

Abstract
The brain tumor glioblastoma remains one of the most aggressive and devastating tumors despite decades of effort to find more effective treatments. A hallmark of glioblastoma is the constitutive activation of the NF-κB signaling pathway, which regulates cell proliferation, inflammation, migration, and apoptosis. The prolyl isomerase Pin1 has been found to bind directly to the NF-κB protein, p65, and cause increases in NF-κB promoter activity in a breast cancer model. We now present evidence that this interaction occurs in glioblastoma and that it has important consequences on NF-κB signaling. We demonstrate that Pin1 levels are enhanced in primary glioblastoma tissues compared to controls, and that this difference in Pin1 expression affects the migratory capacity of glioblastoma-derived cells. Pin1 knockdown decreases the amount of activated, phosphorylated p65 in the nucleus, resulting in inhibition of the transcriptional program of the IL-8 gene. Through the use of microarray, we also observed changes in the expression levels of other NF-κB regulated genes due to Pin1 knockdown. Taken together, these data suggest that Pin1 is an important regulator of NF-κB in glioblastoma, and support the notion of using Pin1 as a therapeutic target in the future.
Keywords: Glioblastoma, Gene Expression, Signal Transduction, NF-κB, Pin1
Introduction
Glioblastoma (GBM) is the most frequent and aggressive primary brain tumor in adults. This tumor is universally fatal, with approximately 60% of patients succumbing within one year of diagnosis and very few surviving 24 months (Stewart, 2002). GBM pathology is defined by cellular pleomorphism, microvascular proliferation, areas of necrosis, and extensive invasion into the surrounding brain tissue (Brandes et al , 2008). While new therapeutic regimens have shown some promise in improving patient lifespans (Stupp et al., 2002), the commonality, lethality, and aggressive nature of GBM warrants further investigation into its underlying biology.
One finding that may lead to novel GBM therapies is the observed activation of the NF-κB signaling pathway in these tumors (Nozell et al., 2008; Yamamoto et al., 2000). The mammalian NF-κB family includes five members: p65 (RelA), RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2). In an unstimulated state, these proteins exist in the cytoplasm as homo- or heterodimers, bound to an inhibitory family of proteins called IκB (Karin and Lin, 2002). Classical or “canonical” signaling of this pathway consists of an extracellular ligand (such as TNFα) binding to its receptor, which, in turn, activates the IκB Kinase (IKK) complex (Ghosh and Karin, 2002). This complex then phosphorylates IκB, which marks it for degradation via the ubiquitin-proteasome pathway. Free NF-κB dimers translocate to the nucleus where they activate a number of pro-inflammatory target genes, including Interleukin-8 (IL-8, CXCL8), CXCL10 (IP-10), Matrix Metalloproteinase-9 (MMP-9) and Interleukin-6 (IL-6) (Karin, 2006). NF-κB proteins undergo post-translational modifications, such as ubiquitination, acetylation, and phosphorylation, resulting in functional changes (Perkins, 2006). Most phosphorylation events of NF-κB appear to be activating modifications. For instance, phosphorylation of p65 at serine residue 276 is critical for protein-protein interactions with CBP/p300 (Zhong et al., 1998), necessary cofactors for NF-κB regulated gene transcription (Hayden and Ghosh, 2004). NF-κB function is also regulated both positively and negatively by cellular proteins (Hayden and Ghosh, 2008). The tumor suppressor ING4 negatively regulates NF-κB signaling (Garkavtsev et al., 2004). We recently demonstrated that ING4 negatively impacts the promoters of NF-κB regulated genes by interrupting interactions between p65 and tri-methylated histone H3 at lysine residue 4, leading to decreased expression of NF-κB regulated genes (Nozell et al., 2008). Of interest is the fact that ING4 expresson is reduced in GBM, and lack of expression is correlated with overexpression of NF-κB regulated genes and tumor progression (Garkavtsev et al., 2004; Nozell et al., 2008)
NF-κB may be one of the long-suspected links between inflammation and cancer (Greten et al., 2004). Constitutive activation of NF-κB is common to most primary tumors and tumor cell lines (Aggarwal, 2004). NF-κB increases the expression of a number of antiapoptotic molecules, while also increasing the expression of angiogenic factors and proinflammatory mediators (Bharti and Aggarwal, 2002). In primary GBM tissue, we recently determined that phosphorylated p65 levels, total p65 levels, and the expression of a number of NF-κB regulated genes are enhanced compared to control tissue (Nozell et al., 2008). Other groups have made similar observations in primary tissue (Wang et al., 2004) and cell culture models (Smith et al., 2007). Furthermore, the invasive capacity of glioblastoma has been linked to aberrantly high constitutive levels of NF-κB activation (Raychaudhuri et al., 2007).
Pin1 is an 18 kDa enzyme with two domains. N-terminal amino acids 1-19 comprise the WW domain which determines substrate specificity, while the C-terminus contains the catalytic PPIase domain (Yeh and Means, 2007). These two domains are connected by a flexible linker peptide. As a member of the PPIase family, Pin1 binds to and cis/trans isomerizes a specific subset of serine-proline or threonine-proline bonds (Ser/Thr-Pro) in its target proteins (Lu and Zhou, 2007). While all members of the familiy exert their enzymatic effects on Ser/Thr-Pro bonds, Pin1 is unique in that it only catalyzes its targets' residues when they are phosphorylated (Lu and Zhou, 2007). Currently, over fifty Pin1 target proteins have been identified (Lu and Zhou, 2007), including the NF-κB family member p65 (Ryo et al., 2003). Many human cancers express significantly higher levels of Pin1 protein than their normal tissue counterparts (Bao et al., 2004), including cancers of the liver (Pang et al., 2006), breast (Ryo et al., 2002; Wulf et al., 2001) and prostate (Ayala et al., 2003; Ryo et al., 2005). Importantly, Pin1 levels in GBM tissue are increased compared to control (Ryo et al., 2007). Since Pin1 expression is correlated with tumor increasing grade, Pin1 may be an excellent target for molecular cancer therapy, as well as a useful prognostic indicator for a variety of human tumors (Finn and Lu, 2008; Lu, 2003).
Ryo and colleagues found that Pin1 binds to the phosphorylated Thr254-pro residue of p65, and through this interaction, stabilizes nuclear p65 (Ryo et al., 2003). They also determined that Pin1 is a positive regulator for NF-κB regulated promoters (Ryo et al., 2003). We were interested in whether Pin1 modulates the expression of NF-κB regulated genes that play a role in the severity of GBM, such as IL-8. IL-8 is a neutrophil and monocyte chemotactic protein, as well as a potent angiogenic factor (Strieter et al., 2006). IL-8 is particularly important in tumor angiogenesis, and studied extensively with respect to human gliomas (Brat et al., 2005; Nozell et al., 2006).
In this study, we examined the effect of Pin1 on the NF-κB signaling pathway and downstream gene expression. Glioma-derived cell lines were created that inducibly express shRNA to Pin1 upon addition of tetracycline (Tet), leading to Pin1 knockdown. Using these cell lines, we demonstrate that TNFα stimulated cells with endogenous Pin1 levels show higher levels of phosphorylation of p65 S276 than those with Pin1 knockdown. We used a microarray screen to compare gene expression in TNFα-stimulated U251.shPin1 cells with endogenous Pin1 levels to those with Pin1 knockdown. Through analysis of these data, we selected IL-8 as a candidate gene. TNFα-induced IL-8 mRNA and protein expression are inhibited by Pin1 knockdown. In addition, recruitment of p65 and CBP to the IL-8 promoter is diminished in Pin1 knockdown cells, resulting in the decrease in IL-8 gene transcription. Collectively, our data demonstrate that Pin1 is a positive regulator of NF-κB signaling, and support the idea of using Pin1 as a therapeutic target in future treatment of GBM.
Results
Pin1 Protein Expression is Enhanced in Human GBM Tissue
A recent study reported increased expression levels of Pin1 in human glioma (Ryo et al., 2007), and we sought to confirm this finding. Immunohistochemistry was performed on ten GBM samples and eight control tissue samples, and representative images from one control (1) and four GBM samples (2-5) are shown in Fig. 1A. Overall, areas of strong Pin1 staining were detected in 8/10 GBM tumor samples and in none of the control samples. Patient Sample 2 is particularly helpful in demonstrating the differences in staining due to the presence of tumor (lower half) and nontransformed brain tissue (upper half) in the same section (Fig. 1A). Tissue integrity was examined with hematoxylin and eosin (H&E) staining (data not shown). These results confirm previous findings of increased Pin1 expression in GBMs.
Figure 1. Pin1 Expression in Primary Human Tissues and Cell Lines.

(A) Immunohistochemistry was performed on control and GBM paraffin-embedded brain tissue. Each section was stained with anti-Pin1 antibody and an IgG control antibody. A total of ten tumor samples and eight control brain samples were analyzed, and representative images from one control sample (1) and four GBM samples (2-5) are shown. (B) U251.shPin1 cells were grown in the absence or presence of Tet (5 μg/ml) for 96 h. Total cell lysates were assayed by immunoblot with anti-Pin1 and anti-GAPDH antibodies. Representative of three experiments.
Establishment of Inducible shPin1 Glioma Cell Lines
The observation of overexpression of Pin1 in primary GBM tissues, coupled with the finding that Pin1 is a positive regulator of p65 (Ryo et al., 2003), led us to hypothesize that Pin1 might be contributing to enhanced NF-κB signaling in GBM. Thus, we generated Tet-regulated U251-MG cell lines that inducibly express shRNA specific for Pin1. In the absence of Tet, endogenous Pin1 was detected (Fig. 1B, lane 1), and in the presence of Tet, shPin1 substantially reduced Pin1 protein levels (lane 2).
Pin1 Binds to p65 in U251-MG Cells
To determine if a p65-Pin1 interaction occurs within U251.shPin1 cells, coimmunoprecipitation experiments were performed. U251.shPin1 cells were stimulated with TNFα for 0 - 120 minutes. Immunoprecipitation was then performed using a Pin1 antibody, followed by immunoblotting for p65 and Pin1 (Fig. 2A). Endogenous Pin1 and p65 show an interaction in the unstimulated state (Fig. 2A, lane 1), which is enhanced upon TNFα stimulation (lanes 3 and 4). To demonstrate the specificity of this interaction, experiments were performed using Tet to knockdown Pin1 (Fig. 2B). A p65-Pin1 interaction was detectable in cells expressing endogenous levels of Pin1 (Fig. 2B, lane 1), which was abolished in cells with Pin1 knockdown (lane 2). Because IκBα associates with p65, the p65-IκBα interaction was examined as a control and was not affected by Pin1 knockdown (lanes 1 and 2). Thus, Pin1 and p65 interact in human GBM cells.
Figure 2. Pin1 Associates with p65 in U251-MG Cells.

(A) U251 shPin1 cells were grown in 150 mm dishes. When cell density reached approximately 80%, the cells were serum-starved for 4 h, treated with TNFα (10 ng/ml) for the indicated times (0-120 min), and lysed in RIPA buffer. Lysate from each sample was then subjected to immunoprecipitation (IP) using antibodies specific for Pin1 or normal rabbit serum (NRS). IP samples were probed with antibodies to p65 to demonstrate a specific interaction between p65 and Pin1. (B) U251.shPin1 cells were grown in 150 mm dishes in the absence or presence of Tet for 72 h. Cells were lysed in RIPA buffer and anti-p65 was used in the subsequent IP along with a normal mouse serum (NMS) control. Immunoblotting was performed using antibodies against Pin1, IκBα and p65. Results shown are representative of three experiments.
Pin1 Knockdown Inhibits Ser276 Phosphorylation of p65
Phosphorylation is important for NF-κB activation, and Serine 276 (S276) is crucial as it allows for optimal binding of the transcriptional coactivator CBP to the promoters of NF-κB regulated genes (Zhong et al., 2002). To determine whether Pin1 knockdown would affect the phosphorylation status of this residue, immunofluorescence (IF) experiments were performed. U251.shPin1 cells were grown in the absence or presence of Tet to modulate endogenous Pin1 levels and were then treated without or with TNFα for 1 h. Levels of total p65 increased substantially in the nucleus after TNFα activation, and this nuclear localization occurred to a similar degree in cells with endogenous Pin1 levels and those with Pin1 knockdown (Fig. 3A, middle row). However, substantial differences were observed in nuclear pS276 p65 (Fig. 3A, top row). In cells with endogenous Pin1 levels, S276 p65 became phosphorylated in the presence of TNFα However, cells with Pin1 knockdown showed lower levels of this phosphoactivation (Fig. 3A, top row), with 92% of these cells showing substantially less phosphorylation (data not shown). Pin1 knockdown was demonstrated in the presence of Tet (Fig. 3A, bottom row). To confirm these IF findings, nuclear extracts were examined. Cells were grown in the absence or presence of Tet and then stimulated with TNFα for up to 2 h. Total nuclear p65 levels were comparable at each time point in cells with endogenous levels of Pin1 versus those with Pin1 knockdown after TNFα stimulation (Fig. 3B, lanes 3-10). However, pS276 levels were decreased in Pin1 knockdown cells at each time point, with a maximal effect seen at 60 min (Fig. 3B, lanes 7-8). Interestingly, Pin1 knockdown did not inhibit two other phosphorylated residues of p65, pS536 and pT254 (data not shown). Cumulatively, these data suggest that Pin1 plays a positive role in the levels of activated nuclear p65 through a particular post-translational modification.
Figure 3. Pin1 Knockdown Inhibits S276 Phosphorylation of p65.

(A) U251 shPin1 cells were grown in the absence or presence of Tet for 72 h. 1 × 104 cells were plated in 8-well chamber slides and allowed to recover overnight. Cells were then serum starved, stimulated with 10 ng/ml of TNFα for 1 h, and assayed by immunofluorescence with the indicated antibodies. Dapi was used to stain nuclei (blue) and the indicated antibodies were used to analyze protein levels and localization (red). Multiple images were taken from each chamber; representative images for each condition from three experiments are shown. (B) 2.7 × 105 U251.shPin1 cells were plated in 100 mm dishes and grown in the absence or presence of Tet. After 72 h, media was changed to serum-free and cells were serum-starved overnight. The next day, cells were stimulated with 50 ng/ml of TNFα for the given timepoints and washed once with PBS. Nuclear (N) and cytoplasmic (C) fractions were separated using the Pierce NE-PER Kit and assayed via immunoblotting with the indicated antibodies. Quantification (Quant) of nuclear pS276 p65 was calculated by dividing the densitometric value of each lane by the corresponding nuclear HDAC1 value. Results are representative of three experiments.
Pin1 Has Differential Effects on NF-κB Regulated Gene Expression
Having seen less p65 activation in Pin1 knockdown cells, we examined the effect of Pin1 knockdown on known NF-κB regulated genes using a microarray-based screen (Gilmore, 2008). Untreated U251.shPin1 cells (UTX), cells treated with TNFα (TNF) for 4 h, and cells pretreated with Tet and then treated with TNFα (TET) for 4 h were screened using the Affymetrix U133 2.0 Plus array. The results were filtered to include only genes which were induced at least four-fold by TNFα and known to be NF-κB regulated (Gilmore, 2008) (Fig. 4A). We then generated a simple ratio to describe the behavior of the filtered gene set by dividing the values from the TET array by the TNF array (TET / TNF). Thus, a number less than 1 represented a decrease of a gene in the Pin1 knockdown conditions, and a number greater than 1 represented an increase in the expression level of a given gene in Pin1 knockdown conditions. IL-8 was selected from this gene set to investigate further (Fig. 4B).
Figure 4. Microarray Data Overview.
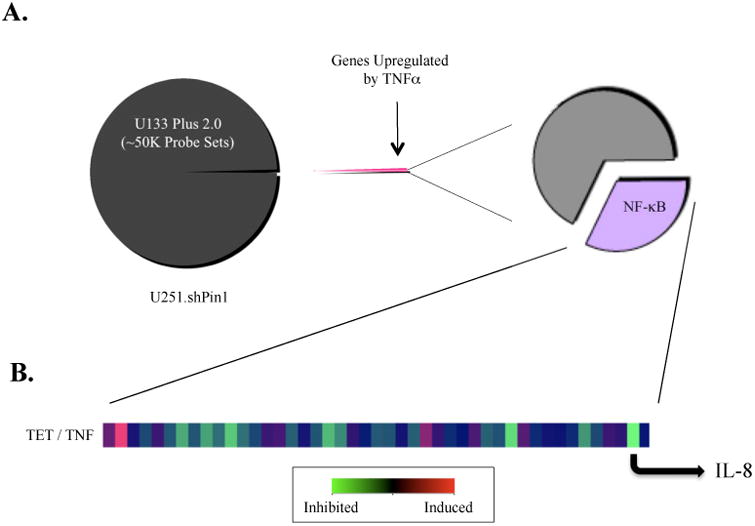
(A) Three conditions were compared: cells with no treatment (UTX), cells treated with TNFα only (TNF), and cells treated with both TNFα and Tet (TET). Approximately 50,000 genes were filtered, allowing only genes that were significantly upregulated (four-fold increase) after a 4 h treatment with TNFα to pass through. This daughter set was cross-referenced with a comprehensive database of NF-κB regulated genes from NF-κB.org, and these NF-κB regulated genes were then analyzed for changes in gene expression due to Pin1 knockdown. (B) A heat-map depicting the ratio TET / TNF shows changes in expression levels of NF-κB regulated genes. IL-8 was selected as a strong candidate for further evaluation due to its inhibition upon depletion of Pin1.
p65 Knockdown Inhibits IL-8 Expression by Decreasing p65 Recruitment to the IL-8 Promoter
To determine the importance of p65 in TNFα-induced IL-8 expression, an inducible p65 knockdown U251-MG-derived cell line, U251-TR.sh-p65 (Nozell et al., 2008), was utilized. U251-TR.sh-p65 cells were grown in the absence or presence of Tet and stimulated with TNFα for 4 h. Tet effectively knocked down levels of p65 mRNA (Fig. 5A, lanes 2 and 4). While TNFα induced IL-8 mRNA expression in the presence of p65 (Fig 5A, lane 3), expression was inhibited in p65 knockdown cells (lane 4). TNFα stimulated the expression of IL-8 protein in the presence of p65, but these levels were significantly diminished in p65 knockdown cells (Fig. 5B). Furthermore, p65 recruitment to the IL-8 promoter was diminished in TNFα-stimulated cells with p65 knockdown (Fig. 5C, lanes 3 and 4).
Figure 5. p65 Knockdown Inhibits IL-8 Expression and Decreases p65 Recruitment to the IL-8 Promoter.

(A) U251-TR.sh-p65 cells were grown in the absence or presence of Tet for 48 h, serum-starved, and then stimulated with TNFα (10 ng/ml) for 4 h. Total RNA was isolated, first-strand cDNA synthesis was performed, and RT-PCR was performed on the samples using primers to specifically detect p65, IL-8 and GAPDH. (B) U251-TR.sh-p65 cells were grown in the absence or presence of Tet for 48 h, serum-starved, and the stimulated with TNFα (10 ng/ml) for 24 h. Supernatant was collected and analyzed by ELISA for IL-8 protein expression (*p<0.05). (C) U251-TR.sh-p65 cells were grown in the absence or presence of Tet for 48 h, serum-starved, and then stimulated with TNFα (10 ng/ml) for 15 min. The samples were then subjected to ChIP assay (see Materials and Methods). Immunoprecipitations were performed with anti-p65 or an IgG control. All results are representative of three experiments.
Pin1 Knockdown Inhibits IL-8 Gene Expression
To confirm the results of the microarray screen, SYBR green quantitative reverse transcriptase PCR (qPCR) was used to analyze the levels of TNFα-induced IL-8 mRNA. In cells with endogenous Pin1 levels (-Tet), IL-8 mRNA was induced by TNFα in a dose-dependent manner (Fig. 6A). At all doses of TNFα, IL-8 mRNA levels were inhibited in Pin1 knockdown cells (Fig. 6A). IL-8 protein expression was also significantly inhibited in TNFα treated Pin1 knockdown cells compared to cells with endogenous Pin1 expression (Fig. 6B).
Figure 6. Pin1 Knockdown Inhibits IL-8 Gene Expression and Decreases p65 Recruitment at the IL-8 Promoter.

(A) Two-step quantitative RT-PCR was performed using SYBR Green. One μg of RNA was reverse transcribed to cDNA as described. Mastermix, primers, and cDNA were combined and run for 50 cycles in an ABI 7500. Ct values for IL-8 qPCR reactions were derived and divided by Ct values for GAPDH. Results are representative of three experiments (*p<0.05). (B) Pin1 knockdown inhibits IL-8 protein expression. Cells were plated at 2.0 × 104 in 12-well plates, grown in the absence or presence of Tet, and stimulated for 24 h with TNFα. Supernatants were analyzed for IL-8 protein expression by ELISA. Results are representative of three experiments (*p<0.05). (C) 3.0 × 106 U251.shPin1 cells were grown in the absence or presence of Tet for 72 h, plated in 150 mm dishes, serum-starved, and stimulated with TNFα. Cells were assayed by ChIP (see Materials and Methods). Immunoprecipitations were performed with antibodies to the given proteins and primers specific to the IL-8 promoter. A PCR reaction using the human IL-8 promoter primers on non-immunoprecipitated DNA (Input) was included as a control for total DNA levels.
Pin1 Knockdown Decreases Activation at the IL-8 Promoter
We speculated that the decrease in IL-8 expression was being mediated through a transcriptional mechanism. As such, ChIP experiments were performed to determine the levels of transcription factors at the IL-8 promoter. Because TNFα-induced S276 p65 phosphorylation occurs rapidly in the U251-MG cells, a 30 min timepoint was used to examine the levels of this particular form of p65 at the IL-8 promoter (Fig 6C, compare lanes 1 and 3). While strong recruitment was observed in TNFα-treated cells with endogenous Pin1 levels, levels of activated p65 were almost completely abolished at the IL-8 promoter in TNFα-treated Pin1 knockdown cells (Fig 6C, compare lanes 3 and 4). A similar pattern was seen when immunoprecipitations for total p65 levels were performed, with Pin1 knockdown cells showing less recruitment of total p65 to the IL-8 promoter (Fig. 6C, compare lanes 3 and 4). Levels of CBP were also diminished at the IL-8 promoter in Pin1 knockdown conditions (Fig. 6C), suggesting that IL-8 inhibition may be partially mediated through inadequate recruitment of CBP/p300 and subsequent failure to fully activate IL-8 transcription. RNA Polymerase II was also immunoprecipitated, as it has also been demonstrated to be a substrate for Pin1 (Xu and Manley, 2007). Pin1 knockdown had little effect on the presence of RNA Polymerase II at the IL-8 promoter (Fig. 6C).
Pin1 Knockdown Decreases the Migration of the U251-MG Cell Line
Many factors that induce cell migration/invasion are regulated by NF-κB, including IL-8. Given our results that Pin1 affects NF-κB activity, scratch assays were performed to assess the migratory capacity of U251-MG cells with endogenous and knockdown Pin1 levels. In serum-free conditions, wells containing endogenous Pin1-expressing cells showed enhanced repopulation of the scratched area compared to those wells with Pin1 knockdown (Figs. 7A and 7B). TNFα treatment yielded similar results, whereby cells with endogenous Pin1 levels demonstrated consistently higher levels of migration than cells with Pin1 knockdown (Figs. 7A and 7B). IL-8 has been suggested to play a role in the invasiveness of GBM (Nagai et al., 2002). Analogously, the migration of U251-MG cells has been shown to increase significantly with the addition of exogenous IL-8 (Wakabayashi et al., 2004). Although we initially suspected that IL-8 might be the key factor in the decreased migration in Pin1 knockdown cells, experiments using exogenous IL-8 demonstrated no significant increase in GBM cell mobility (data not shown). Thus, Pin1 drives the migratory behavior of U251.shPin1 cells, independent of IL-8, and may play a role in the invasive phenotype of GBM.
Figure 7. Pin1 Knockdown Decreases the Migration of U251-MG Cells.
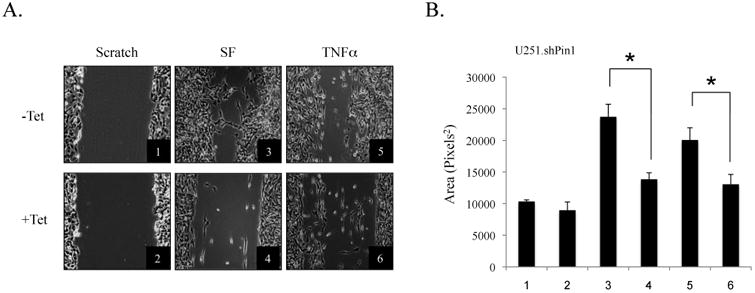
(A) Cells were grown in the absence or presence of Tet (5 μg/ml) for 72 h and plated in 6-well plates. Wells were then scratched with a sterile p200 pipette tip, washed with PBS, and changed to serum-free media (SF) for 48 h. Conditions 5 and 6 were treated with TNFα for 36 h. Conditions were performed in triplicate and three images were taken of each replicate. Representative images from each condition are shown. (B) Images from the scratch assay were quantified using ImageJ software. Results are representative of three experiments (*p<0.05).
Discussion
In this study, we have elucidated the effects of the prolyl isomerase Pin1 on the NF-κB signaling pathway in glioblastoma. In concurrence with one other study (Ryo et al., 2007), we found that Pin1 levels are elevated in GBM compared to control brain tissue. Functionally, these heightened Pin1 levels in GBM have been demonstrated to sensitize tumor cells to oxidative stress-induced apoptosis by promoting the degradation of the pro-apoptotic protein Daxx (Ryo et al., 2007). A host of other Pin1-mediated mechanisms are likely important in GBM, including those regulated by NF-κB.
We describe a mechanism by which Pin1 affects the NF-κB signaling pathway. Activated p65, as measured by S276 phosphorylation, was substantially inhibited by Pin1 knockdown. This was not observed using another marker of NF-κB activation, pS536 p65 (data not shown). A possibility for the specific effect on S276 is this residue's physical proximity to the residue with which Pin1 interacts with p65, pT254. Under this scenario, our results could be explained through two potential mechanisms. The first is that Pin1 acts to facilitate the interaction of a kinase with p65, thereby contributing to its phosphoactivation. Protein Kinase A (PKA) is one candidate, as it has been shown to phosphorylate p65 at S276 (Zhong et al., 1998). Conversely, Pin1 may exert its effect by blocking the access of one or more phosphatases to pS276, leaving this residue more heavily phosphorylated in the presence of Pin1. The phosphatase(s) responsible for p65 pS276 dephosphorylation are currently unknown (Li et al., 2006), so it is not yet possible to test this hypothesis.
One of the key findings in regards to the modulation of gene expression due to Pin1 involves the chemokine IL-8. While IL-8 was the most interesting NF-κB-regulated gene that demonstrated a Pin1 dependent expression pattern in the microarray screen, it was hardly the only one. We found that some NF-κB regulated genes (such as IL-8 and IP-10) were positively regulated by Pin1, others (like FASL and TRAIL) were negatively regulated by Pin1, and still others (such as KIT ligand and IL-15) did not significantly change expression levels with Pin1 knockdown. The mRNA levels of another important NF-κB regulated gene in glioblastoma, MMP-9, were also found to be unchanged in subsequent RT-PCR experiments (data not shown). There are a number of possibilities for these findings. First is the timepoint at which we chose to conduct the screen. Since NF-κB regulated genes demonstrate very specific temporal patterns of expression after activation (Hoffmann et al., 2003; Hoffmann et al., 2002a; Saccani et al., 2001), part of the reason for the variety of expression patterns may have been a suboptimal timepoint for the expression of some of these genes. Another possible reason is the sheer complexity of NF-κB signaling. Although we and others have demonstrated the importance of p65 in IL-8 expression (Harant et al., 1996; Hoffmann et al., 2002b; Nozell et al., 2006), other NF-κB regulated genes are driven through non-canonical NF-κB signaling or rely on non-p65 containing NF-κB dimers. A further possibility is that other NF-κB family members are able to compensate for the diminished p65 activation in Pin1 knockdown cells, thus masking the gene-inhibitory effects of Pin1 knockdown.
This study has attempted to understand the molecular mechanisms responsible for the constitutive increase in NF-κB signaling in GBM. We have shown enhanced levels of a positive regulator of NF-κB signaling, Pin1, in GBM. Conversely, ING4, a negative regulator of NF-κB signaling, is decreased in GBM compared to controls (Nozell et al., 2008). Another negative regulator of NF-κB, PIAS3 (Jang et al., 2004), is also decreased in GBMs (Brantley et al., 2008). However, the importance of Pin1 in GBM signaling defects may not be limited to the NF-κB pathway. Pin1 enhances IL-6 mediated STAT3 signaling in various cancer cell lines (Lufei et al., 2007). We have investigated this effect of Pin1 in our system, however, TNFα is a very poor activator of STAT3, thus no effects were observed (data not shown). Given that overactivation of this pathway has been demonstrated in GBM (Brantley et al., 2008), we speculate that Pin1 may also contribute to aberrant STAT3 signaling in GBM.
One of the most challenging aspects of GBM is the capacity of the tumor to invade deeply into adjacent brain tissue, making full surgical resection impossible (Chandana et al., 2008). Thus, any therapeutic option that would diminish the tumor's capacity for invasion could greatly improve patient lifespans. For this reason, we decided to test whether or not Pin1 knockdown would affect the invasive capacity of malignant glioma cells through an analogous in vitro assay. Indeed, through scratch assays, U251-MG cells with Pin1 knockdown have decreased migratory capacity. We do not know which factors are responsible for this difference, although due to the strong NF-κB activating ability of TNFα, we predict that at least some of these factors are NF-κB regulated. Some possibilities include Intercellular Adhesion Molecule 1 (ICAM-1) and E-selectin. Initially, we speculated that IL-8 was one of the important factors for this difference in migration. Further experiments, however, demonstrated that exogenous IL-8 was unable to restore the migratory capacity of Pin1 knockdown cells (data not shown).
Overall, this study has explored the relevance of the Pin1-p65 interaction in GBM. The interaction between Pin1 and p65 enhances the levels of pS276, increases the migratory capacity of U251-MG cells, and enhances the expression of some NF-κB regulated genes, notably IL-8. Our findings support the notion of using Pin1 as a molecular target for cancer therapy. If this protein can be effectively targeted in cancer cells, GBMs may be less invasive and have lower levels of active NF-κB signaling. This inhibition of NF-κB could be helpful with GBM treatment since inhibiting NF-κB has been shown to be an effective method of retarding tumor growth in a number of different in vitro and in vivo models (Karin et al., 2002). Furthermore, by inhibiting the production of some pro-angiogenic NF-κB regulated genes, we expect that Pin1 inhibition could also help to reduce the blood supply and thus the growth of GBM.
Materials and Methods
Reagents
Recombinant human TNFα was from R&D Systems (Minneapolis, MN). Anti-Pin1 (SC-15340), anti-p65 (SC-8008), anti-IκBα (SC-371), and anti-IKKα (SC-744) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-p65 pS276 (3027) and anti-pIKKα/pIKKβ (2681) antibodies were from Cell Signaling (Danvers, MA). The anti-Pin1 antibody (PC270) used in immunohistochemistry was from Calbiochem/Merck (Darmstadt, Germany), and anti-GAPDH antibody was from Abcam (Cambridge, England). HRP-conjugated secondary mouse (1858413) and rabbit (1858415) antibodies and Protein A/G beads were purchased from Pierce (Rockford, IL).
Plasmids
Plasmids carrying shRNAs specific for either Pin1 or p65 were generated by annealing double-stranded oligonucleotides specific for a 19-bp stretch of the Pin1 ORF or the p65 ORF into the pBABE-HI-TetO plamid, which is under the dual control of the Tet operator and the HI polymerase (Pol) III promoter. Specific sequences are available upon request.
Cell Lines
U251-MG cells were maintained as described previously (Choi et al., 2002). U251-MG cells that stably express the Tet repressor (TetR) protein and inducibly express shRNA specific for Pin1 (shPin1) or p65 (shp65) were generated as previously described (Nozell et al., 2008).
Immunoblotting
Cells were plated at a density of 2.75 × 105 cells per 100 mm dish. After treatment with the appropriate stimuli, cells were lysed, and 35 μg of total protein separated by electrophoresis, as previously described (Baker et al., 2008). After transferring to nitrocellulose paper, blots were blocked in either 5% non-fat milk in TBST for non-phosphorylated protein detection or 5% BSA/TBST for phosphorylated protein detection. Primary antibody incubations using antibodies against Pin1, p65, pS276 p65, IκBα, IKKα, IKKβ, pIKKα/pIKKβ and GAPDH occurred overnight at 4° C. After washing, secondary antibody incubations were performed for 1 h at room temperature. Blots were developed using Supersignal or ECL chemiluminescence systems (Amersham). Representative images from at least 3 separate experiments are shown.
Co-Immunoprecipitation
U251.shPin1-MG cells were grown in the absence or presence of Tet for 72 h, lysed, and subjected to immunopreciptiation overnight using 5 μg of either Pin1 or p65 antibody and protein A/G beads. An IgG control sample for each experiment was included. Beads were then washed and boiled to remove and denature the captured proteins. Samples were analyzed by immunoblotting using antibodies specific for p65, Pin1, and IκBα.
Protein Purification From Human Brain Tissue
Resected glioma and control brain tissue samples were obtained from the UAB Brain Tumor Tissue core facility (IRB X050415007). Samples were processed using the PARIS Protein and RNA extraction system, as previously described (Brantley et al., 2008; Nozell et al., 2008).
Quantitative RT-PCR (qRT-PCR)
SYBR Green qRT-PCR was performed according to the manufacturer's protocol (Applied BioSystems). A positive control sample was selected from the experimental samples, diluted 1:1 in ultra pure water, and used in a series of four serial dilutions for a total of five diluted samples to be used in primer efficiency curves. Appropriate amounts of SYBR Green Mastermix, primers specific for the genes of interest, and sterile water were combined and subsequently added to the wells of a MicroAmp 96-well reaction plate (Applied BioSystems). cDNA from the dilution samples or the experimental samples was then added to the plate and mixed. The 96-well plates were then centrifuged briefly and run for 50 cycles in an ABI 7500 real-time PCR machine. Ct values were derived and then all genes were quantified in relation to an endogenous control, GAPDH. Experimental samples were measured in triplicate reactions, and all primer efficiency curves and non-template controls were performed in duplicate.
RNA Isolation
Cells were lysed in 1 ml of TRIzol® Reagent (Invitrogen) according to the manufacturer supplied protocol and as previously described (Nozell et al., 2004).
Nuclear/Cytoplasmic Extraction
U251.shPin1 cells were grown in the absence or presence of Tet for 72 h. Cells were then lysed and nuclear and cytoplasmic fractions separated, as described previously (Nozell et al., 2004).
Chromatin Immunoprecipitation (ChIP)
ChIP assay was performed as described previously (Nozell et al., 2008; Nozell et al., 2006). U251.shPin1 cells were grown in the absence or presence of Tet for 72 h, serum-starved overnight, and then treated with TNFα for the indicated times and fixed. Nuclei were isolated and sonicated for 50 seconds to shear DNA. All immunoprecipitations were performed overnight using 5 μg of antibody against pS276 p65, total p65, CBP, and RNA Pol II. After immunoprecipitations, beads were washed and cross-linking reversed overnight using 5 M NaCl. DNA was purified and PCR performed using primers specific to the human IL-8 promoter (forward 5′-TTC CTT CCG GTG GTT TCT TC-3′, reverse 5′-GGG CCA TCA GTT GCA AAT C-3′)
Microarray Screen
A screen was performed using the Affymetrix Human Genome U133 Plus 2.0 array, as described previously (Choi et al., 2006). U251.siPin1 cells were grown in the absence or presence of Tet for 72 h, then treated without or with 10 ng/ml of TNFα for 4 h. RNA was extracted and processed for microarray using the UAB Heflin Center for Human Genetics Microarray Core Facility. Bioinformatic analysis was performed using ArrayAssist software (Strategene). A parallel immunoblot was performed to confirm Pin1 knockdown in Tet-treated cells.
Immunofluorescence
Cells were assayed and visualized as described previously (Nozell et al., 2006). U251.shPin1 cells were grown in the absence or presence of Tet for 72 h, plated in 8-well chamber slides and allowed to recover overnight. Cells were then serum starved, stimulated with 10 ng/ml of TNFα for 1 h, and fixed with methanol. Cells were incubated with antibodies against pS276 p65, total p65 and Pin1, washed with PBS, incubated with a fluorochrome-labeled secondary antibody, and visualized using a Leitz Orthoplan upright microscope and IPLab software (BD Biosciences). At least three fields were imaged per condition.
Immunohistochemistry
Human control brain tissue and glioma tissue were obtained from the UAB Brain Tumor Tissue core facility (IRB X050415007) and processed for immunohistochemical analysis as described previously (Brantley et al., 2008; Nozell et al., 2008). Primary antibody incubation for Pin1 was performed overnight at 4°C in blocking buffer. After secondary antibody incubation, the slides were stained with DAB and analyzed in a blinded fashion by a neuropathologist.
ELISA
U251.shPin1 or U251-TR.sh-p65 cells were grown in the absence or presence of Tet for 72 h, and then treated with 10 ng/ml TNFα for 24 h. IL-8 ELISA (Biosource, KHC0082) was performed in triplicate on the supernatants as described previously (Nozell et al., 2006).
Scratch Assay
U251.shPin1 cells were grown in the absence or presence of Tet, then plated in 6-well dishes, and grown to confluence. Each well was scratched using a 200 μl pipette tip, washed with PBS, and incubated for up to 48 h in serum-free media in the absence or presence of TNFα, as previously described (Vila-Carriles et al., 2006). Each condition was represented in triplicate, and at each timepoint 3 images were taken of each well for analysis with IPLab software (BD Biosciences). Representative images are shown from 3 separate experiments. Quantification was performed using ImageJ Software (NIH).
Statistical Analysis
Levels of significance were determined using the Student's t-test, with p values ≤ 0.05 considered significant.
Acknowledgments
We thank Dr. Michael Crowley in the UAB Comprehensive Cancer Center Gene Expression Shared Facility for his assistance with microarray processing, and Dr. Cheryl Ann Palmer (University of Alabama at Birmingham) for providing the brain tumor resections. We also thank Dr. Yancey Gillespie, UAB Brain Tumor Tissue Core, for glioma and control tissue samples, and express our appreciation to the laboratory of Dr. Dale Benos for assistance with the scratch assay. Finally, many thanks to Dr. Rafal Bartoszewski for assistance with qPCR, and to Dr. Gerald Fuller for reading the manuscript.
This work was supported in part by NIH grants CA-97247 (E.N.B.), NS-54158 (E.N.B. and S.E.N.), IRG-60-001-47 from the American Cancer Society and CA-13148-31 from the NCI (S.E.N.). G.P.A. was supported by the UAB Medical Scientist Training Program, and is currently supported by NIH T32-AI0705.
References
- Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell. 2004;6:203–8. doi: 10.1016/j.ccr.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Ayala G, Wang D, Wulf G, Frolov A, Li R, Sowadski J, et al. The prolyl isomerase Pin1 is a novel prognostic marker in human prostate cancer. Cancer Res. 2003;63:6244–51. [PubMed] [Google Scholar]
- Baker BJ, Qin H, Benveniste EN. Molecular basis of oncostatin M-induced SOCS-3 expression in astrocytes. Glia. 2008;56:1250–62. doi: 10.1002/glia.20694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Kimzey A, Sauter G, Sowadski JM, Lu KP, Wang DG. Prevalent overexpression of prolyl isomerase Pin1 in human cancers. Am J Pathol. 2004;164:1727–37. doi: 10.1016/S0002-9440(10)63731-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti AC, Aggarwal BB. Nuclear factor-kappa B and cancer: its role in prevention and therapy. Biochem Pharmacol. 2002;64:883–8. doi: 10.1016/s0006-2952(02)01154-1. [DOI] [PubMed] [Google Scholar]
- Brandes AA, Tosoni A, Franceschi E, Reni M, Gatta G, Vecht C. Glioblastoma in adults. Critical Reviews in Oncology/Hematology. 2008;67:139–152. doi: 10.1016/j.critrevonc.2008.02.005. [DOI] [PubMed] [Google Scholar]
- Brantley EC, Nabors LB, Gillespie GY, Choi YH, Palmer CA, Harrison K, et al. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin Cancer Res. 2008;14:4694–704. doi: 10.1158/1078-0432.CCR-08-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brat DJ, Bellail AC, Van Meir EG. The role of interleukin-8 and its receptors in gliomagenesis and tumoral angiogenesis. Neuro-oncol. 2005;7:122–33. doi: 10.1215/S1152851704001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandana SR, Movva S, Arora M, Singh T. Primary brain tumors in adults. Am Fam Physician. 2008;77:1423–30. [PubMed] [Google Scholar]
- Choi C, Kutsch O, Park J, Zhou T, Seol DW, Benveniste EN. Tumor necrosis factor-related apoptosis-inducing ligand induces caspase-dependent interleukin-8 expression and apoptosis in human astroglioma cells. Mol Cell Biol. 2002;22:724–36. doi: 10.1128/MCB.22.3.724-736.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YH, Bernardi R, Pandolfi PP, Benveniste EN. The promyelocytic leukemia protein functions as a negative regulator of IFN-gamma signaling. Proc Natl Acad Sci U S A. 2006;103:18715–20. doi: 10.1073/pnas.0604800103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn G, Lu KP. Phosphorylation-specific prolyl isomerase Pin1 as a new diagnostic and therapeutic target for cancer. Curr Cancer Drug Targets. 2008;8:223–9. doi: 10.2174/156800908784293622. [DOI] [PubMed] [Google Scholar]
- Garkavtsev I, Kozin SV, Chernova O, Xu L, Winkler F, Brown E, et al. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature. 2004;428:328–32. doi: 10.1038/nature02329. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109 Suppl:S81–96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Gilmore T. (2008). Boston, MA.
- Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Harant H, de Martin R, Andrew PJ, Foglar E, Dittrich C, Lindley IJ. Synergistic activation of interleukin-8 gene transcription by all-trans-retinoic acid and tumor necrosis factor-alpha involves the transcription factor NF-kappaB. J Biol Chem. 1996;271:26954–61. doi: 10.1074/jbc.271.43.26954. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–62. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. Embo J. 2003;22:5530–9. doi: 10.1093/emboj/cdg534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002a;298:1241–5. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Dittrich-Breiholz O, Holtmann H, Kracht M. Multiple control of interleukin-8 gene expression. J Leukoc Biol. 2002b;72:847–55. [PubMed] [Google Scholar]
- Jang HD, Yoon K, Shin YJ, Kim J, Lee SY. PIAS3 Suppresses NF-{kappa}B-mediated Transcription by Interacting with the p65/RelA Subunit. J Biol Chem. 2004;279:24873–24880. doi: 10.1074/jbc.M313018200. [DOI] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Li S, Wang L, Berman MA, Zhang Y, Dorf ME. RNAi screen in mouse astrocytes identifies phosphatases that regulate NF-kappaB signaling. Mol Cell. 2006;24:497–509. doi: 10.1016/j.molcel.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu KP. Prolyl isomerase Pin1 as a molecular target for cancer diagnostics and therapeutics. Cancer Cell. 2003;4:175–80. doi: 10.1016/s1535-6108(03)00218-6. [DOI] [PubMed] [Google Scholar]
- Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–16. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- Lufei C, Koh TH, Uchida T, Cao X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene. 2007;26:7656–64. doi: 10.1038/sj.onc.1210567. [DOI] [PubMed] [Google Scholar]
- Nagai S, Washiyama K, Kurimoto M, Takaku A, Endo S, Kumanishi T. Aberrant nuclear factor-kappaB activity and its participation in the growth of human malignant astrocytoma. J Neurosurg. 2002;96:909–17. doi: 10.3171/jns.2002.96.5.0909. [DOI] [PubMed] [Google Scholar]
- Nozell S, Laver T, Moseley D, Nowoslawski L, Devos M, Atkinson GP, et al. The ING4 tumor suppressor attenuates NF-{kappa}B activity at the promoter of target genes. Mol Cell Biol. 2008;28:6632–45. doi: 10.1128/MCB.00697-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozell S, Laver T, Patel K, Benveniste EN. Mechanism of IFN-beta-mediated inhibition of IL-8 gene expression in astroglioma cells. J Immunol. 2006;177:822–30. doi: 10.4049/jimmunol.177.2.822. [DOI] [PubMed] [Google Scholar]
- Nozell S, Ma Z, Wilson C, Shah R, Benveniste EN. Class II major histocompatibility complex transactivator (CIITA) inhibits matrix metalloproteinase-9 gene expression. J Biol Chem. 2004;279:38577–89. doi: 10.1074/jbc.M403738200. [DOI] [PubMed] [Google Scholar]
- Pang R, Lee T, Man K, Poon R, Fan ST, Kwong YL, et al. PIN1 expression contributes to hepatic carcinogenesis. J Pathol. 2006;210:19–25. doi: 10.1002/path.2024. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25:6717–30. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- Raychaudhuri B, Han Y, Lu T, Vogelbaum MA. Aberrant constitutive activation of nuclear factor kappaB in glioblastoma multiforme drives invasive phenotype. J Neurooncol. 2007;85:39–47. doi: 10.1007/s11060-007-9390-7. [DOI] [PubMed] [Google Scholar]
- Ryo A, Hirai A, Nishi M, Liou YC, Perrem K, Lin SC, et al. A suppressive role of the prolyl isomerase Pin1 in cellular apoptosis mediated by the death-associated protein Daxx. J Biol Chem. 2007;282:36671–81. doi: 10.1074/jbc.M704145200. [DOI] [PubMed] [Google Scholar]
- Ryo A, Liou YC, Wulf G, Nakamura M, Lee SW, Lu KP. PIN1 is an E2F target gene essential for Neu/Ras-induced transformation of mammary epithelial cells. Mol Cell Biol. 2002;22:5281–95. doi: 10.1128/MCB.22.15.5281-5295.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, et al. Regulation of NF-kappaB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell. 2003;12:1413–26. doi: 10.1016/s1097-2765(03)00490-8. [DOI] [PubMed] [Google Scholar]
- Ryo A, Uemura H, Ishiguro H, Saitoh T, Yamaguchi A, Perrem K, et al. Stable suppression of tumorigenicity by Pin1-targeted RNA interference in prostate cancer. Clin Cancer Res. 2005;11:7523–31. doi: 10.1158/1078-0432.CCR-05-0457. [DOI] [PubMed] [Google Scholar]
- Saccani S, Pantano S, Natoli G. Two waves of nuclear factor kappaB recruitment to target promoters. J Exp Med. 2001;193:1351–9. doi: 10.1084/jem.193.12.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D, Shimamura T, Barbera S, Bejcek BE. NF-kappaB controls growth of glioblastomas/astrocytomas. Mol Cell Biochem. 2007;307:141–7. doi: 10.1007/s11010-007-9593-4. [DOI] [PubMed] [Google Scholar]
- Stewart LA. Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet. 2002;359:1011–8. doi: 10.1016/s0140-6736(02)08091-1. [DOI] [PubMed] [Google Scholar]
- Strieter RM, Burdick MD, Mestas J, Gomperts B, Keane MP, Belperio JA. Cancer CXC chemokine networks and tumour angiogenesis. Eur J Cancer. 2006;42:768–78. doi: 10.1016/j.ejca.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Stupp R, Dietrich PY, Kraljevic SO, Pica A, Maillard I, Maeder P, et al. Promising survival for patients with newly diagnosed glioblastoma multiforme treated with concomitant radiation plus temozolomide followed by adjuvant temozolomide. J Clin Onc. 2002;20:1375–1382. doi: 10.1200/JCO.2002.20.5.1375. [DOI] [PubMed] [Google Scholar]
- Vila-Carriles WH, Kovacs GG, Jovov B, Zhou ZH, Pahwa AK, Colby G, et al. Surface expression of ASIC2 inhibits the amiloride-sensitive current and migration of glioma cells. J Biol Chem. 2006;281:19220–32. doi: 10.1074/jbc.M603100200. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Kambe F, Cao X, Murakami R, Mitsuyama H, Nagaya T, et al. Inhibitory effects of cyclosporin A on calcium mobilization-dependent interleukin-8 expression and invasive potential of human glioblastoma U251MG cells. Oncogene. 2004;23:6924–6932. doi: 10.1038/sj.onc.1207778. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang W, Huang HJ, Liao WS, Fuller GN. Analysis of the activation status of Akt, NFkappaB, and Stat3 in human diffuse gliomas. Lab Invest. 2004;84:941–51. doi: 10.1038/labinvest.3700123. [DOI] [PubMed] [Google Scholar]
- Wulf GM, Ryo A, Wulf GG, Lee SW, Niu T, Petkova V, et al. Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. Embo J. 2001;20:3459–72. doi: 10.1093/emboj/20.13.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu YX, Manley JL. Pin1 modulates RNA polymerase II activity during the transcription cycle. Genes Dev. 2007;21:2950–62. doi: 10.1101/gad.1592807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Fukushima T, Hayashi S, Ikeda K, Tsugu H, Kimura H, et al. Correlation of the expression of nuclear factor-kappa B, tumor necrosis factor receptor type 1 (TNFR 1) and c-Myc with the clinical course in the treatment of malignant astrocytomas with recombinant mutant human tumor necrosis factor-alpha (TNF-SAM2) Anticancer Res. 2000;20:611–8. [PubMed] [Google Scholar]
- Yeh ES, Means AR. PIN1, the cell cycle and cancer. Nat Rev Cancer. 2007;7:381–8. doi: 10.1038/nrc2107. [DOI] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–36. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell. 1998;1:661–71. doi: 10.1016/s1097-2765(00)80066-0. [DOI] [PubMed] [Google Scholar]