, but not lipoprotein (a) and thrombophilic polymorphisms, are independently associated with the occurrence of major adverse cardiac events after successful coronary stenting. Heart. 2006;92:377–381. doi: 10.1136/hrt.2005.061895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christ G, Nikfardjam M, Huber-Beckmann R, Gottsauner-Wolf M, Glogar D, Binder BR, Wojta J, Huber K. Predictive value of plasma plasminogen activator inhibitor-1 for coronary restenosis: dependence on stent implantation and antithrombotic medication. J Thromb Haemost. 2005;3:233–239. doi: 10.1111/j.1538-7836.2004.01062.x. [DOI] [PubMed] [Google Scholar]
- 21.Garg N, Goyal N, Strawn TL, Wu J, Mann KM, Lawrence DA, Fay WP. Plasminogen activator Inhibitor-1 and Vitronectin expression level and stoichiometry regulate vascular smooth muscle cell migration through physiological collagen matrices. J Thromb Haemost. 2010;8:1847–1854. doi: 10.1111/j.1538-7836.2010.03907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinkovic A. Prognostic role of plasminogen-activator-inhibitor-1 levels in treatment with streptokinase of patients with acute myocardial infarction. Clin Cardiol. 2000;23:486–9. doi: 10.1002/clc.4960230723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song C, Burgess S, Eicher JD, O'Donnell CJ, Johnson AD. Causal effect of plasminogen activator inhibitor type 1 on coronary heart disease. J Am Heart Assoc. 2017;6. 10.1161/JAHA.116.004918. [DOI] [PMC free article] [PubMed]
- 24.Katsaros KM, Speidl WS, Kastl SP, Zorn G, Huber K, Maurer G, Glogar D, Wojta J, Christ G. Plasminogen activator inhibitor-1 predicts coronary in-stent restenosis of drug-eluting stents. J Thromb Haemost. 2008;6:508–513. doi: 10.1111/j.1538-7836.2007.02884.x. [DOI] [PubMed] [Google Scholar]
- 25.Sakata Y, Murakami T, Noro A, Mori K, Matsuda M. The specific activity of plasminogen activator inhibitor-1 in disseminated intravascular coagulation with acute promyelocytic leukemia. Blood. 1991;77:1949–1957. [PubMed] [Google Scholar]
- 26.Brogren H, Wallmark K, Deinum J, Karlsson L, Jern S. Platelets retain high levels of active plasminogen activator inhibitor 1. PLoS One. 2011;6:e26762. doi: 10.1371/journal.pone.0026762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sancho E, Tonge DW, Hockney RC, Booth NA. Purification and characterization of active and stable recombinant plasminogen-activator inhibitor accumulated at high levels in Escherichia coli. Eur J Biochem. 1994;224:125–134. doi: 10.1111/j.1432-1033.1994.tb20003.x. [DOI] [PubMed] [Google Scholar]
- 28.Hekman CM, Loskutoff DJ. Endothelial cells produce a latent inhibitor of plasminogen activators that can be activated by denaturants. J Biol Chem. 1985;260:11581–11587. [PubMed] [Google Scholar]
- 29.Gils A, Declerck PJ. Modulation of plasminogen activator inhibitor 1 by triton X-100--identification of two consecutive conformational transitions. Thromb Haemost. 1998;80:286–291. doi: 10.1055/s-0037-1615189. [DOI] [PubMed] [Google Scholar]
- 30.Andreasen PA, Egelund R, Jensen S, Rodenburg KW. Solvent effects on activity and conformation of plasminogen activator inhibitor-1. Thromb Haemost. 1999;81:407–414. doi: 10.1055/s-0037-1614487. [DOI] [PubMed] [Google Scholar]
- 31.Scheer FA, Shea SA. Human circadian system causes a morning peak in prothrombotic plasminogen activator inhibitor-1 (PAI-1) independent of the sleep/wake cycle. Blood. 2014;123:590–593. doi: 10.1182/blood-2013-07-517060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Declerck PJ, Alessi MC, Verstreken M, Kruithof EK, Juhan-Vague I, Collen D. Measurement of plasminogen activator inhibitor 1 in biologic fluids with a murine monoclonal antibody-based enzyme-linked immunosorbent assay. Blood. 1988;71:220–225. [PubMed] [Google Scholar]
- 33.Scarabin PY, Aillaud MF, Amouyel P, Evans A, Luc G, Ferrieres J, Arveiler D, Juhan-Vague I. Associations of fibrinogen, factor VII and PAI-1 with baseline findings among 10,500 male participants in a prospective study of myocardial infarction--the PRIME study. Prospective epidemiological study of myocardial infarction. Thromb Haemost. 1998;80:749–756. doi: 10.1055/s-0037-1615353. [DOI] [PubMed] [Google Scholar]
- 34.Simpson AJ, Gray RS, Moore NR, Booth NA. The effects of chronic smoking on the fibrinolytic potential of plasma and platelets. Br J Haematol. 1997;97:208–213. doi: 10.1046/j.1365-2141.1997.d01-2137.x. [DOI] [PubMed] [Google Scholar]
- 35.Byrne CD, Wareham NJ, Martensz ND, Humphries SE, Metcalfe JC, Grainger DJ. Increased PAI activity and PAI-1 antigen occurring with an oral fat load: associations with PAI-1 genotype and plasma active TGF-beta levels. Atherosclerosis. 1998;140:45–53. doi: 10.1016/S0021-9150(98)00108-7. [DOI] [PubMed] [Google Scholar]
- 36.Cooper JA, Nagelkirk PR, Coughlin AM, Pivarnik JM, Womack CJ. Temporal changes in tPA and PAI-1 after maximal exercise. Med Sci Sports Exerc. 2004;36:1884–1887. doi: 10.1249/01.MSS.0000145447.61736.ED. [DOI] [PubMed] [Google Scholar]
- 37.Jung RG, Simard T, Labinaz A, Ramirez FD, Di Santo P, Motazedian P, Rochman R, Gaudet C, Faraz MA, Beanlands RSB, Hibbert B. Role of plasminogen activator inhibitor-1 in coronary pathophysiology. Thrombosis Res. 2018;164:54-62. [DOI] [PubMed]
- 38.Pourdjabbar A, Hibbert B, Simard T, Ma X, O'Brien ER. Pathogenesis of Neointima formation following vascular injury. Cardiovasc Hematol Disord Drug Targets. 2011;11:30–39. doi: 10.2174/187152911795945169. [DOI] [PubMed] [Google Scholar]
- 39.Clowes AW, Reidy MA, Clowes MM. Kinetics of cellular proliferation after arterial injury. I. Smooth muscle growth in the absence of endothelium. Lab Investig. 1983;49:327–333. [PubMed] [Google Scholar]
- 40.Luo M, Ji Y, Luo Y, Li R, Fay WP, Wu J. Plasminogen activator Inhibitor-1 regulates the vascular expression of Vitronectin. J Thromb Haemost. 2017;15:2451-60. [DOI] [PMC free article] [PubMed]
- 41.Czekay RP, Wilkins-Port CE, Higgins SP, Freytag J, Overstreet JM, Klein RM, Higgins CE, Samarakoon R, Higgins PJ. PAI-1: an integrator of cell signaling and migration. Int J Cell Biol. 2011;2011:562481. doi: 10.1155/2011/562481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muldowney JA, 3rd, Stringham JR, Levy SE, Gleaves LA, Eren M, Piana RN, Vaughan DE. Antiproliferative agents alter vascular plasminogen activator inhibitor-1 expression: a potential prothrombotic mechanism of drug-eluting stents. Arterioscler Thromb Vasc Biol. 2007;27:400–406. doi: 10.1161/01.ATV.0000254677.12861.b8. [DOI] [PubMed] [Google Scholar]
- 43.Iacoviello L, Agnoli C, De Curtis A, Cutrone A, Giurdanella MC, Guarrera S, Krogh V, Matullo G, Panico S, Sacerdote C, et al. Type 1 plasminogen activator inhibitor (PAI-1) and risk of acute coronary syndrome in the european prospective investigation into cancer (EPIC)-Italy cohort. Blood Transfus. 2012;10:s37. [Google Scholar]
- 44.Alessi MC, Juhan-Vague I. PAI-1 and the metabolic syndrome: links, causes, and consequences. Arterioscler Thromb Vasc Biol. 2006;26:2200–2207. doi: 10.1161/01.ATV.0000242905.41404.68. [DOI] [PubMed] [Google Scholar]
- 45.Johansson L, Jansson JH, Boman K, Nilsson TK, Stegmayr B, Hallmans G. Tissue plasminogen activator, plasminogen activator inhibitor-1, and tissue plasminogen activator/plasminogen activator inhibitor-1 complex as risk factors for the development of a first stroke. Stroke. 2000;31:26–32. doi: 10.1161/01.STR.31.1.26. [DOI] [PubMed] [Google Scholar]
- 46.Angleton P, Chandler WL, Schmer G. Diurnal variation of tissue-type plasminogen activator and its rapid inhibitor (PAI-1) Circulation. 1989;79:101–106. doi: 10.1161/01.CIR.79.1.101. [DOI] [PubMed] [Google Scholar]
- 47.Chong NW, Codd V, Chan D, Samani NJ. Circadian clock genes cause activation of the human PAI-1 gene promoter with 4G/5G allelic preference. FEBS Lett. 2006;580:4469–4472. doi: 10.1016/j.febslet.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 48.Bae JM. A suggestion for quality assessment in systematic reviews of observational studies in nutritional epidemiology. Epidemiol Health. 2016;38:e2016014. doi: 10.4178/epih.e2016014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lau J, Ioannidis JP, Terrin N, Schmid CH, Olkin I. The case of the misleading funnel plot. BMJ. 2006;333:597–600. doi: 10.1136/bmj.333.7568.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sane DC, Stump DC, Topol EJ, Sigmon KN, Kereiakes DJ, George BS, Mantell SJ, Macy E, Collen D, Califf RM. Correlation between baseline plasminogen activator inhibitor levels and clinical outcome during therapy with tissue plasminogen activator for acute myocardial infarction. Thrombosis and haemostasis. 1991;65:275–279. [PubMed] [Google Scholar]
- 51.Cortellaro M, Cofrancesco E, Boschetti C, Mussoni L, Donati MB, Cardillo M, Catalano M, Gabrielli L, Lombardi B, Specchia G, et al. Increased Fibrin Turnover and High Pai-1 Activity as Predictors of Ischemic Events in Atherosclerotic Patients - a Case-Control Study. Arterioscler Thromb. 1993;13:1412–1417. doi: 10.1161/01.ATV.13.10.1412. [DOI] [PubMed] [Google Scholar]
- 52.Brannstrom M, Jansson JH, Boman K, Nilsson TK. Endothelial Hemostatic Factors May Be Associated with Mortality in Patients on Long-Term Anticoagulant Treatment. Thromb Haemost. 1995;74:612–615. [PubMed] [Google Scholar]
- 53.JuhanVague I, Pyke SDM, Alessi MC, Jespersen J, Haverkate F, Thompson SG. Fibrinolytic factors and the risk of myocardial infarction or sudden death in patients with angina pectoris. Circulation. 1996;94:2057–2063. doi: 10.1161/01.CIR.94.9.2057. [DOI] [PubMed] [Google Scholar]
- 54.Nordt TK, Moser M, Kohler B, Ruef J, Peter K, Kubler W, Bode C. Augmented platelet aggregation as predictor of reocclusion after thrombolysis in acute myocardial infarction. Thromb Haemost. 1998;80:881–886. doi: 10.1055/s-0037-1615382. [DOI] [PubMed] [Google Scholar]
- 55.Alaigh P, Hoffman CJ, Korlipara G, Neuroth A, Dervan JP, Lawson WE, Hultin MB. Lipoprotein(a) level does not predict restenosis after percutaneous transluminal coronary angioplasty. Arterioscler Thromb Vasc Biol. 1998;18:1281–1286. doi: 10.1161/01.ATV.18.8.1281. [DOI] [PubMed] [Google Scholar]
- 56.Moss AJ, Goldstein RE, Marder VJ, Sparks CE, Oakes D, Greenberg H, Weiss HJ, Zareba W, Brown MW, Liang CS, et al. Thrombogenic factors and recurrent coronary events. Circulation. 1999;99:2517–2522. doi: 10.1161/01.CIR.99.19.2517. [DOI] [PubMed] [Google Scholar]
- 57.Redondo M, Carroll VA, Mauron T, Demarmels Biasiutti F, Binder BR, Lammle B, Wuillemin WA. Hemostatic and fibrinolytic parameters in survivors of myocardial infarction: A low plasma level of plasminalpha2-antiplasmin complex is an independent predictor of coronary re-events. Blood Coagulation and Fibrinolysis. 2001;12:17–24. doi: 10.1097/00001721-200101000-00003. [DOI] [PubMed] [Google Scholar]
- 58.Fornitz GG, Nielsen P, Amtorp O, Kassis E, Abildgard U, Sloth C, Winther K, Orskov H, Dalsgard J, Husted S. Impaired fibrinolysis determines the outcome of percutaneus transluminal coronary angioplasty (PTCA) Eur J Clin Invest. 2001;31:586–592. doi: 10.1046/j.1365-2362.2001.00855.x. [DOI] [PubMed] [Google Scholar]
- 59.Bogaty P, Poirier P, Simard S, Boyer L, Solymoss S, Dagenais GR. Biological profiles in subjects with recurrent acute coronary events compared with subjects with long-standing stable angina. Circulation. 2001;103:3062–3068. doi: 10.1161/01.CIR.103.25.3062. [DOI] [PubMed] [Google Scholar]
- 60.Ganti AK, Potti A, Yegnanarayan R. Plasma tissue plasminogen activator and plasminogen activator inhibitor-1 levels in acute myocardial infarction. Pathophysiol Haemost Thromb. 2002;32:80–84. doi: 10.1159/000065080. [DOI] [PubMed] [Google Scholar]
- 61.Lip GYH, Blann AD, Farooqi IS, Zarifis J, Sagar G, Beevers DG. Sequential alterations in haemorheology, endothelial dysfunction, platelet activation and thrombogenesis in relation to prognosis following acute stroke: The West Birmingham Stroke Project. Blood Coagul Fibrinolysis. 2002;13:339–347. doi: 10.1097/00001721-200206000-00010. [DOI] [PubMed] [Google Scholar]
- 62.Inoue T, Yaguchi I, Mizoguchi K, Uchida T, Takayanagi K, Hayashi T, Morooka S, Eguchi Y. Accelerated plasminogen activator inhibitor may prevent late restenosis after coronary stenting in acute myocardial infarction. Clin Cardiol. 2003;26:153–157. doi: 10.1002/clc.4960260311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robinson SD, Ludlam CA, Boon NA, Newby DE. Endothelial fibrinolytic capacity predicts future adverse cardiovascular events in patients with coronary heart disease. Arterioscler Thromb Vasc Biol. 2007;27:1651–1656. doi: 10.1161/ATVBAHA.107.143248. [DOI] [PubMed] [Google Scholar]
- 64.Thogersen AM, Nilsson TK, Weinehall L, Boman K, Eliasson M, Hallmans G, Jansson J-H. Changes in plasma C-reactive protein and hemostatic factors prior to and after a first myocardial infarction with a median follow-up time of 8 years. Blood coagulation & fibrinolysis : an international journal in haemostasis and thrombosis. 2009;20:340–346. doi: 10.1097/MBC.0b013e32832a5fd1. [DOI] [PubMed] [Google Scholar]
- 65.Akkus MN, Polat G, Yurtdas M, Akcay B, Ercetin N, Cicek D, Doven O, Sucu N. Admission Levels of CReactive Protein and Plasminogen Activator Inhibitor-1 in Patients With Acute Myocardial Infarction With and Without Cardiogenic Shock or Heart Failure on Admission. Int Heart J. 2009;50:33–45. doi: 10.1536/ihj.50.33. [DOI] [PubMed] [Google Scholar]
- 66.Pineda J, Marin F, Marco P, Roldan V, Valencia J, Ruiz-Nodar JM, Sogorb F, Lip GYH. Premature coronary artery disease in young (age < 45) subjects: Interactions of lipid profile, thrombophilic and haemostatic markers. Int J Cardiol. 2009;136:222–225. doi: 10.1016/j.ijcard.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 67.Wennberg P, Wensley F, Di Angelantonio E, Johansson L, Boman K, Rumley A, Lowe G, Hallmans G, Danesh J, Jansson JH. Haemostatic and inflammatory markers are independently associated with myocardial infarction in men and women. Thromb Res. 2012;129:68–73. doi: 10.1016/j.thromres.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 68.Yano Y, Hoshide S, Shimada K, Kario K. The Impact of Cigarette Smoking on 24-Hour Blood Pressure, Inflammatory and Hemostatic Activity, and Cardiovascular Risk in Japanese Hypertensive Patients. J Clin Hypertens. 2013;15:234–240. doi: 10.1111/jch.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yano Y, Nakazato M, Toshinai K, Inokuchi T, Matsuda S, Hidaka T, Hayakawa M, Kangawa K, Shimada K, Kario K. Circulating Des-acyl Ghrelin Improves Cardiovascular Risk Prediction in Older Hypertensive Patients. Am J Hypertens. 2014;27:727–733. doi: 10.1093/ajh/hpt232. [DOI] [PubMed] [Google Scholar]
- 70.Knudsen A, Katzenstein TL, Benfield T, Jorgensen NR, Kronborg G, Gerstoft J, Obel N, Kjaer A, Lebech A-M. Plasma plasminogen activator inhibitor-1 predicts myocardial infarction in HIV-1-infected individuals. AIDS (London, England) 2014;28:1171–1179. doi: 10.1097/QAD.0000000000000247. [DOI] [PubMed] [Google Scholar]
- 71.Golukhova EZ, Grigorian MV, Ryabinina MN, Bulaeva NI, Fortmann S, Serebruany VL. Independent Predictors of Major Adverse Events following Coronary Stenting over 28 Months of Follow-Up. Cardiology. 2015;132:176–181. doi: 10.1159/000435909. [DOI] [PubMed] [Google Scholar]
- 72.Shah PK, Amin J. Low high density lipoprotein level is associated with increased restenosis rate after coronary angioplasty. Circulation. 1992;85:1279–1285. doi: 10.1161/01.CIR.85.4.1279. [DOI] [PubMed] [Google Scholar]
- 73.Gray RP, Yudkin JS, Patterson DL. Enzymatic evidence of impaired reperfusion in diabetic patients after thrombolytic therapy for acute myocardial infarction: a role for plasminogen activator inhibitor? British heart journal. 1993;70:530–536. doi: 10.1136/hrt.70.6.530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Malmberg K, Bavenholm P, Hamsten A. Clinical and biochemical factors associated with prognosis after myocardial infarction at a young age. Journal of the American College of Cardiology. 1994;24:592–599. doi: 10.1016/0735-1097(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 75.Brack MJ, More RS, Pringle S, Gershlick AH. Absence of a Prothrombotic State in Restenotic Patients. Coronary Artery Dis. 1994;5:501–506. [PubMed] [Google Scholar]
- 76.Jansson JH, Nilsson TK, Johnson O, et al. Heart (British Cardiac Society) 1998;80:334–337. doi: 10.1136/hrt.80.4.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wiman B, Andersson T, Hallqvist J, Reuterwall C, Ahlbom A, deFaire U. Plasma levels of tissue plasminogen activator/plasminogen activator inhibitor-1 complex and von willebrand factor are significant risk markers for recurrent myocardial infarction in the Stockholm Heart Epidemiology Program (SHEEP) study. Arterioscler Thromb Vasc Biol. 2000;20:2019–2023. doi: 10.1161/01.ATV.20.8.2019. [DOI] [PubMed] [Google Scholar]
- 78.Prisco D, Antonucci E, Fedi S, Margheri M, Giglioli C, Comeglio M, Lombardi A, Chioccioli M, Abbate R, Gensini GF. D-Dimer increase after percutaneous transluminal angioplasty and clinical recurrence after primary revascularization in acute myocardial infarction? A pilot study. Clin Exper Med. 2001;1:219–224. doi: 10.1007/s102380100006. [DOI] [PubMed] [Google Scholar]
- 79.Sargento L, Saldanha C, Monteiro J, Perdigao C, Martins e Silva J. Evidence of prolonged disturbances in the haemostatic, hemorheologic and inflammatory profiles in transmural myocardial infarction survivors. Thrombosis and haemostasis. 2003;89:892–903. doi: 10.1055/s-0037-1613477. [DOI] [PubMed] [Google Scholar]
- 80.Schoebel FC, Peters AJ, Kreis I, Gradaus F, Heins M, Jax TW. Relevance of hemostasis on restenosis in clinically stable patients undergoing elective PTCA. Thromb Res. 2008;122:229–236. doi: 10.1016/j.thromres.2007.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Association between plasminogen activator inhibitor-1 and cardiovascular events: a systematic review and meta-analysis. (DOCX 4410 kb)
Data Availability Statement
All data used for the systematic review and meta-analysis is present in the main manuscript in Tables 1 and 2.