

Abstract
In this study we investigated nanoliposome as an approach to tailoring the pharmacology of cerivastatin as a disease-modifying drug for pulmonary arterial hypertension (PAH). Cerivastatin encapsulated liposomes with an average diameter of 98 ± 27 nm were generated by a thin film and freeze-thaw process. The nanoliposomes demonstrated sustained drug-release kinetics in vitro and inhibited proliferation of pulmonary artery (PA) smooth muscle cells with significantly less cellular cytotoxicity as compared with free cerivastatin. When delivered by inhalation to a rat model of monocrotaline-induced PAH, cerivastatin significantly reduced PA pressure from 55.13 ± 9.82 to 35.56 ± 6.59 mm Hg (P < 0.001) and diminished PA wall thickening. Echocardiography showed that cerivastatin significantly reduced right ventricle thickening (monocrotaline: 0.34 ± 0.02 cm vs. cerivastatin: 0.26 ± 0.02 cm; P < 0.001) and increased PA acceleration time (monocrotaline: 13.98 ± 1.14 milliseconds vs. cerivastatin: 21.07 ± 2.80 milliseconds; P < 0.001). Nanoliposomal cerivastatin was equally effective or slightly better than cerivastatin in reducing PA pressure (monocrotaline: 67.06 ± 13.64 mm Hg; cerivastatin: 46.31 ± 7.64 mm Hg vs. liposomal cerivastatin: 37.32 ± 9.50 mm Hg) and improving parameters of right ventricular function as measured by increasing PA acceleration time (monocrotaline: 24.68 ± 3.92 milliseconds; cerivastatin: 32.59 ± 6.10 milliseconds vs. liposomal cerivastatin: 34.96 ± 7.51 milliseconds). More importantly, the rate and magnitude of toxic cerivastatin metabolite lactone generation from the intratracheally administered nanoliposomes was significantly lower as compared with intravenously administered free cerivastatin. These studies show that nanoliposome encapsulation improved in vitro and in vivo pharmacologic and safety profile of cerivastatin and may represent a safer approach as a disease-modifying therapy for PAH.
Introduction
Pulmonary arterial hypertension (PAH) is a debilitating and life-threatening disease. Idiopathic PAH affects between 140,000 and 200,000 patients in the United States. The complex cellular and molecular pathology of PAH involves endothelial dysfunction, smooth muscle cell proliferation, vasoconstriction, inflammation, nitric oxide deficiency, and genetic mutations such as those observed in the bone morphogenic protein receptor 2 (BMPR2) pathway (Stacher et al., 2012; Guignabert and Dorfmuller, 2013) and Kruppel-like factor 2 (KLF2) (Eichstaedt et al., 2017). These cellular changes contribute to the remodeling of the pulmonary artery (PA) as well as the small vasculature in the lung, ultimately leading to increased pulmonary vascular resistance, right heart failure, and death (Naeije and Manes, 2014).
It is generally recognized that drugs that can target the multifactorial pathology of PAH would have greater probability of reducing disease progression as compared with the current drugs which largely target vasodilation. The current therapies such as prostanoids, endothelin receptor antagonists, or the phosphodiesterase type 5 inhibitors largely impact vasoconstriction pathology, which reduces the symptoms of disease by promoting vasodilation (Yao, 2012). These therapies do not reduce disease progression or produce regression, and only have a limited impact on mortality (Badesch et al., 2010; Benza et al., 2012).
Despite current therapies, the 5-year survival rate of PAH patients remains less than 60% (Farber et al., 2015). Therefore, recent research for new therapeutic modalities for the treatment of PAH has focused on identifying agents that can impact the multifactorial pathophysiology vascular remodeling in PAH.
Statins are one such class of drugs that have been shown to impact several pathologic pathways associated with PAH. Statins inhibit smooth muscle cell proliferation, improve endothelial function, and reduce inflammation and oxidative stress (Wolfrum et al., 2003; Liao and Laufs, 2005). Statins also promote expression of nitric oxide pathway genes (John et al., 2001) and KLF2 (Zhao et al., 2015). Several experimental studies have demonstrated that statins significantly reduce the development and progression of PAH (Nishimura et al., 2002; Sun and Ku, 2008) and reverse the course of disease in preclinical models (Nishimura et al., 2003). However, despite the strong mechanistic and preclinical data, the efficacy of statins in PAH patients was not achieved in clinical trials (Anand et al., 2016).
The observed efficacy of statins in preclinical models occurs at very high doses (Nishimura et al., 2002, 2003; Sun and Ku, 2008). In humans, such high doses would produce liver injury and rhabdomyolysis (Bellosta et al., 2004). Therefore, although the cellular biology and preclinical pharmacology of statins strongly support their novel therapeutic potential as disease-modifying drugs for PAH, the demonstration of efficacy in humans has not been possible using orally administered drugs. Thus, approaches that can modify the pharmacokinetic and pharmacodynamic properties of statins may provide opportunities for their therapeutic actions at sites other than the liver. In this study, we investigated a new approach to circumvent the limitations of developing statins as potential disease modifying therapy for PAH.
Our recent studies demonstrated that, as compared with other statins, cerivastatin was highly effective in reducing inflammatory cytokine monocyte chemoattractant protein-1 (MCP-1) and its receptors, and vascular regulator KLF2 (Zhao et al., 2015). Further cerivastatin was a potent inhibitor of smooth muscle cell proliferation. Previous studies have shown that cerivastatin produces significantly superior efficacy in preclinical models compared with other statins (Leslie et al., 2004; Ohkita et al., 2006). Cerivastatin was also found to produce greater improvement in the cardiac score in clinical trials when compared with other statins (Achenbach et al., 2002). However, cerivastatin has not been previously tested in preclinical models of PAH.
Based on these unique pharmacologic characteristics, we have tested the hypothesis that nanoliposome encapsulation and direct local delivery of cerivastatin may result in a favorable efficacy and metabolic profile with lowers risk of generating toxic lactone metabolites in the liver (Schirris et al., 2015). Our results showed that cerivastatin, and particularly sustained-release liposome-encapsulated cerivastatin, produced significant improvement in key markers of PAH and right heart function in a preclinical model of PAH. Furthermore, liposome delivery produced significantly lower levels of the lactone metabolite that is considered a safety risk at high statin doses (Schirris et al., 2015). These data suggest that direct pulmonary delivery of cerivastatin as a nanoliposome formulation could be a novel disease-modifying therapy to achieve desirable efficacy and safety.
Material and Methods
Materials.
Lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Cerivastatin was purchased from the St. Joseph Translational Research Institute (Atlanta, GA). Cerivastatin lactone metabolite was purchased from Santa Cruz Biotechnology (Dallas, TX). Cellulose acetate syringe filters were purchased from Macherey-Nagel (Düren, Germany). Sephadex G-50 was obtained from GE Healthcare (Chicago, IL). Slide-A-Lyzer MINI dialysis units and LIVE/DEAD fixable green dead cell stain were purchased from Thermo Fisher Scientific (Waltham, MA). Cell culture medium (SmGM-2 BulletKit media) was obtained from Lonza (Basel, Switzerland). RPMI-1640 medium and cell culture reagents such as trypsin and phosphate-buffered saline (PBS) were purchased from Atlanta Biologicals (Flowery Branch, GA). Distilled water was used all through the experiment. All solvents were of analytic grade purchased from commercial sources and used without further purification.
Liposome Preparation and Characterization.
Cerivastatin-loaded liposomes were prepared by the thin-film hydration method. 1,2-Dipalmitoyl-sn-glycero-3-phosphocholine and cholesterol (60 and 40 mol % respectively; 10 mg/ml) were used to prepare liposomes containing cerivastatin for in vitro and in vivo studies. The lipids were dissolved and mixed in chloroform then evaporated using a rotary evaporator to form a lipid film. The lipid film was placed under high vacuum overnight to remove residual organic solvent.
The dried lipid film was hydrated with cerivastatin dissolved in 10 mM HEPES, 300 mM sucrose, pH 7.4 buffer (3 mg/ml) at 55°C and resuspended by frequent vigorous vortexing. The suspension was then transferred to cryogenic vials and subjected to 10 freeze (−196°C)–thaw and (55°C) vortex cycles to enhance hydration and passive encapsulation. The large multilamellar vesicles formed from freeze-thaw were transformed to small, unilamellar vesicles by 10 times extrusion through two-stacks of 100 nm polycarbonate filter at 55°C.
The separation of free cerivastatin from cerivastatin-loaded liposomes was performed by gel filtration using a Sephadex G-50 column. The particle size distribution, polydispersity index, and zeta potential (ζ) of the final product were determined using a dynamic light scattering device (Zetasizer Nano S). All measurements were conducted at 25°C in triplicate and reported as number mean ± S.D.
Electron microscopy of the liposome samples were performed using a Tecnai T20 transmission electron microscope (FEI Company, Hillsboro, OR) operating at an acceleration voltage of 200 kV with LaB6 filament. Samples were deposited on Formvar/carbon film–coated Cu 400-mesh grids and negatively stained with 2% uranyl acetate to enhance the visualization.
Encapsulation Efficiency.
The separation of cerivastatin-loaded liposomes from unencapsulated cerivastatin was performed by gel filtration using a Sephadex G-50 column as described elsewhere (Fresta, 1996). Briefly, the mixture of cerivastatin-loaded liposomes and unencapsulated cerivastatin was added to the Sephadex G-50 column and centrifuged at 1000g for 3 minutes. The high-molecular-size cerivastatin-loaded liposomes were eluted in the collection tube, whereas the small-molecular-size free cerivastatin was left within the Sephadex G-50 resin. The concentration of cerivastatin in the liposome fraction was then determined using an Agilent (Santa Clara, CA) fsSeries 1100 HPLC-UV/Vis system with detection at λ = 257 nm after lysis of liposomes with Triton X-100 (0.1% v/v). The analysis was achieved on an Eclipse XDB-C8 analytical column (3.5 μm, 4.6 × 150 mm) and a solvent system consisting of solvent A (water/acetonitrile/formic acid 94.9/5/0.1) and solvent B (acetonitrile/methanol/formic acid 79.9/20/0.1) at a flow rate of 0.8 ml/min over a 10-minute gradient. The percentage encapsulation efficiency (%EE) was defined by Entrapped drug/Total drug × 100 (Gulati et al., 1998).
In Vitro Release Kinetic Study.
Cerivastatin release kinetics from purified cerivastatin liposome (100 μl) was determined using dialysis (Slide-A-Lyzer MINI, 3500 MWCO) against 10 mM HEPES, 300 mM sucrose, pH 7.4 buffer (5 ml), at room temperature with constant stirring at 100 rpm under sink conditions. At various times, the medium (100 μl) was sampled and replaced with fresh medium (100 μl). The concentration of released cerivastatin was determined using high-performance liquid chromatography with tandem mass spectrometry (HPLC-MS/MS) (Agilent 6460 Triple Quad LC/ESI-MS) equipped with a Zorbax XDB C-18 analytical column (3.5 μm, 2.1 × 50 mm) at a flow rate of 0.3 ml/min over a 10-minute gradient. The analyte (cerivastatin) and internal standard (fluvastatin) were scanned in positive multiple reaction monitoring mode with the detected precursor ion (Q1) and product ion (Q3) observed at m/z 460.40/356.40 (cerivastatin) and 412.20/224.00 (fluvastatin) using a 40 eV collision energy.
Cell Culture.
Human pulmonary artery smooth muscle cells (HPASMC) (Lonza) were cultured in SmGM-2 BulletKit media (Lonza) at 37°C under 5% CO2, and 95% relative humidity atmosphere. Primary cells at the fourth to eighth passages were used for all experiments.
Smooth Muscle Cell Proliferation.
HPASMC were seeded in 24-well plates (50,000 cells per well, in triplicates) in growth medium and allowed to adhere overnight at 37°C. The cells were washed with PBS and incubated with growth medium containing 0, 0.01, 0.03, 0.1, 0.3, and 1 μM cerivastatin (free cerivastatin or liposomal cerivastatin) and growth medium as a control. Cells were washed with PBS, trypsinized, and counted in a Beckman-Coulter FC500 flow cytometer (Brea, CA).
Cell Viability Assay.
Cell viability was measured using LIVE/DEAD fixable green dead cell stains. HPASMC were seeded on 24-well plates at 50,000 cells per well in growth medium and allowed to adhere overnight at 37°C. Cells were washed with PBS and incubated with in growth medium containing 0, 0.01, 0.03, 0.1, 0.3, and 1 μM cerivastatin (free cerivastatin or liposomal cerivastatin) and growth medium as a control for 48 hours. Culture media and trypsinized cells were combined and transferred 200 μl into 96-well plates. Cells were incubated with 0.5 μl cell stain for 20 minutes in the dark. The fluorescence of live/dead cells were analyzed by flow cytometry using the FL1 channel.
Animals.
All experimental procedures were performed in accordance with the Animal Welfare Act and approved by the St. Joseph Translational Research Institute Institutional Animal Care and Use Committee, and Eurofins Institutional Animal Care and Use Committee.
Histologic Studies.
Tissues were fixed in formalin, embedded in paraffin, and stained with H&E. Images were acquired using a Nikon E600 light microscope (Tokyo, Japan) with digital interface and computer imaging software. Immunohistochemical staining for macrophage marker CD68 in paraffin-embedded slides was performed using Vectastain (Vector Laboratories, Burlingame, CA).
PAH Model.
A rat model of monocrotaline-induced PAH was used for in vivo studies. Male Sprague-Dawley rats weighing about 250 g were purchased from Charles River Laboratories (Hollister, CA). PAH was induced by a single subcutaneous injection of 60 mg/kg monocrotaline. Free cerivastatin in saline solution or liposomal cerivastatin (1 mg/kg) were delivered by intratracheal instillation using a microsprayer aerosolizer (model IA-1B; Penn Century, Wyndmoor, PA) to deliver 200 μl, twice a week.
After 6 weeks, the disease development was determined by pulmonary artery pressure (PAP) measurements, echocardiography, and histology. PAP was determined by right heart catheterization using a 1.4-F micromanometer-tipped Millar catheter with fluoroscopy guidance. Transthoracic echocardiography was performed using a GE vivid i with 5.0–13.0 MHz i12L-RS linear array transducer.
Pulmonary artery acceleration time (PAAT) was measured using pulse-wave Doppler echocardiography with the sample volume centrally positioned in the PA distal to the pulmonary valve. M-mode was applied to measure the right ventricular cavity thickness during end diastole using the parasternal long-axis view obtained from the right side of the rat. Tissue and blood samples were collected at termination. Tissues were fixed in 10% formalin, embedded in paraffin, and then processed for histomorphometry.
Pharmacokinetic Studies.
Pharmacokinetic studies were performed at Eurofins Scientific (Taipei, Taiwan). Free cerivastatin solution in PBS or liposomal cerivastatin was administered to rats at 0.5 mg/kg either intravenously or intratracheally, respectively. Blood samples (300–400 μl) were collected by cardiac puncture from sedated rats (3% isoflurane) at various times after administration. At the end of the study period, the rats were decapitated, and whole livers were removed and homogenized in PBS. Each plasma sample and liver homogenate was processed using acetonitrile precipitation for quantitative analysis.
Analysis of Cerivastatin and Its Lactone Metabolite.
Cerivastatin and lactone metabolite levels in the plasma and liver were determined by HPLC-MS/MS. Chromatographic separation was performed using a Eurospher II 100-5 C18 analytical column (4 mm, 2.0 × 50 mm) at a flow rate of 0.5 ml/min over a 4.5-minute gradient (Knauer Wissenschaftliche Geräte, Belin, Germany). A mass spectrometer equipped with an electrospray ionization source was used for the analysis. The analytes (cerivastatin and lactone metabolite) and internal standard (fluvastatin) were scanned in positive multiple reaction monitoring mode, and the detected precursor ion (Q1) and product ion (Q3) at m/z 460.40/356.40 (cerivastatin), 442.30/354.20 (lactone metabolite), and 412.20/224.00 (fluvastatin) were monitored.
Statistical Analysis.
Statistical significance was evaluated by one-way analysis of variance (ANOVA), followed by Dunnett’s test for multiple comparison between the experimental groups. P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Preparation and Characterization of Cerivastatin Nanoliposomes.
Liposome encapsulation of cerivastatin was successfully achieved using thin-film hydration method and sizing the unilamellar vesicles by 10 times extrusion through two stacks of 100-nm polycarbonate filter at 55°C, as described under Materials and Methods. Nanoliposomes were characterized for their morphology, size, and zeta potential (ζ).
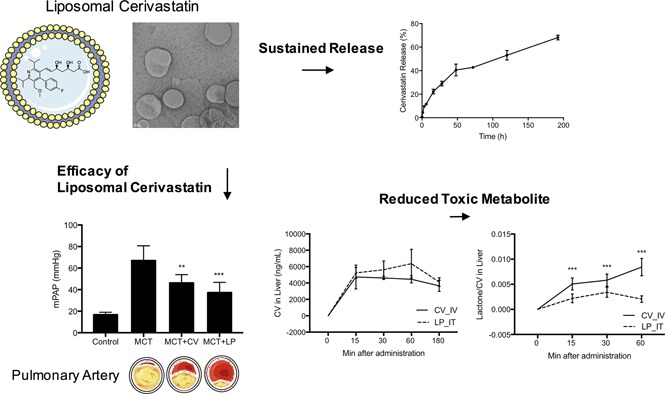
Figure 1, A and B, shows the negative stain transmission electron microscope images of the particles indicating the presence of spherical and generally uniform diameter particles. Dynamic light scattering analysis (Fig. 1C) shows particles with a mean hydrodynamic radius of 98 ± 27 nm, a polydispersity index of 0.08, and a mean ζ of −1.7 ± 6 mV. Quantification of liposome-encapsulated cerivastatin after separation from free cerivastatin using Sephadex G-50 showed that 22.4 ± 7.7 μg/ml cerivastatin was associated with the liposomes from a total of 366.8 ± 13.3 μg/ml loaded onto the column, yielding an encapsulation efficiency of 6.1% ± 1.9% (n = 3). The liposomes were free of unencapsulated cerivastatin as confirmed by the release kinetics experiment where detectable free cerivastatin was not present immediately after suspension of the liposomes in the buffer, but became quantifiable over time as a result of release from the liposomes.
Fig. 1.

Characterization of cerivastatin liposomes. Representative negative stain transmission electron microscope images of cerivastatin-loaded liposomes at (A) original magnification, 5000× and (B) original magnification, 20,000×. (C) The number average hydrodynamic radius of cerivastatin-loaded liposomes as determined by dynamic light scattering. (D) The release kinetic performed using the dialysis method (3500 MWCO) under sink conditions. Samples were collected at different times and the percentage of cerivastatin release were determined by HPLC-MS/MS (positive ion mode electrospray ionization). Data are mean ± S.D. (n = 3).
The kinetics of cerivastatin release from the liposomes was determined using the dialysis method (Chi et al., 2016). Figure 1D shows the biphasic release profile of cerivastatin from the liposomes. A sustained release with higher rate was observed during first 16 hours of dialysis, producing <25% of drug in the external media. The initial release was followed by a slower and sustained release over the subsequent 192 hours. At the last time point, a total of 70% of drug was released from the liposomes. These results show that the release of liposome-encapsulated cerivastatin occurred at a sustained rate, with the drug deployed over more than 192 hours.
Biologic Activity of Liposomal Cerivastatin.
To confirm that liposome-encapsulated cerivastatin retained biologic activity, we tested free cerivastatin and liposome-encapsulated cerivastatin for their effects on HPASMC proliferation. The data in Fig. 2A show that both free and liposome-encapsulated cerivastatin inhibited proliferation of HPASMC. Free cerivastatin was a more potent inhibitor of HPASMC than liposomal cerivastatin. The higher apparent potency of free cerivastatin is attributed to the immediate access of the free drug to the cells, whereas the slow release of cerivastatin from the liposomes would produce a lower extraliposomal concentration of drug that is available to partition into the cells during the 48-hour incubation. These results show that the biologic activity of cerivastatin was preserved during encapsulation and the sustained released cerivastatin was effective in inhibiting HPASMC.
Fig. 2.

Effect of free cerivastatin (CV) and liposomal CV on smooth muscle cell proliferation and cytotoxicity. HPASMC plated at a density of 50,000 cells per well, in triplicates, in 24-well plates. The cells were then treated with the indicated concentrations of free CV (black bar), liposome CV (LP, gray bar), or growth medium (control) for 48 hours. (A) Cell counts after trypsinization were determined by flow cytometry. To determine viability, cells were treated with the indicated concentrations of cerivastatin (CV) or liposome cerivastatin (LP). (B) After 48 hours, viable and dead cell were determined by LIVE/DEAD cell stains using the FL1 channel in flow cytometry. Bars indicate the mean ± S.D. (n = 3). *P < 0.05; **P < 0.01; ***P < 0.001 for control versus different dose with CV. ‡P < 0.01 for control versus different dose with LP. #P < 0.05; ##P < 0.01; ###P < 0.001 for CV versus LP within same dose.
Interestingly, the liposomal cerivastatin did not produce cytotoxicity, whereas free cerivastatin showed a concentration-dependent cytotoxic effect on cells (Fig. 2B). More importantly, liposomal cerivastatin did not produce a cytotoxic effect at the concentrations that inhibited cell growth. This may be due to acute high levels of cerivastatin available to the cells when free cerivastatin was added, whereas the sustained release provided by the liposome formulation may provide adequate cerivastatin to inhibit growth without reaching cytotoxic concentrations. These results may have important implications with regard to the in vivo cytotoxic effect of free statin, which appears to be reduced by liposomal sustained-release formulation.
In Vivo Efficacy of Cerivastatin in a Rat Model of PAH.
Cerivastatin has not been previously tested for efficacy in models of PAH. Therefore, we first investigated the effects of inhaled free cerivastatin on the parameters of PAH in a monocrotaline-induced rat model of PAH. Figure 3B shows that monocrotaline treatment significantly induced intimal hyperplasia of the PA. In cerivastatin treated animals, PA hyperplasia was significantly reduced (Fig. 3, C and D). These results show that inhaled cerivastatin was effective in inhibiting smooth muscle cell hyperplasia in the rat models of PAH.
Fig. 3.

Histology of PA and lung, and immunostaining for macrophage. PAH was induced by a single subcutaneous injection of monocrotaline (MCT, 60 mg/kg) and treatments of saline control and cerivastatin (CV) by intratracheal instillation (1 mg/kg) was initiated on day 1 and twice a week thereafter for 6 weeks. H&E staining of pulmonary artery in rats treated with (A) saline (control), (B) MCT, and (D) MCT and CV (MCT + CV). (D) Bar graph showing PA medial thickness quantified by measuring the intimal area. (E) Histology and immunohistology of the lung in control CV-treated rats show thickening and occlusion of small arterioles (arrow) in the lung of MCT-treated rats. (F) Effect of CV treatment and evidence of blood flow in small vessels (arrows). (G) Immunostaining of lungs for macrophage with CD68 antibodies (arrows) from animals treated with MCT and (H) MCT + CV. Bars indicate the mean ± S.D. (n ≥ 4). ***P < 0.001 when compared with MCT group.
Lung histology showed a significant arterial wall thickening and occlusion of small vessels in the lungs of monocrotaline-treated rats (Fig. 3E). Intratracheal delivery of cerivastatin reduced the occlusion of small arteries, which showed clear evidence of circulating blood (as seen by the presence of red cells in the small arteries, Fig. 3F). Thus, direct delivery of cerivastatin significantly reduced PA wall hyperplasia and occlusion of small vessels in the lungs in the monocrotaline model of PAH.
Immunostaining of lungs with antibodies against macrophage marker CD68 showed a large number of macrophages in the lungs of monocrotaline-treated rats (Fig. 3G). The macrophage population was greatly reduced in the lungs of rats treated with cerivastatin (Fig. 3H). These data show that inhaled cerivastatin reduced inflammation in the lungs, a major hallmark of PAH pathology.
Measurements of PAP showed that the average baseline PAP of 16.68 ± 2.39 mm Hg increased after monocrotaline treatment to 55.13 ± 9.82 mm Hg at the end of 6 weeks (Fig. 4). In the cerivastatin treatment group, the PAP was significantly lower (35.56 ± 6.59 mm Hg). Thus, consistent with PA histology, cerivastatin treatment significant improved PA function.
Fig. 4.

Effect of monocrotaline (MCT) and cerivastatin (CV) on pulmonary artery pressure. PAH was induced by a single subcutaneous injection of MCT (60 mg/kg) and treatments of saline control and CV by intratracheal instillation (1 mg/kg) was initiated on day 1 and twice a week thereafter for 6 weeks. The mean pulmonary artery pressure (mPAP) as measured by right heart catheterization using Millar catheter in control, MCT, and MCT + CV treated rats. Bars indicate the mean ± S.D. (n ≥ 4). ***P < 0.001 when compared with MCT group.
Noninvasive echocardiography after 6 weeks of monocrotaline administration showed that right ventricular thickening was increased from 0.19 ± 0.03 to 0.34 ± 0.02 cm. Treatment with cerivastatin significantly reduced the right ventricular thickening to 0.26 ± 0.02 cm (Fig. 5A). Similarly, PAAT, an indicator of high PAP, was reduced in the monocrotaline group from 34.50 ± 2.53 to 13.98 ± 1.14 milliseconds and was significantly improved in the cerivastatin-treated group to 21.07 ± 2.80 milliseconds (Fig. 5B).
Fig. 5.

Effect of monocrotaline (MCT) and cerivastatin (CV) on (A) right ventricular (RV) thickening and (B) PAAT. PAH was induced by a single subcutaneous injection of MCT (60 mg/kg), and treatments of saline control and CV by intratracheal instillation (1 mg/kg) were initiated on day 1 and twice a week thereafter for 6 weeks. The RV cavity thickening measurements used echocardiography, and PAAT measurement used pulse-wave Doppler for control, MCT treatment, and MCT followed by CV (MCT + CV) treatment groups. The representative pulse wave (PW) images from echocardiography are shown here in the inset. Bars indicate the mean ± S.D. (n ≥ 5). ***P < 0.001 when compared with MCT group.
The inset in Fig. 5B shows the image of systolic notching in the Doppler flow waves. As compared with the normal parabolic waves in control animals, the notching was modified in the monocrotaline group, indicating deceleration of systolic flow across the pulmonary valve and high pulmonary vascular resistance. The notching was partially recovered after cerivastatin treatment, consistent with an enhancement in PAAT and lower pulmonary vascular resistance. These studies for the first time show that cerivastatin delivery by inhalation significantly improved cardiac structure and function in the PAH model.
After having established the effect of cerivastatin on PAP and cardiac function, we investigated and compared the effect of liposomal cerivastatin on PAH parameters in the rat model. As shown in Fig. 6, inhaled liposomal cerivastatin effectively reduced PAP and showed a trend of higher efficacy than free cerivastatin on monocrotaline-induced PAH in rats (monocrotaline: 67.06 ± 13.64 mm Hg; cerivastatin: 46.31 ± 7.64 mm Hg vs. liposomal cerivastatin: 37.32 ± 9.50 mm Hg). Similarly, liposomal cerivastatin reduced right ventricular cavity thickness (monocrotaline: 0.27 ± 0.02 cm; cerivastatin: 0.23 ± 0.02 cm vs. liposomal cerivastatin: 0.22 ± 0.02 cm) and improved PAAT (monocrotaline: 24.68 ± 3.92 milliseconds; cerivastatin: 32.59 ± 6.10 milliseconds vs. liposomal cerivastatin: 34.96 ± 7.51 milliseconds). These results show that a sustained-release delivery of nanoliposome cerivastatin effectively improved and lung and cardiac markers of PAH in the preclinical model.
Fig. 6.

Effect of inhaled cerivastatin (CV) and liposomal CV (LP) on PAH parameters. PAH was induced by a single subcutaneous injection of monocrotaline (MCT, 60 mg/kg) and treatments of saline control and CV by intratracheal instillation (1 mg/kg) was initiated on day 1 and twice a week thereafter for 6 weeks. (A) Mean pulmonary artery pressure (mPAP) as measured by right heart catheterization using Millar catheter. (B) Right ventricle (RV) cavity thickening measurements used echocardiography, and (C) PAAT measurement used pulse-wave Doppler for control, MCT treatment, MCT followed by CV (MCT + CV), and MCT followed by liposomal CV (MCT + LP) treatment groups. Bars indicate the mean ± S.D. (n ≥ 6). *P < 0.05; **P < 0.01; ***P < 0.001 when compared with MCT group.
Generation of Lactone Metabolite from Free and Liposomal Cerivastatin.
We next tested the hypothesis that the slow-release kinetics of liposome-encapsulated cerivastatin would generate lower level of lactone metabolite (which is known to contribute to the adverse effects of statins), thereby providing a superior safety profile. Figure 7, A and B, shows the plasma and liver concentrations, respectively, of cerivastatin produced from intravenously administrated free cerivastatin (0.5 mg/kg) or intratracheally administered liposome-encapsulated cerivastatin (0.5 mg/kg). The plasma levels of total cerivastatin from liposome-encapsulated cerivastatin were higher at early time points (15–30 minutes) compared with free cerivastatin. The liver levels of total cerivastatin from liposome-encapsulated cerivastatin were similar to those of free cerivastatin throughout the studies. These data show that very low plasma levels of free cerivastatin are observed and that cerivastatin was rapidly taken up by the liver after administration, whereas liposome-encapsulated cerivastatin may have higher and longer bioavailability in circulation.
Fig. 7.

Characterization of pharmacokinetics and metabolic profile of free cerivastatin (CV) and liposomal CV. Free CV in PBS or liposome-encapsulated CV was administered to rats at 0.5 mg/kg intravenously and intratracheally, respectively. Plasma samples and liver were collected at the indicated time. Plasma and liver homogenates were processed for quantitative analysis of total CV using HPLC-MS/MS with positive ion mode electrospray ionization (Q1/Q3 at m/z 460.40/356.40). Shown here are total CV levels in (A) plasma and (B) liver from animals that received free CV (solid line) or liposomal CV (dash line). (C) CV lactone metabolite generated from equivalent amount of free CV (solid line) and liposomal CV (dash line) in the liver. Data are mean ± S.D. (n = 4). **P < 0.01; ***P < 0.001 for free CV versus liposomal CV within same time.
Quantification of the cerivastatin metabolite in the liver showed that even though the total cerivastatin concentrations in the liver were similar for the free cerivastatin or liposomal cerivastatin groups, the levels of lactone metabolite was significantly different. The data in Fig. 7C are expressed as the ratio of lactone generated/ng cerivastatin in the liver. At 1 hour after administration, the lactone metabolite levels in the liposome cerivastatin group were 5-fold lower than in the free cerivastatin group. These data demonstrate that even though there were similar total concentrations of cerivastatin in the liver, liposomal cerivastatin was metabolized into lactone at a significantly slower rate. Thus, the generation of this toxic metabolite is greatly reduced as result of the inaccessibility of liposome-encapsulated cerivastatin and its slow release. These data suggest that a liposome delivery may achieve an improved safety profile for cerivastatin.
Discussion
PAH is a highly symptomatic, progressive, debilitating, and life-threatening illness. During the past decade, several vasodilator drugs have been developed, including prostacyclin analogs (treprostinil and epoprostenol), endothelin receptor antagonists (bosentan and ambrisentan), and phosphodiesterase type 5 inhibitors (sildenafil, tadalafil). These drugs primarily reduce vasoconstriction and relieve the symptoms of the disease (Yao, 2012). However, no drug that modifies the course of the disease is currently available. Therefore, considerable morbidity and mortality from PAH remains a clinical problem.
Recent studies have shown that mTOR inhibitors and the antiproliferative platelet-derived growth factor (PDGF) inhibitor imatinib can reduce progression and perhaps reverse the course of the disease in preclinical models (Schermuly et al., 2005). Evidence of clinical efficacy of imatinib has also been achieved. In a phase 2 randomized, double-blind, placebo-controlled trial, imatinib treatment significantly improved the 6-minute walk distance at 24 weeks. The pulmonary vascular resistance was significantly decreased by 379 dyne·s·cm−5 (Hoeper et al., 2013), and noninvasive echocardiographic indices of right ventricular function also improved in the imatinib group (Shah et al., 2015). However, despite these promising clinical benefits, imatinib treatment was associated with substantial side effects, leading to a significant number of patients discontinuing the therapy (27% imatinib vs. 2% placebo, P = 0.005) because of adverse events. Some of the major adverse events included cardiac failure, dyspnea, worsening PAH, syncope, anemia, leukopenia, thrombocytopenia, and device-related infection. In addition, higher incidences of nausea, peripheral edema, diarrhea, vomiting, and periorbital edema—the known side effects of imatinib—were observed. These significant side effects prevented regulatory agency approval of the drug, but these studies demonstrated that efficacy by pharmacologic agents targeted to vascular remodeling is feasible, provided they can be delivered safely.
A variety of studies have shown that the statin class of drugs modulate cellular and molecular pathways that are associated with the multifactorial pathology of PAH. Statins have been shown to prevent the development of PAH, reduce progression, and reverse the course of disease in preclinical models (Nishimura et al., 2002, 2003; Sun and Ku, 2008). However, despite the strong preclinical data, the efficacy of orally delivered statins in patients could not demonstrated.
We hypothesized that the lack of efficacy of orally administered statins in PAH patients may have be due to their rapid uptake and metabolism in the liver. The lung levels of statins after oral administration may not reach levels required for efficacy. The high doses used in animal model studies to demonstrate efficacy are likely to produce toxicity in humans, including liver injury and rhabdomyolysis (Kalaria and Wassenaar, 2002). Thus, modifications in the pharmacodynamics and liver metabolism of statins may provide a new therapeutic strategy for developing statins for the treatment of PAH.
Our and other studies have shown that cerivastatin exhibits unique and differentiating biologic activities relevant to PAH pathology, including inhibition of vascular smooth muscle cell proliferation, inflammation, oxidative stress, and improvement of endothelial function and expression of KLF2 (John et al., 2001; Achenbach et al., 2002; Leslie et al., 2004; Ohkita et al., 2006; Zhao et al., 2015).
To test our hypothesis, we prepared and characterized cerivastatin encapsulated nanoliposomes. Liposomes with an average diameter of 100 nm containing 20 ± 6 μg of cerivastatin per mg of lipids were prepared. The liposome-encapsulated cerivastatin demonstrated slow-release kinetics as determined over a 192-hour incubation in vitro. This release kinetics pattern is favorable in that an initial release of drugs provides a sufficient therapeutic dose shortly after administration, followed by a gradual release over an extended period to sustain the drug at the site of delivery. In vitro studies showed that liposome-encapsulated cerivastatin inhibited proliferation of PA smooth muscle cells. Interestingly, these results showed that, as compared with free cerivastatin, the liposome cerivastatin did not exhibit cytotoxicity at the concentrations that produced antiproliferative effects on human PA smooth muscle cells.
Because the preclinical efficacy of cerivastatin had not been previously investigated, we first tested the in vivo effect of free cerivastatin when delivered to rats by inhalation. These studies for the first time demonstrated that delivery of inhaled cerivastatin significantly reduced PA hyperplasia and occlusion of small arterioles in the lung. This was associated with a reduction in inflammatory macrophages in the lung. Measurements of lung and right heart parameters clearly demonstrated that cerivastatin treatment reduced PAP and improved cardiac function as measured by echocardiography. These data validated the promising pharmacologic activity of cerivastatin in a model of PAH.
We then investigated and compared sustained-release liposomal cerivastatin for efficacy in this model. As shown in Fig. 6, the delivery of liposomal cerivastatin significantly reduced PAP and improved right ventricular function. For each of the parameters, liposomal cerivastatin was more effective at an equivalent dose, although it did not reach statistical significance with the number of animals used in this study. Thus, sustained-release liposome cerivastatin delivered directly to the lung was highly efficacious in the preclinical models of PAH.
Having demonstrated the efficacy of cerivastatin and liposomal cerivastatin, we investigated the potentially safer characteristics of liposomal cerivastatin as compared with free cerivastatin. Previous studies have shown that metabolism of statins to their lactone metabolite may mediate the liver and other toxicities of statins (Schirris et al., 2015). In this regard, the cerivastatin lactone was found to be highly toxic as compared with other statins. Our studies showed that the generation of toxic cerivastatin lactone metabolite in the liver was significantly reduced when the drug was delivered as liposome-encapsulated cerivastatin compared with free cerivastatin. These studies confirm that the slow rate of drug release from liposomes reduces the rate and magnitude of toxic metabolite generation because the encapsulated drug is not accessible for metabolism by cytochrome P450 enzymes in the liver. Thus, studies have demonstrated that higher doses of potential disease-modifying drugs such as statins may be safely delivered using the nanoliposome approach.
In summary, we tested the combination of nanoliposome encapsulation and local delivery to achieve a maximum efficacious and favorable safety profile of cerivastatin for the treatment of PAH. Encapsulated cerivastatin displayed sustained-release kinetics that inhibited the proliferation of PA smooth muscle cells in vitro, without evidence of cytotoxicity. Liposomal cerivastatin was highly effective and showed a trend toward greater effectiveness compared with free cerivastatin in improving lung and cardiac function in the PAH models. Pharmacokinetic and metabolism studies showed that liposome delivery produced a significantly lower level of potentially toxic cerivastatin lactone metabolite in the liver.
These studies suggest that an inhaled delivery of the liposome-encapsulated cerivastatin for patients with PAH could yield a distinctly different risk/benefit balance than that observed after oral administration. Our studies suggest that a combination of unique biologic activities, a novel liposome-encapsulated form, and local delivery of cerivastatin could provide a new therapeutic approach for the development of disease-modifying drugs for PAH.
Abbreviations
- HPASMC
human pulmonary artery smooth muscle cells
- HPLC-MS/MS
high-performance liquid chromatography with tandem mass spectrometry
- KLF2
Kruppel-like factor 2
- PA
pulmonary artery
- PAAT
pulmonary artery acceleration time
- PAH
pulmonary arterial hypertension
- PAP
pulmonary artery pressure
- PBS
phosphate-buffered saline
Authorship Contributions
Participated in research design: Singh, Thompson, Bellamkonda, Lee.
Conducted experiments: Singh, Lee, Pai
Contributed new reagents or analytic tools: Lee, Thompson.
Performed data analysis: Singh, Lee, Pai.
Wrote or contributed to the writing of the manuscript: Singh, Pai, Lee.
Footnotes
This project was supported in part, with support from the Indiana Clinical and Translational Sciences Institute funded, in part from the National Institutes of Health National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award [Grant UL1TR001108], and Georgia Research Alliance.
References
- Achenbach S, Ropers D, Pohle K, Leber A, Thilo C, Knez A, Menendez T, Maeffert R, Kusus M, Regenfus M, et al. (2002) Influence of lipid-lowering therapy on the progression of coronary artery calcification: a prospective evaluation. Circulation 106:1077–1082. [DOI] [PubMed] [Google Scholar]
- Anand V, Garg S, Duval S, Thenappan T. (2016) A systematic review and meta-analysis of trials using statins in pulmonary arterial hypertension. Pulm Circ 6:295–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, et al. (2010) Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest 137:376–387. [DOI] [PubMed] [Google Scholar]
- Bellosta S, Paoletti R, Corsini A. (2004) Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation 109 (23 Suppl 1):III50–III57. [DOI] [PubMed] [Google Scholar]
- Benza RL, Miller DP, Barst RJ, Badesch DB, Frost AE, McGoon MD. (2012) An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. Chest 142:448–456. [DOI] [PubMed] [Google Scholar]
- Chi L, Wu D, Li Z, Zhang M, Liu H, Wang C, Gui S, Geng M, Li H, Zhang J. (2016) Modified release and improved stability of unstable BCS II drug by using cyclodextrin complex as carrier to remotely load drug into niosomes. Mol Pharm 13:113–124. [DOI] [PubMed] [Google Scholar]
- Eichstaedt CA, Song J, Viales RR, Pan Z, Benjamin N, Fischer C, Hoeper MM, Ulrich S, Hinderhofer K, Grünig E. (2017) First identification of Krüppel-like factor 2 mutation in heritable pulmonary arterial hypertension. Clin Sci (Lond) 131:689–698. [DOI] [PubMed] [Google Scholar]
- Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Muros-Le Rouzic E, Romero AJ, Benton WW, Elliott CG, McGoon MD, et al. (2015) Five-year outcomes of patients enrolled in the REVEAL Registry. Chest 148:1043–1054. [DOI] [PubMed] [Google Scholar]
- Fresta M. (1996) Biological effects of CDP-choline loaded long circulating liposomes on rat cerebral post-ischemic reperfusion. Int J Pharm 134:89–97. [Google Scholar]
- Guignabert C, Dorfmuller P. (2013) Pathology and pathobiology of pulmonary hypertension. Semin Respir Crit Care Med 34:551–559. [DOI] [PubMed] [Google Scholar]
- Gulati M, Grover M, Singh M, Singh S. (1998) Study of azathioprine encapsulation into liposomes. J Microencapsul 15:485–494. [DOI] [PubMed] [Google Scholar]
- Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galié N, Gómez-Sánchez MA, Grimminger F, Grünig E, Hassoun PM, et al. (2013) Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 127:1128–1138. [DOI] [PubMed] [Google Scholar]
- John S, Delles C, Jacobi J, Schlaich MP, Schneider M, Schmitz G, Schmieder RE. (2001) Rapid improvement of nitric oxide bioavailability after lipid-lowering therapy with cerivastatin within two weeks. J Am Coll Cardiol 37:1351–1358. [DOI] [PubMed] [Google Scholar]
- Kalaria D, Wassenaar W. (2002) Rhabdomyolysis and cerivastatin: was it a problem of dose? CMAJ 167:737. [PMC free article] [PubMed] [Google Scholar]
- Leslie SJ, Spratt JC, Grieg L, Attina T, Denvir MA, Webb DJ. (2004) The effect of cerivastatin therapy on vascular responses to endothelin antagonists in humans. J Cardiovasc Pharmacol 44 (Suppl 1):S410–S412. [DOI] [PubMed] [Google Scholar]
- Liao JK, Laufs U. (2005) Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 45:89–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeije R, Manes A. (2014) The right ventricle in pulmonary arterial hypertension. Eur Respir Rev 23:476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Faul JL, Berry GJ, Vaszar LT, Qiu D, Pearl RG, Kao PN. (2002) Simvastatin attenuates smooth muscle neointimal proliferation and pulmonary hypertension in rats. Am J Respir Crit Care Med 166:1403–1408. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Vaszar LT, Faul JL, Zhao G, Berry GJ, Shi L, Qiu D, Benson G, Pearl RG, Kao PN. (2003) Simvastatin rescues rats from fatal pulmonary hypertension by inducing apoptosis of neointimal smooth muscle cells. Circulation 108:1640–1645. [DOI] [PubMed] [Google Scholar]
- Ohkita M, Sugii M, Ka Y, Kitamura A, Mori T, Hayashi T, Takaoka M, Matsumura Y. (2006) Differential effects of different statins on endothelin-1 gene expression and endothelial NOS phosphorylation in porcine aortic endothelial cells. Exp Biol Med (Maywood) 231:772–776. [PubMed] [Google Scholar]
- Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, et al. (2005) Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest 115:2811–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirris TJ, Renkema GH, Ritschel T, Voermans NC, Bilos A, van Engelen BG, Brandt U, Koopman WJ, Beyrath JD, Rodenburg RJ, et al. (2015) Statin-induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab 22:399–407. [DOI] [PubMed] [Google Scholar]
- Shah AM, Campbell P, Rocha GQ, Peacock A, Barst RJ, Quinn D, Solomon SD, IMPRES Investigators (2015) Effect of imatinib as add-on therapy on echocardiographic measures of right ventricular function in patients with significant pulmonary arterial hypertension. Eur Heart J 36:623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacher E, Graham BB, Hunt JM, Gandjeva A, Groshong SD, McLaughlin VV, Jessup M, Grizzle WE, Aldred MA, Cool CD, et al. (2012) Modern age pathology of pulmonary arterial hypertension. Am J Respir Crit Care Med 186:261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Ku DD. (2008) Rosuvastatin provides pleiotropic protection against pulmonary hypertension, right ventricular hypertrophy, and coronary endothelial dysfunction in rats. Am J Physiol Heart Circ Physiol 294:H801–H809. [DOI] [PubMed] [Google Scholar]
- Wolfrum S, Jensen KS, Liao JK. (2003) Endothelium-dependent effects of statins. Arterioscler Thromb Vasc Biol 23:729–736. [DOI] [PubMed] [Google Scholar]
- Yao A. (2012) Recent advances and future perspectives in therapeutic strategies for pulmonary arterial hypertension. J Cardiol 60:344–349. [DOI] [PubMed] [Google Scholar]
- Zhao J, Natarajan SK, Chronos N, Singh JP. (2015) Cerivastatin represses atherogenic gene expression through the induction of KLF2 via isoprenoid metabolic pathways. Cell Mol Biol Lett 20:825–839. [DOI] [PubMed] [Google Scholar]