

Abstract
Background:
Mitochondrial dysfunction is increasingly recognized as an important feature of multiple sclerosis (MS) pathology and may be relevant for clinical disease progression. However, it is unknown whether mitochondrial DNA (mtDNA) levels in the cerebrospinal fluid (CSF) associate with disease progression and therapeutic response.
Objectives:
To evaluate whether CSF concentrations of mtDNA in MS patients can serve as a marker of ongoing neuropathology and may be helpful to differentiate between MS disease subtypes. To explore the effect of disease-modifying therapies on mtDNA levels in the CSF.
Methods:
CSF mtDNA was measured using a digital polymerase chain reaction (PCR) CSF mtDNA in two independent MS cohorts. The cohorts included 92 relapsing-remitting multiple sclerosis (RRMS) patients, 40 progressive multiple sclerosis (PMS) patients (27 secondary progressive and 13 primary progressive), 50 various neurologic disease controls, and 5 healthy controls.
Results:
Patients with PMS showed a significant increase in CSF mtDNA compared to non-inflammatory neurologic disease controls. Patients with higher T2 lesion volumes and lower normalized brain volumes showed increased concentration of mtDNA. Patients treated with fingolimod had significantly lower mtDNA copy levels at follow-up compared to baseline.
Conclusion:
Our results showed a non-specific elevation of concentration of mtDNA in PMS patients. mtDNA concentrations respond to fingolimod and may be used to monitor biological effect of this treatment.
Keywords: Multiple sclerosis, biomarkers, mitochondrial DNA, cerebrospinal fluid, digital PCR
Introduction
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disorder of the central nervous system (CNS) and is characterized by a high degree of heterogeneity in progression and treatment response. To date, validated and discriminative body fluid biomarkers reflecting and predicting natural disease progression, crucial for monitoring patients in clinical trials, are lacking.1,2 In addition, a biomarker guiding treatment decisions in early disease would be very useful in daily clinical practice. Cerebrospinal fluid (CSF) is an accessible source of CNS-derived products and its composition can reflect molecular changes occurring in the CNS.
In recent years, impaired mitochondrial function is increasingly recognized as a key pathological hallmark of MS.3,4 Demyelination leads to an increase in energy demand in order to maintain an appropriate intra-axonal ion balance and could thereby affect the number, transport, and activity of mitochondria.5–8 Indeed, the number of mitochondria is highly increased in chronically demyelinated axons as well as in reactive astrocytes5,7 and extensive neuronal mitochondrial DNA (mtDNA) deletions have been observed in MS cortical brain samples.9 Furthermore, significantly higher neuronal mtDNA copy numbers were found in MS normal-appearing gray matter compared to gray matter of non-neurological disease controls.10 Mitochondria contain circular DNA (mtDNA) encoding 37 genes,11 which is more resistant to degradation by nucleases than nuclear DNA and can thereby be detected in CSF. A significant decrease in circulating cell-free mtDNA in CSF samples of both asymptomatic patients at risk of Alzheimer’s disease (AD) and symptomatic AD patients was observed12,13 as well as in Parkinson’s disease (PD) patients.14 In contrast, CSF mtDNA concentration is markedly enhanced in children with traumatic brain injury, suggesting that mtDNA can be released due to acute cellular degeneration.15 So far, it is unknown if CSF mtDNA concentrations are related to MS pathology. Based on the observations that mitochondrial dysfunction plays a crucial role in MS pathology and the possible role of mitochondrial dysfunction in clinical disease progression, we here explored the potential of mtDNA levels in the CSF as a candidate biomarker for identifying patients with progressive disease. We also explored the effect of disease-modifying therapies on free mtDNA levels in longitudinally obtained CSF samples in a Swedish cohort.
Subjects and methods
Patients and controls: Dutch cohort
Patients were recruited either in response to an appeal in the periodical of the Dutch MS society or from patients visiting or admitted at our clinic. Patients were invited to voluntarily undergo a lumbar puncture, magnetic resonance imaging (MRI), and clinical testing in the period from September 2000 to November 2005.16 We included all patients with extensive clinical and radiological data and CSF storage within 2 hours after collection. MS patients were diagnosed according to the Poser criteria.17 Patients were classified as having a relapsing-remitting multiple sclerosis (RRMS), secondary progressive multiple sclerosis (SPMS), or primary progressive multiple sclerosis (PPMS) disease course according to the criteria of Lublin and Reingold.18 In this study, we also combined SPMS and PPMS patients in a progressive MS group (PMS). The control group was divided into non-inflammatory neurologic disease controls (NINDC) or inflammatory neurologic disease controls (INDC).19
Patients and controls: Swedish cohort
The prospective Swedish cohort consisted of longitudinally collected CSF samples in patients on disease-modifying treatment (DMT).
CSF samples were collected during routine visits to the neurology clinic at Karolinska University Hospital beginning from the launch of fingolimod (Novartis Pharma AG, Basel, Switzerland) in Sweden 2011. The sampling period for cases was from April 2011 to May 2015 and for controls from February 2014 to December 2014. The MS cohort consisted of RRMS patients with confirmed disease, where a repeated CSF sampling was indicated, for example, to exclude diseases like Lyme’s disease. In addition, RRMS patients starting fingolimod were invited to donate a CSF sample at baseline and at 6–12 months. All MS patients fulfilled the 2010 revised McDonald criteria.1 There were no significant concomitant diseases, such as infections, and corticosteroids had not been given within 3 months of sampling.
Clinical examination: Dutch and Swedish cohorts
Patients (with MS) underwent clinical examination prior to lumbar puncture, including the Expanded Disability Status Scale (EDSS),20 which was performed by trained medical doctors. All participants provided written informed consent and the local ethics committee approved the study. Laboratory personnel were blinded for clinical and radiological data at the time of CSF analysis.
CSF collection: Dutch cohort
CSF was obtained by lumbar puncture between the L3/L4 and L4/L5 intervertebral space and collected in polypropylene tubes (Sarstedt, Nümbrecht, Germany). CSF samples were centrifuged within 2 hours at 1800g at room temperature (RT) for 10 minutes and stored at −80°C in aliquots of 0.5 or 1 mL in Sarstedt polypropylene tubes according to BioMS-eu guidelines.21
CSF collection: Swedish cohort
Samples were centrifuged immediately after lumbar puncture at 440g for 10 minutes at RT to separate cells from the CSF supernatant. The supernatants were subsequently batched and stored at −80°C until use according to BioMS-eu guidelines.21
MRI acquisition and analysis: Dutch cohort
MRI examination was performed within 3 weeks of CSF collection and only available for the Dutch group. MRI scanning was performed at 1.0 T (Siemens Magnetom Impact, Erlangen, Germany), as earlier described22 (see supplementary text—MRI protocol).
Cell-free mtDNA analysis
The concentration of cell-free circulating mtDNA in CSF was measured directly in 4.5 µL of unpurified CSF by droplet digital polymerase chain reaction (ddPCR) in a QX200 platform (Bio-Rad, Hercules, CA, USA) following the digital PCR MIQE guidelines23 and see the supplementary text about primers. To avoid contamination of CSF with cells that could be a source of cellular mtDNA, we used simultaneous detection of mtDNA and the Bcl-2-associated X (BAX) gene (a single copy nuclear gene) in a multiplex ddPCR reaction. The sequence of the BAX probe is: 6-carboxy-2′,4,4′,5′,7,7′-hexachlorofluorescein HEX-CCCGAGCTGGCCCTGGACCCGGT-BHQ1. Primer sequences targeting BAX gene are Forward: TTCATCCAGGATCGAGCAGG and Reverse: TGAGACACTCGCTCAGCTTC. These primers amplify a single amplicon of 103 base pairs corresponding to apoptosis regulator BAX isoform alpha. We confirmed that either single or multiplex assay of mtDNA-85 and BAX-103 using the procedures described above yields the same results. The ddPCR multiplex assay was performed in a 20 µL reaction volume consisting of 1× ddPCR Supermix for probes (BioRad186-3023), 900 nM forward and reverse primers, 100 nM mtDNA FAM labeled probe, 300 nM BAX HEX labeled probe, and 4.5 µL of unprocessed CSF sample. A volume of double-distilled water equivalent to CSF sample was used as non-template control (NTC). At least two NTCs were included in each ddPCR 96-well plate. If the nuclear BAX gene copies/µL is higher than 1, this indicates contamination of CSF with mtDNA from cells and the CSF samples that contained one or more copies of BAX were discarded from this study. The detection limit of nuclear gene using our ddPCR method is 0.5 cells/µL and more sensitive than CSF cell counts.
Droplet emulsion formation was performed by mixing the 20 µL reaction with 70 µL droplet generation oil using a microfluidic droplet generation cartridge and QX200 Droplet Generator. End point PCR amplifications were performed using C1000 Thermal Cycler with the following conditions: 95°C 10 minutes; 40 cycles of 94°C, 30 seconds and 60°C 1 minute; 98°C 10 minutes. The presence or absence of amplification per droplet was evaluated using QX200 Droplet Reader and analyzed using QuantaSoft protocol. The results were expressed in copies of mtDNA/µL CSF.
The accuracy and linearity of the reaction conditions of this ddPCR method to quantify mtDNA content in CSF has been thoroughly characterized in previous work (linearity of this reaction is 0.999 and the dynamic range is 4–3000 copies/µL).13
Statistical analysis
For statistical analysis, SPSS version 22.0 (Windows) was used. The concentration of mtDNA copies was not normally distributed and therefore a natural logarithm was taken. For correlation analyses, subjects were divided into different groups according to their lesion volumes and normal brain volumes based on a median split. We had to make subgroups for the volume-related comparisons due to non-linearity of the data.
Based on EDSS scores, patients were divided into three categorical groups (EDSS: 0–3.5, 4.0–5.5, 6–10). The three groups largely reflect, respectively, patients with no/limited, moderate, and severe walking impairment.
Differences between the distinct groups were analyzed using linear regression. All results were corrected for disease duration as a confounder. In all statistical analyses, age and disease duration were both relevant confounders, but due to collinearity, only disease duration was included as a covariate (as this was the strongest confounder).
To compare baseline and follow-up mtDNA copies in the fingolimod-RRMS group, a Wilcoxon signed rank test was performed. A p-value <0.05 was considered significant for main effects.
Results
Dutch cohort: mtDNA concentration is significantly increased in progressive MS cases
A total of 120 patients (50 RRMS patients, 40 progressive MS patients (27 SPMS patients, 13 PPMS patients), 23 NIND, and 7 IND controls) were included in the study. The study group consisted of 64 women and 56 men with a mean age of 45.5 ± 11.0 years. The median disease duration, calculated as the time lapse between the onset of neurological symptoms and the time of the lumbar puncture, was 6.5 (interquartile range (IQR), 0.02–38.77) years. See Table 1 for detailed patient characteristics.
Table 1.
Patient characteristics and MRI measures, Dutch cohort.
| All (n = 120) | RR (n = 50) | SP (n = 27) | PP (n = 13) | NINDC (n = 23) | INDC (n = 7) | |
|---|---|---|---|---|---|---|
| M:F | 56:64 | 19:31 | 15:12 | 8:5 | 10:13 | 4:3 |
| Age (years) | 45.5 ± 11.0 | 41.4 ± 9.6 | 49.6 ± 7.6 | 50.4 ± 5.3 | 47.5 ± 15.1 | 43.3 ± 15.1 |
| EDSS | – | 3.0 (2.5–4) | 6.0 (4–7) | 4.0 (3.5–6) | – | – |
| Disease duration (years) | 6.5 (2.3–6.5) | 6.5 (2.9–13.7) | 19.3 ± 8.7 | 10.2 (3.8–19.7) | 1.14 (0.3–2.8) | 0.07 (0.02–2.8) |
| DMT:NODMT | – | 19:31 | 4:23 | 0:0 | – | – |
| T2LV | – | 2.79 (0.99–9.23) | 7.59 (3.67–15.23) | 3.73 (1.19–9.62) | – | – |
| BHLV | – | 0.14 (0.06–0.41) | 0.39 (0.06–1.56) | 0.53 (0.15–2.92) | – | – |
| NBV | – | 1.25 (1.16–1.31) | 1.16 (1.10–1.21) | 1.27 (1.17–1.34) | – | – |
| Gado+ | – | 8:33 | 4:23 | 2:8 | – | – |
All: all subjects; BHLV: black hole lesion volume; MRI: magnetic resonance imaging; DMT: disease-modifying treatment; EDSS: Expanded Disability Status Scale; F: female; Gado+: gadolinium-enhanced lesions; INDC: inflammatory neurologic disease controls; IQR: interquartile range; M: male; n: number of subjects; NBV: normal brain volume; NINDC: non-inflammatory neurologic disease controls; NODMT: no use of disease-modifying treatment; PP: primary progressive; RR: relapsing-remitting; SD: standard deviation; SP: secondary progressive; T2LV: T2 lesion volume.
Values are reported as mean ± SD or median (IQR) (Dutch cohort).
Figure 1 shows the relative increase in the concentration of mtDNA copies for each subtype (Supplementary Figure 1 shows the subtypes with SPMS and PPMS combined as PMS). SPMS and PPMS patients showed a significant increased concentration of circulating cell-free mtDNA in the CSF compared to NINDC (ratio = 3.52, p = 0.01 and ratio = 2.85, p = 0.05, respectively). Patients with RRMS showed a trend toward lower concentrations of mtDNA compared to SPMS patients (ratio = 0.49, p = 0.06; Supplementary Table 1). Patients with progressive MS (PMS) showed a significant increased concentration of circulating cell-free mtDNA in CSF compared to NINDC (ratio = 3.23, p = 0.01). There was a trend for increasing concentrations comparing PMS patients with RRMS patients (ratio = 1.87, p = 0.05). mtDNA concentration did not differ between PMS patients and INDC (ratio = 2.66, p = 0.12; Supplementary Table 2).
Figure 1.

Comparison of mtDNA levels between the subtypes (Dutch cohort).
Scatter plot representing mtDNA concentrations for each individual grouped by diagnosis. The horizontal lines correspond to median and interquartile range. NINDC: median 13 (IQR, 5–35) copies/µL; INDC: median 9 (IQR, 7–42) copies/µL; RRMS: median 22 (IQR, 8–65) copies/µL; SPMS: median 35(IQR, 9–146); PPMS: median 22 (IQR, 7.5–247.5) copies/µL. *p = 0.01, based on linear regression, adjusted for disease duration. **p = 0.05, based on linear regression, adjusted for disease duration. INDC: inflammatory neurologic disease controls; NINDC: non-inflammatory neurologic disease controls; PMS: progressive multiple sclerosis; RRMS: relapsing-remitting multiple sclerosis.
Validation
To validate the ddPCR results, we re-analyzed the high level samples with real-time quantitative polymerase chain reaction (qPCR). The data shown in Supplementary Figure 2A and 2B show a strong significant correlation between ddPCR and real-time qPCR for the quantification of mtDNA in CSF samples from control and the MS subtypes (r = 0.93, p < 0.05).
Moreover, there was no correlation between CSF leukocyte count and mtDNA copies (Spearman’s rho = 0.37, p-value > 0.05), indicating that high levels of mtDNA copies were not a consequence of cell contamination. Finally, previous studies have shown that there was no correlation between mtDNA content in CSF and the presence of 14-3-3 protein, a cytoplasmic protein that would be increased in the CSF if samples were contaminated with cells.13
DMT: Dutch cohort
A total of 19 RRMS patients used DMT (all interferon-β) at the time of lumbar puncture. There was no significant difference in the concentration of mtDNA copies between the groups with and without DMT (ratio = 1.12, p = 0.624). The median CSF mtDNA concentration was 16 copies/µL (IQR, 7.75–65.25) in the group without DMT and 18 copies/µL (IQR, 12–77) in the interferon using groups.
mtDNA copy numbers in relation to EDSS: Dutch cohort
There was no significant correlation between EDSS scores and the number of mtDNA copies/µL, although a trend (p = 0.08) for a positive correlation was observed when dividing patients into different groups (Category 0–3.5 n = 39, category 4–5.5 n = 30, category 6–10 n = 21; see Supplementary Table 3).
mtDNA copy numbers in relation to MRI outcomes in MS patients: Dutch cohort
To study a putative association of mtDNA concentrations and ongoing disease activity, we correlated mtDNA concentration levels in MS patients with radiological parameters, including T2 lesion volume, gadolinium-enhanced lesions, T1-hypointense lesion volumes (black hole lesion volume (BHLV)), and normalized brain volume (NBV) as a measure of atrophy.
The T2 lesion volumes were measured in 78 patients and divided into two groups based on the medians. Group 1 had a median T2 lesion volume of 1.53 (IQR, 0.13–4.30) mL and group 2 had a median T2 lesion volume of 10.53 (IQR, 6.55–16.96) mL. The group with higher T2 lesion volumes showed increased concentrations of mtDNA copies compared to group with lower T2 lesion volumes (ratio = 2.13, p = 0.03).
The cohort was divided into two groups based on the presence (n = 64) or absence (n = 14) of gadolinium-enhanced lesions on MRI. There was no significant difference in mtDNA concentrations between patients with or without gadolinium-enhanced lesions.
Black hole volumes were measured in 66 patients and divided into two groups using the median. Group 1 had a median BHLV volume of 0.063 (IQR, 0.03–0.12) mL and group 2 a median BHLV of 0.89 (IQR, 0.38–2.34) mL. There was no significant difference in mtDNA copy concentrations between these groups (ratio = 0.93, p = 0.85).
For 81 patients, the normal brain volume (NBV) was available and patients were divided into two groups based on the median. Group 1 had a median NBV 1.14 (IQR, 1.09–1.18) L and group 2 had a median NBV 1.29 (IQR, 1.22–1.33) L. Comparing these two groups, the group with higher NBV showed significant lower concentrations of mtDNA (ratio = 0.42, p = 0.02; Table 2).
Table 2.
MRI outcomes (Dutch cohort).
| Ratio | p-value | 95% confidence interval |
||
|---|---|---|---|---|
| Low bound | Upper bound | |||
| T2 lesion volumes | 2.13 | 0.03 | 1.08 | 4.18 |
| G+-enhanced lesions | 1.69 | 0.23 | 0.71 | 4.02 |
| Black hole lesion volumes | 0.93 | 0.85 | 0.44 | 1.96 |
| Normalized brain volumes | 0.42 | 0.02 | 0.24 | 0.90 |
MRI: magnetic resonance imaging; mtDNA: mitochondrial DNA; IQR: interquartile range; BHLV: black hole lesion volumes; NBV: normalized brain volumes.
The concentration of mtDNA copies was not normally distributed and therefore a natural logarithm was taken. For correlation analyses, subjects were divided into different groups according to their lesion volumes and normal brain volumes based on a median split. Using log transformation of the regression coefficients, ratios of the mitochondrial DNA concentrations between the different subgroups were calculated. Analyzing T2 lesion volumes, patients with higher T2 lesion volumes showed 2.13 times higher concentrations of mtDNA copies (p = 0.03). Patients with higher normalized brain volumes showed significant 0.42 times lower concentrations of mtDNA (p = 0.02).
T2 lesion volumes. Comparing group 2 with group 1. Group 1: median T2 lesion volume of 1.53 (IQR, 0.13–4.30) mL. Group 2: median T2 lesion volume of 10.53 (IQR 6.55–16.96) mL. Adjusted for disease duration.
Gadolinium-enhanced lesions. Comparing the group with the presence of gadolinium-enhanced lesions with the group with the absence of gadolinium-enhanced lesions. Adjusted for disease duration.
Black hole lesion volumes. Comparing group 2 with group 1. Group 1: median BHLV volume of 0.063 (IQR, 0.03–0.12) mL. Group 2: a median BHLV of 0.89 (IQR, 0.38–2.34) mL. Adjusted for disease duration.
Normalized brain volumes. Comparing group 2 with group 1. Group 1: median NBV 1.14 (1.09–1.18) L. Group 2: median NBV 1.29 (IQR, 1.22–1.33) L. Adjusted for disease duration and gender.
Analysis of cell-free mtDNA content in CSF in the Swedish cohort
A total of 42 RRMS patients, 20 other neurological disease controls (ONDC), and 5 healthy controls (HC) were included in the study. The RRMS patients were divided into two groups: 23 fingolimod-RRMS patients and 19 differently treated RRMS patients.
The control group consisted of 20 patients undergoing a diagnostic work-up for possible neuroinflammatory disease, but no pathological signs of inflammation were seen on MRI or with established markers of immune activation in the CSF (pleocytosis, oligoclonal bands, albumin quote, and increased IgG index). The diagnoses of this group were paresthesia (n = 6), pain syndromes (n = 5), psychiatric illness (n = 4), vertigo (n = 3), essential tremor (n = 1), Bell’s palsy (n = 1), and dysphasia (n = 1). In addition, five individuals served as HC. See Table 3 for further characteristics of the groups.
Table 3.
Patient characteristics, Swedish cohort.
| Fingolimod-RR (n = 23) | RRMS (n = 19) | ONDC (n = 20) | HC (n = 5) | |
|---|---|---|---|---|
| M:F | 7:16 | 5:14 | 7:13 | 3:2 |
| Age (years) | 42.4 ± 8.1 | 32.4 ± 8.1 | 34.15 ± 11.1 | 29.8 ± 2.9 |
| EDSS | 3.0 (2.0–3.5) | 2.0 (1.5–3.0) | – | – |
| Disease duration (years) | 10.3 ± 6.1 | 5.89 ± 4.3 | – | – |
| DMT:NODMT | 23:0 | 19:0 | – | – |
All: all subjects; EDSS: Expanded Disability Status Scale; F: female; HC: healthy controls; IQR: interquartile range; M: male; n: number of subjects; ONDC: other neurologic disease controls; RR: relapsing-remitting; RRMS: relapsing-remitting multiple sclerosis; DMT: disease-modifying treatment; NODMT: no use of disease-modifying treatment; SD: standard deviation.
Values are reported as mean ± SD or median (IQR) (Swedish cohort).
For fingolimod users, mtDNA copy number concentrations were almost 50% lower on follow-up (median = 9.6 copies/µL) compared to baseline (median = 17.9 copies/µL), z = −2.52, p = 0.012, r = −0.37 (see Figure 2 and see Supplementary Figure 3 for a graph with individual changes).
Figure 2.

The effect of fingolimod on mtDNA concentration levels (Swedish cohort). To compare baseline and follow-up mtDNA copies in the fingolimod relapsing-remitting multiple sclerosis group (n = 23), a Wilcoxon signed rank test was performed. The follow-up moment lies after 6–12 months.
The horizontal lines correspond to median and interquartile range. *p = 0.012 (z = −2.52, r = −0.37).
There was no significant difference in mtDNA copy levels in the fingolimod-RRMS patients (n = 23) compared to the RRMS group using other DMT (dimethyl fumarate: n = 7, interferon-beta: n = 12; ratio = 1.65, p = 0.089).
There were no significant differences in mtDNA copy levels between the RRMS, ONDC, and HC groups in this Swedish cohort (see Figure 3 and Supplementary Table 4).
Figure 3.
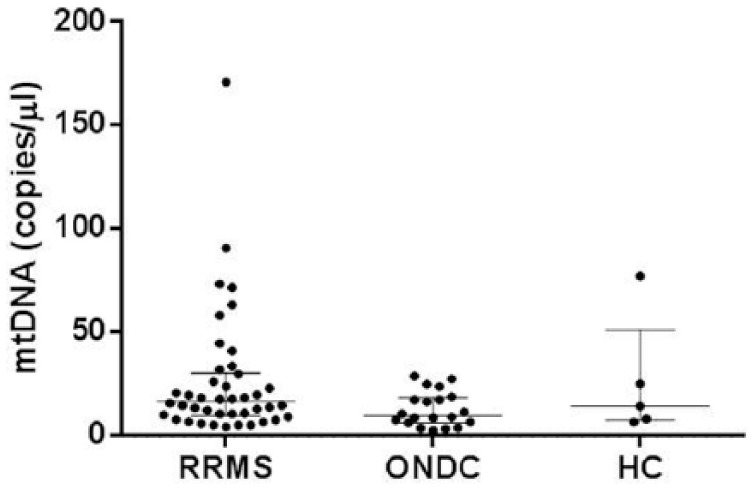
Comparison of mtDNA levels between the subtypes (Swedish cohort). Scatter plot representing mtDNA concentrations for each individual grouped by diagnosis.
The horizontal lines correspond to median and interquartile range. RRMS: median 16.8 (IQR, 9.9–30.5) copies/µL; ONDC: median 9.8 (IQR, 6.4–18.6) copies/µL; HC: median 14.3 (IQR, 7.5–51.3) copies/µL; HC: healthy controls; ONDC: other neurological disease controls; RRMS: relapsing-remitting multiple sclerosis.
Most of the RRMS patients had an EDSS between 0 and 3.5. Therefore, comparing the different EDSS categories, such as performed for the Dutch cohort, was not possible (category 0–3.5 n = 35, category 4–5.5 n = 3, category 6–10 n = 3, missing EDSS n = 1).
Discussion
The major finding of this study is that concentrations of free circulating mtDNA copies are increased in CSF of patients with progressive MS compared with non-inflammatory control patients. Also, there was a trend for a modest positive correlation with EDSS specifically in progressive MS patients. In addition, we showed that patients with a high T2 lesion volume displayed higher mtDNA concentrations compared to patients with a relative low T2 lesion volume. The group with lower normalized brain volumes showed higher mtDNA concentrations compared to patients with higher normal brain volumes, suggesting a positive correlation between the concentration of free circulating mtDNA copies and brain atrophy. Altogether, our data may suggest that increased concentrations of cell-free mtDNA are associated with MS disease activity and progressive disease.
Reduced levels of mtDNA have been reported in both AD and PD cases, and it has been speculated that a decrease in mtDNA might be a common phenomenon observed in neurodegenerative diseases. In contrast to this, elevated mtDNA levels as we found in PMS have also been detected in CSF samples from children with traumatic brain injury and were highly predictive of a poor outcome.15 This might suggest that high values of free circulating mtDNA in CSF can be seen as a potential biomarker of acute cellular and mitochondrial stress. It is nowadays widely accepted that neurodegeneration and concomitant brain atrophy are common pathological features of MS, particularly in the progressive phase of the disease. In MS, demyelination leads to an increase in axonal energy demand, which may superimpose effects of neurodegeneration in MS, which is a possible explanation for the higher concentrations of mtDNA in progressive MS in this study.
Enhanced mtDNA concentrations in the CSF correlated with high T2 lesion volumes and were inversely correlated with normal brain volume suggesting that the increased concentration of mtDNA is due to ongoing neuro-axonal damage which is known to be more extensive in progressive forms of MS.24,25 Even though longitudinal follow-up MRI scans are lacking, the cross-sectional correlations corroborate the finding of higher levels in clinical PMS groups.
Increased mtDNA levels in the CSF might not only reflect disease progression but also contribute to the disease process. mtDNA is a damage-associated molecular pattern (DAMP), which can bind to glial Toll-like receptor-9 and trigger an inflammatory response.26 Hence, it is conceivable that enhanced mtDNA levels in the CSF might elicit a glial immune response; however, additional research is needed to explore the functional effects of increased mtDNA concentrations in the CSF of MS patients.
The cellular origin of enhanced mtDNA levels in the CSF of progressive MS patients is yet unknown; however, it is conceivable that mtDNA is released upon neuro-axonal injury or oligodendrocyte damage, as these are prominent features of progressive MS. Alternatively, mtDNA might be secreted into the extracellular compartment by extracellular vesicles derived from distinct CNS cells, such as reactive astrocytes, particularly in lesions that are packed with mitochondria.
Interestingly, an almost 50% decrease in mtDNA copy number was found upon fingolimod treatment. Fingolimod is a sphingosine-1-phosphate (S1P) receptor modulator that limits the egress of lymphocytes from lymph nodes, preventing them from contributing to autoreactive inflammation in the CNS.27 Fingolimod crosses the blood–brain barrier and can also act on S1P receptors present on oligodendrocytes, oligodendrocyte precursor cells, astrocytes, microglial cells, and neurons.
We observed decreased levels of mtDNA copies in patients using fingolimod (compared to baseline), suggesting a possible role of mtDNA as a biomarker of fingolimod treatment response. It is likely that fingolimod reduces inflammation-mediated cellular damage and subsequent release of mtDNA. However, the precise mechanism underlying reduced mtDNA CSF levels upon fingolimod treatment warrants further investigations. Further comparisons with other disease modulatory treatments are needed in order to understand if this effect is specific for fingolimod or a generic response to reduced inflammation and concomitant CNS cell injury.
Altogether, combining our results of no significant difference between the concentration of mtDNA in CSF between RRMS and SPMS and the significantly decreased levels of mtDNA in CSF in RRMS patients plus the knowledge that fingolimod has been tested in progressive MS trials but with negative outcomes,28 our study showed a non-specific elevation of concentration of mtDNA in PMS patients.
The relatively small sample size and overlap in mtDNA concentrations between patient groups warrants cautious interpretation of our data. It is important to validate our findings in future research, including longitudinal clinical follow-up to further explore the prognostic potential of mtDNA analysis as a tentative biomarker for progressive MS and treatment response.
Supplementary Material
Acknowledgments
J.v.H. and C.E.T. contributed equally to this work.
Footnotes
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: C.E.L., P.P., R.T., L.B., M.D.S., and A.M.: no conflict of interest. F.P. has received unrestricted academic research grants from Biogen, Genzyme, and Novartis, and travel support and/or compensation for lectures and/or participation in advisory boards from Biogen, Merck Serono, Novartis, Genzyme, and Teva, which have been exclusively used for the support of research activities. B.M.J.U. has received personal compensation for consulting from Biogen Idec, Genzyme, Merck Serono, Novartis, Roche, and Teva. J.K. has accepted speaker and consulting fees from Merck Serono, Biogen Idec, Teva, Genzyme, and Novartis. J.v.H. performed contract research for Biogen Idec and GeNeuro. C.E.T. serves on the advisory board of Fujirebio and Roche; received research consumables from Euroimmun, IBL, Fujirebio, Invitrogen, and Mesoscale Discovery; performed contract research for IBL, Shire, Boehringer, Roche, and Probiodrug; and received grants from the European Commission, the Dutch Research Council (ZonMW), Association of Frontotemporal Dementia/Alzheimer’s Drug Discovery Foundation, ISAO, and the Alzheimer’s Drug Discovery Foundation.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work has been funded, in part, by the Ministerio de Economia y Competitividad of Spain (SAF2014-56644) and by CIBERNED grants to R.T. and P.P.
Contributor Information
Cyra E Leurs, Department of Neurology, MS Center Amsterdam, VU University Medical Center, Amsterdam, The Netherlands.
Petar Podlesniy, Institute of Biomedical Research of Barcelona, CSIC-IDIBAPS, CIBERNED, Barcelona, Spain.
Ramon Trullas, Institute of Biomedical Research of Barcelona, CSIC-IDIBAPS, CIBERNED, Barcelona, Spain.
Lisanne Balk, Department of Neurology, MS Center Amsterdam, VU University Medical Center, Amsterdam, The Netherlands.
Martijn D Steenwijk, Departments of Radiology and Nuclear Medicine, VU University Medical Center, Amsterdam, The Netherlands.
Arjan Malekzadeh, Neurochemistry Laboratory and Biobank, Department of Clinical Chemistry, Neuroscience Campus Amsterdam, VU University Medical Center, Amsterdam, The Netherlands.
Fredrik Piehl, Neuroimmunology Unit, Department of Clinical Neuroscience, Karolinska Institutet and Karolinska University Hospital, Stockholm, Sweden.
Bernard MJ Uitdehaag, Department of Neurology, MS Center Amsterdam, VU University Medical Center, Amsterdam, The Netherlands.
Joep Killestein, Department of Neurology, MS Center Amsterdam, VU University Medical Center, Amsterdam, The Netherlands.
Jack van Horssen, Department of Molecular Cell Biology and Immunology, VU University Medical Center, Amsterdam, The Netherlands.
CE Teunissen, Neurochemistry Laboratory and Biobank, Department of Clinical Chemistry, Neuroscience Campus Amsterdam, VU University Medical Center, Amsterdam, The Netherlands.
References
- 1. Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 2011; 69(2): 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014; 83(3): 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Witte ME, Mahad DJ, Lassmann H, et al. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol Med 2014; 20(3): 179–187. [DOI] [PubMed] [Google Scholar]
- 4. Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat Rev Neurol 2012; 8(11): 647–656. [DOI] [PubMed] [Google Scholar]
- 5. Witte ME, Bø L, Rodenburg RJ, et al. Enhanced number and activity of mitochondria in multiple sclerosis lesions. J Pathol 2009; 219(2): 193–204. [DOI] [PubMed] [Google Scholar]
- 6. Zambonin JL, Zhao C, Ohno N, et al. Increased mitochondrial content in remyelinated axons: Implications for multiple sclerosis. Brain 2011; 134(Pt 7): 1901–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mahad DJ, Ziabreva I, Campbell G, et al. Mitochondrial changes within axons in multiple sclerosis. Brain 2009; 132(5): 1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Horssen J, Witte ME, Ciccarelli O. The role of mitochondria in axonal degeneration and tissue repair in MS. Mult Scler 2012; 18(8): 1058–1067. [DOI] [PubMed] [Google Scholar]
- 9. Campbell GR, Ziabreva I, Reeve AK, et al. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann Neurol 2011; 69(3): 481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blokhin A, Vyshkina T, Komoly S, et al. Variations in mitochondrial DNA copy numbers in MS brains. J Mol Neurosci 2008; 35(3): 283–287. [DOI] [PubMed] [Google Scholar]
- 11. DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med 2003; 348(26): 2656–2668. [DOI] [PubMed] [Google Scholar]
- 12. Podlesniy P, Figueiro-Silva J, Llado A, et al. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann Neurol 2013; 74(5): 655–668. [DOI] [PubMed] [Google Scholar]
- 13. Podlesniy P, Llorens F, Golanska E, et al. Mitochondrial DNA differentiates Alzheimer’s disease from Creutzfeldt-Jakob disease. Alzheimers Dement 2016; 12(5): 546–555. [DOI] [PubMed] [Google Scholar]
- 14. Pyle A, Brennan R, Kurzawa-akanbi M, et al. Reduced CSF mitochondrial DNA is a biomarker for early-stage Parkinson’s disease. Ann Neurol 2015; 78: 1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walko TD, Bola RA, Hong JD, et al. Cerebrospinal fluid mitochondrial DNA: A novel DAMP in pediatric traumatic brain injury. Shock 2014; 41(6): 499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eikelenboom MJ, Petzold A, Lazeron RHC, et al. Multiple sclerosis: Neurofilament light chain antibodies are correlated to cerebral atrophy. Neurology 2003; 60(2): 219–223. [DOI] [PubMed] [Google Scholar]
- 17. Poser CM, Paty DW, Scheinberg L, et al. New diagnostic criteria for multiple sclerosis: Guidelines for research protocols. Ann Neurol 1983; 13(3): 227–231. [DOI] [PubMed] [Google Scholar]
- 18. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: Results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996; 46(4): 907–911. [DOI] [PubMed] [Google Scholar]
- 19. Teunissen CE, Menge T, Altintas A, et al. Consensus definitions and application guidelines for control groups in cerebrospinal fluid biomarker studies in multiple sclerosis. Mult Scler 2013; 19(13): 1802–1809. [DOI] [PubMed] [Google Scholar]
- 20. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983; 33(11): 1444–1452. [DOI] [PubMed] [Google Scholar]
- 21. Teunissen CE, Petzold A, Bennett JL, et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 2009; 73(22): 1914–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Teunissen CE, Koel-Simmelink MJ, Pham TV, et al. Identification of biomarkers for diagnosis and progression of MS by MALDI-TOF mass spectrometry. Mult Scler 2011; 17(7): 838–850. [DOI] [PubMed] [Google Scholar]
- 23. Huggett JF, Foy CA, Benes V, et al. The digital MIQE guidelines: Minimum information for publication of quantitative digital PCR experiments. Clin Chem 2013; 59(6): 892–902. [DOI] [PubMed] [Google Scholar]
- 24. Steenwijk MD, Geurts JJG, Daams M, et al. Cortical atrophy patterns in multiple sclerosis are non-random and clinically relevant. Brain 2016; 139: 115–126. [DOI] [PubMed] [Google Scholar]
- 25. Popescu V, Agosta F, Hulst HE, et al. Brain atrophy and lesion load predict long term disability in multiple sclerosis. J Neurol Neurosurg Psychiatry 2013; 84(10): 1082–1091. [DOI] [PubMed] [Google Scholar]
- 26. Zhang Q, Raoof M, Chen Y, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010; 464(7285): 104–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010; 362(5): 387–401. [DOI] [PubMed] [Google Scholar]
- 28. Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis (INFORMS): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016; 387(10023): 1075–1084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.