

Abstract
OXA-239 is a class D carbapenemase isolated from an Acinetobacter baumannii strain found in Mexico. This enzyme is a variant of OXA-23 with three amino acid substitutions in or near the active site. These substitutions cause OXA-239 to hydrolyze late-generation cephalosporins and the monobactam aztreonam with greater efficiency than OXA-23. OXA-239 activity against the carbapenems doripenem and imipenem is reduced approximately 3-fold and 20-fold respectively. Further analysis demonstrated that two of the substitutions (P225S and D222N) are largely responsible for the observed alteration of kinetic parameters, while the third (S109L) may serve to stabilize the protein. Structures of OXA-239 with cefotaxime, doripenem and imipenem bound as acyl-intermediates were determined. These structures reveal that OXA-239 has increased flexibility in a loop that contains P225S and D222N. When carbapenems are bound, the conformation of this loop is essentially identical to that observed previously for OXA-23, with a narrow active site that makes extensive contacts to the ligand. When cefotaxime is bound, the loop can adopt a different conformation that widens the active site to allow binding of that bulky drug. This alternate conformation is made possible by P225S and further stabilized by D222N. Taken together, these results suggest that the three substitutions were selected to expand the substrate specificity profile of OXA-23 to cephalosporins and monobactams. The loss of activity against imipenem, however, suggests that there may be limits to the plasticity of class D enzymes with regard to evolving active sites that can effectively bind multiple classes of β-lactam drugs.
INTRODUCTION
The world-wide spread of Acinetobacter baumannii continues unabated, and the rise of strains resistant to a wide variety of antibiotic classes, including carbapenems, is particularly worrisome [1, 2]. Citing the threat, the World Health Organization has classified this species as “Priority 1: Critical” with regard to the need for research and the development of novel therapeutic agents [3]. A. baumannii becomes resistant to β-lactam antibiotics through the action of native or acquired β-lactamases, deletion of outer membrane porins, addition of efflux pumps, and in some cases, alteration of the cell wall-synthesizing transpeptidases that serve as the target of those agents [4–6]. β-lactamases are by far the most potent threat, however, and A. baumannii has been shown to harbor variants from all four classes of this type of enzyme (A–D) [1]. Class D enzymes are the most abundant, and plasmid-borne genes encoding OXA-23 and OXA-24/40 are found in A. baumannii strains all over the world [7]. When their expression is driven by promoters obtained from mobile insertion elements (e.g. ISAba1), these enzymes confer resistance to penicillins and carbapenems [8]. Two other class D enzymes, OXA-51 and OXA-66, are typically chromosomally-encoded, and are thus thought to be native to the species [7]. These enzymes have modest activity against carbapenems, but only weak activity against penicillins and cephalosporins [9].
The three main classes of class D carbapenemases found in A. baumannii (OXA-23, OXA-24/40 and OXA-51/66) share strong sequence and structural conservation [10–12]. Their structures reveal a deep active site (Figure 1a) with conserved catalytic residues including a nucleophilic serine (Ser79 in OXA-23) that attacks the lactam carbonyl as the first step in an acylation/deacylation covalent catalysis strategy (Figure 2) [13]. The serine is deprotonated and thereby activated by the carbamate of an unusual N-carboxylated lysine (KCX82 in OXA-23), one of the defining features of all class D β-lactamases [14]. There are several non-polar residues that line the active site and a branched aliphatic residue (Val128 in OXA-23; I129 in OXA-51/66) is known to play a key role in the binding of carbapenems (Figure 1b) [15–17]. The active site is surrounded by three loops that play important roles in substrate selection. The region spanned by residues 151–173 is often referred to as the “omega” (Ω) loop, to match a homologous structure in class A β-lactamases [18]. From the Ω loop, the side chain of a highly-conserved leucine (Leu166) projects into the active site toward the previously mentioned Val128 [15]. Another non-polar residue, Trp165, also projects into the active site, where its indole nitrogen stabilizes the carboxy-lysine moiety with a hydrogen bond [18]. Another large loop encompassing residues 93–117 (referred to as the P loop) approaches the active site near the surface of the protein and projects a phenylalanine side chain (Phe111 in OXA-23) across the top of the active site [19]. Lastly, a loop that connects strands β6 and β7 (the β6β7 loop; residues 220–226) lines the opposite side of the active site, and projects a methionine side chain (Met221) across the area of drug binding [15]. Phe111 and Met221 contact each other and thus form a hydrophobic bridge across the top of the active site [12, 15]. These residues are known to interact closely with carbapenem substrates, and greatly increase the affinity of those drugs [15–17]. Their position across the top of the active site, however, is known to hinder the binding of β-lactams with larger side chains (i.e. oxacillin, late-generation cephalosporins, aztreonam) [10, 20].
Figure 1. The structure of OXA-23 with meropenem bound.

(A) OXA-23 wild-type (blue) with meropenem (orange) bound as an acyl-intermediate with the side chain of Ser79 (PDB code: 4JF4). (B) The same protein zoomed in to show key active site residues.
Figure 2. General mechanism for class D β-lactamases.

Class D carbapenemases like OXA-239 use an active site serine to attack the carbonyl carbon on the β-lactam ring, resulting in the drug forming an acyl-intermediate with the serine. This intermediate is subsequently hydrolyzed to release the inactive drug with a broken β-lactam ring.
A large number of clinical variants of OXA-23, OXA-24/40 and OXA-51/66 have been identified with greatly enhanced substrate specificity stretching across all four main classes of β-lactam drugs (penicillins, cephalosporins, carbapenems and monobactams) [9, 10, 16, 20, 21]. Substitutions that affect the orientation of Ile129 in OXA-51/66 (I129L, P130Q, L167V) have been shown to decrease the Km values for carbapenems [9, 16, 21]. Alterations in the β6β7 loop in all three subfamilies leads to a greatly expanded active site and reduced Km values for late-generation cephalosporins and the monobactam aztreonam [10, 20–22]. It thus appears that class D β-lactamases are experiencing the same type of evolutionary adaptation in the face of selective pressure from antibiotic therapy that has long been observed for the class A β-lactamases such as TEM-1 and SHV-1 [23]. Studying these variants is instructive for a number of reasons. First, identification of adaptive mutants reveals the current threats to various classes of antibiotic therapies. Second, studies that illuminate the mechanism by which the variants gain higher activities and/or expanded specificity help us understand the role of particular structural features in general β-lactamase function. Lastly, a thorough understanding of the adaptive plasticity of these enzymes may give insight to the development of novel therapeutic agents. It is with these benefits in mind that we chose to use kinetic analysis and X-ray crystallography to characterize OXA-239, a triple-substitution variant of the Acinetobacter baumannii carbapenemase OXA-23.
The OXA-239 variant was first identified in Mexico City in 2010 [24], and it has been subsequently isolated at a number of different hospitals in Mexico [25]. The sequence alignment in Figure 3 shows that compared to OXA-23, OXA-239 contains three amino acid substitutions (S109L, D222N and P225S), all of which reside in or near the active site [10]. Analysis of such variant β-lactamases from clinical samples can often lead to insights about the structural adaptability of these enzymes in the face of antibiotic chemotherapy. For instance, the P225S substitution is also found in the OXA-23 variant OXA-225, and a homologous substitution (P227S) is found in the OXA-24/40 variant OXA-160 (Figure 3) [26]. We have previously shown that the proline → serine substitution in OXA-225 and OXA-160 imparts hydrolytic activity against late generation cephalosporins and aztreonam, without losing the strong carbapenemase activity of the parent enzymes (OXA-23 and OXA-24/40 respectively) [20]. The presence of P225S and two additional substitutions in OXA-239 led us to hypothesize that the substrate specificity profile of that variant might be further altered. We therefore recreated OXA-239 by introducing the three substitutions into an OXA-23 background, purified the protein and analyzed its enzymatic activities using steady-state kinetics. The results show that the substitutions in OXA-239 do lead to an alteration in activity for a variety of β-lactam substrates, and our structural analysis provides a rational explanation for the role that each of the three substitutions plays in its altered phenotype.
Figure 3. OXA-23 substitutions identified in clinical A. baumannii strains.

Sequence alignments of OXA-23, the single-substitution variant OXA-225, and the triple-substitution variant OXA-239. The related carbapenemase OXA-24/40 and its single-substitution variant OXA-160 are also shown. Stars indicate residues identical between OXA-23 and OXA-24/40.
MATERIALS AND METHODS
Mutagenesis and Protein Purification
The genes for the mature proteolyzed forms of OXA-23 and OXA-225 have previously been cloned into pET24a [20]. Overlap extension PCR [27] was used on these genes to make OXA-239 (OXA-23 S109L/D222N/P225S), the deacylation-deficient mutant OXA-239 K82D, the singly substituted OXA-23 S109L and OXA-23 D222N, and the double mutant OXA-23 D222N/P225S. All mutations were confirmed by Sanger dideoxy sequencing. Plasmids were introduced into BL21(DE3) Escherichia coli and protein was expressed, purified and quantified as previously described [20].
Kinetics
Steady-state kinetic analysis was carried out in 50 mM NaH2PO4, 25 mM NaHCO3, pH 7.40 at 25°C in a Cary 100 UV-Vis spectrophotometer. Reactions with ampicillin were carried out with substrate ranging from 0–500 μM and 5–10 nM enzyme. Reactions with carbapenems were carried out with substrate ranging from 0–100 μM and 0.5–2 μM enzyme. Reactions with cephalosporins and aztreonam were typically carried out with substrate ranging from 0–500 μM and 0.1–5 μM enzyme. In some cases where Km values were very high, substrate concentrations of up to 10 mM were measured using cuvettes with very short path-lengths (2 mm or 0.1 mm). After the addition of enzyme, absorbance was followed for 10–20 seconds, and converted to initial velocity using the following Δε (M−1 cm−1) values: ampicillin, −900 (λ = 235 nm); doripenem, −11,460 (λ = 297 nm); imipenem, −9,000 (λ = 300 nm); cefotaxime, −7,500 (λ = 260 nm); cefepime, −10,000 (λ = 260 nm); ceftazidime, −8,660 (λ = 260 nm); ceftriaxone, −7,700 (λ = 255 nm); and aztreonam, −700 (λ = 320 nm). Values of Km and kcat were determined by nonlinear regression to the Michaelis-Menten equation. Slow substrates with very low Km values were treated as inhibitors in a competition assay. A constant, sub-saturating concentration of the reporter (typically 100 μM ampicillin) was added to the cuvette with various concentrations of inhibitor. Absorption was measured at 235 nm, and velocities were measured in triplicate. In this case, Ks values were determined (instead of Km values) as described previously [20].
Crystallography
Deacylation-deficient OXA-239 K82D was crystallized by the hanging drop method by combining protein (13.6 mg/mL) and well buffer (0.10 M NaH2PO4, 0.10 M sodium citrate, 0.20 M NaCl, 20% PEG 8000, pH 4.2) in a 3:1 ratio, 8 μL drop. The resulting crystals were soaked in 25 mM imipenem, doripenem or cefotaxime (structures shown in Figure 4) and flash-cooled in liquid nitrogen. Diffraction data for all three crystals was collected at LS-CAT at the Advanced Photon Source (Argonne, IL) at 100 K using a MarCCD detector. Reflections were indexed, integrated, and scaled using autoPROC [28]. The OXA-239 K82D structures were determined initially by molecular replacement with Phaser [29] using the model of OXA-23 (PDB code: 4K0X) with all ligand and water molecules removed as the initial phasing model. Refinement and electron density map calculations were carried out with REFMAC5 in the CCP4 program suite [30]. Manual rebuilding of the model was accomplished with Coot [31, 32].
Figure 4. β-lactams used for X-ray crystallography in this study.

(A) imipenem, (B) doripenem and (C) cefotaxime. In the case of cephalosporins, the group that is attached to carbon C7 is referred to as the R1 side chain, and the group attached to carbon C3 is the R2 side chain.
Thermal Denaturation
Stability of all enzymes was determined with a Jasco J-815 spectrometer (Easton, MD) with a Peltier-effect temperature controller, as described previously [20].
RESULTS
Kinetic parameters for OXA-23 and OXA-239 acting on a wide variety of β-lactams are shown in Table 1. Comparison of kcat/Km values for these two enzymes allows us to analyze their relative hydrolytic efficacies, and several broad trends are observed. OXA-239 shows much higher kcat/Km values for cephalosporins and the monobactam aztreonam, ranging from ~ 10-fold higher for cefepime to 220-fold for aztreonam. Conversely, kcat/Km values for the two carbapenems tested went down (~ 3-fold for doripenem, and nearly 25-fold for imipenem). Thus, the effect of the three mutations might be considered a specificity switch that lowers the carbapenemase activity of OXA-23, but increases its cephalosporinase activity.
Table 1.
Kinetic parameters for OXA-23, OXA-239 and other variants
| kcat (s−1) | KM or Ks (μM) | kcat/KM (μM−1·s−1) | |
|---|---|---|---|
|
|
|||
| OXA-23a | |||
| ampicillin | 460 ± 10 | 82 ± 9 | 5.7 ± 0.6 |
| cefotaxime | 5.5 ± 0.1 | 340 ± 30 | 0.016 ± 0.002 |
| cefepime | 20 ± 1 | 1100 ± 200 | 0.018 ± 0.003 |
| ceftriaxone | 0.016 ± 0.001 | 3.7 ± 0.5 | 0.0044 ± 0.0007 |
| ceftazidime | < 0.01 | NA | NA |
| doripenem | 0.028 ± 0.003 | 0.018 ± 0.002b | 1.5 ± 0.3 |
| imipenem | 0.49 ± 0.01 | 0.20 ± 0.02b | 2.4 ± 0.3 |
| aztreonam | 0.24 ± 0.01 | 2400 ± 100 | 0.00010 ± 0.00001 |
| OXA-239 | |||
| ampicillin | 99 ± 3 | 22 ± 3 | 4.5 ± 0.5 |
| cefotaxime | 5.0 ± 0.3 | 12 ± 3 | 0.43 ± 0.10 |
| cefepime | 34 ± 1 | 200 ± 20 | 0.18 ± 0.02 |
| ceftriaxone | 0.047 ± 0.004 | 0.49 ± 0.07b | 0.097 ± 0.016 |
| ceftazidime | 2.8 ± 0.1 | 600 ± 40 | 0.0050 ± 0.0003 |
| doripenem | 0.017 ± 0.002 | 0.036 ± 0.010b | 0.48 ± 0.14 |
| imipenem | 1.3 ± 0.1 | 13 ± 3b | 0.10 ± 0.03 |
| aztreonam | 0.43 ± 0.01 | 20 ± 2 | 0.022 ± 0.002 |
| OXA-23 S109L | |||
| ampicillin | 150 ± 10 | 140 ± 10 | 1.1 ± 0.1 |
| cefotaxime | NA | > 400 | NA |
| doripenem | 0.017 ± 0.001 | < 0.010b | NA |
| imipenem | 0.24 ± 0.01 | 0.23 ± 0.02b | 1.0 ± 0.1 |
| aztreonam | < 0.05 | NA | NA |
| OXA-23 D222N | |||
| ampicillin | 310 ± 10 | 34 ± 2 | 9.0 ± 0.5 |
| cefotaxime | 1.9 ± 0.1 | 33 ± 3 | 0.058 ± 0.005 |
| doripenem | 0.027 ± 0.001 | 0.011 ± 0.001b | 2.5 ± 0.2 |
| imipenem | 1.1 ± 0.1 | 12 ± 3b | 0.098 ± 0.023 |
| aztreonam | 0.43 ± 0.01 | 390 ± 40 | 0.0011 ± 0.0001 |
| OXA-23 P225Sa | |||
| ampicillin | 150 ± 10 | 12 ± 1 | 12 ± 1 |
| cefotaxime | 3.2 ± 0.2 | 100 ± 20 | 0.031 ± 0.006 |
| ceftazidime | 1.4 ± 0.1 | 1400 ± 200 | 0.0010 ± 0.0002 |
| doripenem | 0.026 ± 0.002 | 0.027 ± 0.005b | 1.0 ± 0.2 |
| imipenem | 0.39 ± 0.01 | 0.20 ± 0.02b | 1.9 ± 0.2 |
| aztreonam | 0.79 ± 0.03 | 13 ± 2 | 0.061 ± 0.010 |
| OXA-23 D222N/P225S | |||
| ampicillin | 140 ± 2 | 30 ± 1 | 4.5 ± 0.2 |
| cefotaxime | 1.5 ± 0.1 | 23 ± 3 | 0.066 ± 0.008 |
| doripenem | 0.021 ± 0.001 | 0.071 ± 0.007 | 0.30 ± 0.03 |
| imipenem | 1.1 ± 0.1 | 11 ± 1b | 0.10 ± 0.01 |
| aztreonam | 0.62 ± 0.01 | 21 ± 2 | 0.030 ± 0.002 |
OXA-23 and OXA-225 values (except for cefepime) were previously reported in Mitchell et al. Biochemistry. 54, 1976–1987b
For variants with very low Km values, Ks values were calculated by competition with a reporter substrate
NA: not applicable
For most of these substrates, the largest factor driving the change in kcat/Km is a significant alteration in Km. Of particular note is the drop in Km for aztreonam (~ 120-fold), cefotaxime (~ 30-fold) and ceftazidime. For ceftazidime, OXA-23 has no hydrolytic activity, but OXA-239 displays modest turnover and a Km of 600 ± 40 μM. With regard to imipenem, the three substitutions increase Km over 60-fold. In contrast, the very low Km observed for doripenem and OXA-23 is only increased 2-fold for OXA-239.
The increased hydrolytic efficiency displayed by OXA-239 for cephalosporins and aztreonam is reminiscent of similar effects observed for OXA-225, which also contains P225S (but not S109L or D222N). In several cases however, kcat/Km values were significantly higher or lower for OXA-239 than OXA-225, giving insight into the potential effect of the additional two substitutions. For instance, the kcat/Km value for OXA-239 acting on cefotaxime was approximately 14-fold higher than for OXA-225, and in the case of ceftazidime, it was ~ 5-fold higher. In both cases, the increase was at least partly due to a decrease in Km values. OXA-239, on the other hand, had lower kcat/Km values compared to OXA-225 for aztreonam (~3-fold), imipenem (~ 20-fold) and doripenem (2-fold). Thus the addition of the S109L and D222N substitutions seemed to favor hydrolysis of some cephalosporins, but not aztreonam or carbapenems.
In order to determine the contribution of S109L and D222N to these effects, we made a series of single and double substitutions for kinetic analysis (OXA-23 S109L, OXA-23 D222N and OXA-23 D222N/P225S). These results (Table 1) show that the addition of S109L to OXA-23 on its own had a generally negative effect on the hydrolytic parameters for ampicillin, cefotaxime and aztreonam. Conversely, D222N alone increased activity by lowering the Km for cefotaxime (~ 10-fold), for ampicillin (~ 2.5-fold) and for aztreonam (~ 6-fold). Interestingly, the D222N substitution raises the imipenem Km as much as the three substitutions of OXA-239. Taken together, these observations suggest that D222N is responsible for most of the kinetic changes—whether gain-of-function or loss-of-function—that are observed between OXA-225 and OXA-239. In support of this, The OXA-23 D222N/P225S double mutant was largely similar to OXA-239 (the triple mutant) for all drugs tested, except cefotaxime, for which the former possessed an approximately 3-fold lower kcat.
Our previous kinetic and structural studies of OXA-225 (OXA-23 P225S) revealed that the loss of the proline at position 225 allowed the β6β7 loop (residues 220–226) to adopt a new conformation that is more amenable to the binding of substrates that possess bulky side chain substituents (e.g. cefotaxime and aztreonam) [20]. Given the interesting substrate switch observed for OXA-239, we sought to use X-ray crystallography to assess the structural impact of the two additional substitutions in that variant (S109L and D222N). In order to increase the likelihood of capturing relevant ligands bound in the active site, we introduced an additional substitution (K82D) into the OXA-239 background. This substitution of the active site carboxy-lysine general base allows substrate acylation, but greatly slows deacylation [33]. Protein crystals were grown using the hanging drop vapor diffusion method under the same conditions used previously for OXA-225. We soaked doripenem, imipenem or cefotaxime into the crystals, flash-cooled them in liquid nitrogen, and successfully collected high-resolution data sets (one for each ligand).
All three structures diffracted in the P21212 space group and generated datasets with resolution in the range of 1.8–1.9 Å (Table 2) and all had two monomers in the asymmetric unit. Main chain 2Fo-Fc electron density contoured to 1.0 σ was clear for the entire protein from residue 31–273 for monomer A in all three structures. In monomer B, residues in part of the P loop had lower quality electron density and evidence of multiple conformations. Difficulty in modeling the backbone in this region led us to delete residues 103–107 from all of the monomer B structures (and additionally residue 102 from the imipenem-bound structure). For further analysis in this study we used the model for monomer A from all three structures, unless otherwise noted. The quality of all three structures was analyzed using Molprobity, and greater than 97% of residues were in the favored Ramachandran regions. For OXA-239 K82D/imipenem, there was one Ramachandran outlier, while both OXA-239 K82D/imipenem and OXA-239 K82D/cefotaxime had no outliers.
Table 2.
Crystallography statistics
| OXA-239 K82D/doripenem (PDB 5WI7) | OXA-239 K82D/imipenem (PDB 5WIB) | OXA-239 K82D/cefotaxime (PDB 5WI3) | |
|---|---|---|---|
| Wavelength (Å) | 0.97872 | 0.97872 | 1.07814 |
| Cell Constants | 99.0 Å, 143.9 Å, 44.6 Å | 98.4 Å, 144.7 Å, 44.0 Å | 98.1 Å, 143.0 Å, 44.3 Å |
| Space Group | P21212 | P21212 | P21212 |
| Resolution (Å)a | 81.57–1.86 (1.867–1.861) | 81.38–1.87 (1.875–1.868) | 98.10 – 1.81( 1.817–1.811) |
| No. of reflections (Unique) | 54,365 | 53,055 | 57,689 |
| No. of reflections (Total) | 330,254 | 319,458 | 449,890 |
| Redundancya | 6.1 (6.0) | 6.0 (6.1) | 7.8 (8.2) |
| Rmerge (%)a | 10.2 (78.4) | 5.1 (86.6) | 8.4 (94.9) |
| CC(1/2)a | 99.4 (76.7) | 99.9 (78.1) | 99.8 (87.3) |
| % Data Completeness (%)b | 99.9 (99.5) | 100.0 (100.0) | 99.9 (99.8) |
| <I/σ(I)> | 10.2 (2.1) | 18.7 (1.9) | 13.1 (2.1) |
| Resolution range for refinement | 81.57–1.86 | 81.38–1.87 | 80.9–1.81 |
| No. of protein atoms | 3790 | 3767 | 3792 |
| No. of water atoms | 278 | 235 | 240 |
| RMSD bond lengths (Å) | 0.009 | 0.009 | 0.014 |
| RMSD bond angles (°) | 1.35 | 1.23 | 1.65 |
| R-factor, Rfree (%)c | 20.0, 23.3 | 18.6, 22.8 | 18.0, 20.5 |
| Ave B-factor Protein | 40.0 | 47.3 | 25.2 |
| Ave B-factor Water | 42.7 | 51.5 | 46.3 |
Values in parentheses are for the highest-resolution shell.
Fraction of theoretically possible reflections observed.
Rfree was calculated with 5% of reflections set aside randomly
In all three structures, strong electron density (Fo-Fc contoured at 3.0 σ) was observed extending from the nucleophilic serine side chain (Ser79) into the active site. Attempts to fit the acyl-ligand structures were successful, and the subsequent models fit the density well (Figure S1). In both monomers, all three structures show continuous electron density between the Oγ of Ser79 and the carbonyl carbon of the substrate indicating a covalent acyl linkage. All three acyl-ligands adopt orientations that match “canonical” β-lactam/β-lactamase interactions: First, the carbonyl oxygens of the acyl linkage are embedded in the so-called oxyanion hole formed by the main chain amide hydrogens of Ser79 and Trp219. Second, the carboxylate that extends from the penem ring (on the carbapenems) or the cephem ring (on cefotaxime) makes a salt bridge with a highly-conserved active site arginine (Arg259). Beyond these common interactions, the three acyl ligands show very interesting deviations from each other.
The structure of acyl-doripenem shows clear density for the hydroxyethyl group, the penem ring, the exocyclic sulfur attached to it, and the pyrrolidine ring, but not the sulfonamide at the terminus of the side chain. As in other structures of doripenem bound to class D carbapenemases, the hydroxyethyl group is in close contact with active site hydrophobic residues (3.3 Å from the methyl group of doripenem’s hydroxyethyl to Cγ2 of Val128 and 3.5 Å from the same methyl group to Cδ2 of Leu166) [16, 17]. The methyl group attached to carbon C4 of doripenem makes contacts with Met221 (Cε, 3.8 Å) and Trp219 (Cβ, 4.6 Å). The structure of acyl-imipenem makes similar contacts to Val128 and Leu166, but its lack of a methyl group attached to C4 means it is missing the contacts to Met221 and Trp219 that doripenem makes. The interaction of doripenem and imipenem with key active site residues can be seen in Figures 5a and 5b.
Figure 5. Active site environments for OXA-239 with three different ligands bound.

(A) Acyl-doripenem, (B) acyl-imipenem, and (C) acyl-cefotaxime bound in the active site of OXA-239. Key residues are shown to illustrate differences in how each drug interacts with the active site.
The electron density for acyl-imipenem terminates after the exocyclic sulfur extending from the penem in one monomer (A) and extends for the full side chain in the other monomer (B). Previous studies have shown that carbapenems can undergo tautomerization after the intial acylation event (Figure S2a). We attempted to fit both the Δ2 (the initial, untautomemerized form) and Δ1 (tautomerized) structures into the density, but only the Δ1 fit well. Figure S3 shows that in both monomers, the electron density for the sulfur places it clearly out of the plane of the ring, confirming that imipenem has transformed from the Δ2 state to the Δ1 state and therefore C3 has become sp3-hybridized. The electron density for doripenem is less conclusive about its tautomer state; attempts to fit the Δ2 and Δ1 of that drug led to equally good fits to the density. It has been noted previously that doripenem makes much more substantial contacts with the phenylalanine side of the bridge (Phe110 in OXA-23/Tyr112 in OXA-24/40) than with the methionine side (Met221/Met223) [17]. This again appears to be true, with the pyrrolidine ring stacked entirely under Phe110 of OXA-239 (Figure 5a). In monomer B of the acyl-imipenem structure however, the alkyl group of the side chain is in a different area and makes much less extensive contacts with Phe110. This potentially weakens the interaction of imipenem with the “bridge” residues of OXA-239. The leucine side chain created by the S109L substitution in OXA-239 projects toward the active site, parallel to the phenyl ring of Phe110. Despite this, it makes little significant contact with either acyl-imipenem, or acyl-doripenem.
Cefotaxime, a third-generation cephalosporin, is much more bulky than the carbapenems, and makes significantly different contacts with the active site of OXA-239 compared to imipenem or doripenem. All major portions of acyl-cefotaxime are represented well by the Fo-Fc electron density maps (contoured at 3.0 σ), except one portion of the thiazole ring of the R1 side chain (Figure S1c). There is no density extending beyond the exocyclic carbon that is attached to carbon C3 of the cephem ring, consistent with the occurrence of an electronic rearrangement after cleavage of the lactam ring that leads to the elimination of that acetyl group (Figure S2b) [20, 34, 35]. As seen with the carbapenems, the acyl-carbonyl oxygen of acyl-cefotaxime is in hydrogen bonding distance to the main chain nitrogens of Ser79 and Trp219 (Figure 5c). The drug’s carboxylate interacts with Arg259 (3.1 Å), Ser126 (2.9 Å) and Thr217 (2.5 Å). The thiazine ring of acyl-cefotaxime (particularly carbon C4) makes contact with the Cζ2 of Trp113 (3.8 Å) and the Cε2 and Cζ carbons of Phe110 (3.6 Å, 4.1 Å).
The large R1 side chain of cefotaxime projects toward the edge of the active site in the area of the β6β7 loop (residues 220–226) and also toward the Ω loop (Figure 5c). This R1 side chain is oriented in a manner such that the methyl group attached to the oxyimino moiety (for reference, see Figure 4c) makes close contacts with Leu166 (3.9 Å with Cδ2 and 3.6 Å with Cβ). The thiazole group that makes up the other branch interacts closely with Trp219 by partially stacking underneath its indole side chain (3.4 Å between S16 and Cζ2 of Trp219). The nitrogen attached to the thiazole ring is in hydrogen bonding distance to the main chain carbonyl of Met221, an interaction that would not be possible without a large conformational change in the loop that contains Met221 (see below). In adopting this orientation, the R1 side chain of cefotaxime is rotated ~180° compared to the similarly-shaped side chain of ceftazidime bound to OXA-160 and OXA-225. In the latter two structures, the thiazole ring projects toward Leu166, and the carboxylpropyl group that makes up the oxyimino branch is directed toward Trp219. In considering the potential implications of the S109L substitution of OXA-239, we also note that the thiazole ring of cefotaxime is within van der Waals contact distance of carbon Cδ2 of Leu109 (4.1 Å).
The overall fold of OXA-239 is quite similar to OXA-23, and the presence of various acyl ligands causes few major changes in the backbone. When the three acylated OXA-239 proteins are aligned with the apo OXA-23 structure, the overall RMSD for Cα carbons is 0.321 Å, 0.454 Å and 0.384 Å for the imipenem-acyl, doripenem-acyl and cefotaxime-acyl structures of OXA-239 K82D respectively. The overlap of key secondary structures for the three OXA-239 structures is visually apparent in Figure 6, with one key exception in the β6β7 loop encompassing residues 220–226. When either imipenem or doripenem is present, this loop maintains roughly the same structure that is has in both the apo [10] and meropenem-bound [15] forms of OXA-23 (Figure 7a,b). The structural conservation is evidenced by the low RMSD values obtained (0.395 Å for OXA-239 K83D/imipenem and 0.538 Å for OXA-239 K83D/doripenem) when all atoms in the loop (main chain and side chain) are aligned with the same region of apo OXA-23 (PDB code: 4K0X). The presence of cefotaxime in the active site as an acyl-intermediate, however, leads to a major alteration of the loop (Figure 7c), and an RMSD of 2.723 Å when compared to apo OXA-23. The bulky R1 side chain of this 3rd generation cephalosporin causes the loop’s main chain, starting at residue 221, to deviate from the structure it takes in OXA-23. Instead of looping in toward the active site, strand β6 continues straight toward the surface of the enzyme. This new conformation pulls both the main chain and side chain of Met221 away from cefotaxime, and thus makes room for the thiazole ring of the drug. Residues 222–225 (Asn-Ala-Ile-Ser) form a β-turn before entering the core of the protein again as strand β7. In all three ligand-bound structures, the average B-factors for the β6β7 loop is significantly higher than the average for the entire protein (54.6 vs 40.1 Å for doripenem, 73.6 vs 47.8 Å for imipenem and 42.4 vs 25.5 Å for cefotaxime), consistent with the idea that this loop has increased flexibility.
Figure 6. Superposition of three acylated OXA-239 structures with OXA-23.
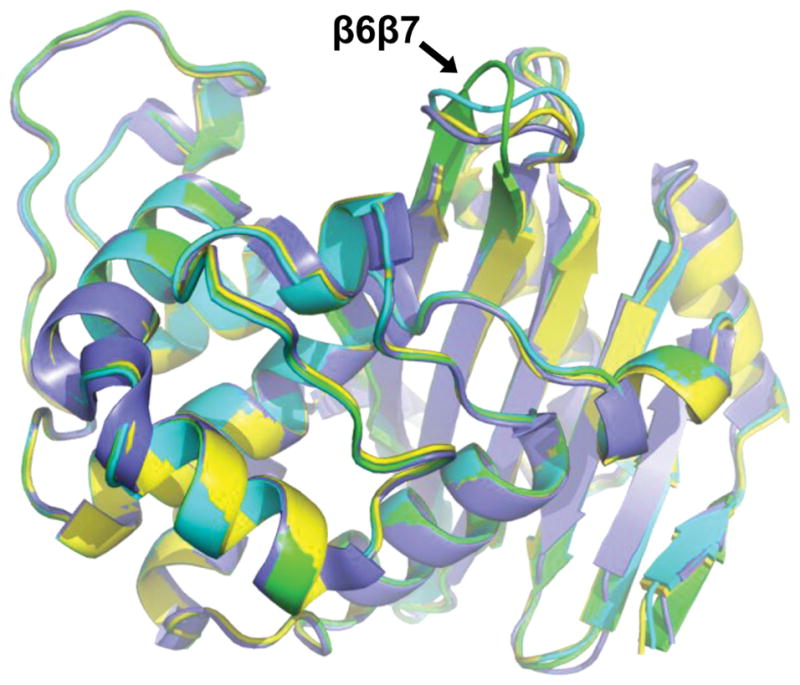
Cartoon structures of OXA-23 wild-type (blue; PDB code: 4K0X), OXA-239 K82D/imipenem (yellow; PDB code: 5WIB), OXA-239 K82D/doripenem (cyan; PDB code: 5WI7), OXA-239 K82D/cefotaxime (green; PDB code: 5WI3). The structures overlay closely except in the area of the β6β7 loop where the cefotaxime-bound structure adopts a different conformation.
Figure 7. Stereoview showing the effect of three different β-lactam ligands on the upper active site of OXA-239.

Active site overlays for OXA-23 (blue; PDB code: 4K0X) and OXA-239 K82D with (A) acyl-doripenem (cyan; PDB code: 5WI7), (B) acyl-imipenem (yellow; PDB code: 5WIB) and (C) acyl-cefotaxime (green; PDB code: 5WI3).
The area of this deviation between OXA-239 and OXA-23 contains two of the three amino acid differences (D222N and P225S) between those two proteins. The way this loop expands the size of the active site and extends further out from the enzyme is reminiscent of how a similar mutation (P227S) alters the β6β7 loop in OXA-160 to allow aztreonam and ceftazidime to bind [20]. In that case, the substitution allows Ser227 to adopt a Φ torsion angle that would not be allowed for proline. To see if this was the case again, we measured the Φ angles for both Ser225 (in OXA-239 K82D/cefotaxime) and Pro225 in apo OXA-23 (PDB code: 4K0X). For apo OXA-23, the Φ angle of Pro225 (−66.7°) falls within the narrow allowable band for that residue. The substituted residue Ser225 in OXA-239 however, adopts a Φ angle (−135.4°) that is acceptable for serine, but not for proline. This suggests that main chain torsion flexibility at position 225 is a significant factor in expanding the OXA-239 active site similar to the way it is in OXA-160.
The other substitution that is found in the β6β7 loop of OXA-239 is D222N. On the surface, this seems to be a much more subtle change, with polarity, size and shape highly conserved. Comparison of the orientation of the side chain for residue 222 between the three structures reveals a major alteration in its position, however. In OXA-239 K82D/imipenem and OXA-239 K82D/doripenem, Asp222 is pointing across the top of the active site, in a similar orientation as “bridge” residue Met221 (Figure 7a,b). In this position, the aspartate side chain makes no contacts with the rest of the protein, or the bound ligands. Figure 7c shows that in the OXA-239 K82D/cefotaxime structure, Asn222 flips its orientation and points away from the active site toward the β-turn that is formed when the β6β7 loop undergoes its conformational change (noted above). Figure 8 shows that the carbonyl oxygen of the asparagine is in hydrogen-bonding distance to the main chain atoms of the next three residues (Ile223, Ala224 and Ser225). In forming the hydrogen bond with the main chain nitrogen of Ile223, Asn222 forms a 10-membered ring referred to as an Asx-turn [36]. These motifs mimic the geometry of a β-turn, but with the side chain of an aspartate or asparagine taking the place of the main chain peptide carbonyl of the first residue in the turn. In addition to these stabilizing interactions, the side chain amide nitrogen of Gln226 is hydrogen bonded to the main chain carbonyls of Asn222 and Ile223. Lastly, the carbonyl oxygen of Asn222 aligns with the carbonyl carbon of its own main chain carbonyl (3.0 Å). This type of dipole-dipole interaction has been shown to have a stabilizing energy equivalent to a hydrogen bond [37]. Interestingly, it is noted that these types of side chain/main chain dipoles occur much more often with asparagine than with aspartate, suggesting that the latter may be more energetically favorable [37].
Figure 8. Stabilizing interactions in the β6β7 loop of OXA-239 K82D/cefotaxime.
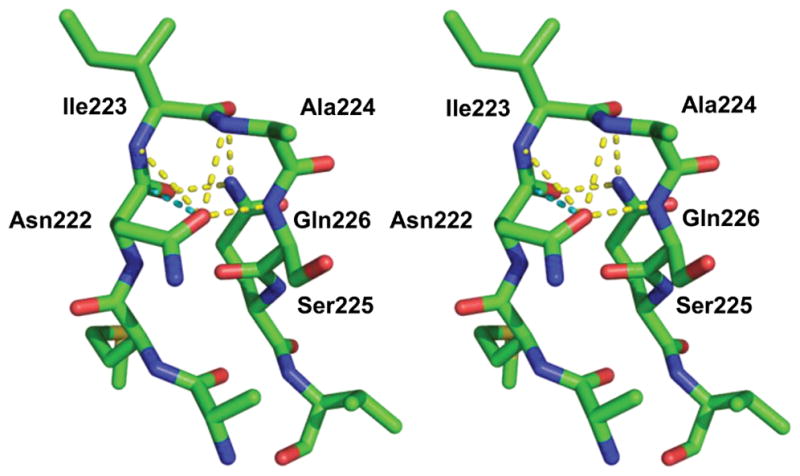
(PDB code: 5WI3). Stereoview of the alternate conformation formed by the β6β7 loop in OXA-239 when cefotaxime is bound as an acyl-intermediate. Stabilizing hydrogen bonds are shown as yellow dashed lines, and the side chain/main chain dipole-dipole interaction for Asn222 is shown as a cyan dashed line.
It has been observed previously that substitutions in β-lactamases that generate greater activity by increasing flexibility sometimes suffer loss of overall stability as well [38, 39]. It has also been shown that secondary mutations sometimes arise to restore protein stability while maintaining the gain-of-function effects of the primary mutation [40]. To see if these effects might be playing a role in OXA-239 structure/function, we used thermal denaturation to determine the stability of OXA-23 and OXA-239, as well as the single- and double-substitution variants that we had tested for kinetic effects. Figure 9 shows that the triple substitution OXA-239 was much less stable than OXA-23, with a decrease in melting temperature (Tm) of −4.6° (Table 3). Individually, the D222N and P225S substitutions decreased stability, but less so than the triple mutant. The effect of P225S on Tm (−3.2°) was approximately twice as large as that observed for D222N (−1.4°). When put together, these two substitutions (P225S and D222N) showed the largest destabilization effect of any combination tested (−5.2°). In contrast to the effects of these two substitutions, the S109L change stabilized the protein modestly (+1.6°). It should be noted that the slightly higher Tm for the triple P225S/D222N/S109L mutant (i.e. OXA-239) compared to that for the P225S/D222N double mutant is consistent with a stabilizing effect of S109L.
Figure 9. Thermal denaturation profiles for OXA-23, OXA-239 and other variants.

Thermal denaturation profiles show that OXA-239 (S109L/D222N/P225S; green) is much less stable than OXA-23 (purple). The S109L substitution on its own gives a higher stability, while D222N and P225S, individually or in combination with each other, destabilize the protein.
Table 3.
Thermal denaturation parameters
| Enzyme | Tm (ºC) | ΔHVH (kcal·mol−1) | ΔTm (ºC)a |
|---|---|---|---|
| OXA-23 | 60.4 | 180 ±16 | - |
| OXA-239 | 56.0 | 240 ± 30 | −4.4 |
| OXA-23 S109L | 62.0 | 260 ± 59 | +1.6 |
| OXA-23 D222N | 59.0 | 164 ± 15 | −1.4 |
| OXA-23 P225S | 57.2 | 204 ± 20 | −3.2 |
| OXA-23 D222N/P225S | 55.2 | 226 ± 13 | −5.2 |
ΔTm = Tvariant-TWT are relative to OXA-23
DISCUSSION
Hydrolysis of carbapenems by class D β-lactamases is greatly enhanced by a particular conformation of the β6β7 loop in which a residue on that loop (Met221 in OXA-23) extends over the active site and helps stabilize the bound carbapenem substrate [12]. This conformation, because of its narrowing of the active site, precludes the binding of β-lactams with bulky side chains such as those found in oxacillin, 3rd and 4th generation cephalosporins (e.g. cefotaxime) and the monobactam aztreonam. The binding of such drugs therefore requires an alternative conformation that enlarges the active site. The data we present in this manuscript suggest that the OXA-23 subfamily variant OXA-239 has the ability to adopt multiple conformations. Our data further suggest that the P225S substitution enhances main chain flexibility in the β6β7 loop, and thus permits an alternate conformation to form, enlarging the active site. A more flexible β6β7 loop could potentially raise the affinity for bulky drugs, an explanation consistent with the lower Km values for OXA-239 that we observed for cephalosporins such as cefotaxime. As Waley notes however, interpretation of kinetic changes observed after mutagenesis of β-lactamases should be approached with caution, as scenarios exist in which no change in affinity can lead to lower Km values [41]. Without knowledge of binding, acylation and deacylation rate constants, it is difficult to exclude the possibility that changes in the deacylation rate alone, for instance, are responsible for lower Km values. The D222N substitution appears to encourage the formation of the alternate conformation by adopting an Asx-turn motif that is further stabilized by a side chain/main chain dipole interaction that is more stable for asparagine than aspartate [37].
Despite these effects, the β6β7 loop is still capable of forming the carbapenem-stabilizing trajectory observed in the OXA-23/meropenem structure (PDB code 4JF4), as evinced by its presence in the acyl-imipenem and acyl-doripenem structures of OXA-239. It is intriguing that the three substitutions in OXA-239 appear to cause a large increase of imipenem Km, but much less so for doripenem. There are two main structural differences between these two carbapenems: an additional methyl group on doripenem attached to carbon 4 and a larger side-chain on the same drug (Figure 4). These differences necessarily lead to the altered kinetic values, but absent a more detailed analysis, it is difficult to be certain how they do so.
The purpose of a third substitution, S109L, may be two-fold. Its modest stabilizing effect (revealed by thermal denaturation experiments) could counteract the destabilizing effect of the other two activity-enhancing substitutions (D222N and P225S). The transformation of a short, polar serine side chain into a longer, non-polar leucine would be expected to stabilize the phenylalanine side chain of the adjacent residue against which it packs (Phe111). This stabilization would be particularly important upon the loss of Met221 as a “bridge” partner for F111 upon rearrangement of the β6β7 loop. In addition to stabilizing OXA-239, there is some indication that the S109L substitution may have a positive kinetic effect as well. The effect of S109L alone on the hydrolysis of various β-lactams ranges from neutral to negative, but the addition of the same substitution in the presence of P225S and D222N lowers the Km of some cephalosporins significantly. Our observation of a potentially stabilizing non-polar contact between Leu109 and the thiazole ring of acyl-cefotaxime could lead to tighter affinity, which would be one possible explanation for a lower Km. This interaction would not be possible in the absence of one or both of the other substitutions that allow the drug to bind in the first place.
While the clinical scenario that resulted in the transformation of OXA-23 to OXA-239 by the accumulation of the three substitutions is not clear, the recent identification of OXA-469, a single-substitution variant of OXA-239 (M250I), suggests this adaptive accumulation of substitutions may have proceeded another step [25]. While it is not known what effect the M250I substitution has on hydrolytic efficiency or stability, it is intriguing that residue 250 occupies a position directly underneath the β6β7 loop, the structure which is so highly impacted by both P225S and D222N.
The mechanism by which clinical substitutions allow class D carbapenemases to expand their substrate specificity into new classes of β-lactams has been useful in understanding the basic functions of various structural features of those enzymes. Given the on-going efforts to develop novel therapeutic agents that target A. baumannii, these observations may also serve a clinically-relevant harbinger role. While cephalosporins and monobactams are not currently used against A. baumannii, novel agents such as the cephalosporin cefiderocol and monobactam BAL30072 show great potential against carbapenem-resistant strains of that species [42–44]. Enhanced activity against advanced generation cephalosporins and aztreonam has now been observed for at least five unique variants of the OXA-23, OXA-24/40 and OXA-51/66 subfamilies of class D β-lactamases [9, 10, 20, 21]. These variants all achieve that enhanced activity with just 1–3 substitutions (or in one case, a single amino acid duplication) compared to their parental enzymes. Given that these parental enzymes are so common in A. baumannii lineages across the world, it will be important to consider the potential effect of these variants on any novel agents.
In summary, we have investigated the kinetic and structural properties of the class D carbapenemase OXA-239, which differs from OXA-23 by just three amino acid substitutions. The substrate specificity of OXA-239 is altered compared to OXA-23, with higher kcat/Km ratios for a number of advanced generation cephalosporins and aztreonam, but slightly lower activity against carbapenems. Structures of OXA-239 with of doripenem, imipenem and cefotaxime bound as acyl-intermediates indicate that the structure of a key active site loop adopts a different conformation when carbapenems are bound compared to when cefotaxime is bound. We propose that two of the substitutions (P225S and D222N), which are found in this active site loop, may increase its flexibility and thereby give it the plasticity needed to accept multiple different β-lactam structures.
Supplementary Material
Acknowledgments
Funding Information
This research was supported by National Institutes of Health grants 1R15AI082416 (D.A.L.) and R15AI094489 (R.A.P). Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Abbreviations
- ISAba1
Insertion sequence Acinetobacter baumannii 1
- KCX
carboxy-lysine
- PEG
polyethylene glycol
- RMSD
root mean square deviation
Footnotes
Declarations of Interest
The Authors declare that there are no competing interests associated with the manuscript.
Author Contribution Statements
Protein purification and kinetics assays were performed by T.M.H. under the supervision of D.A.L. Structure determination by X-ray crystallography was carried out by T.M.H and C.M.J. under the supervision of R.A.P. Thermal denaturation experiments were performed by M.A.T. under the supervision of R.A.B. The final version of this manuscript was written by C.M.J. and D.A.L. and discussed and revised in agreement with all authors.
References
- 1.Evans BA, Hamouda A, Amyes SG. The rise of carbapenem-resistant Acinetobacter baumannii. Curr Pharm Des. 2013;19:223–238. [PubMed] [Google Scholar]
- 2.Higgins PG, Dammhayn C, Hackel M, Seifert H. Global spread of carbapenem-resistant Acinetobacter baumannii. J Antimicrob Chemother. 2010;65:233–238. doi: 10.1093/jac/dkp428. [DOI] [PubMed] [Google Scholar]
- 3.WHO Priority List for Pathogens List for R&D of New Antibiotics. 2017 http://www.who.int/medicines/publications/global-priority-list-antibiotic-resistant-bacteria/en/
- 4.Gehrlein M, Leying H, Cullmann W, Wendt S, Opferkuch W. Imipenem resistance in Acinetobacter baumanii is due to altered penicillin-binding proteins. Chemotherapy. 1991;37:405–412. doi: 10.1159/000238887. [DOI] [PubMed] [Google Scholar]
- 5.Hu WS, Yao SM, Fung CP, Hsieh YP, Liu CP, Lin JF. An OXA-66/OXA-51-like carbapenemase and possibly an efflux pump are associated with resistance to imipenem in Acinetobacter baumannii. Antimicrob Agents Chemother. 2007;51:3844–3852. doi: 10.1128/AAC.01512-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rumbo C, Gato E, López M, Ruiz de Alegría C, Fernández-Cuenca F, Martínez-Martínez L, Vila J, Pachón J, Cisneros JM, Rodríguez-Baño J, Pascual A, Bou G, Tomás M. Contribution of efflux pumps, porins, and β-lactamases to multidrug resistance in clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother. 2013;57:5247–5257. doi: 10.1128/AAC.00730-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans BA, Amyes SG. OXA β-Lactamases. Clin Microbiol Rev. 2014;27:241–263. doi: 10.1128/CMR.00117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turton JF, Ward ME, Woodford N, Kaufmann ME, Pike R, Livermore DM, Pitt TL. The role of ISAba1 in expression of OXA carbapenemase genes in Acinetobacter baumannii. FEMS Microbiol Lett. 2006;258:72–77. doi: 10.1111/j.1574-6968.2006.00195.x. [DOI] [PubMed] [Google Scholar]
- 9.Mitchell JM, Leonard DA. Common clinical substitutions enhance the carbapenemase activity of OXA-51-like class D β-lactamases from Acinetobacter spp. Antimicrob Agents Chemother. 2014;58:7015–7016. doi: 10.1128/AAC.03651-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaitany KC, Klinger NV, June CM, Ramey ME, Bonomo RA, Powers RA, Leonard DA. Structures of the class D carbapenemases OXA-23 and OXA-146: mechanistic basis of activity against carbapenems, extended-spectrum cephalosporins, and aztreonam. Antimicrob Agents Chemother. 2013;57:4848–4855. doi: 10.1128/AAC.00762-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith CA, Antunes NT, Stewart NK, Frase H, Toth M, Kantardjieff KA, Vakulenko S. Structural Basis for Enhancement of Carbapenemase Activity in the OXA-51 Family of Class D β-Lactamases. ACS Chem Biol. 2015;10:1791–1796. doi: 10.1021/acschembio.5b00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santillana E, Beceiro A, Bou G, Romero A. Crystal structure of the carbapenemase OXA-24 reveals insights into the mechanism of carbapenem hydrolysis. Proc Natl Acad Sci USA. 2007;104:5354–5359. doi: 10.1073/pnas.0607557104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leonard DA, Bonomo RA, Powers RA. Class D β-Lactamases: A Reappraisal after Five Decades. Acc Chem Res. 2013;46:2407–2415. doi: 10.1021/ar300327a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Golemi D, Maveyraud L, Vakulenko S, Samama JP, Mobashery S. Critical involvement of a carbamylated lysine in catalytic function of class D β-lactamases. Proc Natl Acad Sci USA. 2001;98:14280–14285. doi: 10.1073/pnas.241442898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith CA, Antunes NT, Stewart NK, Toth M, Kumarasiri M, Chang M, Mobashery S, Vakulenko SB. Structural basis for carbapenemase activity of the OXA-23 β-lactamase from Acinetobacter baumannii. Chem Biol. 2013;20:1107–1115. doi: 10.1016/j.chembiol.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.June CM, Muckenthaler TJ, Schroder EC, Klamer ZL, Wawrzak Z, Powers RA, Szarecka A, Leonard DA. The structure of a doripenem-bound OXA-51 class D β-lactamase variant with enhanced carbapenemase activity. Protein Sci. 2016;25:2152–2163. doi: 10.1002/pro.3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider KD, Ortega CJ, Renck NA, Bonomo RA, Powers RA, Leonard DA. Structures of the class D carbapenemase OXA-24 from Acinetobacter baumannii in complex with doripenem. J Mol Biol. 2011;406:583–594. doi: 10.1016/j.jmb.2010.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maveyraud L, Golemi D, Kotra LP, Tranier S, Vakulenko S, Mobashery S, Samama JP. Insights into class D β-lactamases are revealed by the crystal structure of the OXA10 enzyme from Pseudomonas aeruginosa. Structure. 2000;8:1289–1298. doi: 10.1016/s0969-2126(00)00534-7. [DOI] [PubMed] [Google Scholar]
- 19.Szarecka A, Lesnock KR, Ramirez-Mondragon CA, Nicholas HB, Wymore T. The Class D β-lactamase family: residues governing the maintenance and diversity of function. Protein Eng Des Sel. 2011;24:801–809. doi: 10.1093/protein/gzr041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell JM, Clasman JR, June CM, Kaitany KC, LaFleur JR, Taracila MA, Klinger NV, Bonomo RA, Wymore T, Szarecka A, Powers RA, Leonard DA. Structural basis of activity against aztreonam and extended spectrum cephalosporins for two carbapenem-hydrolyzing class D β-lactamases from Acinetobacter baumannii. Biochemistry. 2015;54:1976–1987. doi: 10.1021/bi501547k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schroder EC, Klamer ZL, Saral A, Sugg KA, June CM, Wymore T, Szarecka A, Leonard DA. Clinical Variants of the Native Class D β-Lactamase of Acinetobacter baumannii Pose an Emerging Threat through Increased Hydrolytic Activity against Carbapenems. Antimicrob Agents Chemother. 2016;60:6155–6164. doi: 10.1128/AAC.01277-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cayô R, Merino M, Ruiz Del Castillo B, Cano ME, Calvo J, Bou G, Martínez-Martínez L. Antimicrob Agents Chemother. 2014. OXA-207, a novel OXA-24 variant with reduced catalytic efficiency against the carbapenems in Acinetobacter pittii from Spain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher JF, Meroueh SO, Mobashery S. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chemical Reviews. 2005;105:395–424. doi: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 24.Tamayo-Legorreta EM, Garza-Ramos U, Barrios-Camacho H, Sanchez-Perez A, Galicia-Paredes A, Meza-Chavez A, Silva-Sanchez J. Identification of OXA-23 carbapenemases: novel variant OXA-239 in Acinetobacter baumannii ST758 clinical isolates in Mexico. New Microbes New Infect. 2014;2:173–174. doi: 10.1002/nmi2.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gonzalez-Villoria AM, Tamayo-Legorreta E, Garza-Ramos U, Barrios H, Sanchez-Pérez A, Rodríguez-Medina N, Uribe-Aviña N, Cevallos MA, Silva-Sanchez J CRAB Study Group. A Multicenter Study in Mexico Finds Acinetobacter baumannii Clinical Isolates Belonging to Clonal Complexes 636B (113B) and 92B Harboring OXA-72, OXA-239, and OXA-469. Antimicrob Agents Chemother. 2016;60:2587–2588. doi: 10.1128/AAC.02042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tian GB, Adams-Haduch JM, Bogdanovich T, Pasculle AW, Quinn JP, Wang HN, Doi Y. Identification of diverse OXA-40 group carbapenemases, including a novel variant, OXA-160, from Acinetobacter baumannii in Pennsylvania. Antimicrob Agents Chemother. 2010;55:429–432. doi: 10.1128/AAC.01155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Research. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, Womack T, Bricogne G. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider KD, Bethel CR, Distler AM, Hujer AM, Bonomo RA, Leonard DA. Mutation of the active site carboxy-lysine (K70) of OXA-1 β-lactamase results in a deacylation-deficient enzyme. Biochemistry. 2009;48:6136–6145. doi: 10.1021/bi900448u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Powers RA, Caselli E, Focia PJ, Prati F, Shoichet BK. Structures of ceftazidime and its transition-state analogue in complex with AmpC β-lactamase: implications for resistance mutations and inhibitor design. Biochemistry. 2001;40:9207–9214. doi: 10.1021/bi0109358. [DOI] [PubMed] [Google Scholar]
- 35.Birck C, Cha JY, Cross J, Schulze-Briese C, Meroueh SO, Schlegel HB, Mobashery S, Samama JP. X-ray crystal structure of the acylated β-lactam sensor domain of BlaR1 from Staphylococcus aureus and the mechanism of receptor activation for signal transduction. Journal of the American Chemical Society. 2004;126:13945–13947. doi: 10.1021/ja044742u. [DOI] [PubMed] [Google Scholar]
- 36.Duddy WJ, Nissink JW, Allen FH, Milner-White EJ. Mimicry by asx- and ST-turns of the four main types of β-turn in proteins. Protein Sci. 2004;13:3051–3055. doi: 10.1110/ps.04920904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deane CM, Allen FH, Taylor R, Blundell TL. Carbonyl-carbonyl interactions stabilize the partially allowed Ramachandran conformations of asparagine and aspartic acid. Protein Eng. 1999;12:1025–1028. doi: 10.1093/protein/12.12.1025. [DOI] [PubMed] [Google Scholar]
- 38.Beadle BM, Shoichet BK. Structural bases of stability-function tradeoffs in enzymes. Journal of Molecular Biology. 2002;321:285–296. doi: 10.1016/s0022-2836(02)00599-5. [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Minasov G, Shoichet BK. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. Journal of Molecular Biology. 2002;320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 40.Marciano DC, Pennington JM, Wang X, Wang J, Chen Y, Thomas VL, Shoichet BK, Palzkill T. Genetic and structural characterization of an L201P global suppressor substitution in TEM-1 β-lactamase. Journal of Molecular Biology. 2008;384:151–164. doi: 10.1016/j.jmb.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Waley SG. β-Lactamase: mechanism of action. In: Page MI, editor. The Chemistry of β-Lactams. Springer; Netherlands, Dordrecht: 1992. pp. 198–228. [Google Scholar]
- 42.Falagas ME, Skalidis T, Vardakas KZ, Legakis NJ, Group HCS. Activity of cefiderocol (S-649266) against carbapenem-resistant Gram-negative bacteria collected from inpatients in Greek hospitals. J Antimicrob Chemother. 2017;72:1704–1708. doi: 10.1093/jac/dkx049. [DOI] [PubMed] [Google Scholar]
- 43.Kohira N, West J, Ito A, Ito-Horiyama T, Nakamura R, Sato T, Rittenhouse S, Tsuji M, Yamano Y. In Vitro Antimicrobial Activity of a Siderophore Cephalosporin, S-649266, against Enterobacteriaceae Clinical Isolates, Including Carbapenem-Resistant Strains. Antimicrob Agents Chemother. 2016;60:729–734. doi: 10.1128/AAC.01695-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mushtaq S, Woodford N, Hope R, Adkin R, Livermore DM. Activity of BAL30072 alone or combined with β-lactamase inhibitors or with meropenem against carbapenem-resistant Enterobacteriaceae and non-fermenters. Journal of Antimicrobial Chemotherapy. 2013;68:1601–1608. doi: 10.1093/jac/dkt050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.