

Abstract
Aim
Osteoarthritis (OA) is caused by complex interactions between genetic and environmental factors. Epigenetic mechanisms control the expression of genes and are likely to regulate the OA transcriptome. We performed integrative genomic analyses to define methylation-gene expression relationships in osteoarthritic cartilage.
Patients and Methods
Genome-wide DNA methylation profiling of articular cartilage from five patients with OA of the knee and five healthy controls was conducted using the Illumina Infinium HumanMethylation450 BeadChip (Illumina, San Diego, California). Other independent genome-wide mRNA expression profiles of articular cartilage from three patients with OA and three healthy controls were obtained from the Gene Expression Omnibus (GEO) database. Integrative pathway enrichment analysis of DNA methylation and mRNA expression profiles was performed using integrated analysis of cross-platform microarray and pathway software. Gene ontology (GO) analysis was conducted using the Database for Annotation, Visualization and Integrated Discovery (DAVID).
Results
We identified 1265 differentially methylated genes, of which 145 are associated with significant changes in gene expression, such as DLX5, NCOR2 and AXIN2 (all p-values of both DNA methylation and mRNA expression < 0.05). Pathway enrichment analysis identified 26 OA-associated pathways, such as mitogen-activated protein kinase (MAPK) signalling pathway (p = 6.25 × 10-4), phosphatidylinositol (PI) signalling system (p = 4.38 × 10-3), hypoxia-inducible factor 1 (HIF-1) signalling pathway (p = 8.63 × 10-3 pantothenate and coenzyme A (CoA) biosynthesis (p = 0.017), ErbB signalling pathway (p = 0.024), inositol phosphate (IP) metabolism (p = 0.025), and calcium signalling pathway (p = 0.032).
Conclusion
We identified a group of genes and biological pathwayswhich were significantly different in both DNA methylation and mRNA expression profiles between patients with OA and controls. These results may provide new clues for clarifying the mechanisms involved in the development of OA.
Cite this article: A. He, Y. Ning, Y. Wen, Y. Cai, K. Xu, Y. Cai, J. Han, L. Liu, Y. Du, X. Liang, P. Li, Q. Fan, J. Hao, X. Wang, X. Guo, T. Ma, F. Zhang. Use of integrative epigenetic and mRNA expression analyses to identify significantly changed genes and functional pathways in osteoarthritic cartilage. Bone Joint Res 2018;7:343–350. DOI: 10.1302/2046-3758.75.BJR-2017-0284.R1.
Keywords: Osteoarthritic cartilage, DNA methylation profiles, mRNA expression profiles, Integrative analysis
Article focus
We explored the relationship between DNA methylation and mRNA expression profiles of osteoarthritic cartilage from the entire genome perspective. To what extent are aberrant genes of DNA methylation consistent with those of mRNA expression profiles?
Which biological functions do identified significant common genes in our study involve?
Key messages
A total of 145 common aberrant genes for DNA methylation and mRNA expression profiles, and 26 pathways were detected for osteoarthritis. It has been suggested that some of the identified genes and pathways, such as DLX5 and mitogen-activated protein kinase signalling pathway, are associated with the development of osteoarthritis.
These results yielded new insights into the pathogenesis of osteoarthritis.
Strengths and limitations
This study was unique in that it combined genome-wide DNA methylation and gene expression profiles of osteoarthritic cartilage to identify significantly changed genes and functional pathways.
Considering the sample size, further biological studies are required to validate these findings.
The pathogenesis of osteoarthritis (OA) remains unclear. Many factors have been implicated in its development, including age, gender, obesity, physical activity and genetic factors.1-4 The prevalence of OA remains high due to the ageing population and rising levels of obesity,3 and its prevention and treatment involves a huge burden on publica health resources.5-7 High-throughout analysis, such as genomics and transcriptomics, has been used in pathogenetic studies of OA.8-11 For example, a gene expression study observed specific profiles in damaged and undamaged cartilage and determined the relevant genes and pathways in the progression of OA.10 A genome-wide DNA methylation study of osteoarthritic cartilage identified epigenetic dysregulations of a host of genes and pathways associated with OA.9 However, the molecular mechanisms of OA are very complicated, involving abnormal alterations of multilevel biological macromolecules. Therefore, single-omics studies cannot reveal the complicated pathogenesis of OA. In order to address this issue, multi-omics integrative analysis is currently receiving much attention12-14 and has been used in pathogenetic studies of many conditions.15-17
Moreover, as one of the major epigenetic modifications, the methylation of DNA plays an important role in the regulation of gene expression. Several authors have investigated the relationship between levels of gene expression and DNA methylation status in osteoarthritic chondrocytes.18-20 For example, downregulation of collagen type IX alpha 1 chain (COL9A1) expression was associated with hypermethylation in osteoarthritic cartilage.19 The loss of methylation in cytosine-phosphate-guanine (CpG) sites in the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) enhancer elements of inducible nitric oxide synthase (NOS) is responsible for gene induction in human articular chondrocytes.18 Nevertheless, previous studies only focused on specific genes, rather than the whole genome. Integrating genome-wide DNA methylation and mRNA expression profiles has the potential to detect more comprehensive aberrant changes related to OA.
In this study, we aimed to explore the relationships between DNA methylation and gene expression in osteoarthritic cartilage, and to identify significantly changed genes and functional pathways in patients with OA, hoping that the findings would provide new clues about the pathogenesis of OA.
Methods and Materials
Ten specimens of knee cartilage were used, five specimens from patients with primary OA who had undergone total knee arthroplasty (TKA) and five normal specimens from healthy patients who had undergone amputation following a road traffic accident. All patients were Han Chinese and were collected at the Xi'an Red Cross Hospital. They came from the Xi’an City of Shaanxi Province and were genetically unrelated. The basic characteristics of the patients for genome-wide DNA methylation profiling are shown in Table I. Nurse-administered questionnaires were used to obtain general clinical data, including ethnicity, lifestyle characteristics, health status and family and medical histories. Each patient underwent clinical and radiological examination. Primary OA of the knee was diagnosed according to the Western Ontario and McMaster Universities OA Index (WOMAC) for pain, stiffness, function, and clinical and radiological assessment.21 We excluded those who were obese or who had bone or cartilage disease, rheumatoid arthritis, or any other skeletal disorder.
Table I.
Basic characteristics of the patients for genome-wide DNA methylation profiling
| Osteoarthritis |
Normal |
||
|---|---|---|---|
| Gender | Age (yrs) | Gender | Age (yrs) |
| Female | 58 | Female | 47 |
| Female | 62 | Female | 62 |
| Male | 72 | Male | 55 |
| Male | 66 | Male | 46 |
| Male | 65 | Male | 45 |
The specimens of articular cartilage were harvested from the same anatomical areas of the femoral condyles. For genome-wide DNA methylation profiling, the specimens from the ten patients were dissected and rapidly frozen in liquid nitrogen. The DNA was then extracted using QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) following the standard protocol. Agarose gel electrophoresis was used to evaluate the quality of extracted DNA.
Genome-wide DNA methylation profiling
Genome-wide DNA methylation profiling of the articular cartilage from the specimens was performed using the Illumina Infinium HumanMethylation450 BeadChip (Illumina, San Diego, California), covering more than 485 000 sites of methylation across the entire genome. Bisulfite treatment of 500 ng DNA specimens was performed using the EZ DNA Methylation Kit (Zymo Research Corp., Irvine, California). The bisulfite-converted DNA specimens were then amplified, hybridized to HumanMethylation450 array, stained and washed following a standard protocol. The raw image intensities were scanned using the iScan SQ Scanner (iScan System, Illumina) and the data were processed by GenomeStudio Software (Illumina). The mean percentage of methylated cytosine at a given CpG site was expressed as a β value, varying from 0 (completely unmethylated) to 1 (completely methylated).
Differential methylation analysis
The empirical Bayes moderated t-test of limma package22 in Bioconductor was used to identify differentially-methylated CpG sites. The Benjamini-Hochberg method was used to calculate a false discovery rate (FDR) adjusted p-value for each CpG site. Significant sites were defined as: (1) FDR adjusted p-value (Padj) ⩽ 0.05; (2) β-value difference (Δβ)| ⩾ 0.2. The sites with missing values, or detection p-values > 0.05 in more than 90% of cartilage specimens were eliminated. These parameters are widely used quality control parameters for DNA methylation analysis, and have been used in previous methylation profile studies.23-25 As a result, 618 of the CpG probe sites were eliminated, which did not result in significant information missing.
mRNA expression profiles
Microarray data used here have been deposited in the National Centre for Biotechnology Information Gene Expression Omnibus (GEO) and are accessible through GEO series accession number GSE16464. Chondrocytes cultured in monolayer (passage 2) were subjected to gene expression profiling using genome-wide oligonucleotide microarrays HGU133plus2.0 (Affymetrix, Santa Clara, California) according to the manufacturer's recommendations. Detailed information about the patients, cell culture, and microarray analysis have been previously reported.26
Statistical analysis
Significantly common altered genes were screened by comparing genes with p-value < 0.05 identified by analysis of genome-wide DNA methylation profiling and mRNA expression profiles. The results of single gene analysis were further subjected to gene ontology (GO) enrichment analysis to identify OA-associated GO terms by means of the Database for Annotation, Visualization and Integrated Discovery 6.7. Integrative pathway enrichment analysis of DNA methylation and mRNA expression profiles was conducted by analysis of cross-platform microarray and pathway (InCroMAP) (Fig. 1). InCroMAP can perform pathway enrichment analysis using heterogeneous, cross-platform data sets. We used Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. Following the parameters recommended by the developers of InCroMAP, we used a threshold for each data set, that is, “p-value < 0.05” for DNA methylation profiles and “fold-change > 1.5” for mRNA expression profiles. A p-value was calculated by InCroMAP for each analyzed pathway.
Fig. 1.
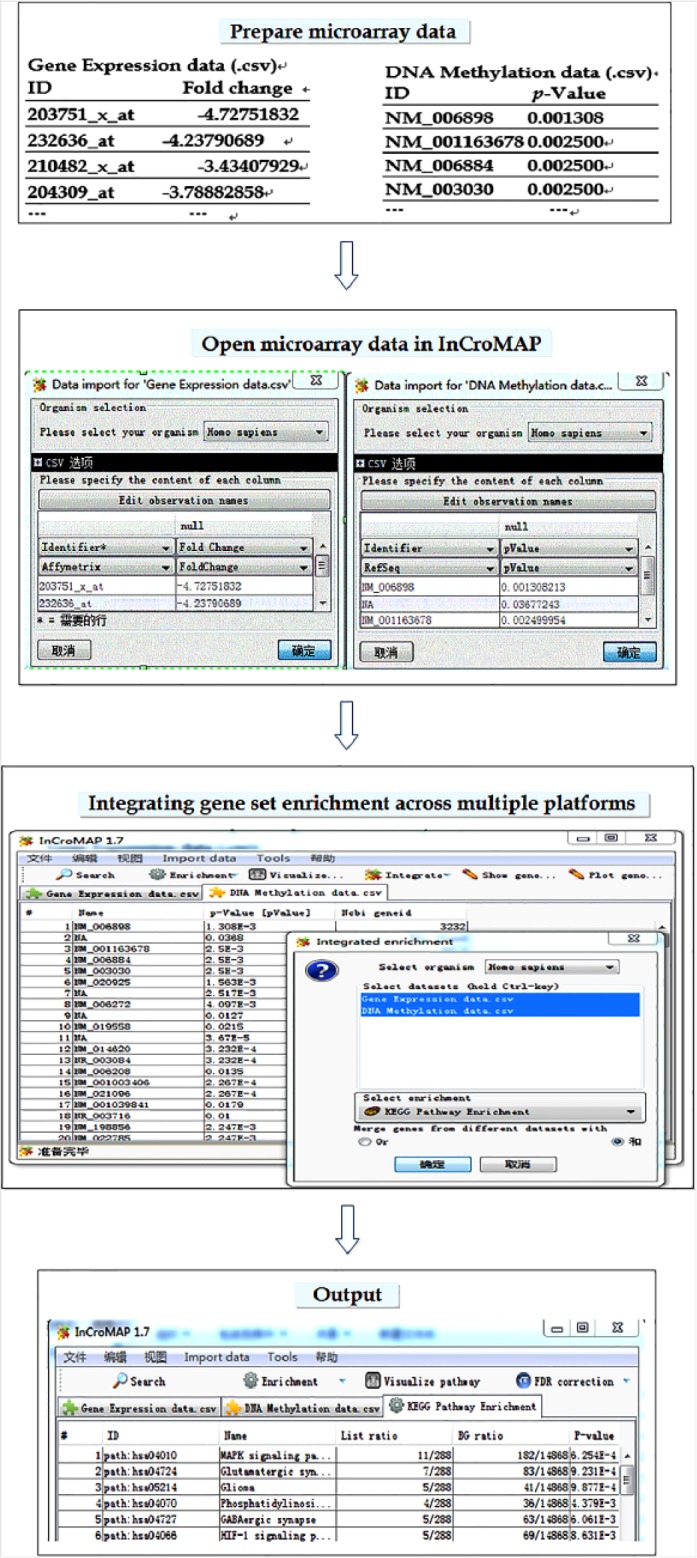
Integrative pathway enrichment analysis of DNA methylation and mRNA expression profiles using integrated analysis of cross-platform microarray and pathway (InCroMap).
Results
Single gene analysis
We identified 1265 differentially methylated genes, including 678 methylated and 587 demethylated genes. Integrating the DNA methylation of mRNA expression profiles detected 145 genes significantly altered in both (Supplementary Table i and Fig. 2). Among these, 48 were significantly hypermethylated and downregulated genes from the methylated group, and 24 were hypomethylated and upregulated genes the from the demethylated group.
Fig. 2.

A scatter plot of the 145 common aberrant genes of DNA methylation and mRNA expression profiles.
GO enrichment analysis
Further enrichment analysis of the 145 genes identified 127 significant GO terms for biological process, 11 significant GO terms for cell component, and 50 significant GO terms for molecular function, such as cytoskeleton organization (p-value = 1.47 × 10-11, false discovery rate (FDR) = 2.47×10-8), F-actin capping protein complex (p-value = 9.20 × 10-10, FDR = 1.20×10-6) and skeletal system development (p-value = 9.88 × 10-10, FDR = 1.72 × 10-5). More detailed results of GO enrichment analysis with FDR < 0.01 are shown in Supplementary Table ii.
Pathway enrichment analysis
KEGG pathway enrichment analysis identified 26 significant biological pathways (Supplementary Table iii), such as the mitogen-activated protein kinase (MAPK) signalling pathway (p = 6.25 × 10-4, Fig. 3), phosphatidylinositol (PI) signalling system (p = 4.38 × 10-3), hypoxia-inducible factor 1 (HIF-1) signalling pathway (p = 8.63 × 10-3), pantothenate and coenzyme A (CoA) biosynthesis (p = 0.017), ErbB signalling pathway (p = 0.024), inositol phosphate (IP) metabolism (p = 0.025), and calcium signalling pathway (p = 0.032).
Fig. 3.

Diagram of the mitogen-activated protein kinase signalling pathway. Rectangular nodes correspond to genes or gene families and their colour indicates mRNA expression (blue means downregulated, red means upregulated). The box to the left of those nodes indicates promoter DNA methylation. The largest peak is shown and an empty box indicates no differential methylation. The bar to the left indicates hypomethylation and the bar which goes from the middle of the box to the right indicates hypermethylation. Small rectangular boxes below the mRNA nodes indicate protein and protein modification expression. Each of those boxes is red, which indicates upregulation.
Discussion
mRNA expression profiling is often used to identify disease-related genes and gene sets. DNA methylation can regulate gene expression by recruiting proteins involved in gene repression or by inhibiting the binding of transcription factor(s) to DNA. Through integrative analysis of genome-wide DNA methylation profiles and mRNA expression profiles of osteoarthritic cartilage, we can learn more about the significant changes that occur between them, and how they play a role in the development of OA. In this study, we first identified 145 common aberrant genes in both DNA methylation and mRNA expression levels, and 26 associated biological pathways in osteoarthritic cartilage.
Some of the 145 common aberrant genes are thought to be associated with OA. For example, distal-less homeobox 5 (DLX5) is a transcription factor, which, it has been suggested, not only controls skeletal development, but also participates in ectopic chondrocyte hypertrophy during the development of OA.27 Furthermore, overexpression of DLX5 in chicken or mouse cartilage promoted chondrocyte hypertrophy, while DLX5 deficiency resulted in delayed hypertrophy.28-30 It was also found that DLX5 could induce the expression of runt related transcription factor 2 (RUNX-2) in animal models by means of specifically trans-activating the RUNX-2 P1 promoter,31 and thus influence matrix metallopeptidase 13 (MMP-13) expression in osteoarthritic chondrocytes,32 which is associated with abnormal gene expression and cartilage degeneration processes connected with OA. Nuclear receptor corepressor 2 (NCOR2), a transcription factor, was significantly associated with three OA-related traits (presence/absence of joint space narrowing, presence/absence of osteophytes, and Kellgren-Lawrence score).33 In this study, we observed that NCOR2 was significantly hypermethylated and downregulated in osteoarthritic cartilage, which was consistent with a previous study.34 The mechanism of NCOR2 implicated in the pathogenesis of OA is largely unknown and needs to be studied in greater detail. Axis Inhibition Protein 2 (AXIN2) is another interesting gene identified in this study. Previous authors observed that disruption of AXIN2 expression resulted in the accelerated maturation of chondrocytes.35 Furthermore, AXIN2 in the Wnt pathway was upregulated in osteoarthritic bone, and involved not only in the distortion of cartilage, but also in changes in the subchondral bone.36
GO enrichment analysis of 145 common affected genes also identified a group of OA-related GO terms, functionally involved in apoptosis, the development of the skeletal system and ossification. Previous authors reported similar results.37-40 Embryonic organ and skeletal system morphogenesis have been reported to be major pathways involved in the development of OA.37 Several biomolecules participated in the progress of apoptosis and embryonic skeletal growth of osteoarthritic chondrocytes. For example, insulin-like growth factor 1 (IGF-1) may downregulate the expression of programmed cell death 5 (PDCD5) and thus inhibit apoptosis of osteoarthritic chondrocytes.41 casein kinase 2 (CK2) downregulation facilitates TNF-a-mediated chondrocyte death through apoptosis and autophagy.42 Hypoxia-inducible factor 2 alpha (HIF-2α) was essential for the endochondral ossification of cultured chondrocytes and embryonic skeletal growth in mice.43
Integrative pathway enrichment analysis identified several significant functional pathways for OA, of which the MAPK signalling pathway is the most significant. The MAPK cascade is a highly conserved module that is involved in various cellular functions, including proliferation, differentiation and migration. Specifically, inhibition of the p38-MAPK signalling pathway could suppress the apoptosis and expression of proinflammatory cytokines in human osteoarthritic chondrocytes.44 This pathway is implicated in the production of matrix metalloproteinases and the regulation of biological responses of apoptosis, fatalization, calcification and proliferation of cartilage cells in OA.45,46 Matrix metalloproteinases (MMPs), in particular, can accelerate the degradation of articular cartilage.45
The PI signalling system belongs to a class of environmental information processing or signal transduction and includes various molecular and signalling pathways, some of which have been shown to be related to the development of OA. For examplee, it has been shown that the disruption of phosphoinositide-specific phospholipase γ1 (PLCγ1) contributes to the synthesis of extracellular matrix (ECM) in human osteoarthritic chondrocytes through regulating the expression of ECM-related signalling molecules, including matrix metalloproteinase 13 (MMP-13), type II collagen (Col II), tissue inhibitor of metalloproteinase 1 (TIMP1), sex determining region Y- box 9 (Sox-9), and a negatively charged proteoglycan (AGG). Furthermore, the PLCγ1/IP3/Ca2+/CaMK II signalling axis regulated the ECM synthesis of human chondrocytes by triggering the mTOR/P70S6K/S6 pathway.47 Another study found that through the phosphoinositide 3‑kinase (PI3K)/AKT and extracellular signal‑regulated kinase 1/2 pathway, osteopontin (OPN) could upregulate the expression of vascular endothelial growth factor (VEGF).48 The expression of OPN and VEGF were associated with the severity of the destruction of osteoarthritic cartilage.49-51 Based on previous results and our own, we suspect that the PI signalling system is involved in the development of OA. More functional studies are needed to confirm our findings.
Hypoxia-inducible factor 1 is a transcription factor that functions as a master regulator of oxygen homeostasis. Despite its name, HIF-1 is induced not only in response to the reduced availability of oxygen, but also by other stimulants, such as nitric oxide or various growth factors. It exhibits protective activity in chondrocytes during the development of OA.52 It has been shown that HIF-1 is important for the formation and maintenance of cartilage tissue in conditional knockout mice, in which deletion of the oxygen-sensitive subunit HIF-1α severely interfered with skeletal development and led to massive cell death within the centre of the forming cartilaginous elements.53 However, HIF-1α and -2α exert differing, even opposite, effects in chondrocytes and cartilage. For example, HIF-2α acts as a catabolic factor in the destruction of cartilage by regulating the expression of target genes in chondrocytes.54 Our results further confirm the importance of HIF-1 signalling in the development of OA.
We used five patients with OA and five healthy controls for genome-wide DNA methylation profiling. This sample size is relatively small. Articular cartilage specimens were collected from patients with advanced OA who had undergone TKA, and from healthy controls who had undergone amputation following a road traffic accident. Thus, it was difficult to use a large sample for DNA methylation profiling. However, the sample size is generally acceptable for microarray-based studies and has been used in recent studies on OA.55-57 The DNA methylation and mRNA expression profiles were also derived from different cartilage specimens. This may affect the results, which should be interpreted with caution. Further studies are needed to confirm our findings, and reveal the potential roles of identified genes and pathways in the development of OA.
In summary, we conducted an integrative analysis of genome-wide DNA methylation and gene expression profiles of osteoarthritic cartilage. Compared with a single-omics study, an integration study can provide more detailed information to detect common defective functional genes or pathways related to OA, especially those in which genes are low-expressed because of underlying DNA methylation, or over-expressed due to DNA demethylation. In addition, not all of these DNA methylations are correlated with altered mRNA transcription.58,59 Further biological studies are required to validate these results.
Acknowledgments
The authors wish to thank the Gene Expression Omnibus database for making their processed data publicly available. This study was approved by the Human Ethics Committees of Xi’an Jiaotong University. Inform-consent documents were signed by all participants.
F. Zhang takes responsibility for the integrity of the work as a whole, from inception to the finished manuscript.
Footnotes
Author Contributions: A. He, Collection and assembly of data, Analysis and interpretation of the data, Drafting and final approval of the manuscript.
Y. Ning, Collection and assembly of data, Drafting and final approval of the manuscript.
Y. Wen, Collection and assembly of data.
Y. Cai, Collection and assembly of data.
K. Xu, Collection and assembly of data.
Y. Cai, Collection and assembly of data.
J. Han, Drafting and final approval of the manuscript.
L. Liu, Collection and assembly of data.
Y. Du, Collection and assembly of data.
X. Liang, Collection and assembly of data.
P. Li, Analysis and interpretation of data.
Q. Fan, Analysis and interpretation of data.
J. Hao, Analysis and interpretation of data.
X. Wang, Drafting and final approval of the manuscript.
X. Guo, Conception and design of the study.
T. Ma, Drafting and final approval of the manuscript.
F. Zhang, Contributed to conception and design, drafting and final approval of the manuscript.
Conflict of Interest Statement: None declared
Follow us @BoneJointRes
Supplementary material
Tables providing further information on 145 significantly common changed genes and their gene ontology enrichment analysis results, together with integrative pathway enrichment results of DNA methylation and mRNA expression profiles by integrated analysis of cross-platform microarray and pathway.
Funding Statement
The study was supported by the National Natural Scientific Fund of China (81472925, 81673112), and the Special Key Project for Intergovernmental Innovation and Cooperation of National Key Research and Development Plan (2016YFE0119100), and the Science and Technology Research and Development Program of in Shaanxi Province of China (2013KJXX-51).
References
- 1. Chapman K, Valdes AM. Genetic factors in OA pathogenesis. Bone 2012;51:258-264. [DOI] [PubMed] [Google Scholar]
- 2. Felson DT, Lawrence RC, Dieppe PA, et al. Osteoarthritis: new insights. Part 1: the disease and its risk factors. Ann Intern Med 2000;133:635-646. [DOI] [PubMed] [Google Scholar]
- 3. Neogi T, Zhang Y. Epidemiology of osteoarthritis. Rheum Dis Clin North Am 2013;39:1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yuan Y, Zhang GQ, Chai W, et al. Silencing of microRNA-138-5p promotes IL-1β-induced cartilage degradation in human chondrocytes by targeting FOXC1: miR-138 promotes cartilage degradation. Bone Joint Res 2016;5:523-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Felson DT, Lawrence RC, Hochberg MC, et al. Osteoarthritis: new insights. Part 2: treatment approaches. Ann Intern Med 2000;133:726-737. [DOI] [PubMed] [Google Scholar]
- 6. Papandony MC, Chou L, Seneviwickrama M, et al. Patients’ perceived health service needs for osteoarthritis (OA) care: a scoping systematic review. Osteoarthritis Cartilage 2017;25:1010-1025. [DOI] [PubMed] [Google Scholar]
- 7. Yelin E, Murphy L, Cisternas MG, et al. Medical care expenditures and earnings losses among persons with arthritis and other rheumatic conditions in 2003, and comparisons with 1997. Arthritis Rheum 2007;56:1397-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fernández-Tajes J, Soto-Hermida A, Vázquez-Mosquera ME, et al. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann Rheum Dis 2014;73:668-677. [DOI] [PubMed] [Google Scholar]
- 9. Jeffries MA, Donica M, Baker LW, et al. Genome-wide DNA methylation study identifies significant epigenomic changes in osteoarthritic cartilage. Arthritis Rheumatol 2014;66:2804-2815. [DOI] [PubMed] [Google Scholar]
- 10. Snelling S, Rout R, Davidson R, et al. A gene expression study of normal and damaged cartilage in anteromedial gonarthrosis, a phenotype of osteoarthritis. Osteoarthritis Cartilage 2014;22:334-343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Fukui N, Yahata M, et al. Genome-wide DNA methylation profile implicates potential cartilage regeneration at the late stage of knee osteoarthritis. Osteoarthritis Cartilage 2016;24:835-843. [DOI] [PubMed] [Google Scholar]
- 12. Bonnet E, Calzone L, Michoel T. Integrative multi-omics module network inference with Lemon-Tree. PLOS Comput Biol 2015;11:e1003983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fernandez-Banet J, Esposito A, Coffin S, et al. OASIS: web-based platform for exploring cancer multi-omics data. Nat Methods 2016;13:9-10. [DOI] [PubMed] [Google Scholar]
- 14. Kim M, Rai N, Zorraquino V, Tagkopoulos I. Multi-omics integration accurately predicts cellular state in unexplored conditions for Escherichia coli. Nat Commun 2016;7:13090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Castro-Vega LJ, Letouzé E, Burnichon N, et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun 2015;6:6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen L, Ge B, Casale FP, et al. Genetic Drivers of Epigenetic and Transcriptional Variation in Human Immune Cells. Cell 2016;167:1398-1414.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thaiss CA, Levy M, Korem T, et al. Microbiota Diurnal Rhythmicity Programs Host Transcriptome Oscillations. Cell 2016;167:1495-1510.e12. [DOI] [PubMed] [Google Scholar]
- 18. de Andrés MC, Imagawa K, Hashimoto K, et al. Loss of methylation in CpG sites in the NF-κB enhancer elements of inducible nitric oxide synthase is responsible for gene induction in human articular chondrocytes. Arthritis Rheum 2013;65:732-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Imagawa K, de Andrés MC, Hashimoto K, et al. Association of reduced type IX collagen gene expression in human osteoarthritic chondrocytes with epigenetic silencing by DNA hypermethylation. Arthritis Rheumatol 2014;66:3040-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takahashi A, de Andrés MC, Hashimoto K, et al. DNA methylation of the RUNX2 P1 promoter mediates MMP13 transcription in chondrocytes. Sci Rep 2017;7:7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stucki G, Sangha O, Stucki S, et al. Comparison of the WOMAC (Western Ontario and McMaster Universities) osteoarthritis index and a self-report format of the self-administered Lequesne-Algofunctional index in patients with knee and hip osteoarthritis. Osteoarthritis Cartilage 1998;6:79-86. [DOI] [PubMed] [Google Scholar]
- 22. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hernandez-Vargas H, Lambert MP, Le Calvez-Kelm F, et al. Hepatocellular carcinoma displays distinct DNA methylation signatures with potential as clinical predictors. PLoS One 2010;5:e9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Marsit CJ, Christensen BC, Houseman EA, et al. Epigenetic profiling reveals etiologically distinct patterns of DNA methylation in head and neck squamous cell carcinoma. Carcinogenesis 2009;30:416-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang R, Pfütze K, Zucknick M, et al. DNA methylation array analyses identified breast cancer-associated HYAL2 methylation in peripheral blood. Int J Cancer 2015;136:1845-1855. [DOI] [PubMed] [Google Scholar]
- 26. Dehne T, Karlsson C, Ringe J, Sittinger M, Lindahl A. Chondrogenic differentiation potential of osteoarthritic chondrocytes and their possible use in matrix-associated autologous chondrocyte transplantation. Arthritis Res Ther 2009;11:R133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Solomon LA, Bérubé NG, Beier F. Transcriptional regulators of chondrocyte hypertrophy. Birth Defects Res C Embryo Today 2008;84:123-130. [DOI] [PubMed] [Google Scholar]
- 28. Bendall AJ, Hu G, Levi G, Abate-Shen C. Dlx5 regulates chondrocyte differentiation at multiple stages. Int J Dev Biol 2003;47:335-344. [PubMed] [Google Scholar]
- 29. Chin HJ, Fisher MC, Li Y, et al. Studies on the role of Dlx5 in regulation of chondrocyte differentiation during endochondral ossification in the developing mouse limb. Dev Growth Differ 2007;49:515-521. [DOI] [PubMed] [Google Scholar]
- 30. Ferrari D, Kosher RA. Dlx5 is a positive regulator of chondrocyte differentiation during endochondral ossification. Dev Biol 2002;252:257-270. [DOI] [PubMed] [Google Scholar]
- 31. Lee MH, Kim YJ, Yoon WJ, et al. Dlx5 specifically regulates Runx2 type II expression by binding to homeodomain-response elements in the Runx2 distal promoter. J Biol Chem 2005;280:35579-35587. [DOI] [PubMed] [Google Scholar]
- 32. Orfanidou T, Iliopoulos D, Malizos KN, Tsezou A. Involvement of SOX-9 and FGF-23 in RUNX-2 regulation in osteoarthritic chondrocytes. J Cell Mol Med 2009;13:3186-3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Valdes AM, Hart DJ, Jones KA, et al. Association study of candidate genes for the prevalence and progression of knee osteoarthritis. Arthritis Rheum 2004;50:2497-2507. [DOI] [PubMed] [Google Scholar]
- 34. Alvarez-Garcia O, Fisch KM, Wineinger NE, et al. Increased DNA Methylation and Reduced Expression of Transcription Factors in Human Osteoarthritis Cartilage. Arthritis Rheumatol 2016;68:1876-1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dao DY, Yang X, Flick LM, et al. Axin2 regulates chondrocyte maturation and axial skeletal development. J Orthop Res 2010;28:89-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Velasco J, Zarrabeitia MT, Prieto JR, et al. Wnt pathway genes in osteoporosis and osteoarthritis: differential expression and genetic association study. Osteoporos Int 2010;21:109-118. [DOI] [PubMed] [Google Scholar]
- 37. Aref-Eshghi E, Zhang Y, Liu M, et al. Genome-wide DNA methylation study of hip and knee cartilage reveals embryonic organ and skeletal system morphogenesis as major pathways involved in osteoarthritis. BMC Musculoskelet Disord 2015;16:287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Frank S, Peters MA, Wehmeyer C, et al. Regulation of matrixmetalloproteinase-3 and matrixmetalloproteinase-13 by SUMO-2/3 through the transcription factor NF-κB. Ann Rheum Dis 2013;72:1874-1881. [DOI] [PubMed] [Google Scholar]
- 39. Yang B, Kang X, Xing Y, et al. Effect of microRNA-145 on IL-1β-induced cartilage degradation in human chondrocytes. FEBS Lett 2014;588:2344-2352. [DOI] [PubMed] [Google Scholar]
- 40. Rufino AT1, Rosa SC, Judas F, et al. Expression and function of K(ATP) channels in normal and osteoarthritic human chondrocytes: possible role in glucose sensing. J Cell Biochem 2013;114:1879-1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yi C, Ma C, Xie Z, et al. Down-regulation of programmed cell death 5 by insulin-like growth factor 1 in osteoarthritis chondrocytes. Int Orthop 2013;37:937-943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee SW, Song YS, Lee SY, et al. Downregulation of protein kinase CK2 activity facilitates tumor necrosis factor-α-mediated chondrocyte death through apoptosis and autophagy. PLoS One 2011;6:e19163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Saito T, Fukai A, Mabuchi A, et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat Med 2010;16:678-686. [DOI] [PubMed] [Google Scholar]
- 44. Sun H-Y, Hu K-Z, Yin Z-S. Inhibition of the p38-MAPK signaling pathway suppresses the apoptosis and expression of proinflammatory cytokines in human osteoarthritis chondrocytes. Cytokine 2017;90:135-143. [DOI] [PubMed] [Google Scholar]
- 45. Gao S-C, Yin H-B, Liu H-X, Sui YH. Research progress on MAPK signal pathway in the pathogenesis of osteoarthritis. Zhongguo Gu Shang 2014;27:441-444. (Article in Chinese) [PubMed] [Google Scholar]
- 46. Huang D, Ding Y, Luo W-M, et al. Inhibition of MAPK kinase signaling pathways suppressed renal cell carcinoma growth and angiogenesis in vivo. Cancer Res 2008;68:81-88. [DOI] [PubMed] [Google Scholar]
- 47. Zeng G, Cui X, Liu Z, et al. Disruption of phosphoinositide-specific phospholipases Cγ1 contributes to extracellular matrix synthesis of human osteoarthritis chondrocytes. Int J Mol Sci 2014;15:13236-13246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xu J, Yi Y, Li L, Zhang W, Wang J. Osteopontin induces vascular endothelial growth factor expression in articular cartilage through PI3K/AKT and ERK1/2 signaling. Mol Med Rep 2015;12:4708-4712. [DOI] [PubMed] [Google Scholar]
- 49. Gao SG, Li KH, Zeng KB, et al. Elevated osteopontin level of synovial fluid and articular cartilage is associated with disease severity in knee osteoarthritis patients. Osteoarthritis Cartilage 2010;18:82-87. [DOI] [PubMed] [Google Scholar]
- 50. Jansen H, Meffert RH, Birkenfeld F, Petersen W, Pufe T. Detection of vascular endothelial growth factor (VEGF) in moderate osteoarthritis in a rabbit model. Ann Anat 2012;194:452-456. [DOI] [PubMed] [Google Scholar]
- 51. Lingaraj K, Poh CK, Wang W. Vascular endothelial growth factor (VEGF) is expressed during articular cartilage growth and re-expressed in osteoarthritis. Ann Acad Med Singapore 2010;39:399-403. [PubMed] [Google Scholar]
- 52. Sartori-Cintra AR, Mara CS, Argolo DL, Coimbra IB. Regulation of hypoxia-inducible factor-1α (HIF-1α) expression by interleukin-1β (IL-1 β), insulin-like growth factors I (IGF-I) and II (IGF-II) in human osteoarthritic chondrocytes. Clinics (Sao Paulo) 2012;67:35-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schipani E, Ryan HE, Didrickson S, et al. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev 2001;15:2865-2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bohensky J, Terkhorn SP, Freeman TA, et al. Regulation of autophagy in human and murine cartilage: hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Rheum 2009;60:1406-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Díaz-Prado S, Cicione C, Muiños-López E, et al. Characterization of microRNA expression profiles in normal and osteoarthritic human chondrocytes. BMC Musculoskelet Disord 2012;13:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Glossop JR, Haworth KE, Emes RD, et al. DNA methylation profiling of synovial fluid FLS in rheumatoid arthritis reveals changes common with tissue-derived FLS. Epigenomics 2015;7:539-551. [DOI] [PubMed] [Google Scholar]
- 57. Taylor SEB, Li YH, Wong WH, Bhutani N. Genome-wide mapping of DNA hydroxymethylation in osteoarthritic chondrocytes. Arthritis Rheumatol 2015;67:2129-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Blair JD, Yuen RK, Lim BK, et al. Widespread DNA hypomethylation at gene enhancer regions in placentas associated with early-onset pre-eclampsia. Mol Hum Reprod 2013;19:697-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chu T, Bunce K, Shaw P, et al. Comprehensive analysis of preeclampsia-associated DNA methylation in the placenta. PLoS One 2014;9:e107318. [DOI] [PMC free article] [PubMed] [Google Scholar]