

Abstract
Background
Safety data is continuously evaluated throughout the life cycle of a medical product to accurately assess and characterize the risks associated with the product. The knowledge about a medical product’s safety profile continually evolves as safety data accumulate.
Methods
This paper discusses data sources and analysis considerations for safety signal detection after a medical product is approved for marketing. This manuscript is the second in a series of papers from the American Statistical Association Biopharmaceutical Section Safety Working Group.
Results
We share our recommendations for the statistical and graphical methodologies necessary to appropriately analyze, report, and interpret safety outcomes, and we discuss the advantages and disadvantages of safety data obtained from passive post-marketing surveillance systems compared to other sources.
Conclusions
Signal detection has traditionally relied on spontaneous reporting databases which have been available worldwide for decades. However, current regulatory guidelines and ease of reporting have increased the size of these databases exponentially over the last few years. With such large databases, data mining tools using disproportionality analysis and helpful graphics are often used to detect potential signals. Although the data sources have many limitations, analyses of these data have been successful at identifying safety signals post-marketing. Experience analyzing these dynamic data is useful in understanding the potential and limitations of analyses with new data sources such as social media, claims or electronic medical records data.
Keywords: Adverse events, Post-market Surveillance, Passive surveillance, Signal Detection, Data Mining
1 Introduction
Signal detection for safety is one of the oldest and systematically used post-market risk evaluations of medical products in the real-world setting. In this paper, medical product refers to a drug, biologic or a vaccine. At marketing approval, non-clinical and clinical data demonstrated that the medical product’s benefits outweighed its risks in the intended indicated population. Although non-clinical studies can be sufficient to support medical product safety prior to first use in humans, these studies are not predictive of all potential safety outcomes in humans. Moreover, clinical trials primarily designed to assess efficacy have limitations for safety evaluation. They are mainly powered for efficacy and are not typically designed to address specific safety questions. Patient population in these trials is usually a selected sample with some entry criteria excluding vulnerable patients such as pregnant women, children, the elderly or patients with severe comorbidities. These eligibility criteria may limit generalizability of the safety findings from the trials to the whole indicated population for the medical product. Another limitation relates to the low statistical power of finding a rare safety issue if it exists due to limited sample sizes and short trial duration. Thus, there is a need for post-marketing surveillance to monitor the safety of a medical product after it has been released on the market and potentially used by a large population.
The fundamental goal of safety surveillance is detection of any safety signals after a medical product has been on the market. A signal could be for an adverse event (AE) that was not previously known to be associated with a medical product or it could be for an AE that is known to be associated with a medical product but with a number of reported cases higher than expected in everyone or in a vulnerable population. This signal could come as a result of data mining of the literature or, as we discuss in this paper, data mining of passive surveillance systems maintained by regulatory agencies. Passive surveillance systems receive spontaneous reports from the public and manufacturers of possible safety outcomes or AEs observed in users or medical products.
Signal detection is one building block in post-market safety assessment. More specifically, if a signal is flagged with passive surveillance, this signal is evaluated and further refined with a literature review, a meta-analysis of published evidence, a newly designed epidemiological study or a randomized clinical trial. Any new safety data, including a post-market surveillance signal can lead to regulatory actions such as changing the label, issuing a safety communication, requiring implementation of a risk evaluation and mitigation program, or even pulling a medical product from the market if its benefits no longer outweigh its risks. Post-market signal detection is also one building block in the overall safety assessment of a new medical product throughout its development life-cycle. Because evaluating safety is important throughout the pre-market development culminating in the marketing application, discovering a safety signal that would completely change the benefit-risk profile for a newly marketed medical product is expected to be rare.
A few medical products have been withdrawn from the market because of AEs identified post-market that were unknown or not fully characterized when it was given marketing approval. This occurs mainly because the benefit no longer outweighs the updated risk information in the indicated population. Moreover, approval of use of any medical product on the basis of evidence from just a few thousand patients and short term trials is risky, as illustrated by the experience with cerivastatin and several other recently withdrawn drugs. Cerivastatin is a statin marketed in the late 1990s to lower cholesterol and to prevent cardiovascular disease. Post-marketing surveillance identified 52 deaths attributed to drug-related rhabdomyolysis that lead to kidney failure [1]. In addition, surveillance found 385 nonfatal cases of rhabdomyolysis, most of whom required hospitalization, among the estimated 700,000 users in the United States. The risk was found to be higher among patients who received the full dose (0.8 mg/day) and those who received gemfibrozil concomitantly. This put the risk of this rare complication at 5 to 10 times that of the other statins. On the basis of the finding of a markedly increased reporting rate of fatal rhabdomyolysis in association with cerivastatin, the drug manufacturer, with the concurrence of the FDA, withdrew cerivastatin from the U.S. market in August 2001 [2]. The cerivastatin experience supports the position that unexpected serious adverse events (SAEs) may not be detected until a large number of patients have been exposed for an extended period of time.
This manuscript is the second in a series of papers from the American Statistical Association (ASA) Biopharmaceutical Section Safety Working Group to examine various sources of safety data and the statistical strategies for appropriate design and analysis. First, we describe the data sources available for passive post-market surveillance of medical products. We also summarize the existing statistical methods for signal detection and visualization. Finally, we discuss the advantages and disadvantages of safety data obtained from passive post-marketing surveillance systems compared to other sources.
2 Data Sources for Surveillance
Post-marketing drug surveillance for AEs has typically relied on spontaneous reporting to surveillance systems that are usually maintained by a country’s or a region’s regulatory agency. We review some of these systems below. Note that the information in different systems is not identical but can substantially overlap. For example, the US reporting system collects reports for all products marketed in the US no matter where the product is used or where the event occurs. Thus, this surveillance system receives worldwide reports and the same spontaneous reports can be found in surveillance systems hosted in different regions.
2.1 Adverse Event Reporting Databases at the Food and Drug Administration (FDA): FAERS and VAERS
The main sources FDA uses for signal detection of safety events of medical products post-marketing are spontaneous reporting databases. Those are FDA Adverse Event Reporting System (FAERS) and the Vaccine Adverse Event Reporting System (VAERS). These databases contain information submitted to FDA on AEs for medical products (drugs and therapeutic biologic, or vaccines).
The reports are submitted by manufacturers, consumers, or healthcare professionals and they are either mandatory or voluntary. More specifically, several regulations mandate manufacturers to report AEs occurring anywhere in the world for medical products marketed in the US. However, reports from patients and medical professionals are voluntary and can be done through VAERS for vaccines and MedWatch program for drugs and non-vaccine biologics. While FDA is the sole sponsor for FAERS, VAERS is co-sponsored with the Center of Disease Control and Prevention.
Although FAERS and VAERS were implemented at different times and are governed by different sets of regulations, they share many similarities. They contain safety data on medical products for over 20 years and as far back as 1969 for some medical products. The systems have millions of reports to date and grow by at least 1.5 million reports a year across medical products[3]. The number of reports increased dramatically in the last decade for several reasons including increase use and availability of medical products, ease of voluntary reporting and mandatory reporting requirements for manufacturers. For example, FAERS received less than 500,000 reports in 2006 but over a million reports in 2014. Although the majority of the reports every year are from the US, about one third of the reports are from foreign sources.
The FDA makes some de-identified information on these reports freely available to the public on a regular basis, quarterly for FAERS [4] and monthly for VAERS [5]. AEs in these data are coded at the preferred term level using Medical Dictionary for Regulatory Activities (MedDRA)[6]. The report can also include demographic and administrative information, medical products information, patient outcome, and source information. Reporters of AEs fill this information in the individual case safety report form (ICSR) with instructions to follow standardized terminology recommended in the ICH E2BM specifications[7].
These freely available data files are not statistical analysis ready and users need to be familiar with creation of relational databases prior to use. Some free tools and several commercial software for data-mining include important data curating steps such as flagging duplicate records, and mapping standardized terminology versions over time as medical product dictionaries and MedDRA versions change. In an effort to make the data more user friendly to the public, FDA is developing several interactive, open-source applications for data mining and visualization of reports at OpenFDA[8].
For the past 10 years, the Food and Drug Administration has taken several initiatives to implement its new authority to regulate safety post-marketing (Food and Drug Administration Amendments Act 2007 (FDAAA) (ref Title IX, Section 915, Section 921 amends the Federal Food, Drug and Cosmetic Act (FDCA) to add new subsections to section 505 (21 U.S.C. 355)). Among those initiatives regarding safety surveillance is to post quarterly reports on the adverse event reporting system website of any new safety information on drugs or post safety evaluation 18 months after a new biologic product approval [9]. Any new safety information is also publically accessible for patients and providers on a website [10].
2.2 Adverse Event Reporting Databases at the European Medicines Agency (EMA): EudraVigilance and VAESCO
2.2.1 EMA EudraVigilance database
The European Medicines Agency (EMA) European Union Drug Regulating Authorities Pharmacovigilance (EudraVigilance)[11] is the European data processing network and management system for reporting and evaluation of suspected AEs during the development of new drug and after its marketing authorization in the European Economic Area (EEA). Using the system is mandatory for marketing authorization holders and sponsors for clinical trials. It has a fully automated safety and message-processing mechanism using XML-based messaging and a large pharmacovigilance database with query and tracking functions.
The EudraVigilance system deals with the electronic exchange of ICSR: a) EudraVigilance Clinical Trial Module (EVCTM) for reporting Suspected Unexpected Serious Adverse Reactions (SUSARs); b) EudraVigilance Post-Authorization Module (EVPM) for post-authorization ICSRs. It supports safe and effective use of medicines by facilitating the electronic exchange of ICSRs among EMA, national competent authorities, marketing authorization holders and sponsors of clinical trials in the EEA and early detection and evaluation of possible safety signals.
The EMA and national competent authorities are responsible for regularly reviewing and analyzing EudraVigilance data to detect safety signals. The Pharmacovigilance Risk Assessment Committee (PRAC) evaluates the safety signals detected in EudraVigilance and may recommend regulatory action as a result.
2.2.2 EU Vaccine Adverse Event Surveillance and Communication (VAESCO)
The European Union (EU) Vaccine Adverse Event Surveillance and Communication (VAESCO)[12] project aims to establish a European collaborative network of regulatory agencies, public health institutes and academia responsible and able to collect and collate information on AEs following immunization in Europe. VAESCO aims to provide information about the safety of immunizations using the most rigorous scientific basis possible, since safety concerns may lead to the modification of the pertinent recommendations or possibly the withdrawal of a product from the market should a safety signal be confirmed, or even loss of public confidence.
2.3 WHO safety database (VigiBase)
VigiBase[13] is a World Health Organization’s (WHO) global ICSR database that contains reports submitted by the participating member states enrolled under WHO’s international drug monitoring program. It is the single largest drug safety data repository in the world which includes reports from the data sources described earlier in this paper: FAERS, VAERS, EudraVigilance and VASCO. It is a computerized pharmacovigilance system that records information in a structured, hierarchical form to allow for easy and flexible retrieval and analysis of the data. The ICSR in the WHO database do not identify the patient or reporter. It includes linked databases containing medical and drug classifications such as the WHO-ART/MedDRA, WHO ICD, and WHO Drug Dictionary. The program now has 110 member countries from all parts of the world contributing ICSRs.
The rationale for bringing spontaneous reports into one international database was to enable the earliest possible detection of medical product related problems, and one of the primary tasks at the outset of the WHO program was to develop an international signaling system. The goal was to help prevent drug disasters like the devastating fetal malformations caused by thalidomide in the early 1960s.
2.4 New Data Sources for Signal Detection
In addition to the above mentioned databases of spontaneous reporting of AEs and or ADRs with use of drugs and vaccines there are other sources for drug safety surveillance, including disease registries and registries of patients treated with a particular medication, and electronic health data or secondhand databases for pharmacovigilance. These data sources are more traditionally used for epidemiological studies designed to answer pre-specified hypotheses and we refer to the third paper in this series “Sources of Safety Data and Statistical Strategies for Design and Analysis: Real World Insights” for a detailed review of these data sources.
FDA uses electronic medical records in vaccine data link (VSD) (started in 1990) and claims data in the Sentinel System (created under FDAAA in 2007) for active signal detection and routine review of new vaccines. One of the most prominent efforts is in vaccine safety and the work of the Sentinel Post-Licensure Rapid Immunization System (PRISM)[13, 14] which led to incorporating use of signal detection from electronic health data in routine review work in the US. Similar work of active signal detection with these electronic data sources for drugs and biologic is in the pilot stages[15]. The Observational Medical Outcomes Partnership (OMOP), the Pharmacoepidemiological Research on Outcomes of Therapeutics by a European Consortium (PROTECT) and now Observational Health Data Sciences and Informatics (OHDSI) have also explored use of claims data and European registries for signal detection[16–18]. The same statistical methods for signal detection used in spontaneous reporting databases, discussed in the next section of this paper, have been applied to electronic health records. However, contrary to spontaneous reporting databases, electronic health records data can be used to derive prevalence rates (i.e. denominators exists from the same data source) or control for some confounding for causal inference.
3 Statistical Methods for Signal Detection and Visualization
3.1 Methods
Traditional drug safety surveillance uses large spontaneous reporting systems, which include thousands of drugs and AEs. In these databases, the proportion of reports for a particular drug that are linked to a specific AE and the same proportion for all other drugs in the database can be laid out as in a 2 × 2 table format. The commonly used disproportionality measures are the proportional reporting ratio (PRR) [19], reporting odds ratio (ROR) [20], and information component (IC) [21]. PRR is defined as the ratio of the reporting ratio of a drug for one AE divided by the reporting ratio of all other drugs in the database, while ROR is defined as the ratio between the odds of reporting a specific AE for a drug divided by the corresponding odds for all other drugs. IC is defined as the logarithm of the ratio of the observed rate of a specific AE to the expected rate of AE under the null hypothesis of no drug-AE association. PRR method has been implemented by UK Medicines and Healthcare Products Regulatory Agency.
One approach for generating signals with a disproportionality measures uses the normal approximation to derive confidence intervals for these measures. Then, a drug-AE pair signals when the lower bound of the confidence interval exceed a pre-specified threshold [19]. A second approach is to use chi-square tests to signal for those drug-AE pairs with p-value lower than a pre-specified significance level. A third approach to control family-wise type I error and avoid a large number of false signals is to use the likelihood ratio test (LRT) based method [22]. These LRT methods assume that the number of reports follows a Poisson distribution with mean proportional to the unknown reporting rate of the drug-AE pair. They use likelihood ratio measures (or the log-scale) to make inferences based on the empirical distribution [22–26].
In addition to frequentist approaches described above, there are many Bayesian methods. Among these, the two commonly used ones are Bayesian Component Propagation Neural Network (BCPNN) method and multi-item Gamma Poisson shrinkage (MGPS, or GPS) method. BCPNN computes IC and its interval estimates for each drug-AE combination. With a beta-binomial distribution prior, IC is estimated using the posterior mean and variance from a fully Bayesian model specification. Then, this method generates a signal when a credible interval for IC exceeds a pre-specified threshold [21, 27–32]. MGPS assumes that the number of reports for a particular drug-AE pair follows a Poisson distribution and that the Poisson means arise as a mixture of two gamma distributions. MGPS adopts an empirical Bayes approach to maximize the marginal likelihood and calculates two summary statistics: the geometric mean of the posterior distribution for each Empirical Bayes Geometric Mean (EBGM) or the fifth percentile of the posterior (EB05) [33–36]. EBGM can be seen as a ‘shrinkage’ estimate of the true relative risk for a particular drug-event combination. If the observed or expected number of AE of a drug is large, then EBGM will be close to the relative risk, but otherwise EBGM will be shrunk towards the null value of 1. These two methods have been implemented by the WHO Drug Monitoring Centre in Uppsala and FDA for pharmacovigilance surveillance.
With the increasing use of observational health care databases for real-time active surveillance, there are also several methods proposed for signal detections in those environments, which update the signals longitudinally at different looks as the evidence accumulates. Commonly used sequential methods include, maximum sequential probability ratio test (MaxSPRT) [15, 37–39], conditional sequential sampling procedure with stratification [40], and generalized estimating equation regression approach using permutation tests [41]. These signal detection methods, for passive and active surveillance, along with others, have been reviewed in several papers [42–47].
3.2 Data Visualization
Due to the large number of drug-event pairs possible in a post market setting, data visualization is the key to effective summary and communication of important safety signals[3]. Example figures below are generated using the 2016 fourth quarter FAERS data files, with effort taken to remove duplicate instances of drug names and events reported within Primary IDs [48]. Because these figures summarize a single quarter of data, any findings observed here should be viewed with caution. Preferred terms were merged with system organ classes (SOCs) from MedDRA 19.1.
A heat map (Figure 1) uses color to summarize the frequency of events for the twenty-five most reported drugs and the fifty most common gastrointestinal, psychiatric, and nervous system disorders, with darker red indicating a drug-event combination that occurs with greater frequency. Reddish rows highlight events that are common across drugs (headache, dizziness, vomiting, nausea, and diarrhea) while columns with numerous red cells exhibit an excess of numerous events (e.g. Otezla®, apremilast). The grouping of events along the y-axis by SOCs can further highlight body systems affected by particular drugs. Note that in lieu of the event frequency, other variables such as a particular disproportionality estimate or the lower limit of the 95% confidence or credible interval of that estimate can be summarized using a heat map. Heat maps can also be summarized by important covariates to assess their effect on signals.
Figure 1. Heat Map of Common Drugs and Disorders.
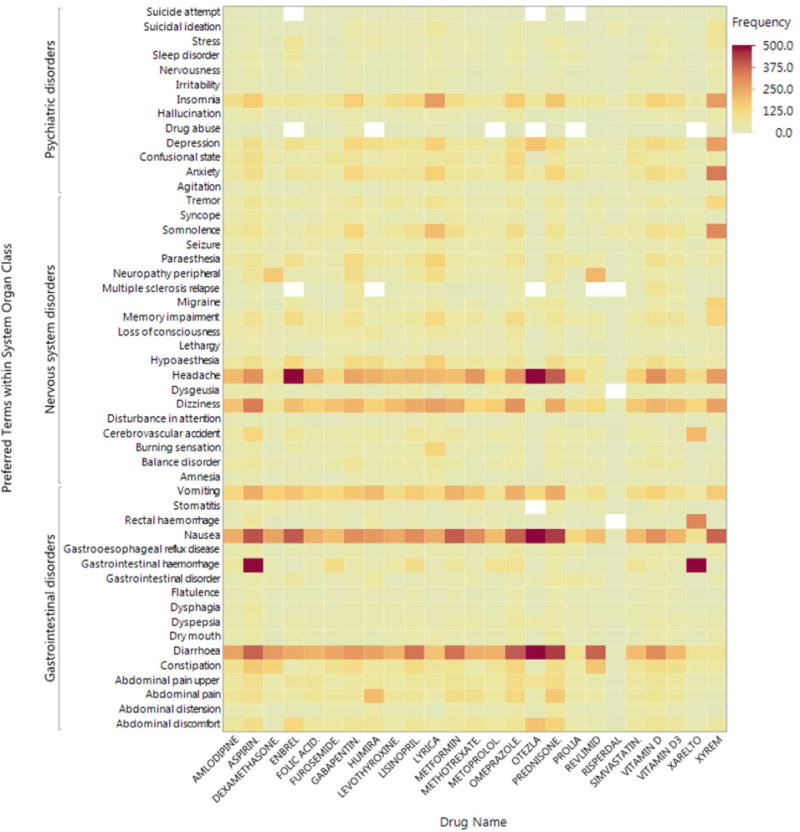
Figure 1 includes the twenty-five most reported drugs and the fifty most common gastrointestinal, psychiatric, and nervous system disorders from the 2016 fourth quarter FAERS data files [1]. Darker red indicates a greater frequency for a particular drug-event combination. Though 500 is listed as the maximum frequency in the legend, 6 drug-event combinations exceed 500, with a maximum of 1584 for gastrointestinal haemorrhage for Xarelto. The legend was truncated to provide greater contrast in the figure.
Tree maps can be used to summarize hierarchical relationships and use area and color to summarize numerical quantities. Figure 2 summarizes the 10 most reported nervous system disorders within the ten most reported drugs. Here, the size of each area is proportional to the frequency of drug-event pairs, and color is used to convey the magnitude of the ROR. For example, somnolence had a strong ROR and was frequently reported for Xyrem (sodium oxybate). In lieu of disproportionality estimates, the lower limits of the confidence or credible intervals of disproportionality measures could be summarized for a more conservative analysis. Covariates, such as gender, can be used as levels within the tree map hierarchy.
Figure 2. Tree Map of Common Drugs and Nervous System Disorders.
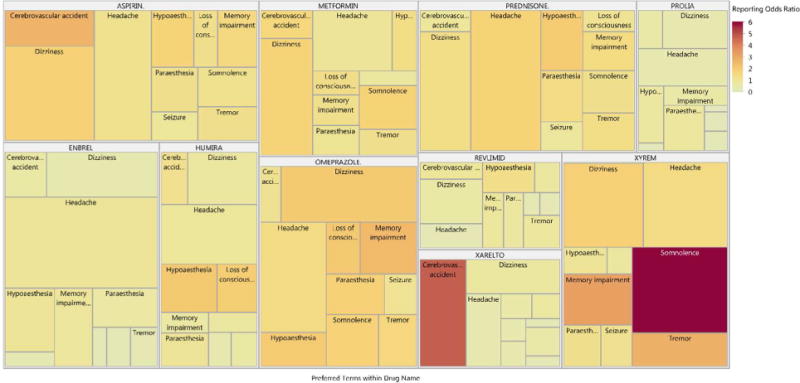
Figure 2 includes the ten most reported drugs and nervous system disorders from the 2016 fourth quarter FAERS data files [1]. Tree maps use area and color to display numerical quantities. In this example, the size of each area is proportional to the frequency of drug-event pairs, and color is used to convey the magnitude of the reporting odds ratio. Note that exceptionally long labels were suppressed for small cells to improve presentation.
Finally, network plots can be used to summarize the strength of relationships between events reported in the same Primary IDs. Figure 3 summarizes the 12 most common musculoskeletal and connective tissue disorders for Primary IDs listing Enbrel (etanercept), with bubbles sized according to the frequency of IDs reporting a particular event. The edges connect pairs of events where the pairwise odds ratio between events was considered significant according to unadjusted 95% confidence intervals. Larger odds ratios indicate a greater likelihood of events being reported together within a primary ID; arthralgia occurs frequently with joint swelling and musculoskeletal pain. In practice, bubbles can be colored to communicate other characteristics of the data, such as SOC for events occurring across numerous body systems or the magnitude of other covariates. In a similar manner, the co-occurrence of drugs could be summarized for primary IDs experiencing a particular event.
Figure 3. Network Plot of Common Musculoskeletal and Connective Tissue Disorders for Primary IDs listing Etanercept.
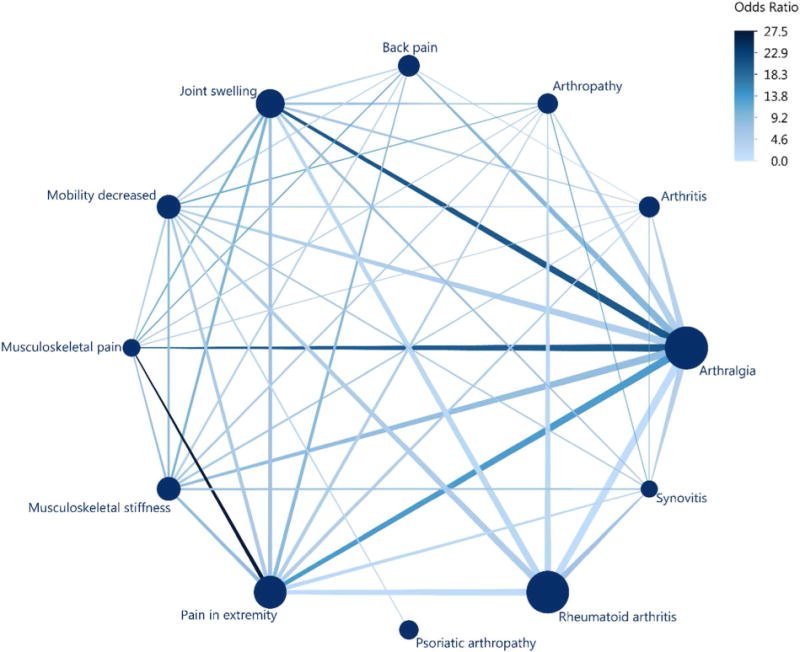
Figure 3 includes primary IDs listing Embrel (etanercept) for the 12 most common musculoskeletal and connective tissue disorders from the 2016 fourth quarter FAERS data files [1]. Bubbles sized according to the frequency of primary IDs reporting a particular event, ranging from 128 for synovitis to 931 for arthralgia. Edges connect pairs of events where the pairwise odds ratio between events was considered significant according to unadjusted 95% confidence intervals. Larger odds ratios indicate a greater likelihood of events being reported together within a primary ID.
4 Discussion
Although our paper focuses on spontaneous reports of adverse events for drugs, vaccines and other biologics, spontaneous reports exists for injury, death or malfunctions after use of a medical device. Spontaneous reports on devices are collected by MAUDE[49] and MedSun[50] at US FDA, EUDAMED [11, 51] in EU and is part of Vigibase in WHO. Although spontaneous reports for devices are regulated differently than for drugs and biologics, all systems share many similarities in data collection, strengths and limitations. The device systems do not encompass all devices or device related safety adverse events. In addition, use of data-mining methods discussed in this paper for devices is not as well established as for drugs, biologics and vaccines.
Post-market safety surveillance is a key component of lifecycle management of medical product safety. There are two main important advantages of surveillance system based on spontaneous reports: they potentially maintain ongoing surveillance of all patients and they are relatively inexpensive [52]. These data sources have been established worldwide and have been the primary source of regulatory post-marketing safety surveillance for decades. They collect data on patients exposed to the medical product, including those who would not normally be included in clinical trials. The wide coverage of these systems makes it possible to identify cluster of rare events that may be associated with the use of the product. These databases are especially well suited for detecting unexpected rare AEs with acute onset after exposure to medical product.
With the strengthened regulatory measures and increased awareness of importance of safety surveillance, the amount of AE reported to such systems has increased dramatically over recent years. They can include detailed narrative information about patients and medical products and use relatively standardized data capture and coding which make them easy to aggregate and mine for signals. Use of automated text mining methods in narratives, as in [53], could augment results of established quantitative disproportionality measures and further exploit the richness of the reports while reducing the time-intensive human review of narratives.
In addition to spontaneous reporting systems, other promising data sources are also evaluated for post-marketing safety monitoring. For example, electronic health data for drug safety risk assessment has substantial growth potential. The FDA’s Sentinel Initiative and OMOP and EMA’s 5-year project PROTECT were among the most visible activities. Leveraging information from social media for text mining and signal detection is also a growing area. However, using electronic health data or social media data for active safety surveillance is not standard regulatory practice and has practical limitations. It may require substantial financial resources to acquire and conduct systematic mining over time and a lot of work is needed to address the shortcomings in specificity, reliability, reproducibility, statistical standards and interpretability[54].
There are many limitations of surveillance system data mainly related to reporting bias and data quality. Spontaneous reports from patients and physicians are voluntary. Thus, reporters are neither a random sample nor a census of the population using medical products.
On one hand, underreporting of AE has been a major concern. For example, experts believe that FAERS includes an estimated 1 to 10 percent of experienced adverse reactions [55]. A recent study indicates that serious adverse drug events reporting rates in FAERS vary widely by indication, with reporting rates ranging from 0.7% for type 2 diabetes mellitus to 47.3% for multiple sclerosis [56]. Some AEs or usage errors that occur with a product can be underreported if the AE is expected or the product is well known and has been on the market for a long time. Conversely, underreporting could occur for latent or hidden AEs.
On the other hand, overreporting of AEs is also a concern for a newly marketed product less familiar to health practitioners or patients. News stories or public discussion of the AE or product can also stimulate reporting. Because of this reporting bias, as well as the lack of information on product utilization and/or the extent of consumption (i.e. a denominator), the spontaneous reporting system data cannot be used to calculate the incidence of an adverse event or usage error.
Although many reporting forms are standardized, data quality can still be an issue. Reporters may fill information inconsistently, may have difficulty with adverse event recognition, may mention only one product when they used multiple concomitant products and they either miss reporting important information or have uninformative narratives. The databases can also have multiple duplicates from the same report (e.g. one report from physician and another report from a manufacturer). Finally, reports do not require that a causal relationship between a product and event be proven and reports do not always contain enough details to properly evaluate causality. The inherent structure of spontaneous reporting systems means that no reference populations with same underlying condition but not being treated by the medical product could be constructed from such databases [57]. Thus, signals from spontaneous reports are most often hypothesis generating rather than confirmatory of a causal relationship between an AE and a drug. Most often, one needs supportive information from other data sources to establish causality (e.g. biologic link, findings from controlled studies).
Although spontaneous reporting systems have been available for decades, review of these reports was for a long time considered more qualitative than quantitative. It is not until recent years that the sheer sizes of the databases have made it possible to use sophisticated statistical tools for data mining. Data mining algorithms enable a systematic screening for safety signals. However, they may result in a large number of statistical signals that require additional evaluation, which could be resource intensive. Many of these data mining methods are used by manufacturers and in regulatory practice; their advantages and limitations were compared in many review papers including the work of these authors [3, 47, 58–60]. Work is still underway to make criteria to sift through the signals more efficient, determine which signals need further evaluation, and level of evidence needed for updating labeling or communication of risk to patients and practitioners.
The same statistical methods used for data mining AE spontaneous reporting systems are being used in new data sources with less reporting bias issues such as electronic healthcare data. Lessons learned from spontaneous reports and methods developed can also be applied to data mining of newer sources of health information from social media.
Despite its limitations, the spontaneous reporting system is an extremely valuable mechanism for safety monitoring. Regulatory agencies use spontaneous reporting system as an early warning system for emerging safety issues in the real world. In recent years, with efforts from regulatory agencies, pharmaceutical industry and the medical community, the amount, quality and timely reporting of AEs to the spontaneous reporting systems have greatly improved. The analytic tools for signal detection and results interpretation also became more mature. These advances increased the value of spontaneous reporting systems for post-marketing pharmacovigilance purpose. Other uses of spontaneous reporting systems such as FAERS have been explored, for disease surveillance, guiding the development of expensive epidemiological studies and identifying new opportunities in translational medicine and regulatory science [61]. With the continuous improvement of reporting and data quality, augmented with data from other sources, such as electronic health database, the spontaneous reporting systems will remain an important component of lifecycle management of medical product safety.
Acknowledgments
The authors would like to thank Olga Marchenko, Qi Jiang and especially Estelle Russek-Cohen for constructive comments and suggestions which improved the content of this manuscript.
Footnotes
Disclosures: Dr. Izem, Dr. Sanchez-Kam, Dr. Zink and Dr. Zhao have nothing to disclose. Dr. Ma is an employee of Amgen Inc.
Publisher's Disclaimer: Disclaimer: This article reflects the views of the authors and should not be construed to represent FDA’s views or policies.
References
- 1.Furberg CD, Pitt B. Withdrawal of cerivastatin from the world market. Curr Control Trials Cardiovasc Med. 2001;2(5):205–207. doi: 10.1186/cvm-2-5-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Staffa JA, Chang J, Green L. Cerivastatin and reports of fatal rhabdomyolysis. N Engl J Med. 2002;346(7):539–40. doi: 10.1056/NEJM200202143460721. [DOI] [PubMed] [Google Scholar]
- 3.Duggirala H, et al. Data Mining at the Food and Drug Administration. 2015:24. [Google Scholar]
- 4.Food and Drug Administration. FDA - FAERS - Quarterly Report [cited. 2017 Apr; Available from: https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/AdverseDrugEffects/ucm082196.htm#QuarterlyReports.
- 5.Health and Human Services. VAERS Data [cited. 2017 Apr; Available from: https://vaers.hhs.gov/data/data.
- 6.Medical Dictionary for Regulatory Activities. MedDRA home page. [cited 2017 April], Available from: https://www.meddra.org/
- 7.Food and Drug Administration. Food and Drug Administration-Individual Case Safety Reports. [cited 2017 April]; Available from: https://www.fda.gov/ForIndustry/DataStandards/IndividualCaseSafetyReports/default.htm.
- 8.Food and Drug Administration. OpenFDA public website. [cited 2017 April]; Available from: https://open.fda.gov/research/
- 9.Food and Drug Administration. FDA, Post-Market Drug and Biologic Evaluation. [cited 2017 April]; Available from: https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Surveillance/ucm204091.htm.
- 10.Food and Drug Administration. FDA Post-Market Safety Information for Patients and Providers. [cited 2017 April]; Available from: https://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/default.htm.
- 11.European Medicines Agency. European Medicines Agency: Eudravigilance. [cited 2017 April]; Available from: http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000679.jsp&mid=WC0b01ac05800250b5.
- 12.European Center for Disease Prevention and Control. Vaccine Adverse Event Surveillance and Communication. [cited 2017 April]; Available from: http://vaesco.net/vaesco.html.
- 13.World Health Organization. WHO: VigiBase. [cited 2017 April]; Available from: https://www.who-umc.org/vigibase/vigibase/
- 14.FDA-PRISM. Public Workshop: The Sentinel Post-Licensure Rapid Immunization Safety Monitoring (PRISM) System. Silver Spring, MD: 2016. [Google Scholar]
- 15.Kulldorff M, Fang ZX, Walsh SJ. A tree-based scan statistic for database disease surveillance. Biometrics. 2003;59(2):323–331. doi: 10.1111/1541-0420.00039. [DOI] [PubMed] [Google Scholar]
- 16.Coloma PM, et al. Postmarketing safety surveillance : where does signal detection using electronic healthcare records fit into the big picture? Drug Saf. 2013;36(3):183–97. doi: 10.1007/s40264-013-0018-x. [DOI] [PubMed] [Google Scholar]
- 17.Patadia VK, et al. Using real-world healthcare data for pharmacovigilance signal detection - the experience of the EU-ADR project. Expert Rev Clin Pharmacol. 2015;8(1):95–102. doi: 10.1586/17512433.2015.992878. [DOI] [PubMed] [Google Scholar]
- 18.Schuemie MJ, et al. Using electronic health care records for drug safety signal detection: a comparative evaluation of statistical methods. Med Care. 2012;50(10):890–7. doi: 10.1097/MLR.0b013e31825f63bf. [DOI] [PubMed] [Google Scholar]
- 19.Evans SJ, Waller PC, Davis S. Use of proportional reporting ratios (PRRs) for signal generation from spontaneous adverse drug reaction reports. Pharmacoepidemiol Drug Saf. 2001;10(6):483–6. doi: 10.1002/pds.677. [DOI] [PubMed] [Google Scholar]
- 20.Rothman KJ, Lanes S, Sacks ST. The reporting odds ratio and its advantages over the proportional reporting ratio. Pharmacoepidemiol Drug Saf. 2004;13(8):519–23. doi: 10.1002/pds.1001. [DOI] [PubMed] [Google Scholar]
- 21.Bate A, et al. A Bayesian neural network method for adverse drug reaction signal generation. Eur J Clin Pharmacol. 1998;54(4):315–21. doi: 10.1007/s002280050466. [DOI] [PubMed] [Google Scholar]
- 22.Huang L, Zalkikar J, Tiwari RC. A Likelihood Ratio Test Based Method for Signal Detection With Application to FDA’s Drug Safety Data. Journal of the American Statistical Association. 2011;106(496):1230–1241. [Google Scholar]
- 23.Huang L, Zalkikar J, Tiwari RC. Likelihood ratio test-based method for signal detection in drug classes using FDA’s AERS database. J Biopharm Stat. 2013;23(1):178–200. doi: 10.1080/10543406.2013.736810. [DOI] [PubMed] [Google Scholar]
- 24.Huang L, et al. Zero-inflated Poisson model based likelihood ratio test for drug safety signal detection. Stat Methods Med Res. 2017;26(1):471–488. doi: 10.1177/0962280214549590. [DOI] [PubMed] [Google Scholar]
- 25.Zhao Y, Yi M, Tiwari RC. Extended likelihood ratio test-based methods for signal detection in a drug class with application to FDA’s adverse event reporting system database. Stat Methods Med Res. 2016 doi: 10.1177/0962280216646678. [DOI] [PubMed] [Google Scholar]
- 26.Nam K, et al. Logistic Regression Likelihood Ratio Test Analysis for Detecting Signals of Adverse Events in Post-market Safety Surveillance. J Biopharm Stat. 2017:1–19. doi: 10.1080/10543406.2017.1295250. [DOI] [PubMed] [Google Scholar]
- 27.Bate A. Bayesian confidence propagation neural network. Drug Saf. 2007;30(7):623–5. doi: 10.2165/00002018-200730070-00011. [DOI] [PubMed] [Google Scholar]
- 28.Bate A, I, Edwards R. Data mining in spontaneous reports. Basic & Clinical Pharmacology & Toxicology. 2006;98(3):324–330. doi: 10.1111/j.1742-7843.2006.pto_232.x. [DOI] [PubMed] [Google Scholar]
- 29.Bate A, Evans SJ. Quantitative signal detection using spontaneous ADR reporting. Pharmacoepidemiol Drug Saf. 2009;18(6):427–36. doi: 10.1002/pds.1742. [DOI] [PubMed] [Google Scholar]
- 30.Bate A, et al. A data mining approach for signal detection and analysis. Drug Safety. 2002;25(6):393–397. doi: 10.2165/00002018-200225060-00002. [DOI] [PubMed] [Google Scholar]
- 31.Orre R, et al. A Bayesian recurrent neural network for unsupervised pattern recognition in large incomplete data sets. International Journal of Neural Systems. 2005;15(3):207–222. doi: 10.1142/S0129065705000219. [DOI] [PubMed] [Google Scholar]
- 32.Orre R, et al. Bayesian neural networks with confidence estimations applied to data mining. Computational Statistics & Data Analysis. 2000;34(4):473–493. [Google Scholar]
- 33.Almenoff JS, et al. Disproportionality analysis using empirical Bayes data mining: a tool for the evaluation of drug interactions in the post-marketing setting. Pharmacoepidemiol Drug Saf. 2003;12(6):517–21. doi: 10.1002/pds.885. [DOI] [PubMed] [Google Scholar]
- 34.Almenoff JS, et al. Novel statistical tools for monitoring the safety of marketed drugs. Clin Pharmacol Ther. 2007;82(2):157–66. doi: 10.1038/sj.clpt.6100258. [DOI] [PubMed] [Google Scholar]
- 35.Curtis JR, et al. Adaptation of Bayesian data mining algorithms to longitudinal claims data: coxib safety as an example. Med Care. 2008;46(9):969–75. doi: 10.1097/MLR.0b013e318179253b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DuMouchel W. Bayesian data mining in large frequency tables, with an application to the FDA spontaneous reporting system. American Statistician. 1999;53(3):177–190. [Google Scholar]
- 37.Kulldorff M, et al. Drug safety data mining with a tree-based scan statistic. Pharmacoepidemiol Drug Saf. 2013;22(5):517–23. doi: 10.1002/pds.3423. [DOI] [PubMed] [Google Scholar]
- 38.Kulldorff M, et al. A Maximized Sequential Probability Ratio Test for Drug and Vaccine Safety Surveillance. Sequential Analysis-Design Methods and Applications. 2011;30(1):58–78. [Google Scholar]
- 39.Silva IR, Kulldorff M. Continuous versus group sequential analysis for post-market drug and vaccine safety surveillance. Biometrics. 2015;71(3):851–8. doi: 10.1111/biom.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li L, Kulldorff M. A conditional maximized sequential probability ratio test for pharmacovigilance. Stat Med. 2010;29(2):284–95. doi: 10.1002/sim.3780. [DOI] [PubMed] [Google Scholar]
- 41.Cook AJ, et al. Group sequential method for observational data by using generalized estimating equations: application to Vaccine Safety Datalink. Journal of the Royal Statistical Society Series C-Applied Statistics. 2015;64(2):319–338. [Google Scholar]
- 42.Chan KA, Hauben M. Signal detection in pharmacovigilance: empirical evaluation of data mining tools. Pharmacoepidemiol Drug Saf. 2005;14(9):597–9. doi: 10.1002/pds.1128. [DOI] [PubMed] [Google Scholar]
- 43.Zhao SS, et al. Statistical performance of group sequential methods for observational post-licensure medical product safety surveillance: A simulation study. Statistics and Its Interface. 2012;5(4):381–390. [Google Scholar]
- 44.Cook AJ, et al. Statistical approaches to group sequential monitoring of postmarket safety surveillance data: current state of the art for use in the Mini-Sentinel pilot. Pharmacoepidemiol Drug Saf. 2012;21(Suppl 1):72–81. doi: 10.1002/pds.2320. [DOI] [PubMed] [Google Scholar]
- 45.Huang L, Zalkikar J, Tiwari R. Likelihood ratio based tests for longitudinal drug safety data. Statistics in Medicine. 2014;33(14):2408–2424. doi: 10.1002/sim.6103. [DOI] [PubMed] [Google Scholar]
- 46.Nelson JC, et al. Methods for observational post-licensure medical product safety surveillance. Stat Methods Med Res. 2015;24(2):177–93. doi: 10.1177/0962280211413452. [DOI] [PubMed] [Google Scholar]
- 47.Banks D, et al. Comparing data mining methods on the VAERS database. Pharmacoepidemiol Drug Saf. 2005;14(9):601–9. doi: 10.1002/pds.1107. [DOI] [PubMed] [Google Scholar]
- 48.Food and Drug Administration. FDA Adverse Event Reporting System (FAERS): Latest Quarterly Data Files, October – December 2016. [cited 2017 April]; Available from: https://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/surveillance/adversedrugeffects/ucm082193.htm.
- 49.Food and Drug Administration. Manufacturer and User Facility Device Experience Database - (MAUDE) [cited 2017 April]; Available from: https://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/PostmarketRequirements/ReportingAdverseEvents/ucm127891.htm.
- 50.Food and Drug Administration. Medsun: Medical Product Safety Network. [cited 2017 April]; Available from: https://www.fda.gov/MedicalDevices/Safety/MedSunMedicalProductSafetyNetwork/default.htm.
- 51.European Commission. EUDAMED: European Database on Medical Devices. [cited 2017 April]; Available from: http://ec.europa.eu/idabc/en/document/2256/5637.html.
- 52.Goldman SA, et al. In: A MedWatch Continuing Education Article. C.f D.E.a R Staff College, Food and Drug Administration, editor. 1996. [Google Scholar]
- 53.Botsis T, et al. Vaccine adverse event text mining system for extracting features from vaccine safety reports. J Am Med Inform Assoc. 2012;19(6):1011–8. doi: 10.1136/amiajnl-2012-000881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moore TJ, Furberg CD. Electronic Health Data for Postmarket Surveillance: A Vision Not Realized. Drug Safety. 2015;38(7):601–610. doi: 10.1007/s40264-015-0305-9. [DOI] [PubMed] [Google Scholar]
- 55.Heinrich J. (GAO report Before the Committee on Health, Education, Labor, and Pensions, U.S Senate).Substantial Problem but Magnitude Uncertain. 2000 [Google Scholar]
- 56.Dimbil M, C D, Erdman CB, Dmakas A, Kyle RF. Adverse Drug Event Reporting Rates: Comparing FAERS to Clinical Trials; Presented at AMPC Annual Meeting.2017. [Google Scholar]
- 57.Sharrar RG, Dieck GS. Monitoring product safety in the postmarketing environment. Therapeutic advances in drug safety. 2013;4(5):211–219. doi: 10.1177/2042098613490780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Boer A. When to publish measures of disproportionality derived from spontaneous reporting databases? British journal of clinical pharmacology. 2011;72(6):909–911. doi: 10.1111/j.1365-2125.2011.04087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Montastruc JL, et al. Benefits and strengths of the disproportionality analysis for identification of adverse drug reactions in a pharmacovigilance database. British journal of clinical pharmacology. 2011;72(6):905–908. doi: 10.1111/j.1365-2125.2011.04037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sakaeda T, et al. Commonality of drug-associated adverse events detected by 4 commonly used data mining algorithms. International journal of medical sciences. 2014;11(5):461. doi: 10.7150/ijms.7967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fang H, et al. Exploring the FDA Adverse Event Reporting System to Generate Hypotheses for Monitoring of Disease Characteristics. Clinical Pharmacology & Therapeutics. 2014;95(5):496–498. doi: 10.1038/clpt.2014.17. [DOI] [PMC free article] [PubMed] [Google Scholar]