

SUMMARY
Heterotrimeric G proteins (Gαβγ) are essential transducers in G protein signaling systems in all eukaryotes. In yeast, G protein signaling differentially activates mitogen-activated protein kinases (MAPKs)—Fus3 and Kss1—a phenomenon controlled by plasma membrane (PM) association of the scaffold protein Ste5. Here, we show that phosphorylation of the yeast Gγ subunit (Ste18), together with Fus3 docking on Ste5, controls the rate and stability of Ste5/PM association. Disruption of either element alone by point mutation has mild but reciprocal effects on MAPK activation. Disabling both elements results in ultra-fast and stable bulk Ste5/PM localization and Fus3 activation that is 6 times faster and 4 times more amplified compared to wild-type cells. These results further resolve the mechanism by which MAPK negative feedback phosphorylation controls pathway activation and provides compelling evidence that Gγ subunits can serve as intrinsic regulators of G protein signaling.
In Brief
Choudhury et al. show that Gγ subunits, besides acting as anchors for their obligate Gβ subunits, have more complex roles in regulating G protein signaling. Furthermore, they show that this tuning of G protein signaling by the phosphorylated Gγ N-terminal tail is achieved by altering the interaction between Gβγ and downstream effectors in a PTM-dependent manner.
INTRODUCTION
Canonical G protein signaling systems—including 7-transmembrane G-protein-coupled receptors (GPCRs), heterotrimeric G-proteins (Gαβγ), and diverse effector proteins—constitute a highly conserved system enabling the transduction of extracellular signaling molecules such as hormones, neurotransmitters, and chemokines (Cabrera-Vera et al., 2003; Wettschureck and Offermanns, 2005). As one of the primary signal transduction mechanisms in eukaryotes, G protein signaling pathways control a wide range of processes in both single-cell and multi-cellular organisms such as yeast and humans (Bargmann, 2006; Fan et al., 1997; Kaul et al., 2001; Rockman et al., 2002; Rosenbaum et al., 2009). Owing to their high degree of structural conservation, fundamental mechanisms underlying the regulation of G protein signaling have emerged from empirical studies conducted across widely diverse organisms from yeast to human. Such attributes have also helped to solidify their prominent role as pharmaceutical targets for the treatment of human disease (Hauser et al., 2017).
The yeast model system for G protein signaling remains one of the most well-characterized signaling pathways in history and has been instrumental in the discovery of G protein regulatory mechanisms, including regulators of G protein signaling (RGS) proteins and post-translational-modification (PTM)-based regulators (Cappell et al., 2010; Clement et al., 2013; Deflorio et al., 2013; Dohlman et al., 1991, 1996; Dohlman and Thorner, 2001; Isom et al., 2013; Li et al., 1998; Stone et al., 1991; Wang et al., 2005). In budding yeast Saccharomyces cerevisiae, a single canonical G protein signaling pathway controls the process of mating, wherein two yeasts of opposite and complementary mating types, MATa and MATα, fuse to form an a/α diploid cell (Dohlman and Thorner, 2001). As in the case of multicellular organisms, including mammals, G protein signaling in yeast is initiated by agonist-dependent activation of a GPCR at the cell surface (Dohlman and Thorner, 2001). In MATa cells, a single peptide mating pheromone (α factor) serves as the agonist of the pheromone GPCR, Ste2, and upon binding promotes the exchange of GDP for GTP on the Gα subunit (Gpa1). GTP binding stabilizes a conformational change in Gα and dissociation of the heterotrimer into Gα and Gβγ (Ste4/Ste18) components (Dohlman and Thorner, 2001; Sprang, 1997). In yeast, Gβγ serves as the primary activator of the mating pathway, whereas Gα serves primarily to sequester Gβγ (Dohlman and Thorner, 2001). When not sequestered, Gβγ nucleates the formation of a protein complex at the plasma membrane that includes a p21-activated protein kinase (PAK; Ste20) and a protein scaffold (Ste5) in complex with a mitogen-activated protein kinase (MAPKKK) (Ste11), MAPKK (Ste7), and mitogen-activated protein kinase (MAPK) (Fus3), as well as additional proteins important for cell polarization (Arkowitz, 2009; Nern and Arkowitz, 1999). Activation of the pheromone response pathway through this process drives a phosphorylation cascade resulting in double phosphorylation (referred to here as di-phosphorylation or activation) of Kss1T183,Y185 and Fus3T180,Y182—highly conserved orthologs of the human MAPKs Erk1 and Erk2—which phosphorylate several targets necessary for a complete mating response. Once activated, Fus3 translocates to the nucleus, wherein it phosphorylates the transcription factor Ste12 and other proteins necessary to drive a gene transcription program resulting in morphological change, cell-cycle arrest, and eventual mating (van Drogen et al., 2001; Elion et al., 1993). Signaling is terminated by hydrolysis of GTP to GDP on the Gα subunit and re-association of the heterotrimeric complex, a process that is accelerated by the RGS protein (Sst2) (Dohlman et al., 1996).
Several aspects of pheromone pathway behavior—such as switch-like versus graded dose-responsiveness and differential activation of MAPKs Fus3 and Kss1—have been linked directly to the Ste5 scaffold, which plays a major role in nearly all aspects of pathway control. Ste5 contains multiple domains that effectively tune the pheromone activation profile of Fus3, which binds to Ste5, distinctly from Kss1, which does not (Figure 1). The Fus3-binding domain (FBD) plays a critical role in this process by allosterically activating the autophosphorylation of Fus3Y182 (referred to here as mono-phosphorylation) in a manner independent of pheromone stimulation (Bhattacharyya et al., 2006). Mono-phosphorylated Fus3 phosphorylates Ste5T287 and three other phosphosites proximal to the RING domain, which functions to inhibit full activation of Fus3 and the mating pathway (Good et al., 2009). Inhibition is relieved in part by the action of the pheromone-dependent phosphatase, Ptc1, which competes with Fus3 for FBD binding and dephosphorylates Ste5, failure of which (ptc1Δ) prevents activation of Fus3 (Malleshaiah et al., 2010).
Figure 1. Working Model of the Mating Pathway of Saccharomyces cerevisae.

To facilitate clarity, a contemporary model of the pheromone pathway is shown here, which is inferred from prior studies described in the text. The mating pathway of Saccharomyces cerevisae is triggered in response to pheromone-dependent activation of the G-protein-coupled receptor, Ste2, and subsequent recruitment of Ste5 to the plasma membrane (PM), mediated by Ste5PM and Ste5PH domains as well as the Ste5RING domain that binds directly to free Gβ/Ste4. Under resting conditions, Ste5/PM association and MAPK activation is disfavored due to hyper-phosphorylation at positions surrounding Ste5PM (sites marked as sticks), auto-inhibition of the Ste5PH domain through a competitive interaction with Ste5VWA, and the lack of free Gβ/Ste4 that is sequestered by GDP-bound Gα/Gpa1. Pathway activation is also inhibited by the Ste5FBD, which allosterically activates Fus3 to promote phosphorylation of Ste5T287 and three other phosphosites proximal to Ste5FBD and Ste5RING (P-lollipops). Upon pheromone stimulation, cascade activation of MAP kinases relies on PM recruitment and stable association of Ste5 that is concomitant with several state changes, including (but not limited to): (1) upregulated expression of the phosphatase Ptc1, which competes with Fus3 for binding to Ste5FBD and promotes Ste5 de-phosphorylation; (2) a large conformational change in Ste5 accompanied by de-inhibition of Ste5VWA; and (3) MAPK cascade activation of Fus3 and Kss1. For simplicity, interactions involving proteins such as Ste20, Far1, Cdc42, Cdc24, and many more involved in shmoo formation are not shown. Central components of the model discussed extensively here as a framework for this work include the Ste5FBD and phosphoregulatory control by Fus3 and Ptc1, captured in part by Bhattacharyya et al. (2006), Coyle et al. (2013), and Malleshaiah et al. (2010); conformational dynamics and PM association of Ste5, captured in part by Takahashi and Pryciak (2008) and Zalatan et al. (2012); and the role of Gβγ/Ste5RING as an essential feature for Ste5/PM association, captured in part by Winters et al. (2005). Additional, but not all, contributions have been cited in the Introduction, Discussion, and Supplemental Information.
Within the context of G protein signaling, Gγ subunits have been largely understood as membrane anchors for Gβ subunits and rarely have been studied in any other context. Phosphorylation of Gγ12 has been shown to enhance the affinity of Gβγ for Gα subunits in vitro (Morishita et al., 1995) and impact the activity of the Gβγ12 dimers toward specific effectors (Yasuda et al., 1998). Recently, we showed that Gγ subunits across all eukaryotes harbor at least 2 phosphorylation sites in their intrinsically disordered N-terminal tails, many of which have also been observed as phosphorylated in high-throughput proteomics screens (Dewhurst et al., 2015). Here, we show that the N-terminal tail of the yeast Gγ subunit, Ste18, together with the Fus3/Ste5FBD docking interaction, constitute a negative regulatory system that synergistically controls the activation of Fus3 and Kss1 through a dynamic phospho-inhibitory mechanism that prevents rapid and stable association of bulk Ste5 at the plasma membrane. Disruption of either side alone (by point mutation of Ste18Nt or Ste5FBD) results in minor but significant and reciprocal changes to the rate and amplitude of Fus3 activation and Ste5/PM recruitment, whereas combined disruption of both elements results in ultra-rapid and robust Fus3 activation and Ste5/PM recruitment. We show that negative regulation is facilitated by weakened binding between Gβγ and Ste5 when both proteins are phosphorylated—a mechanism that may have emerged through co-evolution of the two distinct phospho-regulatory elements. Taken together, these data reveal a way in which Gβγ/effector binding can be modulated to control signaling output through post-translational modification.
RESULTS
The N-Terminal Tail of Ste18 Is Rapidly Phosphorylated in Response to GPCR Activation
Like other Gγ subunits throughout Eukarya, the terminal ends of Ste18, representing 20% of its residue content, are intrinsically disordered—most of which correspond to the N-terminal tail (Ste18Nt). This region in Ste18 (residues 1–13) harbors phosphorylation sites at Thr-2, Ser-3, and Ser-7, specifically (Dewhurst et al., 2015; Soufi et al., 2009). Phosphorylation of Ste18Nt produces a distinctive electrophoretic mobility shift in response to pheromone stimulation, which can be detected by SDS-PAGE and immunoblotting (Dewhurst et al., 2015). Treatment with alkaline phosphatase eliminates the mobility shift in a manner that depends on the presence or absence of phosphatase inhibitors (Figure S1A). Moreover, phospho-null mutations (Ste18T2A, S3A, S7A; referred to here as Ste183A) and phospho-mimic mutations (Ste18T2E, S3E, S7E; referred to here as Ste183E) eliminate or restore the mobility shift, respectively (Dewhurst et al., 2015). To determine a precise estimate of phosphorylation kinetics early and late after receptor activation, we surveyed the phosphorylation-dependent mobility shift of Ste18 in both long and short time course experiments (Figures 2A and S1B). We found that phosphorylation occurs almost instantaneously after receptor activation and is readily detectable within 30 s (Figures 2B and S1B). Maximum phosphorylation is achieved at ~80% of total Ste18 levels within 3 min, with time at half maximum (t1/2) of 1.25 min (Figure 2B, inset). Phosphorylation remains stable for the duration of pheromone exposure up to 90 min. Activation profiles for Fus3 and Kss1 MAPKs were consistent with their expected patterns of activation at 3-μM pheromone concentrations (Figure 2C) (Hao et al., 2008).
Figure 2. Ste18Nt Is Rapidly Phosphorylated in Response to GPCR Activation.

(A) Immunoblots of HA-Ste18, activated MAPKs (ppKss1 and ppFus3), and a protein loading control (LC) in wild-type cells treated with 3 μM pheromone (α-F) for the indicated time (long time course).
(B) Quantification of pHA-Ste18 over short (black circles) and long (gray circles) time course periods in wild-type cells. Data correspond to the percent abundance of phosphorylated HA-Ste18-Nt (pHA-Ste18) relative to total HA-Ste18 (mean ± SD; n = 12). Short and long time course data were normalized to each other at 5 min.
(C) MAPK activation profile in wild-type cells from the long time course experiment shown in (A).
See also Figures S1 and S2.
Surmising that Ste18 phosphorylation may depend on a kinase within the pheromone pathway, we monitored Ste18 phosphorylation in cells lacking single components of the pathway, including: Ste20, Ste11, Ste7, Fus3, and Kss1, in addition to the MAPK scaffold Ste5. We found that all kinases upstream of and including the scaffolded MAPK complex were necessary for robust phosphorylation of Ste18Nt in response to pheromone (Figures S1C and S1D). Kss1, which is rapidly phosphorylated after receptor activation but is not scaffolded by Ste5, is not required for pheromone-dependent Ste18 phosphorylation. Since Ste18 phosphorylation should not be lost in fus3Δ cells if MAPKs upstream of Fus3 are responsible for phosphorylation, these data suggest that Fus3, but not upstream MAPKs, are necessary. Other genes involved in the pheromone pathway that either regulate pheromone dependent kinase activation or are themselves pheromone-activated kinases had little to no effect when deleted from the genome (Figure S2). Taken together, these data demonstrate that Ste18 is rapidly phosphorylated in response to GPCR activation in a manner that requires Fus3.
Phosphorylation of Ste18Nt and Ste5 Function Synergistically to Delay Fus3 Peak Activation in Response to Pheromone
Considering that Ste18 is rapidly phosphorylated within seconds of receptor stimulation, we hypothesized that it must be a prerequisite for proper pathway activation. Consistent with this hypothesis, we found that preventing phosphorylation of Ste18 (Ste183A) resulted in a significant shift in peak activation of Fus3, which occurred twice as fast (15 min earlier) than in wild-type cells (Figures S3A and S3C). In contrast, activation of Fus3 and Kss1 in cells harboring the phosphomimic form, Ste183E, was no different from that in wild-type cells, as would be expected if phosphorylation is a prerequisite feature required for normal pathway activation (Figures S3A and S3B). Thus, Ste18 phosphorylation is required for delayed peak activation of the scaffolded MAPK Fus3 but has no effect on the activation of the un-scaffolded MAPK, Kss1.
Negative feedback phosphorylation of Ste5, like phosphorylation of Ste18, occurs early/before the mating response and also requires Fus3. Therefore, we hypothesized that both elements may function in concert to delay Fus3 peak activation. This hypothesis was inspired, in part, by two points of indirect evidence that converge on Fus3 as a negative regulator of the pheromone response. First, rapid receptor-activated phosphorylation of Ste18Nt requires Fus3 (Figure S1), and activated Fus3 has been previously shown to counteract the stability of Ste5/PM association during the pheromone response (Yu et al., 2008b). Second, Fus3-mediated negative feedback phosphorylation on Ste5 dampens the intensity of Fus3 activation in response to pheromone (Dowell et al., 1998; Hao et al., 2008; Inouye et al., 1997). Additionally, we considered as further evidence that stable Ste5/PM association is facilitated in part by the interaction of the Ste5RING domain (Ste5138–214) with residues in Ste4 (Ste449–65) that are located in the coiled-coil structure formed by both Gβ (Ste4) and Gγ (Ste18) subunits and in very close proximity to where phosphorylation occurs in Ste18Nt (Dowell et al., 1998).
To test the hypothesis, we monitored phosphorylation of Ste18, Kss1, and Fus3 in cells that endogenously express different combinations of Ste18 phosphosite and Ste5FBD docking-site mutants. Precise mutation of multiple sites within two essential binding surfaces (A and B) in the Ste5FBD (a.k.a. Ste5ND) prevents Fus3 docking, allosteric activation, and negative feedback phosphorylation of Ste5 (Bhattacharyya et al., 2006). Interestingly, phosphorylation of Ste18Nt was unaffected by Ste5ND, suggesting that Fus3 docking to Ste5 is not necessary for Ste18 phosphorylation (Figures 3A and 3B). As observed in previous experiments, peak activation of Fus3 occurred significantly early in Ste183A compared to Ste183E or wild-type cells (Figures 3C and 3E), whereas Kss1 activation was unaffected (Figures 3C and 3D). In contrast, we found that peak activation of Fus3 (at 30 min) and Kss1 (between 5 and 30 min) was significantly elevated, but not early, in Ste5ND cells harboring the wild-type form of Ste18, an observation that is nearly identical with previously reported evidence (Figures 3C–3E) (Hao et al., 2008).
Figure 3. Phosphorylation on Ste18 and Ste5 Cooperate to Prevent Early and Maximal Fus3 Activation.

Cells harboring the indicated combination of wild-type or mutant versions of Ste18 and Ste5 were stimulated with 3 μM α-F followed by quantitative immunoblot analysis of HA-Ste18 or activated Fus3 and Kss1.
(A) HA-Ste18 immunoblot in cells harboring wild-type (WT/WT) or Ste5ND (WT/non-docking [ND]).
(B) Quantitative comparison of pHA-Ste18 from (A) (n = 4).
(C) Representative immunoblot for activated Kss1 and Fus3.
(D) Quantitative comparison of activated Kss1 relative to wild-type peak activation at 5 min from immunoblots shown in (C).
(E) Quantitative comparison of activated Fus3 relative to wild-type peak activation at 30 min from immunoblots shown in (C). Data represent mean ± SD; n = 12. SE, short exposure; LE, long exposure; LC, loading control.
See also Figures S3, S4, and S5 and Table S2.
We made several observations with yeast harboring both Ste18 and Ste5 mutations in combination. First, peak activation of Fus3 occurs rapidly in Ste183A/Ste5ND cells, wherein neither protein can be phosphorylated (Figure 3E; Table S2). Significantly, this response was 25 min earlier and nearly 1.5-fold greater in amplitude than observed for Ste5ND alone and 3.6-fold greater than the response in wild-type cells (Figure 3E; Table S2). Neither this nor any other mutant showed abrupt differences in the pattern of pheromone-dependent Fus3 expression level compared to wild-type cells, suggesting that differences in the intensity of activated Fus3 at early time points is due to Fus3 phosphorylation and independent of Fus3 expression (Figure S4A). Interestingly, Ste183A/Ste5ND, but not other cell types, also exhibited morphological defects in the absence of pheromone, showing pronounced elongation in multiple cases (Figure S4B). Second, in cells harboring the combination of Ste183E and Ste5ND, Fus3 peak activation occurs early (15 min) and with no change in amplitude—nearly identical to the response in Ste183A/Ste5WT cells and indicative that phosphorylation restricted to either Ste18 or Ste5 is sufficient to elicit an equivalent outcome in Fus3 peak activation (Figure 3E; Table S2). Complementary phos-tag analysis of Fus3 phosphorylation further suggests that either Ste18Nt phosphorylation or Ste5 phosphorylation (controlled by Fus3 docking on Ste5) can effectively inhibit the aberrant di-phosphorylation of Fus3 in the absence of pheromone (Figure S5). Moreover, preventing such control on both proteins together results in aberrant Fus3 di-phosphorylation in the absence of pheromone. Unexpectedly, activation of Kss1 was also affected by combined disruption of Ste18/Ste5. However, in contrast to Fus3, Kss1 becomes hyper-activated in Ste183E/Ste5ND cells (Figure 3D). Further confirmation of these results was achieved by endpoint immunoblot assays conducted within the first 15 min of pheromone stimulation (Figures S4C–S4E).
Evidence from MAPK activation analysis suggested that Ste18 and Ste5 phosphorylation behave synergistically so that the combined effect of both produces an effect that is greater than the sum of their separate effects. Indeed, a quantitative test for synergy revealed that phosphorylated Ste18 and Ste5 are synergistic in their control of Fus3 peak activation time but additive in their control of Fus3 peak amplitude, with an overall synergistic impact that is ~4× greater than the sum of effects contributed by Ste18 or Ste5 alone (Table S2). Taken together, these data suggest that the phosphorylation of Ste18 and MAPK docking/phosphorylation of Ste5 (referred to here as the Ste18/Ste5 phospho-inhibitory system) synergize to delay the activation and repress the amplitude of Fus3 activation in response to pheromone.
Together, Ste18/Ste5 Phosphorylation Delays Bulk Recruitment Rate and Reduces the Duration of Ste5/PM Association
Activation of Fus3 requires recruitment and stable association of Ste5 with the plasma membrane, and consequently, mutations that disrupt or enhance Ste5/PM association should be reflected by the kinetics and amplitude of Fus3 activation. To test this hypothesis, we monitored Ste5/PM association by fluorescence microscopy before, during, and after pheromone stimulation of yeast cells. We were unable to detect Ste5-GFP at the PM in any cell that was not treated with pheromone (Figure 4A, top). However, in as few as 23–26 min after pheromone stimulation, we observed robust PM accumulation of Ste5-GFP in all cell types, most noticeably in Ste183A/Ste5ND cells (Figure 4A, bottom). Consistent with our hypothesis, preventing phosphorylation on both Ste18 and Ste5 (Ste183A/Ste5ND) resulted in the robust accumulation of Ste5-GFP at the PM after 25 min of pheromone stimulation, reaching a level nearly 7-fold greater than that of wild-type cells measured at the same time (Figure 4B). However, no differences were observed in the protein levels of Ste5-GFP (Figure S6). Thus, the Ste5-GFP signal at the membrane reflects altered Ste5/PM accumulation rather than protein stability. Furthermore, these data suggest that the bulk recruitment rate of Ste5 to the PM—reflected in the number of Ste5-GFP molecules that accumulate at the PM per unit time (i.e., total fluorescence intensity/time)—is significantly faster when phosphorylation on both proteins is prevented.
Figure 4. Phosphorylation on Ste18/Ste5 Regulates the Rate and Duration of Ste5 Association at the Plasma Membrane.

Cells expressing phosphorylation mutants of Ste18 with either Ste5-GFP or Ste5ND-GFP were treated with α-factor to examine Ste5 localization by fluorescence microscopy (see Experimental Procedures).
(A) Representative fluorescent images showing cells with localized GFP signal at the membrane before (top) or 23–26 min post-pheromone treatment (bottom).
(B) Quantification of total Ste5-GFP fluorescence at the shmoo tip in cells treated with pheromone.
(C) Time-resolved percentage of the cell population with Ste5-GFP localized at the plasma membrane in response to pheromone stimulation. Inset is indicated with a dashed line.
(D) Zoomed view of the first 60 min from (C, inset). Time required before 100% of all cells display Ste5-GFP at the shmoo tip is indicated.
(E) Mean duration of Ste5-GFP/PM association (at the site of an emerging or extant mating projection).
(F) Immunoblot showing coIP of HA-Ste18 with Ste5-GFP from pheromone-treated cells. The lysate shown is 4% of the total lysate used for coIP (Experimental Procedures).
(G) Quantitative analysis of immunoblots from coIP in (F). Bars represent fold enrichment of total HA-Ste18 relative to wild-type.
Data represent mean ± SD; n = 3. All microscopy data represent GFP signal scored in 8–14 cells, with error bars depicting SEM in (B).
See also Figure S6.
To compare the relative rates and stability of Ste5-GFP/PM accumulation at the population level, we quantified the percentage of the cell population with Ste5-GFP localized at the PM as a function of time. Measured in this way, Ste5/PM accumulation data are more comparable to MAPK activation data, which is a population-based analysis. Preventing phosphorylation on both Ste18 and Ste5 (Ste183A/Ste5ND) significantly enhanced the rate at which the population of cells exhibited Ste5-GFP/PM accumulation, where 100% of the cells were responsive by 14 min (Figures 4C and 4D). This effect was reverted to a wild-type state in Ste183E/Ste5ND cells, mimicking the effects of reciprocal mutations on MAPK activation seen earlier. In contrast, Ste5ND cells exhibited an intermediate phenotype. As expected, mutations to Ste18 alone had mild average effects on Ste5-GFP/PM accumulation.
Lastly, we also observed that Ste5/PM association was noticeably more stable in Ste183A/Ste5ND cells compared to all other strains, whereby the population of Ste183A/Ste5ND cells had maximal association for the longest time (141 min) compared to that of wild-type or Ste183E/Ste5ND cells (Figure 4E). In support of the hypothesis that this is promoted by more stable association between Ste4/18 and Ste5, co-immunoprecipitation (coIP) experiments revealed significantly more (2×–5×) Ste18 immunopreciptated with Ste5-GFP in Ste183A/Ste5ND cells compared to all other cell types (Figures 4F and 4G). We conclude that rapid bulk recruitment of Ste5 to the PM and subsequent Fus3 activation in Ste183A/Ste5ND cells can be attributed in part to greater physical association between the two proteins in their non-phosphorylated states and support evidence that phosphorylation of Ste18Nt and Ste5, together, control the bulk rate and stability of Ste5/PM association and MAPK activation.
Ste183E Partially Restores a Switch-like Mating Response in Ste5ND Cells
Ste5 has been shown previously to be an important control point for the switch-like morphological response, which, in wild-type cells, is controlled by competitive Ste5FBD docking by Fus3 and the phosphatase Ptc1 (Malleshaiah et al., 2010). Consequently, non-docking Ste5ND mutants, which cannot bind to either Fus3 or Ptc1, exhibit a graded morphological dose-response. We hypothesized that Ste18 phosphorylation might also be involved in controlling the switch-like mating decision. To test this hypothesis, we performed microscopy-based dose-response experiments to quantify shmoo formation as a function of pheromone dose. Modeling the data with a sigmoidal dose-response curve with variable slope, we confirmed that wild-type cells exhibit switch-like dose-responsiveness with a hill slope well above 1 (hill coefficient [nH]WT = 6.2) (Figure 5A; Table S3). Similarly, Ste183A and Ste183E cells were also switch-like (nH3A = 6.3; nH3E = 3.7). We observed that Ste5ND cells are graded rather than switch-like (nHND = 0.97), consistent with previous reports (Coyle et al., 2013; Malleshaiah et al., 2010) (Figure 5B; Table S3). Disabling both regulatory elements (Ste183A/Ste5ND) produced a slightly more graded response (nH3A/ND = 0.85), exhibiting a hill slope lower than that for Ste5ND. Not surprisingly, Ste183A/Ste5ND cells also exhibited an elevated basal response that was ~3-fold greater than that for wild-type cells, consistent with earlier results showing elevated Fus3 activation in these cells (Figures 3, S4B, S4C, and S5). Expression of the phosphomimic form of Ste18 in the Ste5ND background (Ste183E/Ste5ND) resulted in a reversion of the analog response in Ste5ND cells back to a switch-like state (nH3E/ND = 2.7) (Figure 5B; Table S3). Taken together, these data suggest that phosphorylation on Ste18 regulates the morphological mating switch in cells when Ste5 is incapable of docking with Fus3. Furthermore, these data suggest that Ste18/Ste5 phosphorylation regulates the switch-like mating decision in yeast.
Figure 5. The Switch-like Morphological Response to Pheromone Is Regulated by Ste18-Nt Phosphorylation When Fus3 Cannot Bind to Ste5.

The morphological dose-response to mating pheromone represented by the cellular formation of a mating projection (i.e., shmoo) was quantified as a percentage of total cells in a population by DIC microscopy. Data were fit to a sigmodal dose-response curve with variable slope.
(A) Effect of individual Ste18Nt phosphorylation mutations on the mating response.
(B) Effect of Ste18Nt phosphorylation mutations in strains exclusively expressing the Fus3 non-docking mutant Ste5ND.
Error bars represent SEM across ≥200 cells per experiment.
See also Table S3.
Phosphorylation of the Gγ Subunit and MAPK Docking on Ste5 Appear Simultaneously in the Phylogeny of Yeasts
Recent phylogenetic and experimental evidence has revealed that the Ste5 FBD arose ~130 million years ago and is almost exclusively found in the clade to which Saccharomyces cerevisiae belongs but is also partially extant in V. polyspora species, which contain a partial FBD that is moderately active (Coyle et al., 2013). Having determined that the phosphorylation of the N-terminal tail of Ste18 and the Ste5FBD function synergistically to control signaling, we asked whether these two structural elements may have co-evolved. Comparison of the phylogeny of Ste18 and Ste5 fungal orthologs revealed compelling evidence in support of this hypothesis. Indeed, the appearance of the phosphorylation sites in the Ste18 intrinsically disordered region (phospho-IDR) coincides with the appearance of Ste5FBD (Figures 6A and S7A). Furthermore, all extant members of the clade retain nearly 100% identity for sites of phosphorylation and MAPK binding that are essential to either element (Figure 6B). A somewhat similar putative phospho-IDR was also found in the Ste18 ortho-log from S. castellii and the distantly related Y. lipolytica, both species of which harbor 2 of the 3 phosphosites observed in S. cerevisiae but exhibit dramatically different N-terminal IDR lengths (Figure S7B). When looking strictly within the phylogeny of Ste5-containing yeast species (excluding A. gossypii, which harbors an extraordinarily long N-terminal IDR), we observed a progressive lengthening over time of the N-terminal IDRs of Ste18 orthologs, culminating in a dramatic increase of 8–10 amino acid residues that appeared coincidently with the Ste5FBD (Figure S7C). Taken together, these data suggest that Ste18 and Ste5 phospho-regulatory elements arose at similar times and appear only once in the evolution of budding yeast.
Figure 6. Coordinated Phospho-regulation of Ste18 and Ste5 Evolved at the Same Time.

(A) Bootstrapped phylogenetic tree of Ste18 orthologs from the Ascomycota phylum showing the evolutionary co-occurrence of regulatory phosphorylation sites on the N-terminal tail of the Gγ subunit and the FBD of orthologous Ste5 scaffolds. The presence of Ste5 orthologous proteins, functional Ste5-like FBDs, and N-terminal Gγ phospho-regulatory intrinsically disordered regions (IDRs) are shown next to yeast species harboring each element.
(B) Species that contain the synergistic regulatory element (Ste18Nt and Ste5FBD) are nearly 100% identical at phosphorylation site alignment positions in Ste18 and MAPK binding sites reported previously in Ste5 (Coyle et al., 2013).
See also Figure S7.
DISCUSSION
Gγ subunits are recognized as having limited function as membrane anchors for Gβ subunits in heterotrimeric G protein signaling systems (Cook et al., 1998; Kisselev et al., 1995; Muntz et al., 1992). As such, their role in signal transduction beyond this function is generally perceived as benign. We previously revealed that Gγ N-terminal tails are prominent targets of phosphorylation, indicating that they might also possibly be important signal regulators (Dewhurst et al., 2015). Here, we have demonstrated that phosphorylation of the Gγ subunit in yeast regulates G protein signaling through a feedback phosphorylation mechanism that synergizes with MAPK-docking-dependent phosphorylation on the protein scaffold Ste5.
New Insights into the Regulation of Ste5/PM Association and MAPK Activation
For good reason, understanding the mechanism by which Ste5 coordinates mating pathway output has been a long-standing endeavor with many revisions over the past 10–15 years. The primary function of Ste5 is to properly translate stimulus dose to MAPK response signals—a function that is facilitated by scaffolding of the MAPK cascade module (MAPKKK Ste11, MAPK Ste7, and MAPK Fus3). In resting cells, Ste5/MAPK and Ste5 inter-domain interactions, as well as phosphorylation, prevent signaling from occurring spontaneously. Under such conditions, Ste5 is distributed between the cytoplasm and the nucleus—a phenotype that is cell-cycle regulated in part (Maeder et al., 2007; Strickfaden et al., 2007). Cytoplasmic Ste5 can still interact with all three components of the MAPK cascade module (Takahashi and Pryciak, 2008). However, Ste5 is incapable of facilitating MAPK activation in this state due to inter-domain binding between Ste5PH and Ste5VWA domains (Zalatan et al., 2012) as well as inhibitory phosphorylation mediated by Fus3 and Ste5FBD interactions (Bhattacharyya et al., 2006). Relief of inter-domain binding inhibition is mediated by pheromone-dependent accumulation of PIP2 in the PM, which, when stimulated, promotes Ste5PH/PM association, and de-inhibition of Ste5VWA allows for a 3-fold increase in Fus3 phosphorylation over the inhibited state in vitro (Zalatan et al., 2012).
Our evidence suggests that Ste5/PM association is also modulated by phosphorylation on Ste18 in addition to Ste5, both of which function synergistically to control Fus3 activation output. In a wild-type cell, phosphorylation of both proteins slows the rate and restricts the amplitude of Fus3 activation, resulting in 30-min peak activation (Ste18WT/Ste5WT). Engaging either only one element (Ste183E/Ste5ND) or the other (Ste183A/Ste5WT) produces an equivalent enhancement in peak activation and amplitude that is 2 times faster and 1.5 to 2 times more intense than in wild-type cells (Figure 3E). Disabling both elements (Ste183A/Ste5ND) results in ultra-fast and intense Fus3 activation that is 6 times faster and 3–4 times greater than the wild-type response. We show further that the response in MAPK activation for these mutants correlates very well with the bulk rate and duration of Ste5/PM association, indicating that it is the dominant control factor for the Fus3 activation profiles observed in each mutant.
Considering our body of evidence in light of previous work by others, we propose a model in which the activation/deactivation of the Ste18/Ste5 phospho-inhibitory system is regulated differentially (Figure 7A). In a pre-stimulated state, Fus3 is mono-phosphorylated and, in this state, is capable of phosphorylating Ste5 at up to 4 positions near Ste5FBD (Figure S5; Malleshaiah et al., 2010), while Ste18 is also basally phosphorylated (Figures 2 and S1). This suggests that inhibition is partially engaged even before a pheromone stimulus. Upon the addition of pheromone, the Ste18/Ste5 inhibitory elements become fully activated, as both Ste18 and Ste5 become hyper-phosphorylated, demonstrated herein directly for Ste18 (Figure 2) and presumed by an in vitro Fus3 kinase assay with Ste5 (see Figure S9 in Malleshaiah et al., 2010). We propose that this constitutes the fully activated inhibitory element that is responsible for bulk inhibition of Ste5/PM association early in the pheromone response, as determined from the relative amounts of Ste5-GFP/PM accumulation observed in Ste183A/Ste5ND compared to other cells (Figure 4). Simultaneously, pheromone stimulates recruitment of Ptc1 phosphatase to Ste5FBD, which outcompetes Fus3 binding within 2 min post-stimulus and dephosphorylates the inhibitory feedback phosphosites on Ste5 (see Figure S18 in Malleshaiah et al., 2010). Since as few as one occupied Ste5 phosphosite leads to the inhibition of Fus3 di-phosphorylation (Malleshaiah et al., 2010), it is presumed that all four sites must be completely dephosphorylated. We propose that this constitutes a release by partial de-inhibition of the Ste18/Ste5 inhibitory element, which leaves the Ste18 element intact (phosphorylated), as can be seen by the fact that Ste18 phosphorylation remains unchanged during this time (Figure 3B). We propose that, as a consequence of having one phospho-inhibitory element still intact (phospho-Ste18), the bulk recruitment of Ste5 to the membrane is permitted but relatively slow, and the activation of Fus3 is delayed, not reaching peak amplitude until ~30 min post-stimulus (Figures 2C, 3E, and 4). This half-activated state of the Ste18/Ste5 system also controls the degree to which Ste18 co-immunoprecipitates with Ste5 and the degree to which Ste5/PM association occurs (Figure 4G)—observations that correlate very well with Fus3 activation.
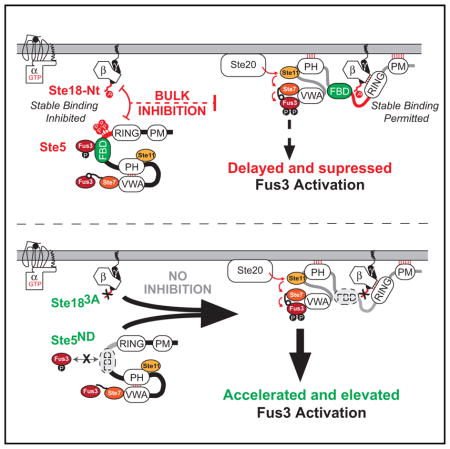
Figure 7. Phosphorylated Ste18Nt and Ste5FBD Constitute a Dynamic Phosphoregulatory System for Pheromone Signaling.

(A) In response to pheromone, Ste18 is rapidly phosphorylated at its N-terminal tail (P-lollipop). Ste5 is simultaneously phosphorylated via negative feedback controlled by Fus3/Ste5FBD docking (P-lollipops). Together, this constitutes a phospho-inhibitory system that prevents otherwise rapid Ste5/PM association. While not shown outright here, previous work implicates pheromone-stimulated expression of Ptc1 phosphatase and removal of inhibitory phosphorylation on Ste5 as the inhibition release (Malleshaiah et al., 2010) (dashed orange box). Consequently, the mating pathway is activated with a kinetic delay, as evident by the slower rate of Ste5 association at the membrane and delayed peak activation of Fus3.
(B) Cells engineered to prevent activation of the Ste18/Ste5 system (Ste183A/Ste5ND) respond ~6 times faster with ~4 times greater intensity than observed in wild-type cells—a response that demonstrates synergy between the two phospho-regulatory elements (phosphorylated Ste18 and Ste5) in the system.
In preventing phosphorylation on Ste18 and Ste5 (Ste183A/Ste5ND), we show that this system is necessary to delay what is otherwise ultra-fast and intense Fus3 activation (Figure 7B). Consequently, Fus3 activation in this state appears to match the profile of Kss1 activation, which is not scaffolded (Figures 3D and 3E). Again, the rapid bulk recruitment of Ste5 under these conditions (Figure 4) can explain this effect, since rapid Ste5/PM association is known to drive rapid de-inhibition of the Ste5VWA domain by promoting the association of Ste5PH with the plasma membrane—a mechanism that has been demonstrated previously by the Wendell Lim lab as an essential step to drive phosphorylation of Fus3 by Ste7 (Zalatan et al., 2012). Switch-like behavior of the pathway is also modulated, in part, by the Ste18/Ste5 phospho-inhibitory system, which controls Ste5/PM association, an observation that is consistent with previous reports demonstrating that Ste5FBD and Ste5/PM association regulate hill slope (Coyle et al., 2013; Takahashi and Pryciak, 2008).
Several pieces of evidence, both from our work and others, suggest that Fus3 is the major (if not primary) kinase for activating the phospho-inhibitory system. Fus3 phosphorylates proline +1 sites on Ste5 (Bhattacharyya et al., 2006) and on Ste18Nt (Figures S3C and S3D)—of which, both sites emerged at the same time in the evolution of budding yeast (Figure 6). Second, Ste5/PM association is enhanced in fus3Δ compared to FUS3 cells treated with pheromone (Yu et al., 2008a)—a phenotype that we also observe for the phospho-null mutant Ste183A/Ste5ND (Figure 4). Third, inhibition of an analog-sensitive form of Fus3 (fus3-as2) serves to stabilize Ste5/PM association upon pheromone stimulation (Yu et al., 2008b). Consequently, a Fus3-specific phosphatase such as Msg5, which has been suggested to synergize with Ste5 to repress Fus3 activation (Nagiec et al., 2015), will alter the ability of Fus3 to function as an activator of the system. Indeed, deletion of the Msg5 phosphatase in Ste5ND cells (Ste5ND/msg5Δ) results in more intense, but not faster, Fus3 activation compared to wild-type or either mutation alone (Nagiec et al., 2015), consistent with long-standing evidence that deletion of Fus3-specific phosphatases permits intense and prolonged Fus3 activation (Zhan et al., 1997). Thus, modulation of Fus3 activity necessarily imparts both positive (gene transcription) and negative (Ste18/Ste5) feedback on the pheromone pathway, which will be affected by inhibition or deletion of the kinase.
Gγ N-Terminal Tails as Regulators of G Protein Signaling—from Yeast to Humans
Our data reveal the potential for Gγ subunits to play more intricate roles in regulating G protein signaling, effective through PTM-altered protein interactions. As stated earlier, most eukaryotic Gγ subunits are phosphorylated in their N-terminal tails, and all Gγ subunits have the potential to be phosphorylated, since serine and threonine are ubiquitous in eukaryotic Gγ subunit N-terminal tails (based on data curated and reviewed by UniProt) (Dewhurst et al., 2015). In yeast, we find that phosphorylation plays a major role in effector recruitment, but we do not observe evidence that it impacts heterotrimer association, which can be sensitively detected by monitoring Fus3 activation in yeast. This is consistent with previous reports that negative feedback by Fus3 does not impact heterotrimeric G protein dissociation (Yu et al., 2008b). Thus, functional, GPCR-activated phosphorylation of Gγ N-terminal tails is not a process restricted to yeast and, as we have shown here, can regulate G protein signaling output by modulating Gβγ effector binding.
EXPERIMENTAL PROCEDURES
Yeast Strains and Plasmids
Standard methods for cell growth, maintenance, and transformation of yeast and for the manipulation of DNA were used throughout. Strain BY4741 (MATa leu2Δ met15Δ his3Δ ura3Δ)- and BY4741-derived mutants were used. Details of strains used are listed in Table S1. Strains were constructed using the two-step dellitto perfetto mutagenesis method (Storici and Resnick, 2006) and confirmed by dideoxy sequencing. LRB341 and LRB345 strains harboring yck1Δ and temperature-sensitive yck2ts alleles were graciously provided by Dr. Lucy Robinson (Robinson et al., 1993). Plasmids used in this study for kinase deletion screening (pRS316-CUP1-HA-STE18) were graciously provided (a gift from T. Chernova). The detailed method of strain construction is provided in the Supplemental Information.
Yeast Cell Culture and Treatments
Yeast strains were grown in YPD growth medium (yeast extract, peptone, 2% dextrose media), unless otherwise noted. All experiments were conducted with log-phase cells with an optical density at 600 nm (OD600) between 0.75 and 0.85 and stimulated with α-factor peptide hormone (GenScript) at a 3-μM final concentration, if required. Cells were harvested with trichloroacetic acid (5% final v/v) and frozen at −80°C.
For kinase screening, deletion strains carrying the pRS316-CUP1-HA-STE18 plasmid were grown in synthetic media lacking uracil and other appropriate amino acids as necessary. Expression of HA-STE18 was induced by 100 μM copper sulfate, and cells were stimulated as described earlier. The detailed procedure is provided in the Supplemental Information.
Cell Extracts and Immunoblotting
Proteins were extracted by glass bead lysis in trichloroacetic acid (TCA) as previously described (Lee and Dohlman, 2008). Protein concentration was determined by the DC Protein Assay (Bio-Rad). Protein extracts were resolved by 7.5% or 12.5% SDS-PAGE and immunoblotted with epitope-specific antibodies (see entire list in the Supplemental Information). Note that the current lot of Phospho-p44/42 MAPK antibody (Cell Signaling Technologies catalog #9101) exhibits reduced detection sensitivity for activated Fus3 compared to activated Kss1, which affects the ability to detect Fus3 activation in the very early stages of a pheromone response (i.e., before 1 min). Horseradish-peroxidase (HRP)-conjugated secondary antibodies (goat-anti-rabbit, goat-anti-mouse, or rabbit-anti-goat) were used for the detection of reactant bands by chemiluminescence with ECL reagent (PerkinElmer catalog #NEL 104001EA). Immunoblots were quantified by high-resolution scanning and pixel densitometry using ImageJ software (Schindelin et al., 2015).
Morphological Response Assay
The morphological response of STE18/STE5 mutants to α-factor was measured as described previously (Coyle et al., 2013; Malleshaiah et al., 2010) and detailed in the Supplemental Information. The morphology of the cells was determined, 3 hr post-pheromone stimulation, by differential interference contrast (DIC) confocal microscopy using a PerkinElmer UltraVIEW spinning disk confocal microscope. The number of cells with mating projections was counted as a percentage of total.
Ste5-GFP Localization Assay
Live cells endogenously expressing either Ste5-GFP or Ste5ND-GFP were visualized by microscopy. To ensure visibility across all strains, we exposed cells to 10 μM α-factor followed immediately by deposition onto agar pads saturated with 30 μM α-factor. Once deposited, cells were monitored using a PerkinElmer UltraVIEW VoX spinning disk confocal microscope. A detailed procedure for image acquisition and quantification is provided as Supplemental Information.
CoIP
Cells endogenously expressing different combinations of wild-type or mutant hemagglutinin epitope (HA)-STE18 and STE5-GFP were treated with 10 μM pheromone for 20 min followed by cell lysis and coIP using anti-GFP monoclonal antibody (mAb)-agarose (MBL International, #D153-8). Eluted proteins were separated by SDS-PAGE and immunoblotting with anti-GFP (Invitrogen, A11122) and anti-HA (Supplemental Information).
Phylogenetic Analysis
Ascomycota protein sequences were retrieved from the Broad Institute Fungal Orthogroups Repository (https://portals.broadinstitute.org/regev/orthogroups/). Multiple sequence alignments were achieved using MUSCLE with default parameters (Edgar, 2004). Phylogenetic and graphical analyses of Ste18 and Ste5 sequence alignments were achieved using Unipro UGENE software (Okonechnikov et al., 2012). Bootstrap consensus trees were prepared using 100 (shown) and 500 (not shown) iterations. Presence or absence of Ste5 or the Ste5FBD was determined from a combination of sequence alignment homology and previous phylogenetic analyses of Ste5 (Coyle et al., 2013).
Statistical Analysis
Statistical analysis for quantifying immunoblots and microscopy data was achieved using Prism software, v6/7 (GraphPad Software). Statistical significance was determined by ANOVAs.
Supplementary Material
Highlights.
Yeast Gγ/Ste18 phosphorylation in response to GPCR activation requires Fus3
Ste18/Ste5 phosphorylation represses the rate and amplitude of Fus3 activation
Ste18/Ste5 phosphorylation controls the bulk rate of Ste5/plasma membrane association
Ste18 and Ste5 phosphorylation, together, control switch-like mating in yeast
Acknowledgments
We would like to give special thanks to Aaron Lifland and the GT EBB1 microscopy core facility for support with confocal microscopy experiments; Prof. Kirill Lobachev and Ziwei Sheng for graciously providing yeast deletion strains; Joseph Lachance for guidance in phylogenetic analysis; Dr. Tatiana Chernova for the CUP1 yeast expression plasmid; and Dr. Lucy Robinson for temperature-sensitive YCK1/2 yeast strains. This work was supported by NIH grants R01GM117400 and R00GM094533 to M.P.T. and by startup funds to M.P.T. provided by the Georgia Institute of Technology.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.135.
AUTHOR CONTRIBUTIONS
S.C. conducted experiments; S.C. and P.B.-M. constructed plasmids and yeast strains; and M.P.T. and S.C. wrote the manuscript.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Arkowitz RA. Chemical gradients and chemotropism in yeast. Cold Spring Harb Perspect Biol. 2009;1:a001958. doi: 10.1101/cshperspect.a001958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI. Comparative chemosensation from receptors to ecology. Nature. 2006;444:295–301. doi: 10.1038/nature05402. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya RP, Reményi A, Good MC, Bashor CJ, Falick AM, Lim WA. The Ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science. 2006;311:822–826. doi: 10.1126/science.1120941. [DOI] [PubMed] [Google Scholar]
- Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE. Insights into G protein structure, function, and regulation. Endocr Rev. 2003;24:765–781. doi: 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- Cappell SD, Baker R, Skowyra D, Dohlman HG. Systematic analysis of essential genes reveals important regulators of G protein signaling. Mol Cell. 2010;38:746–757. doi: 10.1016/j.molcel.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement ST, Dixit G, Dohlman HG. Regulation of yeast G protein signaling by the kinases that activate the AMPK homolog Snf1. Sci Signal. 2013;6:ra78. doi: 10.1126/scisignal.2004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook LA, Schey KL, Wilcox MD, Dingus J, Hildebrandt JD. Heterogeneous processing of a G protein gamma subunit at a site critical for protein and membrane interactions. Biochemistry. 1998;37:12280–12286. doi: 10.1021/bi980230e. [DOI] [PubMed] [Google Scholar]
- Coyle SM, Flores J, Lim WA. Exploitation of latent allostery enables the evolution of new modes of MAP kinase regulation. Cell. 2013;154:875–887. doi: 10.1016/j.cell.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deflorio R, Brett ME, Waszczak N, Apollinari E, Metodiev MV, Dubrovskyi O, Eddington D, Arkowitz RA, Stone DE. Phosphorylation of Gβ is crucial for efficient chemotropism in yeast. J Cell Sci. 2013;126:2997–3009. doi: 10.1242/jcs.112797. [DOI] [PubMed] [Google Scholar]
- Dewhurst HM, Choudhury S, Torres MP. Structural analysis of PTM hotspots (SAPH-ire)–a quantitative informatics method enabling the discovery of novel regulatory elements in protein families. Mol Cell Proteomics. 2015;14:2285–2297. doi: 10.1074/mcp.M115.051177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohlman HG, Thorner JW. Regulation of G protein-initiated signal transduction in yeast: paradigms and principles. Annu Rev Biochem. 2001;70:703–754. doi: 10.1146/annurev.biochem.70.1.703. [DOI] [PubMed] [Google Scholar]
- Dohlman HG, Thorner J, Caron MG, Lefkowitz RJ. Model systems for the study of seven-transmembrane-segment receptors. Annu Rev Biochem. 1991;60:653–688. doi: 10.1146/annurev.bi.60.070191.003253. [DOI] [PubMed] [Google Scholar]
- Dohlman HG, Song J, Ma D, Courchesne WE, Thorner J. Sst2, a negative regulator of pheromone signaling in the yeast Saccharomyces cerevisiae: expression, localization, and genetic interaction and physical association with Gpa1 (the G-protein alpha subunit) Mol Cell Biol. 1996;16:5194–5209. doi: 10.1128/mcb.16.9.5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowell SJ, Bishop AL, Dyos SL, Brown AJ, Whiteway MS. Mapping of a yeast G protein betagamma signaling interaction. Genetics. 1998;150:1407–1417. doi: 10.1093/genetics/150.4.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elion EA, Satterberg B, Kranz JE. FUS3 phosphorylates multiple components of the mating signal transduction cascade: evidence for STE12 and FAR1. Mol Biol Cell. 1993;4:495–510. doi: 10.1091/mbc.4.5.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- Good M, Tang G, Singleton J, Reményi A, Lim WA. The Ste5 scaffold directs mating signaling by catalytically unlocking the Fus3 MAP kinase for activation. Cell. 2009;136:1085–1097. doi: 10.1016/j.cell.2009.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao N, Nayak S, Behar M, Shanks RH, Nagiec MJ, Errede B, Hasty J, Elston TC, Dohlman HG. Regulation of cell signaling dynamics by the protein kinase-scaffold Ste5. Mol Cell. 2008;30:649–656. doi: 10.1016/j.molcel.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye C, Dhillon N, Durfee T, Zambryski PC, Thorner J. Mutational analysis of STE5 in the yeast Saccharomyces cerevisiae: application of a differential interaction trap assay for examining protein-protein interactions. Genetics. 1997;147:479–492. doi: 10.1093/genetics/147.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom DG, Sridharan V, Baker R, Clement ST, Smalley DM, Dohlman HG. Protons as second messenger regulators of G protein signaling. Mol Cell. 2013;51:531–538. doi: 10.1016/j.molcel.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–994. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kisselev O, Ermolaeva M, Gautam N. Efficient interaction with a receptor requires a specific type of prenyl group on the G protein gamma subunit. J Biol Chem. 1995;270:25356–25358. doi: 10.1074/jbc.270.43.25356. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Dohlman HG. Coactivation of G protein signaling by cell-surface receptors and an intracellular exchange factor. Curr Biol. 2008;18:211–215. doi: 10.1016/j.cub.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li E, Cismowski MJ, Stone DE. Phosphorylation of the pheromone-responsive Gbeta protein of Saccharomyces cerevisiae does not affect its mating-specific signaling function. Mol Gen Genet. 1998;258:608–618. doi: 10.1007/s004380050774. [DOI] [PubMed] [Google Scholar]
- Maeder CI, Hink MA, Kinkhabwala A, Mayr R, Bastiaens PI, Knop M. Spatial regulation of Fus3 MAP kinase activity through a reaction-diffusion mechanism in yeast pheromone signalling. Nat Cell Biol. 2007;9:1319–1326. doi: 10.1038/ncb1652. [DOI] [PubMed] [Google Scholar]
- Malleshaiah MK, Shahrezaei V, Swain PS, Michnick SW. The scaffold protein Ste5 directly controls a switch-like mating decision in yeast. Nature. 2010;465:101–105. doi: 10.1038/nature08946. [DOI] [PubMed] [Google Scholar]
- Morishita R, Nakayama H, Isobe T, Matsuda T, Hashimoto Y, Okano T, Fukada Y, Mizuno K, Ohno S, Kozawa O, et al. Primary structure of a gamma subunit of G protein, gamma 12, and its phosphorylation by protein kinase C. J Biol Chem. 1995;270:29469–29475. doi: 10.1074/jbc.270.49.29469. [DOI] [PubMed] [Google Scholar]
- Muntz KH, Sternweis PC, Gilman AG, Mumby SM. Influence of gamma subunit prenylation on association of guanine nucleotide-binding regulatory proteins with membranes. Mol Biol Cell. 1992;3:49–61. doi: 10.1091/mbc.3.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagiec MJ, McCarter PC, Kelley JB, Dixit G, Elston TC, Dohlman HG. Signal inhibition by a dynamically regulated pool of monophosphorylated MAPK. Mol Biol Cell. 2015;26:3359–3371. doi: 10.1091/mbc.E15-01-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nern A, Arkowitz RA. A Cdc24p-Far1p-Gbetagamma protein complex required for yeast orientation during mating. J Cell Biol. 1999;144:1187–1202. doi: 10.1083/jcb.144.6.1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M UGENE team. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012;28:1166–1167. doi: 10.1093/bioinformatics/bts091. [DOI] [PubMed] [Google Scholar]
- Robinson LC, Menold MM, Garrett S, Culbertson MR. Casein kinase I-like protein kinases encoded by YCK1 and YCK2 are required for yeast morphogenesis. Mol Cell Biol. 1993;13:2870–2881. doi: 10.1128/mcb.13.5.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman HA, Koch WJ, Lefkowitz RJ. Seven-transmembrane-spanning receptors and heart function. Nature. 2002;415:206–212. doi: 10.1038/415206a. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev. 2015;82:518–529. doi: 10.1002/mrd.22489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi B, Kelstrup CD, Stoehr G, Fröhlich F, Walther TC, Olsen JV. Global analysis of the yeast osmotic stress response by quantitative proteomics. Mol Biosyst. 2009;5:1337–1346. doi: 10.1039/b902256b. [DOI] [PubMed] [Google Scholar]
- Sprang SR. G protein mechanisms: insights from structural analysis. Annu Rev Biochem. 1997;66:639–678. doi: 10.1146/annurev.biochem.66.1.639. [DOI] [PubMed] [Google Scholar]
- Stone DE, Cole GM, de Barros Lopes M, Goebl M, Reed SI. N-myristoylation is required for function of the pheromone-responsive G alpha protein of yeast: conditional activation of the pheromone response by a temperature-sensitive N-myristoyl transferase. Genes Dev. 1991;5:1969–1981. doi: 10.1101/gad.5.11.1969. [DOI] [PubMed] [Google Scholar]
- Storici F, Resnick MA. The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 2006;409:329–345. doi: 10.1016/S0076-6879(05)09019-1. [DOI] [PubMed] [Google Scholar]
- Strickfaden SC, Winters MJ, Ben-Ari G, Lamson RE, Tyers M, Pryciak PM. A mechanism for cell-cycle regulation of MAP kinase signaling in a yeast differentiation pathway. Cell. 2007;128:519–531. doi: 10.1016/j.cell.2006.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Pryciak PM. Membrane localization of scaffold proteins promotes graded signaling in the yeast MAP kinase cascade. Curr Biol. 2008;18:1184–1191. doi: 10.1016/j.cub.2008.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Drogen F, Stucke VM, Jorritsma G, Peter M. MAP kinase dynamics in response to pheromones in budding yeast. Nat Cell Biol. 2001;3:1051–1059. doi: 10.1038/ncb1201-1051. [DOI] [PubMed] [Google Scholar]
- Wang Y, Marotti LA, Jr, Lee MJ, Dohlman HG. Differential regulation of G protein alpha subunit trafficking by mono- and poly-ubiquitination. J Biol Chem. 2005;280:284–291. doi: 10.1074/jbc.M411624200. [DOI] [PubMed] [Google Scholar]
- Wettschureck N, Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- Winters MJ, Lamson RE, Nakanishi H, Neiman AM, Pryciak PM. A membrane binding domain in the ste5 scaffold synergizes with gbetagamma binding to control localization and signaling in pheromone response. Mol Cell. 2005;20:21–32. doi: 10.1016/j.molcel.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Lindorfer MA, Myung CS, Garrison JC. Phosphorylation of the G protein gamma12 subunit regulates effector specificity. J Biol Chem. 1998;273:21958–21965. doi: 10.1074/jbc.273.34.21958. [DOI] [PubMed] [Google Scholar]
- Yu L, Qi M, Sheff MA, Elion EA. Counteractive control of polarized morphogenesis during mating by mitogen-activated protein kinase Fus3 and G1 cyclin-dependent kinase. Mol Biol Cell. 2008a;19:1739–1752. doi: 10.1091/mbc.E07-08-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RC, Pesce CG, Colman-Lerner A, Lok L, Pincus D, Serra E, Holl M, Benjamin K, Gordon A, Brent R. Negative feedback that improves information transmission in yeast signalling. Nature. 2008b;456:755–761. doi: 10.1038/nature07513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalatan JG, Coyle SM, Rajan S, Sidhu SS, Lim WA. Conformational control of the Ste5 scaffold protein insulates against MAP kinase misactivation. Science. 2012;337:1218–1222. doi: 10.1126/science.1220683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan XL, Deschenes RJ, Guan KL. Differential regulation of FUS3 MAP kinase by tyrosine-specific phosphatases PTP2/PTP3 and dual-specificity MSG5 in Saccharomyces cerevisiae. Genes Dev. 1997;11:1690–1702. doi: 10.1101/gad.11.13.1690. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.