

Abstract
Background
Congenital heart disease (CHD) is the leading non-infectious cause of death in infants. Monozygotic (MZ) twins share nearly all of their genetic variants before and after birth. Nevertheless, MZ twins are sometimes discordant for common complex diseases. The goal of this study is to identify genomic and epigenomic differences between a pair of twins discordant for a form of congenital heart disease, double outlet right ventricle (DORV).
Results
A monoamniotic monozygotic (MZ) twin pair discordant for DORV were subjected to genome-wide sequencing and methylation analysis. We identified few genomic differences but 1566 differentially methylated regions (DMRs) between the MZ twins. Twenty percent (312/1566) of the DMRs are located within 2 kb upstream of transcription start sites (TSS), containing 121 binding sites of transcription factors. Particularly, ZIC3 and NR2F2 are found to have hypermethylated promoters in both the diseased twin and additional patients suffering from DORV.
Conclusions
The results showed a high correlation between hypermethylated promoters at ZIC3 and NR2F2 and down-regulated gene expression levels of these two genes in patients with DORV compared to normal controls, providing new insight into the potential mechanism of this rare form of CHD.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4814-7) contains supplementary material, which is available to authorized users.
Keywords: CHD, MZ twins, ZIC3, NR2F2, DNA methylation, RRBS, WGS
Background
Congenital heart disease (CHD) is the leading non-infectious cause of death in infants. In Asia, CHD occurs in 9.3 per 1000 live births [1]. Double outlet right ventricle (DORV), defined when both great arteries originate from the morphological right ventricle in a heart, is a rare form of congenital heart disease, accounting for 1–3% of all CHD cases [2–4]. Multiple factors have been identified in contributing to the disease, of which both genetic and epigenetic changes and the interplay between them and the related environment play a key role in the pathogenesis [5, 6].
During the normal development of the heart, the outflow tract initially connects exclusively with the primitive right ventricle and must remodel to divide into a separate pulmonary artery and aorta; subsequently, there is continued remodeling to establish direct continuity from the left ventricle to the aorta [4]. In double outlet right ventricle, drainage of the left ventricle is commonly achieved through a ventricular septal defect (VSD) at different locations and with varying relations to the pulmonary and aortic outflow tract. An insufficient mitral valve and an atrial septal defect (ASD) can be found in DORV cases [3]. Since temporal and spatial expression of transcription factors (TFs) is a major determinant of cell lineage specification and patterning of the heart [7–10], mutations or expression dysregulation in them can impair cardiac development and lead to congenital heart malformations and dysfunction [7]. Changes of epigenetic modifications also affect the expression patterns of these TFs [11]. Environmental alterations or stresses including drugs may perturb the TFs-related transcriptional and epigenetic programs in the process of cardiac development and give rise to CHD [12–14].
Cytosine methylation, an epigenetic modification, is essential in mammalian development and particularly in cell-lineage specification [15, 16]. Although each individual’s genome is fixed throughout life and across cell types, epigenetic modifications are plastic and influence the temporal and spatial pattern of gene expression [14]. Cell type-specific DNA methylation patterns emerge during development and play a role in gene expression by directing chromatin activation or interfering with the binding of TFs [17]. Emerging evidence suggests that DNA methylation is responsive to both physical and social environments during pregnancy and early life [14, 18].
Monozygotic (MZ) twins share nearly all of their genetic variants before and after birth. Nevertheless, MZ twins are often discordant for common complex diseases, such as type 1 diabetes (T1D; 61%), type 2 diabetes (41%) [19], autism (58 to 60%) [20, 21], schizophrenia (58%), and different types of cancer (up to 16%) [22, 23]. These observations support the model that for many complex traits, genotype alone may not fully determine phenotypic variation, and the interplay between genes and environment needs to be considered and epigenetics has been proposed to be one of the main mediators of this interaction [24–26]. Therefore, disease-discordant MZ twin pairs provide an ideal model for examining epigenetic functions in diseases due to their shared genetic and environmental factors during the pregnancy [24]. Analysis of discordant MZ twins has been successfully used to study epigenetic mechanisms in aging, cancer, autoimmune disease, and psychiatric, neurological and other traits [20, 21, 23]. However, comprehensive analysis of genome-wide DNA methylation in a MZ twin pair discordant for double outlet right ventricle (DORV) is lacking.
In this study, we dissected the contributions of DNA methylation pattern to the pathogenesis of DORV through DNA methylation profiling at single nucleotide level by utilizing the whole-blood-DNA derived from a MZ twin pair discordant for DORV. The two years and two months old Chinese girl, given a diagnosis of DORV at birth, has a healthy MZ twin sister. Their parents have no past or family history of heart diseases and are not consanguineous. Thus, we hypothesize that genome-wide analysis in this phenotype-discordant twins will provide insights into genetic and epigenetic factors affecting normal heart development.
Results
Genome sequence differences in MZ twins discordant for DORV
We first tried to identify de novo genomic sequence variation through whole genome sequencing. DNA extracted from whole blood samples of the MZ twins discordant for DORV (septal-defect heart (D3) and normal heart (D4) was sequenced by Illumina HiSeq X-Ten (150 bp paired-end reads) and aligned to the hg19 reference human genome. The average sequencing depth of D3 and D4 were 31.9 and 28.5 respectively, and 4-fold coverage represented more than 91% of the hg19 human reference genome (Additional file 1: Table S1). Single-nucleotide variations (SNVs) and short insertions/deletions (InDels) were identified using SAMtools [27] and filtered by Varscan [28]. More than 99.9% SNVs shared between the MZ twins (Fig. 1a). Three hundred and sixteen SNVs and 114 InDels were identified specifically in D3. After filtering by the dbSNP (Version 138) database, forty-one SNVs and 71 InDels in D3 remained (Additional file 2: Table S2). There was one deletion located in non-coding RNA (ANKRD30BP2, pseudogene) and one synonymous SNV in the exon of DSPP gene, but none of the SNVs or InDels altered proteins (Fig. 1b & Additional file 2: Table S2). No pathogenic variation specific to D3 was detected when filtered by the Online Mendelian Inheritance in Man (OMIM) and Human Genetic Mutation Database (HGMD). Sequence variants in promoter of GATA4 and TBX1, which were closely related to VSD [29, 30], were found in both of our samples, indicating that the sequences variants in the promoters of these two transcription factor may not contribute to the pathogenesis of DORV in D3 (Additional file 3: Table S3).
Fig. 1.

Overview of whole genome sequencing. a The numbers of SNVs and InDels detected in D3 and D4, 99.9% loci shared between the MZ twins. b There are 1736 SNVs or InDels specific in the disease sample D3, of which only 430 loci showing high confidence confirmed by Varscan are regarded as potential de novo SNVs/InDels. All loci are classified into 7 categories (upstream, downstream, intergenic, exonic, intronic, UTR5, UTR3) according to the relative position of nearby genes. The pie plot shows the number of loci and proportion of each category. c Normalized copy number profiles of D3 and D4. Each point shows a 5 kb window (chromosome 1) of sequencing reads normalized by GC-content and mappability using Control-FREEC
Copy number variants (CNVs) are deletions or amplifications of DNA segments that arise from inappropriate chromatid recombination or segregation during cell division. As CNVs alter the gene expression dosage of contiguous genes, they may result in syndromic CHDs [31]. Using Control-FREEC [32], we detected 15 focal CNVs; however, these CNVs did not pass CNVnator confirmation criteria [33], suggesting the absence of bona fide CNVs (Fig. 1c; Additional file 4: Figure S1 & Additional file 5: Table S4). Structural variation (SV), typically a large insertion/deletion, inversion or translocation affecting a sequence length from 1 kb to 3 Mb, was not detected to be different between the two samples using the CREST software (Additional file 6: Figure S2). Together, the genomic analyses suggest that the DORV in D3 is not likely caused by genome alterations.
DNA methylation differences between the MZ twin pair
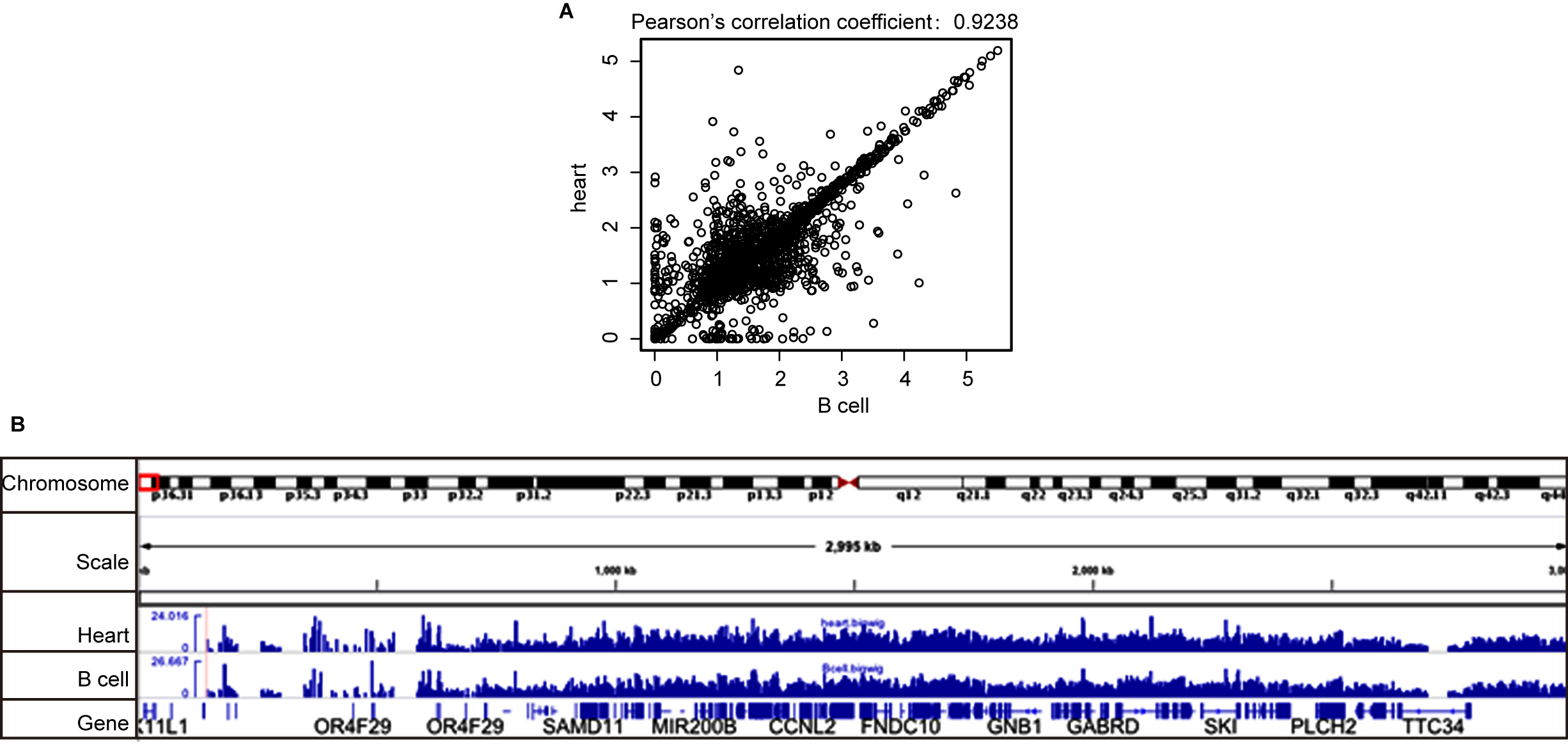
DNA methylation is involved in multiple processes, including regulation of gene expression, silencing of retrotransposons, genomic imprinting, X-chromosome inactivation and occurrence of various diseases [34, 35]. We next sought to compare genome-scale DNA methylation profiles between the MZ twins at nucleotide resolution. DNA samples were extracted from whole blood acquired before heart surgery, and gel-free reduced representation bisulfite sequencing (RRBS) libraries were constructed as previously reported [36] and then sequenced on Illumina HiSeq2000 platform (100 bp paired-end reads). After aligning the reads to the hg19 reference genome, we obtained 56 million and 48 million high-quality, 100-nucleotide, uniquely mapped reads from the D3 and D4 samples, respectively. The two samples had very similar sequencing-depth patterns of cytosine sites and covered more than 34 million C sites with sequencing depth at least 5 fold (Additional file 7: Table S5). More than 6 million high-quality CpG dinucleotides were supported by at least by 5 reads (depth ≥ 5×), which covered 10% of all CpG sites (Fig. 2a). In addition, we checked the methylation status of samples from blood cell and heart tissue in ENCODE project (Accession ID: ENCFF388NTJ, ENCFF107RMQ). We found a high correlation between B cell and heart with Pearson’s correlation coefficient equal to 0.9238, and visualizing the genome-wide methylation between them in IGV showed similar patterns (Additional file 8: Figure S3), suggesting that the DNA methylation pattern in blood correlate with that in heart.
Fig. 2.

Comparison of CpG methylomes between two samples. a Overall reads coverage of reduced representation bisulfite sequencing (RRBS) at CpGs between the twins. The RRBS strategy shows a high covered rate of CpGs in two samples. 10.88% and 10.72% of all human CpGs are sequenced by more than 5 reads (5×) in D3 and D4, respectively. b Overall distribution of CpGs’ methylated levels in D3 and D4. This violin plot shows that most CpGs are 100% methylated or 0% methylated, and the global methylation level is unchanged between the pair (Wilcoxon test non-significant). c Average CpGs’ methylation profile in gene body, upstream (− 2 kb of TSS) and downstream (+ 2 kb of TTS). d Circos plot shows the distribution of the DMRs along the genome. The tracks from outsider to inner: CpG islands downloaded from UCSC (green); the –log10 p-value of detected DMRs (blue); the average methylation levels per 1 kb in D3 (red); the average methylation levels per 1 kb in D4 (green). e Scatter diagram of all CpGs’ methylation levels calculated by RRBS data between two samples, D3 and D4, showing a similar overall CpGs methylation levels with Pearson correlation coefficient equal to 0.9613
The methylation levels of CpG dinucleotides in both samples showed a bimodal distribution with most CpG sites being unmethylated or extensively methylated (Fig. 2b). As expected, genome-wide patterns of DNA methylation (CpG and non-CpG) were highly correlated between the MZ twin pair (Figs. 2c-e & Additional file 9: Figure S4), suggesting that DORV is not associated with a global reprogramming of methylation.
Next, we sought to identify differentially methylated regions (DMRs) between the twin pair using a window sliding strategy to reduce the sampling variation of individual CpG sites (see Methods). We identified 1566 significant DMRs between the MZ twin pair (Fig. 3a). Hypermethylation and hypomethylation DMRs in D3 showed similar genomic distributions, with 80% (1254/1566) of the DMRs distributed in gene bodies or intergenic regions and 20% (312/1566) of the DMRs located within 2 kb upstream of TSS (Fig. 3b and Additional file 10: Table S6). We concluded that DMRs between the MZ twins may be involved in the pathogenesis of DORV.
Fig. 3.
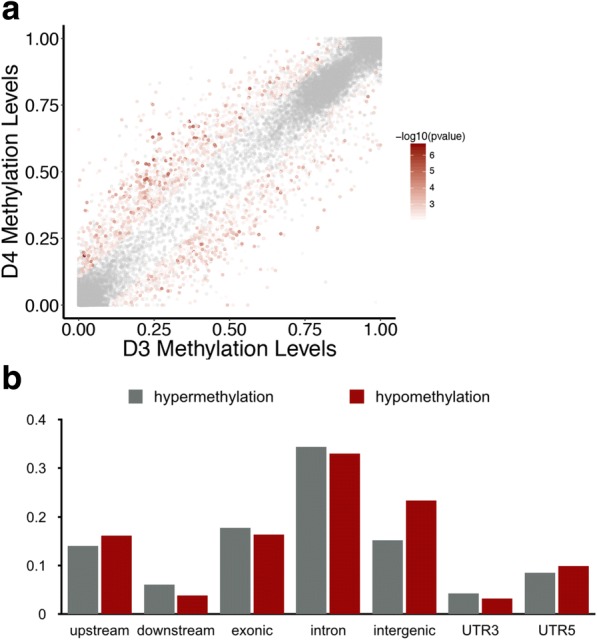
Distribution of D3 specific DMRs. a The scatter plot shows the found DMRs, the red color indicates the p-value, and non-significant regions are showed in gray. b The distribution of significantly DMRs. All DMRs are classified into 2 main categories (hypermethylation and hypomehtylation), and each category is further classified into 7 classes (upstream, downstream, intergenic, exonic, intronic, UTR5, UTR3) according to the relative position of nearby genes. The bar plot shows the proportion of each class. There are 294 genes that have a differentially methylated region in upstream (2 kb) of transcription start site
Gene ontology (GO) and transcription factor binding sites enriched in DMRs
We analyzed the genes that contained DMRs within 2 kb upstream of their TSS. In total, three hundred and twelve DMRs were located in the upstream of 621 TSSs (belonging to 294 genes annotated in Refseq) (Fig. 3b & Additional file 11: Table S7). These DMR-associated genes were then subjected to KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway and Gene Ontology (GO) analysis using the Enrichr and DAVID Web servers [37–39]. GO analysis revealed that genes associated with cardiolipin acyl-chain remodeling, cardiac muscle tissue regeneration, vascular function and organ growth are enriched in DMR-associated genes (Fig. 4a & Additional file 11: Table S7), suggesting that DMRs may be associated with the abnormal heart development of D3.
Fig. 4.

Gene Ontology (GO) and KEGG analysis of D3 specific DMRs. Gene Ontology (GO) and pathway enrichment analysis of differentially methylated regions using Enrichr and DAVID under default parameters. The bar charts show the most relevant and significantly enriched terms. Terms that are highly related to CHD are marked in blue. The x-axis represents the –log10 of the enrichment p-value. The y-axis represents the enriched terms in GO or KEGG databases. a GO enrichment analysis of genes associated with differentially methylated regions in 2 kb upstream of genes. b GO enrichment analysis of genes associated with differentially methylated regions in gene body. c GO enrichment analysis of TFs whose binding sites are differentially methylated. d KEGG pathway enrichment analysis of TFs whose binding sites are differentially methylated
We next analyzed the 851 genes containing DMRs in their gene bodies. These genes were also enriched in several biological processes that are involved in various stages of cardiovascular development (Fig. 4b and Additional file 12: Table S8). Furthermore, multiple signaling-pathways involved in heart development were also enriched, including regulation of BMP signaling and TGF beta receptor signaling (Fig. 4b and Additional file 12: Table S8). These results suggest that gene regulatory networks during the heart development of D3 may be affected by the DMRs.
Moreover, DMRs can influence gene expression through directly altering the binding of transcription factors to their targets. We analyzed the TF binding motifs in DMRs by using ANNOVAR (using tfbsConsSites database downloaded from UCSC). We identified 138 TFs (Additional file 10: Table S6) that belong to GATA family and NKX family, which are essential for cardiac development (Additional file 13: Table S9). GO analysis showed that many of these TFs were involved in heart development, muscle organ development, regulation of muscle cell differentiation, and blood vessel development (Fig. 4c and Additional file 13: Table S9). These TFs were also enriched in KEGG signaling pathways including MAPK signaling, Wnt signaling, TGF beta signaling, and VEGF signaling pathway (Fig. 4d and Additional file 13: Table S9), which were all involved in heart development [40–45]. Taken together, these results imply that DMRs in TFs binding sites may contribute to the DORV pathogenesis.
Aberrant promoter methylation of ZIC3 and NR2F2 in the diseased twin
Since promoter hypermethylation is typically associated with the repression of gene transcription [44], we then focused on promoter DMRs for further analysis. We found that DMRs existed in the upstream of multiple genes whose family members have been reported to be involved in pathogenesis of CHDs, and their normal functions are critical in morphogenesis and establishment of the cardiovascular system during cardiac development [46–48]. These genes include CITED1 (member of CREB-binding protein/p300-interacting transactivator with Asp/Glu-rich C-terminal domain (CITED) family of proteins, hypomethylated), GATA2 (member of the GATA family of zinc-finger TFs, hypermethylated), SOX3 (member of the SOX (SRY-related HMG-box) family of transcription factors, hypomethylated) (Additional file 14: Figure S5, Additional file 15: Figure S6 and Additional file 16: Figure S7).
Fig. 5.

Aberrant methylation in the upstream regions of ZIC3 and NR2F2, visualized in UCSC genome browser. DMRs are indicated by light blue bar; methylated levels in the twins are showed in blue (D3) and red (D4) bars. Transcription factor binding sites are also showed in zooming-in panels, which are indicated by black bars with names marked in front. An arrow gives TSS and transcriptional orientation. Two genes, ZIC3 (a) and NR2F2 (b), are showing differentially methylated in the upstream of TSS, and these two genes are known be associated with CHD. Transcription factor binding sites analyses are performed by ChIP-seq data of RNA Pol II and TBP derived from ENCODE. Bisulfite sequencing data (c and d) show methylation status of the same region of ZIC3 and NR2F2 as in (a) and (b). Methylated and unmethylated CpG sites are shown as black and white circles, respectively
We also found that genes encoding important epigenetic factors contained DMRs in their upstream of TSS, implying that these factors may play roles in causing CHDs through regulating expression of genes implicated in heart development. The genes include NSD1 (Nuclear Receptor Binding SET Domain Protein 1, which preferentially methylates ‘Lys-36’ of histone H3 and ‘Lys-20’ of histone H4, hypomethylated), MTA2 (Metastasis Associated 1 Family, Member 2, a component of NuRD, hypomethylated), MECP2 (Methyl CpG Binding Protein 2, hypermethylated) and SUV39H1 (Suppressor of Variegation 3–9 Homolog 1, a histone methyltransferase that trimethylates lysine 9 of histone H3, hypomethylated) (Additional file 17: Figure S8, Additional file 18: Figure S9, Additional file 19: Figure S10 and Additional file 20: Figure S11). We also utilized the public expression profiling data of embryo and adult heart from ENCODE (Accession ID: ENCFF704AHC, ENCFF199GQY, ENCFF987YOV) to analyze the possible contribution of these genes to DORV. We found that these genes were expressed in both embryo and heart (Additional file 21: Table S10), suggesting that these genes may play important roles in embryonic and cardiac development and dysregulated expression of them may contribute to CHD such as DORV.
Moreover, by scrutinizing all the DMRs located in gene promoter regions, we noticed that two genes, ZIC3 and NR2F2, encode TFs annotated with CHD in OMIM (300,265 and 107,773) [49–51]. In the DORV diseased twin D3, the upstream of ZIC3 (harboring P300 and HNF1 binding sites) was hypermethylated, corresponding to a region with high Pol II binding density in hESC cells (ENCODE data) (Fig. 5a and c). Similarly, hypermethylation of NR2F2 was detected in the upstream of the TSS (harboring an IRF2 binding site) of the shortest NR2F2 transcript variant, corresponding to a region with high intensity of TBP and Pol II binding in K562 cells (ENCODE data) (Fig. 5b and d). These results suggested a possible association of hypermethylated promoters of ZIC3 and NR2F2 and their functions during the heart development of DORV patients.
Hypermethylation and dysregulation of ZIC3 and NR2F2 in additional DORV patients
In order to further confirm the dysregulation of ZIC3 and NR2F2 in DORV pathogenesis, we collected twenty DNA samples of whole blood from normal individuals and clinical DORV patients. In order to guarantee the similarity in age and in gender ratio between the normal group and the patient cases, the samples included five controls with normal heart development (aged 0.8–3.8 years; 3 males, 2 females) and fifteen cases with DORV diagnosis (aged 1–3.5 years; 9 males, 6 females). Using bisulfite sequencing, we confirmed hypermethylated promoters of both ZIC3 (Fig. 6a and b) and NR2F2 (Fig. 6c and d) in twelve of fifteen DORV patients compared to normal subjects (Additional file 22: Figure S12, Additional file 23 Figure S13). The association of DORV and hypermethylated ZIC3 and NR2F2 promoters showed a significant correlation by Fisher’s exact test (p values are both 0.0036) (Fig. 6a and c). Correspondingly, samples harboring hypermethylated promoters of the two genes have comparatively lower gene expression levels (Fig. 6e and f), and the methylation and gene expression levels of these two genes were negatively correlated (Fig. 6g and h). These results confirm that lower gene expression levels of ZIC3 and NR2F2 are associated with promoter hypermethylation of these two genes in normal individuals and DORV patients.
Fig. 6.

DNA methylation and gene expression detection of ZIC3 and NR2F2 from clinical cases. (a and c) Statistical summaries about DNA methylation status of DMRs in ZIC3 and NR2F2 in 20 clinical samples, consisting of five normal providers and fifteen DORV patients. (b and d) Diagrams exhibiting average methylated levels of individual CpG sites in DMRs of ZIC3 and NR2F2 from the indicated groups, respectively. (e and f) Histograms showing relative gene expression levels of ZIC3 and NR2F2 in different groups of specimens. (g and h) Scatterplots showing the gene expression levels of ZIC3 (g) and NR2F2 (h) are negatively correlated with their promoter methylation status. Pearson’s correlation coefficient and p-values were listed above the plot
Discussion
This study represents the first analysis of genome and epigenome profiling of MZ twins discordant for DORV, and provides the evidence for the presence of epigenetic differences between the twin pair. Genetic variations between twins can affect proteins coding, gene transcription and epigenetic modifications [52]. We therefore first performed genomic sequence variation detection and revealed some differences between the twin pair, but stringent filtering analyses of CNVs, SNVs and short InDels failed to identify genomic differences that may contribute to pathogenic DORV. Even though sequence variants within the promoter regions of the GATA4 and TBX1 were reported to contribute to congenital heart disease by altering their gene expression [10, 29, 30], our results showed the sequence variants existed in both the normal and diseased twins, indicating that the pathogenesis of DORV may not be due to these differences. It is possible that the shared sequence variants showed different penetrance, which may cause the DORV in one of the MZ twins. Notably, sequences alignment in our study covered 91% of the hg19 human reference genome, leaving the rest of the genome not assessed. Therefore, we cannot rule out the possibility that genome sequence differences are involved in DORV of this twin pair.
Epigenetic variation at specific genomic regions has high heritability, and MZ twins typically share similar epigenetic profiles [25]. However, a set of factors including dietary components, physical changes, psychological states and environmental changes could affect epigenomes of the two years and two months old twins after birth [53]. In this study, many DMRs-related genes (within upstream or gene body) are enriched in pathways that contribute to cardiac development. Most importantly, we found that ZIC3 and NR2F2, which are annotated with CHD in OMIM database, were hypermethylated at the TSS upstream region of the diseased twin compared to the normal heart sample. ZIC3 is a member of the ZIC family of C2H2-type zinc finger proteins, which functions as a TF in early stages of left-right body axis formation and heart development [53]. ZIC3 acts in organizer formation by inhibiting the canonical Wnt signaling pathway, and its expression is regulated by determinants of the early neural fate specification and dorsal-ventral (D-V) axis formation, including BMP, FGF, and Nodal signaling pathways [54]. Mutations in ZIC3 result in heterotaxy or isolated CHD (phenotypes including DORV, ASD and VSD [49, 50]. In addition, the ZIC3 gene is located in X chromosome, so it may contribute to the pathogenesis of CHD in male and female differently. NR2F2 was identified to be a member of the steroid thyroid hormone superfamily of nuclear receptors, involving in the regulation of many different genes in development [55, 56]. In human cases and mouse models, NR2F2 has been crucially implicated in angiogenesis and heart development, and abnormal expression or depletion of NR2F2 leads to AVSD (atrioventricular septal defect) and VSD (ventricular septal defects) [51]. Consistently, we also found DMRs in BMP and Wnt signaling pathway-related genes, indicating that the regulation of these signaling pathways by ZIC3 or NR2F2 may be critical for normal heart development. Using additional normal and clinical DORV samples, we confirmed promoter hypermethylation of ZIC3 and NR2F2 in DORV patients and the anti-correlation between their methylation and gene expression. Taken together, aberrant methylation at promoter regions of ZIC3 and NR2F2 and their dysregulated gene transcription levels, may contribute to DORV in human heart development. However, the decisive conclusion needs further investigations, since epigenetic changes in blood may not be able to fully reflect the causative basis of the disease due to lack of more DORV cases in twins and difficulty in obtaining heart samples.
DMRs were also found in the upstream of CITED1, GATA2, SOX3 and some important epigenetic genes, including MTA2, NSD1, MECP2 and SUV39H1, indicating that they might also contribute to DORV. The MZ twins shared the similar but not exactly the same environment, especially the postnatal environment. Thus, we suggest that the non-shared environmental and stochastic factors, including physical changes, chemical pollutants, dietary components, temperature changes and other external stresses during pregnancy [6, 57, 58], may contribute to the pathogenesis of DORV through the mediation of epigenetic changes.
Conclusions
In conclusion, disease-discordant MZ twin pairs are outstanding subjects to study epigenetic mechanisms driving a number of pathologies. Here, using DNA methylation profiling technology to analyze genome-wide DNA methylation, we described differentially methylated regions in a DORV-discordant MZ twin pair. A limitation to our study is that we only obtained one MZ twin pair discordant for DORV, and the present results call for more DORV discordant twins and extensive tests for the generalizability of our findings. Nevertheless, our results provide new insights into the mechanism of DORV, a rare disease that has been less studied by genomic and epigenomic approaches.
Methods
Patients and materials
Genomic DNA was extracted from the donated whole blood samples of DORV patients and normal people by using the DNeasy Blood & Tissue Kit (Qiagen, Cat no. 69504). This study was conducted in accordance with the principles of the Declaration of Helsinki and has been reviewed and approved by the Medical Ethics Committee of Fuwai Hospital. Written informed consent was obtained from the twins’ parents and mentioned samples’ providers.
Reduced representation bisulfite sequencing (RRBS)
MspI-digested RRBS library was prepared as published [36], one hundred bp paired-end reads were generated from Illumina Hiseq2000 platform (BIOPIC, Peking University, Beijing). Raw reads were trimmed adapters and low quality bases using trim_galore in RRBS mode. Human genome (hg19) was indexed with bismark_genome_preparation (a script from bismark mapping package), and then, all clean reads aligned to indexed human genome using bismark (−-bowtie2). To extract the methylation information for individual cytosines, bismark_methylation_extractor (−p --cytosine_report --CX --no_overlap) in paired-end mode was applied, and the output CX_report file was sorted by chromosomes using linux shell commands (awk). The sorted CX_report files were then used for downstream analysis.
DNA methylation detection and quantitative RT-PCR
To monitor CpG methylation of screened DMRs in promoters, genomic DNA was treated with sodium bisulfite using EpiTect Bisulfite Kit (Qiagen, USA). The converted DNA was then amplified by PCR with specific primers (ZIC3: Forward: 5′-GAGTGATTGATTTTATTAGTTTAAGGATAT-3’Reverse: 5’-AACCAAAAAACTCCCTAAATACC-3′; NR2F2: Forward: 5′-GAAGTAGGAAAGGGTGGG-3’ Reverse: 5’-CGAACCCAAACTATTATCTAAC-3′), PCR products were purified, ligated into pEasy-T5 vector (Transgene, China) and then transduced into competent Escherichia.coli. When bacteria colonies appeared on the plate, at least 20 independent clones were selected and sequenced. The sequenced results were analyzed by BiQ analyzer (Max Plank Institute, Germany). Total RNA was extracted with RNAliquid Kit (Aidlab, China) and mRNA expression levels were detected with one-step RT-PCR kit (Takara, China) on lightcycler (Roche, USA). RT-qPCR primers were listed as follows: ZIC3: Forward: 5′- GGCGCTCAGTTTCCTAACTAC-3’Reverse: 5′- CTGCCGCATATAACGGAAGAA-3′; NR2F2: Forward: 5′- AACCAGCCGACGAGATTCG-3’ Reverse: 5′- CCCGGATGAGGGTTTCGATG-3′.
DMRs detection
Differentially methylated regions (DMRs) were detected based on a windows swiping method. We used a 100 bp window sliding on the genome at a 50 bp step to find differentially methylated windows (DMWs) between two samples, and the neighboring windows were joined together as a DMR. Only >10X CpG sites were used to calculate DMWs. To test the different average methylations in a window between two samples, Wilcoxon test was applied, and p-value < 0.01 was then considered as the DMW. A significant DMW was discarded if less than five CpG sites in the window and average methylation levels between two samples were less than 10%. Finally, adjacent DMWs were joined together as DMRs using BEDtools (intersectBed). DMRs were annotated by BEDtools (intersectBed) and ANNOVAR [59].
Gene ontology (GO) and pathway enrichment
Annotated DMRs were separated into 3 subsets: gene upstreams (2 kb in front of TSS), gene bodies and transcript factor binding sites. Genes which had DMRs in upstreams and gene bodies were submitted to Database for Enrichr and Annotation, Visualization and Integrated Discovery (DAVID) respectively, GO enrichments used GOTERM_BP_FAT database under default parameters, and pathway enrichments used Kyoto Encyclopedia of Genes and Genomes (KEGG) database. DMRs which annotated as transcript factors binding sites by ANNOVAR (using tfbsConsSites database downloaded from UCSC [60]) were considered to influence the TFs function, so we analyzed those TFs using DAVID. GO (biological process) and pathway enrichments were obtained to understand those DMRs’ biological meanings.
Additional files
Table S1. Overview of WGS data. (XLSX 39 kb)
Table S2. D3 specific SNVs and short InDels (XLSX 82 kb)
Table S3. Sequence variations reported related to VSD. (XLSX 30 kb)
Figure S1. Comparison of copy number profiles of 22 pairs of autochromosomes and X chromosome between twin pair. Normalized copy number profiles of D3 and D4. Each point shows a 5 kb windows (all chromosomes) of sequencing reads normalized by GC-content and map-ability using Control-FREEC. (PDF 1756 kb)
Table S4. Analysis of CNVs two samples. (XLSX 46 kb)
Figure S2. D3 specific structure variations (SVs) analysis. Alignments of reads in both D3 (top panel) and D4 (bottom panel) at each break points detected by CREST. The header lines in blue given the detail information of each structure variation (columns meanings: left_chr, left_pos, left_strand, number of left soft-clipped reads, right_chr, right_pos, right_strand, number right soft-clipped reads, SV type, coverage at left_pos, coverage at right_pos, assembled length at left_pos, assembled length at right_pos, average percent identity at left_pos, percent of non-unique mapping reads at left_pos, average percent identity at right_pos, percent of non-unique mapping reads at right_pos, start position of consensus mapping to genome, starting chromosome of consensus mapping, position of the genomic mapping of consensus starting position, end position of consensus mapping to genome, ending chromosome of consensus mapping, position of genomic mapping of consensus ending position, and consensus sequences). (PDF 626 kb)
Table S5. Overview of RRBS data. (XLS 26 kb)
{kind=link}
Figure S3. Analysis of WGBS data of B cell and heart tissue from the ENCODE project. (A) Scatter plot and Pearson’s correlation analysis of DNA methylation of B cell and heart. Pearson’s correlation coefficient was listed above the plot. (B) Visualization of the DNA methylation status of B cell and heart in chr1:1-3 M by IGV. (JPG 500 kb)
Figure S4. Comparison of systemic changes of methylome between two samples. (A) Cumulative depth distribution of RRBS data. The x-axis represents the depth of cytosine, and the y-axis represents the fraction of cytosine ≤ depth. (B) Methylation levels distribution in two samples of three different kinds of cytosine (CG, CHG, CHH). The x-axis is the methylation level; y-axis shows the log2 counts of the cytosine under a methylated level. (C) CHH methylation profile in the gene body, upstream and downstream. (D) CHG methylation profile in the gene body, upstream and downstream. (PDF 891 kb)
Table S6. DMRs between two samples. (XLSX 580 kb)
Table S7. GO and KEGG analysis of DMRs located in upstream region. (XLSX 43 kb)
Table S8. GO and KEGG analysis of DMRs located in gene body. (XLSX 99 kb)
Table S9. GO and KEGG analysis of DMRs located in TF binding sites. (XLSX 80 kb)
Figure S5. Aberrant methylation in the upstream regions of CITED1. Visualizing the methylation levels of DMRs near CITED1 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. Arrows give TSSs and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 910 kb)
Figure S6. Aberrant methylation in the upstream regions of GATA2. Visualizing the methylation levels of DMRs near GATA2 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 767 kb)
Figure S7. Aberrant methylation in the upstream regions of SOX3. Visualizing the methylation levels of DMRs near SOX3 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 590 kb)
Figure S8. Aberrant methylation in the upstream regions of NSD1. Visualizing the methylation levels of DMRs near NSD1 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 781 kb)
Figure S9. Aberrant methylation in the upstream regions of MTA2. Visualizing the methylation levels of DMRs near MTA2 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 633 kb)
Figure S10. Aberrant methylation in the upstream regions of MECP2. Visualizing the methylation levels of DMRs near MECP2 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. Arrows give TSSs and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 969 kb)
Figure S11. Aberrant methylation in the upstream regions of SUV39H1. Visualizing the methylation levels of DMRs near SUV39H1 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 748 kb)
Table S10. Expression profiling data of embryo and adult heart. (XLSX 9 kb)
Figure S12. DNA methylation detection of ZIC3 from clinical samples. Bisulfite sequencing tested DNA methylation status of DMRs in ZIC3 in 20 clinical samples, five normal providers (1–5) and fifteen DORV patients (6–20). Methylated and unmethylated CpG sites are indicated as respective black and white circles. (PDF 5226 kb)
Figure S13. DNA methylation detection of NR2F2 from clinical samples. Bisulfite sequencing detected DNA methylation status of DMRs in NR2F2 in 20 clinical samples, five normal providers (1–5) and fifteen DORV patients (6–20). Methylated and unmethylated CpG sites are indicated as black and white circles, respectively. (PDF 5463 kb)
Acknowledgements
We are grateful to participants for kindly providing us with clinical samples. We are thankful to Fuwai Hospital for taking care of the participants. We thank the Bioinformatics Core, School of Life Sciences, Peking University for providing bioinformatics analysis, and the reviewers for critical comments.
Funding
This work was supported by 2014 China Postdoctoral Science Foundation 55th General Financial Grant (No. 2014 M550007), 2013 Postdoctoral Fellowship of Peking University-Tshinghua University Center for Life Sciences, the National Natural Science Foundation of China (NSFC, Grant No. 31471205, 31671426 and 91219101) and the National Basic Research Program of China (973 Program, Grant No. 2010CB529500 and 2013CB530700). These funding bodies had no role in the design of the study, collection, analysis, and interpretation of data, nor in writing the manuscript.
Availability of data and materials
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Abbreviations
- ASD
Atrial septal defect
- CHD
Congenital heart disease
- CITED1
Cbp/p300 interacting transactivator with Glu/Asp rich carboxy-terminal domain 1
- CNVs
Copy number variations
- DAVID
Database for Annotation, Visualization and Integrated Discovery
- DMRs
Differentially methylated regions
- DMWs
Differentially Methylated Windows
- DORV
Double outlet right ventricle
- GATA2
GATA-binding protein 2
- GATA4
GATA-binding factor 4
- HGMD
Human Genetic Mutation Database
- HNF1
Hepatocyte nuclear factor 1
- InDels
Insertions/deletions
- IRF2
Interferon regulatory factor 2
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- MAPK
Mitogen activated protein kinase
- MECP2
Methyl CpG binding protein 2
- MTA2
Metastasis associated 1 family member 2
- MZ
Monozygotic
- NR2F2
Nuclear receptor subfamily 2 group F member 2
- NSD1
Nuclear receptor binding SET domain protein 1
- OMIM
Online Mendelian Inheritance in Man
- P300
Histone acetyltransferase p300
- Pol II
RNA polymerase II
- RRBS
Reduced representation bisulfite sequencing
- SNVs
Single nucleotide variations
- SOX3
SRY-box 3
- SUV39H1
Suppressor of variegation 3–9 homolog 1
- T1D
Type 1 diabetes
- TBP
TATA-box binding protein
- TBX1
T-box transcription factor 1
- TFs
Transcription factors
- TGF
Transforming growth factor
- TSS
Transcription start sites
- VEGF
Vascular endothelial growth factor
- VSD
Ventricular septal defect
- ZIC3
Zic family member 3
Authors’ contributions
GLL and TL designed and performed molecular experiments, CZ contributed to bioinformatics analysis, they contributed equally to this work; RL helped with collections of clinical blood samples, CL, LZ, YTG, XKH, LS, LH and LJZ contributed to data analysis; WT, CL and YN conceived the project and designed experiments; TL, GLL, CZ, CL and WT wrote the manuscript. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
This study was conducted in accordance with the principles of the Declaration of Helsinki and has been reviewed and approved by the Medical Ethics Committee of Fuwai Hospital (Approval no. 229). We confirm the informed written consent was obtained from all participants.
Consent for publication
For investigations involving clinical subjects, informed written consent has been obtained from the participants involved. The participants consented to publish all of the sequencing data.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Guoliang Lyu, Chao Zhang and Te Ling contributed equally to this work.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4814-7) contains supplementary material, which is available to authorized users.
Contributor Information
Guoliang Lyu, Email: guolianglyu@pku.edu.cn.
Chao Zhang, Email: zhangchao3@hotmail.com.
Te Ling, Email: teling@pku.edu.cn.
Rui Liu, Email: fuwailiurui@126.com.
Le Zong, Email: zongle@pku.edu.cn.
Yiting Guan, Email: yt_guan@pku.edu.cn.
Xiaoke Huang, Email: huangxk@pku.edu.cn.
Lei Sun, Email: sunlei7486@163.com.
Lijun Zhang, Email: lj_zhang@pku.edu.cn.
Cheng Li, Email: cheng_li@pku.edu.cn.
Yu Nie, Email: nieyuniverse@126.com.
Wei Tao, Email: weitao@pku.edu.cn.
References
- 1.Fahed AC, Gelb BD, Seidman J, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112(4):707–720. doi: 10.1161/CIRCRESAHA.112.300853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McMahon CJ, Breathnach C, Betts DR, Sharkey FH, Greally MT. De novo interstitial deletion 13q33. 3q34 in a male patient with double outlet right ventricle, microcephaly, dysmorphic craniofacial findings, and motor and developmental delay. Am J Med Genet A. 2015;167(5):1134–1141. doi: 10.1002/ajmg.a.36978. [DOI] [PubMed] [Google Scholar]
- 3.Hartge DR, Niemeyer L, Axt-Fliedner R, Krapp M, Gembruch U, Germer U, Weichert J. Prenatal detection and postnatal management of double outlet right ventricle (DORV) in 21 singleton pregnancies. J Matern Fetal Neonatal Med. 2012;25(1):58–63. doi: 10.3109/14767058.2011.561387. [DOI] [PubMed] [Google Scholar]
- 4.Obler D, Juraszek A, Smoot LB, Natowicz MR. Double outlet right ventricle: aetiologies and associations. J Med Genet. 2008;45(8):481–497. doi: 10.1136/jmg.2008.057984. [DOI] [PubMed] [Google Scholar]
- 5.Ordovás JM, Smith CE. Epigenetics and cardiovascular disease. Nat Rev Cardiol. 2010;7(9):510. doi: 10.1038/nrcardio.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vecoli C, Pulignani S, Foffa I, Grazia Andreassi M. Congenital heart disease: the crossroads of genetics, epigenetics and environment. Curr Genomics. 2014;15(5):390–399. doi: 10.2174/1389202915666140716175634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruneau BG. Signaling and transcriptional networks in heart development and regeneration. Cold Spring Harb Perspect Biol. 2013;5(3):a008292. doi: 10.1101/cshperspect.a008292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatcher CJ, Basson CT. Specification of the cardiac conduction system by transcription factors. Circ Res. 2009;105(7):620–630. doi: 10.1161/CIRCRESAHA.109.204123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kathiriya IS, Nora EP, Bruneau BG. Investigating the transcriptional control of cardiovascular development. Circ Res. 2015;116(4):700–714. doi: 10.1161/CIRCRESAHA.116.302832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stefanovic S, Christoffels VM. GATA-dependent transcriptional and epigenetic control of cardiac lineage specification and differentiation. Cell Mol Life Sci. 2015;72(20):3871–3881. doi: 10.1007/s00018-015-1974-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu L, Jin G, Zhou X. Modeling the relationship of epigenetic modifications to transcription factor binding. Nucleic Acids Res. 2015;43(8):3873–3885. doi: 10.1093/nar/gkv255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13(2):97. doi: 10.1038/nrg3142. [DOI] [PubMed] [Google Scholar]
- 13.Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123(19):2145–2156. doi: 10.1161/CIRCULATIONAHA.110.956839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szyf M. The early life social environment and DNA methylation: DNA methylation mediating the long-term impact of social environments early in life. Epigenetics. 2011;6(8):971–978. doi: 10.4161/epi.6.8.16793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 16.Hodges E, Molaro A, Dos Santos CO, Thekkat P, Song Q, Uren PJ, Park J, Butler J, Rafii S, McCombie WR. Directional DNA methylation changes and complex intermediate states accompany lineage specificity in the adult hematopoietic compartment. Mol Cell. 2011;44(1):17–28. doi: 10.1016/j.molcel.2011.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunner AL, Johnson DS, Kim SW, Valouev A, Reddy TE, Neff NF, Anton E, Medina C, Nguyen L, Chiao E. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009;19(6):1044–1056. doi: 10.1101/gr.088773.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanherkar RR, Bhatia-Dey N, Csoka AB. Epigenetics across the human lifespan. Frontiers in cell and developmental biology. 2014;2:49. doi: 10.3389/fcell.2014.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan W, Xia Y, Bell CG, Yet I, Ferreira T, Ward KJ, Gao F, Loomis AK, Hyde CL, Wu H. An integrated epigenomic analysis for type 2 diabetes susceptibility loci in monozygotic twins. Nat Commun. 2014;5:5719. doi: 10.1038/ncomms6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong C, Meaburn EL, Ronald A, Price T, Jeffries AR, Schalkwyk L, Plomin R, Mill J. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. 2014;19(4):495. doi: 10.1038/mp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arora M, Reichenberg A, Willfors C, Austin C, Gennings C, Berggren S, Lichtenstein P, Anckarsäter H, Tammimies K, Bölte S. Fetal and postnatal metal dysregulation in autism. Nat Commun. 2017;8:15493. doi: 10.1038/ncomms15493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caramori ML, Kim Y, Moore JH, Rich SS, Mychaleckyj JC, Kikyo N, Mauer M. Gene expression differences in skin fibroblasts in identical twins discordant for type 1 diabetes. Diabetes. 2012;61(3):739–744. doi: 10.2337/db11-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Castillo-Fernandez JE, Spector TD, Bell JT. Epigenetics of discordant monozygotic twins: implications for disease. Genome Med. 2014;6(7):60. doi: 10.1186/s13073-014-0060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bell JT, Saffery R. The value of twins in epigenetic epidemiology. Int J Epidemiol. 2012;41(1):140–150. doi: 10.1093/ije/dyr179. [DOI] [PubMed] [Google Scholar]
- 25.Bell JT, Spector TD. A twin approach to unraveling epigenetics. Trends Genet. 2011;27(3):116–125. doi: 10.1016/j.tig.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papadopoulos GK, Wijmenga C, Koning F. Interplay between genetics and the environment in the development of celiac disease: perspectives for a healthy life. J Clin Invest. 2001;108(9):1261–1266. doi: 10.1172/JCI14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25(17):2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomita-Mitchell A, Maslen C, Morris C, Garg V, Goldmuntz E. GATA4 sequence variants in patients with congenital heart disease. J Med Genet. 2007;44(12):779–783. doi: 10.1136/jmg.2007.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang H, Chen D, Ma L, Meng H, Liu Y, Xie W, Pang S, Yan B. Genetic analysis of the TBX1 gene promoter in ventricular septal defects. Mol Cell Biochem. 2012;370(1–2):53–58. doi: 10.1007/s11010-012-1397-5. [DOI] [PubMed] [Google Scholar]
- 31.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, Barillot E. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2011;28(3):423–425. doi: 10.1093/bioinformatics/btr670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–984. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oda M, Yamagiwa A, Yamamoto S, Nakayama T, Tsumura A, Sasaki H, Nakao K, Li E, Okano M. DNA methylation regulates long-range gene silencing of an X-linked homeobox gene cluster in a lineage-specific manner. Genes Dev. 2006;20(24):3382–3394. doi: 10.1101/gad.1470906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 36.Gu H, Smith ZD, Bock C, Boyle P, Gnirke A, Meissner A. Preparation of reduced representation bisulfite sequencing libraries for genome-scale DNA methylation profiling. Nat Protoc. 2011;6(4):468. doi: 10.1038/nprot.2010.190. [DOI] [PubMed] [Google Scholar]
- 37.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016;45(D1):D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(suppl_2):W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–W97. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Rath N, Hannenhalli S, Wang Z, Cappola T, Kimura S, Atochina-Vasserman E, Lu MM, Beers MF, Morrisey EE. GATA and Nkx factors synergistically regulate tissue-specific gene expression and development in vivo. Development. 2007;134(1):189–198. doi: 10.1242/dev.02720. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007;116(12):1413–1423. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rose BA, Force T, Wang Y. Mitogen-activated protein kinase signaling in the heart: angels versus demons in a heart-breaking tale. Physiol Rev. 2010;90(4):1507–1546. doi: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madonna R, De Caterina R. VEGF receptor switching in heart development and disease. Cardiovasc Res. 2009;84(1):4–6. doi: 10.1093/cvr/cvp270. [DOI] [PubMed] [Google Scholar]
- 44.Dor Y, Camenisch TD, Itin A, Fishman GI, McDonald JA, Carmeliet P, Keshet E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 2001;128(9):1531–1538. doi: 10.1242/dev.128.9.1531. [DOI] [PubMed] [Google Scholar]
- 45.Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V. Signaling via the Tgf-β type I receptor Alk5 in heart development. Dev Biol. 2008;322(1):208–218. doi: 10.1016/j.ydbio.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bamforth SD, Bragança J, Farthing CR, Schneider JE, Broadbent C, Michell AC, Clarke K, Neubauer S, Norris D, Brown NA. Cited2 controls left-right patterning and heart development through a nodal-Pitx2c pathway. Nat Genet. 2004;36(11):1189. doi: 10.1038/ng1446. [DOI] [PubMed] [Google Scholar]
- 47.Connelly JJ, Wang T, Cox JE, Haynes C, Wang L, Shah SH, Crosslin DR, Hale AB, Nelson S, Crossman DC. GATA2 is associated with familial early-onset coronary artery disease. PLoS Genet. 2006;2(8):e139. doi: 10.1371/journal.pgen.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Paul MH, Harvey RP, Wegner M, Sock E. Cardiac outflow tract development relies on the complex function of Sox4 and Sox11 in multiple cell types. Cell Mol Life Sci. 2014;71(15):2931–2945. doi: 10.1007/s00018-013-1523-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ware SM, Peng J, Zhu L, Fernbach S, Colicos S, Casey B, Towbin J, Belmont JW. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004;74(1):93–105. doi: 10.1086/380998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cowan J, Tariq M, Ware SM. Genetic and functional analyses of ZIC3 variants in congenital heart disease. Hum Mutat. 2014;35(1):66–75. doi: 10.1002/humu.22457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Al Turki S, Manickaraj AK, Mercer CL, Gerety SS, Hitz M-P, Lindsay S, D’Alessandro LC, Swaminathan GJ, Bentham J, Arndt A-K. Rare variants in NR2F2 cause congenital heart defects in humans. Am J Hum Genet. 2014;94(4):574–585. doi: 10.1016/j.ajhg.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X, Kuja-Halkola R, Rahman I, Arpegård J, Viktorin A, Karlsson R, Hägg S, Svensson P, Pedersen NL, Magnusson PK. Dominant genetic variation and missing heritability for human complex traits: insights from twin versus genome-wide common SNP models. Am J Hum Genet. 2015;97(5):708–714. doi: 10.1016/j.ajhg.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102(30):10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fujimi TJ, Hatayama M, Aruga J. Xenopus Zic3 controls notochord and organizer development through suppression of the Wnt/β-catenin signaling pathway. Dev Biol. 2012;361(2):220–231. doi: 10.1016/j.ydbio.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 55.Mendoza-Villarroel RE, Robert NM, Martin LJ, Brousseau C, Tremblay JJ. The nuclear receptor NR2F2 activates star expression and steroidogenesis in mouse MA-10 and MLTC-1 Leydig cells. Biol Reprod. 2014;91(1) [DOI] [PubMed]
- 56.Hubert MA, Sherritt SL, Bachurski CJ, Handwerger S. Involvement of transcription factor NR2F2 in human trophoblast differentiation. PLoS One. 2010;5(2):e9417. doi: 10.1371/journal.pone.0009417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cortessis VK, Thomas DC, Levine AJ, Breton CV, Mack TM, Siegmund KD, Haile RW, Laird PW. Environmental epigenetics: prospects for studying epigenetic mediation of exposure–response relationships. Hum Genet. 2012;131(10):1565–1589. doi: 10.1007/s00439-012-1189-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hogenson TL. Epigenetics as the underlying mechanism for monozygotic twin discordance. Medical Epigenetics. 2013;1(1):3–18. doi: 10.1159/000353688. [DOI] [Google Scholar]
- 59.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karolchik D, Hinrichs AS, Kent WJ: The UCSC genome browser. Current protocols in bioinformatics 2012, 40(1):1.4. 1–1.4. 33. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Overview of WGS data. (XLSX 39 kb)
Table S2. D3 specific SNVs and short InDels (XLSX 82 kb)
Table S3. Sequence variations reported related to VSD. (XLSX 30 kb)
Figure S1. Comparison of copy number profiles of 22 pairs of autochromosomes and X chromosome between twin pair. Normalized copy number profiles of D3 and D4. Each point shows a 5 kb windows (all chromosomes) of sequencing reads normalized by GC-content and map-ability using Control-FREEC. (PDF 1756 kb)
Table S4. Analysis of CNVs two samples. (XLSX 46 kb)
Figure S2. D3 specific structure variations (SVs) analysis. Alignments of reads in both D3 (top panel) and D4 (bottom panel) at each break points detected by CREST. The header lines in blue given the detail information of each structure variation (columns meanings: left_chr, left_pos, left_strand, number of left soft-clipped reads, right_chr, right_pos, right_strand, number right soft-clipped reads, SV type, coverage at left_pos, coverage at right_pos, assembled length at left_pos, assembled length at right_pos, average percent identity at left_pos, percent of non-unique mapping reads at left_pos, average percent identity at right_pos, percent of non-unique mapping reads at right_pos, start position of consensus mapping to genome, starting chromosome of consensus mapping, position of the genomic mapping of consensus starting position, end position of consensus mapping to genome, ending chromosome of consensus mapping, position of genomic mapping of consensus ending position, and consensus sequences). (PDF 626 kb)
Table S5. Overview of RRBS data. (XLS 26 kb)
Figure S3. Analysis of WGBS data of B cell and heart tissue from the ENCODE project. (A) Scatter plot and Pearson’s correlation analysis of DNA methylation of B cell and heart. Pearson’s correlation coefficient was listed above the plot. (B) Visualization of the DNA methylation status of B cell and heart in chr1:1-3 M by IGV. (JPG 500 kb)
Figure S4. Comparison of systemic changes of methylome between two samples. (A) Cumulative depth distribution of RRBS data. The x-axis represents the depth of cytosine, and the y-axis represents the fraction of cytosine ≤ depth. (B) Methylation levels distribution in two samples of three different kinds of cytosine (CG, CHG, CHH). The x-axis is the methylation level; y-axis shows the log2 counts of the cytosine under a methylated level. (C) CHH methylation profile in the gene body, upstream and downstream. (D) CHG methylation profile in the gene body, upstream and downstream. (PDF 891 kb)
Table S6. DMRs between two samples. (XLSX 580 kb)
Table S7. GO and KEGG analysis of DMRs located in upstream region. (XLSX 43 kb)
Table S8. GO and KEGG analysis of DMRs located in gene body. (XLSX 99 kb)
Table S9. GO and KEGG analysis of DMRs located in TF binding sites. (XLSX 80 kb)
Figure S5. Aberrant methylation in the upstream regions of CITED1. Visualizing the methylation levels of DMRs near CITED1 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. Arrows give TSSs and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 910 kb)
Figure S6. Aberrant methylation in the upstream regions of GATA2. Visualizing the methylation levels of DMRs near GATA2 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 767 kb)
Figure S7. Aberrant methylation in the upstream regions of SOX3. Visualizing the methylation levels of DMRs near SOX3 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 590 kb)
Figure S8. Aberrant methylation in the upstream regions of NSD1. Visualizing the methylation levels of DMRs near NSD1 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 781 kb)
Figure S9. Aberrant methylation in the upstream regions of MTA2. Visualizing the methylation levels of DMRs near MTA2 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 633 kb)
Figure S10. Aberrant methylation in the upstream regions of MECP2. Visualizing the methylation levels of DMRs near MECP2 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. Arrows give TSSs and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 969 kb)
Figure S11. Aberrant methylation in the upstream regions of SUV39H1. Visualizing the methylation levels of DMRs near SUV39H1 with UCSC genome browser. Methylated levels in the twins are showed in blue (D3) and red (D4). Transcription factor binding sites are also showed in zooming-in panels, which indicated by black bars with names marked in front. An arrow gives TSS and transcriptional orientation. Transcription factor binding sites, Pol II ChIP-seq and TBP ChIP-seq data from ENCODE. (PDF 748 kb)
Table S10. Expression profiling data of embryo and adult heart. (XLSX 9 kb)
Figure S12. DNA methylation detection of ZIC3 from clinical samples. Bisulfite sequencing tested DNA methylation status of DMRs in ZIC3 in 20 clinical samples, five normal providers (1–5) and fifteen DORV patients (6–20). Methylated and unmethylated CpG sites are indicated as respective black and white circles. (PDF 5226 kb)
Figure S13. DNA methylation detection of NR2F2 from clinical samples. Bisulfite sequencing detected DNA methylation status of DMRs in NR2F2 in 20 clinical samples, five normal providers (1–5) and fifteen DORV patients (6–20). Methylated and unmethylated CpG sites are indicated as black and white circles, respectively. (PDF 5463 kb)
Data Availability Statement
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.