

Abstract
During a heart attack loss of blood flow and oxygen to regions of the heart results in the massive death of myocardial cells. Since, terminally differentiated adult myocardial cells proliferate at a very low rate the mammalian heart is unable to recover from the massive cell loss. This is in contrast to other animals such as adult zebrafish, axolotl, newts and mammalian neonates in which de-differentiation and proliferation facilitates regeneration after injury. Thus, a lack of myocardial plasticity and proliferation within adult mammalian myocardial cells results in insufficient repair after a myocardial infarction. Conversely, de-differentiation appears to be associated with the pathology of many chronic cardiomyopathies. Here we highlight studies from the developmental, regenerative and clinical cardiac fields which together provide insight into the molecular and conceptual underpinning of myocardial plasticity.
Introduction
Myocardial lineage and differentiation
Despite constituting only ~30% of heart, myocardial cells are the primary source of cardiac contractile function resulting in the efficient pumping of blood throughout the body [1,2]. The different myocardial sub-types are broadly divided into working myocardium responsible for blood flow (i.e. atrial, ventricular myocytes) and non-working myocardium responsible for the efficiency of blood flow (i.e. outflow tract, inflow tract and conduction myocytes) [3,4].
Myocardial cells are generated from a diverse set of lineages (for an in depth review see [3]). Two waves of mesodermal-derived cells during gastrulation, named first heart field (FHF) and second heart field (SHF) constitute the majority of the left ventricle (FHF), right ventricle (SHF), right and left atria (FHF/SHF) and outflow/inflow tracts (SHF)[5–8]. These contributions occur sequentially with myocardial cells derived from the FHF forming the outer layer of the primitive heart tube, which is then further expanded by the addition of late differentiating SHF cells [9,10]. A third wave closely associated with the SHF forms the Sino-atrial node (pacemaker) [11]. These cells are supplemented by a set of neural crest derived cells, which contribute to the septation of the heart [12]. Each lineage contributes to a specific anatomical set of myocardial cells. Yet, they contribute to an overlapping variety of myocardial sub-types, which are indistinguishable histologically[3,13].
Myocardial cells must maintain appropriate contractile function throughout the entire development and morphogenesis of the heart. As the embryo develops and grows into an adult, the requirements on the heart change dramatically requiring alterations in the number, size and efficiency of the myocardial muscle. As a result, myocardial cells dynamically differentiate during embryonic and fetal development to meet these functional demands, which include a metabolic switch from glycolysis to fatty acid metabolism, increases in cell size, diminished proliferative capacity, and changes in protein isoforms [14–23]
Many of these changes, such as loss of proliferative capacity in the adult mammalian heart, become detrimental during acute heart disease when there is a large loss of myocardial cells. In other animals such as the adult zebrafish, myocardial cells are able to respond to a similar injury by de-differentiating and proliferating [24]. De-differentiation has also been observed during chronic cardiac disease where it is associated with the progression of disease [25]. In this review, we bring together studies from cardiac development, regeneration and disease to summarize the role of myocardial plasticity in adaptive responses such as during regeneration as well as maladaptive responses such as during chronic cardiac disease.
(Each of these fields is extensive, thus we apologize in advance to our colleagues whose valuable work has been omitted due to space constraints. Further, we point the reader to more extensive reviews focused on each individual topic [3,25–29].)
Restriction of atrial and ventricular myocardial identity during development
Atrial and ventricular myocytes display dramatic biological differences; yet a series of genetic studies reveals that these differentiated myocytes are able to exchange their identities. Differences between atrial and ventricular myocytes can be observed in structural, physiological and molecular myocyte properties. Ventricular myocytes are rod-like in morphology with highly organized sacromeric structures, including specialized T-tubule sub-structures and also exhibit a flat action potential plateau. In contrast, atrial myocytes are more squamous with poorly developed, disorganized sacromeres and display a triangular shaped action potential with a shorter contraction and relaxation period [30–38].
These genetic studies have identified a set of counterbalancing transcription factors that maintain the identity of differentiated chamber myocytes by repressing the gene program of the opposing chamber (atrial or ventricular) and promoting their own chamber-specific gene program. For example, COUP-TFII is an ophraned nuclear hormone receptor that is expressed in atrial but not ventricular myocytes[39] where it functions to maintain atrial identity. Studies in mouse by Wu et al. show that conditional removal of COUP-TFII in the atrium using the Myh6:cre results in atria containing myocytes with ventricular identity. Conversely, forced ventricular expression of COUP-TFII, results in ectopic atrial fates [40**]. Thus, COUP-TFII is involved in restricting ventricular identity and promoting atrial identity. Microarray and ChiP-seq studies further revealed that COUP-TFII controls a large range of genes important for both atrial and ventricular identity including Tbx5 (important for promoting atrial identity[41–43]) and hey2 (important for promoting ventricular identity [44]). Thus, COUP-TFII may act by repressing the ventricular transcriptional network while promoting the atrial expression program. Intriguingly, COUP-TFII restricts this atrial plasticity until e15.5, after which the COUP-TFII gene program is no longer needed to maintain chamber identity [40].
Conversely, the transcription factors Nkx2.5, Hey2 and Irx4 are important for maintaining ventricular chamber identity. Nkx2.5 is expressed throughout the heart and is required for a range of functions during heart development including cardiac differentiation, conduction specification and septation [45–47] (reviewed-[48,49]). A recent set of studies in zebrafish have identified a new role for Nkx2.5 in restricting the plasticity of ventricular cells. In the absence of Nkx2.5 and its ortholog Nkx2.7, ventricular cells trans-differentiate into atrial cells leading to an enlarged atrium and a small ventricle without change in overall cardiac cell number [50,51]. These studies correspond with recent studies identifying plasticity in atrial lineages after Amhc expression [52] and with ventricular chamber defects in mammalian studies of Nkx2.5 mutants [53–55].
Irx4 and Hey2 are restricted to the ventricle, where loss and gain-of-function studies in the mouse reveal a similar exchange of ventricular identity [44,56–58]. However, unlike Nkx2.5 mutants in zebrafish, distinct chambers are maintained despite ectopic expression of the atrial gene program within the ventricle of Irx4 or Hey2 mutants. The expression of Irx4 and Hey2 are lost in Nkx2.5 mutants suggesting a hierarchy in which Nkx2.5 regulates Irx4 and Hey2 [50,59]. Furthermore, Irx4 was shown to bind to RxRa and inhibit its binding to the Myh6 (atrial specific myosin) promoter [60,61]. These results suggest that plasticity may be restricted by interacting with inductive pathways, such as Retinoic acid signaling. Atrial-ventricular antagonism also appears to occur at the level of myosin expression. As demonstrated by the ectopic ventricular expression of MLC2a in MLC2v mutants [62*]. Intriguingly, these studies of chamber plasticity are similar to Ebstein’s anamoly a rare congenital heart defect in which a portion of the right ventricule is “atrialized” and to which mutations in Nkx2.5 have been linked [63].
Separate from atrial-ventricular identity, another set of studies revealed that overexpression of the Notch-intercellular domain (NICD) or Tbx18 in adult working myocardial cells can transform them into non-working conduction myocytes [64,65]. Whether there exists a regulatory network to suppress conduction identity in mature working myocytes or whether these experiments represent reprogramming events remains unclear.
Together these studies reveal a genetic network that regulates the ability of mature myocytes to exchange identities. Interestingly, these genes do not regulate all possible forms of plasticity but rather the transformation between atrial and ventricular states. The existence of a genetic network that regulates a specific identity transformation is similar to function of Scl/Tal1 which inhibits endothelial cells from trans-differentiating to a myocardial lineage [66–68**]. The close proximity of these lineages (atrial/ventricular, endothelial/myocardial) and the existence of genetic programs that restrict their ability to trans-differentiate during development into each other suggests a possible fundamental property of proximal lineages. Indeed a system-level analysis of C. elegans lineages [69*], confirms the existence of regulatory mechanisms that restrict the plasticity of proximal lineages to exchange identities. However, further experimentation is necessary to confirm this relationship and to elucidate the underlying mechanism of plasticity.
Myocardial plasticity during cardiac regeneration
While mammalian adult cardiac tissue does not undergo regeneration after injury[70,71*], the hearts of several amphibians and fish species have been shown to regenerate. Utilizing histological and SEM, classical studies in these animals observed cardiac cells near the site of injury which appear to undergo a process of de-differentiation (reduced sarcomere structure) and proliferation (increased incorporation of tritiated thymidine/BrdU and mitoses) [72–75*] [76*] similar to the phases of regeneration in other organs [77]. However, the question of whether new myocardial tissue is generated from an unknown stem cell population, the transdifferentiation of other cell types, or un-injured mature myocardial cells was unknown. Using cre-lox lineage tracing to mark and track myocardial cells, studies of adult zebrafish after ventricular injury revealed that new myocardial cells are generated almost exclusively from pre-existing differentiated myocardial cells[78,79**], which undergo a de-differentiation and proliferative response. During de-differentiation, myocardial cells display disorganized sarcomeric structures [80] as shown in earlier SEM studies and also express genes from embryonic development including fetal myosin genes and transcription factors such as Gata4, Tbx20, Tbx5, Nkx2.5 and Hand2 [79,81,82].
Many of these transcription factors are important for cardiogenesis during development. For example, the Gata 4-6 and Hand transcription factors are essential for de-novo cardiac differentiation [83–91] and are part of the cocktail of factors sufficient to reprogram fibroblasts into cardiomyocytes [92–96]. Inhibition of gata4 during cardiac injury in the adult zebrafish, reduces the generation of new myocardial cells leading to scar formation[97]. Similarly, over-expression of Hand2 during cardiac injury increases myocardial proliferation after injury. [81]. However, their exact role in the processes of de-differentiation and re-differentiation remain to be elucidated.
A number of studies have begun to elucidate upstream signals responsible activating myocardial de-differentiation (reviewed here [28]). NfKb, BMP, RA, Shh, Pdgf, reactive oxygen, Paxillin have all been shown to be required for the activation of gata4 and other embryonic genes [98–106]. Intriguingly, some signals (Notch, Igf) appear to be required for proliferative activity, but not the re-expression of this embryonic cardiac program [107,108], suggesting these signals may act in parallel or downstream. Of note are the studies focusing on Neuregulin signaling, which is upregulated in perivascular cells upon injury. Gemberling et al. show that Neuregulin signaling is not only necessary but also sufficient to trigger de-differentiation and proliferation, suggesting it provides an instructive signal to stimulate a regenerative response in myocardial cells upon injury [109**].
Intriguingly, studies of zebrafish larval hearts revealed a trans-differentiation response within atrial cells after ventricular injury. Larval hearts contain differentiated cardiomyocytes with highly organized sarcomeric structures [17,110]. When larval ventricular myocytes are ablated using nitroreductase and metrodinazole [111], a de-differentiation and proliferative response is induced in both ventricular and atrial myocytes. Genetic marking of differentiated atrial cells revealed that atrial cells de-differentiate, migrate and then re-differentiate as ventricular myocyte, helping to repopulating the injured ventricle. Although, the molecular mechanisms underlying this atrial response still remain to be fully elucidated, initial studies indicate a requirement for Notch signaling within the endocardium [110**].
This trans-differentiation response of larval atrial cardiomyocytes to ventricular injury contrasts with adult injury, in which atrial cardiomyocytes have not been shown to contribute to the ventricle. Indeed the atrial trans-differentiation response diminishes with age [110]. However, adult atrial myocytes do retain a degree of plasticity; the adult zebrafish atrium regenerates after injury and adult newt and rat atrial cells re-enter the cell cycle upon ventricular injury [80,112–115]. Future investigations into the intrinsic and extrinsic differences between adult and larval atrial cells are likely to be informative in understanding the regulation of myocardial plasticity.
Neo-natal Mammalian Regeneration
Despite the intransgience of mammalian adult hearts to regenerate, observations from human case studies [116–122] and mouse studies of cardiac damage during development [123–125] have suggested that embryonic and neonatal mammalian hearts might possess regenerative potential. This hypothesis has been recently tested, with several researchers showing that neonatal hearts (P1) are able to regenerate upon injury but fetal (>P7) and adult hearts can not [123,126–129] (further reviewed here [24,29]). These studies reveal striking parallels between neonatal heart regeneration and adult zebrafish heart regeneration, including the generation of new myocytes from the de-differentiation and proliferation of pre-existing mature myocytes [129] and the critical role of the re-expression of embryonic transcription factors such as Gata4 [130]. Additionally, Neuregullin signaling plays a pivotal role in both adult zebrafish and neonatal mouse regeneration. Addition of neuregulin stimulates proliferation in neonatal cardiomyocytes, but not after P7 due to the downregulation of the neuregulin receptor, Erbb2. However, a transient ectopic Erbb2 signaling pulse in juvenile animals is sufficient to stimulate de-differentiation and proliferation [131**]. Further studies exploring whether changes to Erbb2 expression is the only event that mediates the regenerative capacity of P1 compared to P7 animals will help to elucidate what portions of the regenerative program remain poised in adult cardiomyocytes.
Myocardial plasticity during pathological cardiac remodeling
De-differentiation has also been shown to occur during acute and chronic cardiac disease (reviewed here [25]). For example, de-differentiation is observed in the ventricle of human patients after acute myocardial injury [132,133] and in animal models of cardiac injury, such as pressure-overload in rats and rabbits [132,134]. Chronic cardiac diseases in humans such as dilated cardiomyopathies and mitral valve disease [135–137] as well as animal models such as MCP-1 also feature myocardial de-differentiation [138].
Myocardial de-differentiation during cardiac disease is characterized by changes from a highly organized striated sarcomeric structure to a less dense disorganized complex, [137] along with metabolic and molecular changes reminiscent of fetal cardiomyocytes. For example, myocardial cells in a diseased hearts switch from fatty acid metabolism, used almost exclusively in adult myocytes, to the glycolytic pathway used in fetal myocytes [139]. Furthermore, fetal gene isoforms expressed during development are re-expressed during cardiac disease [137,140–142] as are transcription factors associated with early cardiac development, such as Gata4, Nkx2.5 and Mef2c [143–147] (reviewed here [27,148]).
Several studies have begun to investigate the role embryonic cardiac transcription factors have on disease progression. For example, loss of Gata4 in adult cardiomyocytes attenuates TAC-induced hypertrophy, indicating an essential role for Gata4 in a pathological hypertrophic response[149]. Gata4 is expressed in adult cardiomyocytes where its activity is upregulated in response to pathological hypertrophy[150]. Loss of Gata4 as well as gain of Gata4 activity in adult cardiomyocytes leads to pathological hypertrophy indicating that a proper balance of Gata4 activity is required to limit hypertrophy [149,151]. Similarly, Mef2 activity is upregulated during pressure overload [147,152–154], where it is implicated in promoting hypertrophy [155,156]. These studies indicate an integral role for de-differentiation in the pathology of cardiac remodeling. Future studies investigating how de-differentiation is triggered and how it contributes to cardiac disease are likely to be highly informative for therapeutic interventions.
One of the mechanisms through which de-differentiation could be triggered is through inflammatory cytokines which are prevalent during cardiac disease. Expression of the inflammatory cytokine, oncostatin M (OSM) occurs in both patients who have suffered a myocardial infarct (MI) or who have dilated cardiomyopathy (DCM)[157**]. Studies by Kubin et al. show that exposure of adult rat myocardial cells to OSM in vivo and in vitro induces MAPK-dependent de-differentiation and EdU-incorporation. Interestingly, OSM was found to have opposing affects on models of MI and DCM. OSM-addition increased survival upon MI, but decreased survival in the chronic MCP-1 model of DCM [158][157]. Additionally, Neuregulin signaling has also been shown to stimulate de-differentiation and proliferation in adult cardiomyocytes [131,159], indicating that multiple signaling sources may stimulate de-differentiation during cardiac disease.
Future perspectives
Despite the identification of multiple factors that trigger or restrict myocardial plasticity, we still lack a complete understanding of how this plasticity is regulated or its role in cardiac disease. Epigenetic regulation such as chromatin remodeling represents a logical hypothesis for how myocardial plasticity is restricted and reactivated. Chromatin remodeling factors have been shown to be essential for cardiac differentiation [91,160], reprogramming [96,161] and cardiac disease[162,163]. Thus, identification of chromatin states and chromatin modulators essential for plasticity processes such as de-differentiation and trans-differentiation is likely to be essential for understanding these changes. These ongoing studies of myocardial plasticity represent the forefront of an exciting cross-disciplinary field important for a fundamental understanding of cell identity and for developing reparative therapies.
Figure 1. Restriction of atrial and ventricular identity.
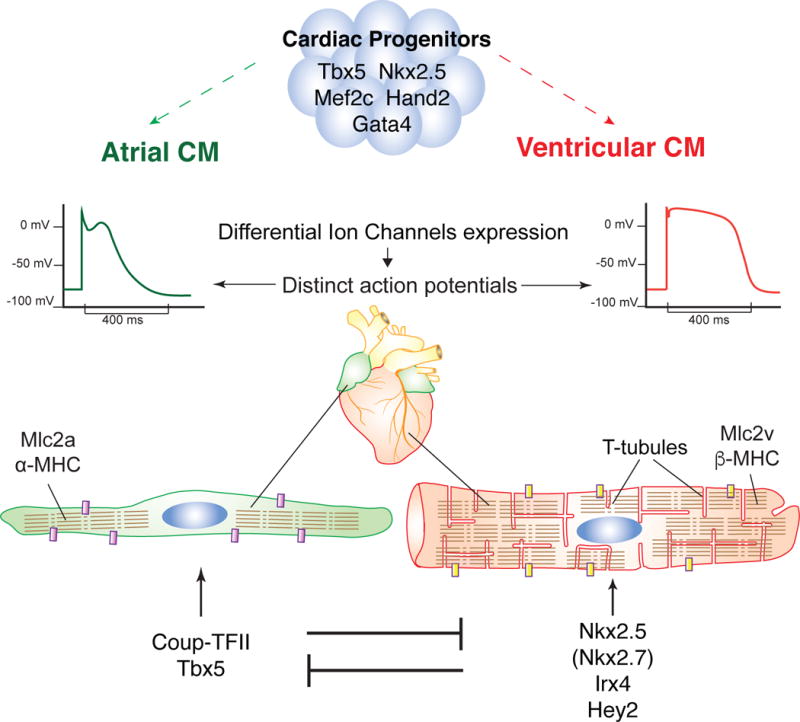
(A) Atrial (green) and ventricular (red) cells arise from distinct yet overlaping sets of cardiac progenitors, characterized by the expression of transcription factors such as Tbx5, Nkx2.5, Mef2c, Hand2 and Gata4. (B) Atrial and ventricular myocytes display distinct physiological, structural and molecular differences. For example, ventricular cardiomyocytes display a flatter action potential plateau, have specialized T-tubule structures and express different gene variants e.g. Mlc2v/Mlc2a, β-MHC/α-MHC. (C) Atrial and ventricular myocytes each express a mutually exclusively transcriptional program that continues to promote their own identity and suppresses the other.
CM: Cardiomyocyte, Purple/Yellow block: different ion channels, Segmented lines represent sacromeres.
Figure 2. Myocardial changes after cardiac injury in larval and adult zebrafish.
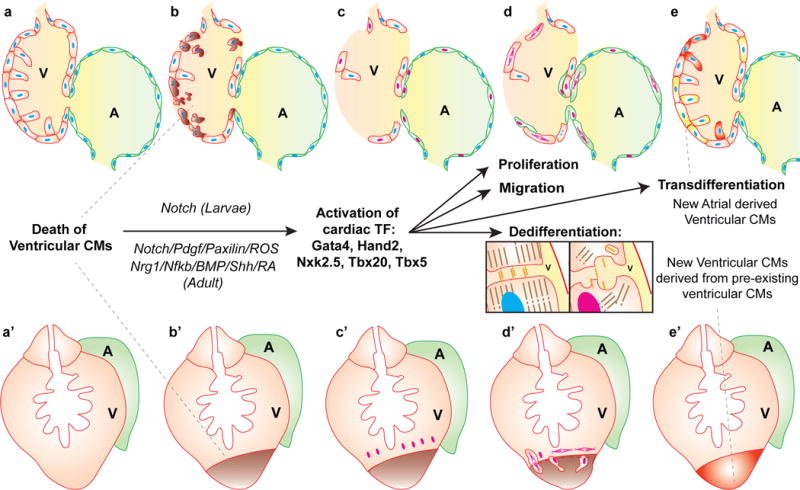
Larval (a-e) and adult (a′-e′) zebrafish hearts consist of a single ventricular (red) and atrial (green) chamber. After injury (b, b′) pre-existing mature cardiomyocytes de-differentiate, re-activate earlier cardiac transcription factors (red nuclei) (c, c′) and proliferate (d, d′) to contribute new cardiomyocytes to the injured area (e,e′). In larvae atrial cardiomyocytes also respond to injury by dedifferentiating, migrating and then transdifferentiating to a ventricular fate (Yellow cells, c-e). De-differentiation involves the disassembly of sacromeres and cell-cell contacts (inset).
V: Ventricle, A: Atrium, Blue and red nuclei: differentiated and de-differentiated nuclei, respectively.
Figure 3. Schematic contrasting the different properties of de-differentiated and mature cardiomyocytes.
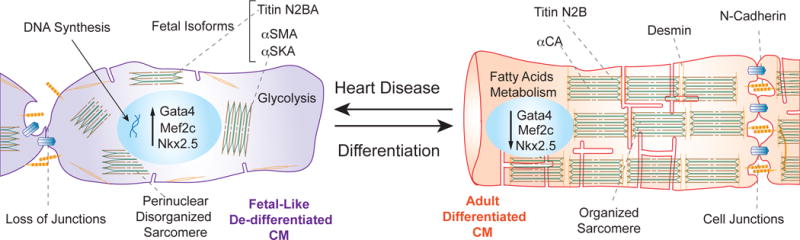
De-differentiated cardiomyocytes (purple) associated with cardiac disease display fetal-like characteristics compared to a mature adult cardiomycoyte (red). These fetal-like characteristics include a metabolic change to glycolysis, the activation of early cardiac transcription factors such as Gata4, Mef2c, Nkx2.5 and the activation of fetal specific isoforms such as Titin N2BA (green), αSMA, αSKA. Additionally, Desmin (orange) is re-localized in de-differentiated cardiomyocyctes and sarcomeres are not as highly organized.
CM: Cardiomyocytes,
Acknowledgments
Bart Weijts
References
References and recommended reading:
Papers of particular interest, published within the period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, et al. Revisiting Cardiac Cellular Composition. Circ Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nag AC. Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;28:41–61. [PubMed] [Google Scholar]
- 3.Evans SM, Yelon D, Conlon FL, Kirby ML. Myocardial lineage development. Circ Res. 2010;107:1428–1444. doi: 10.1161/CIRCRESAHA.110.227405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Solloway MJ, Harvey RP. Molecular pathways in myocardial development: a stem cell perspective. Cardiovasc Res. 2003;58:264–277. doi: 10.1016/s0008-6363(03)00286-4. [DOI] [PubMed] [Google Scholar]
- 5.Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–440. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- 6.Waldo KL, Kumiski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, Kirby ML. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128:3179–3188. doi: 10.1242/dev.128.16.3179. [DOI] [PubMed] [Google Scholar]
- 7.Meilhac SM, Esner M, Kelly RG, Nicolas JF, Buckingham ME. The clonal origin of myocardial cells in different regions of the embryonic mouse heart. Dev Cell. 2004;6:685–698. doi: 10.1016/s1534-5807(04)00133-9. [DOI] [PubMed] [Google Scholar]
- 8.Mjaatvedt CH, Nakaoka T, Moreno-Rodriguez R, Norris RA, Kern MJ, Eisenberg CA, Turner D, Markwald RR. The outflow tract of the heart is recruited from a novel heart-forming field. Dev Biol. 2001;238:97–109. doi: 10.1006/dbio.2001.0409. [DOI] [PubMed] [Google Scholar]
- 9.Lescroart F, Chabab S, Lin X, Rulands S, Paulissen C, Rodolosse A, Auer H, Achouri Y, Dubois C, Bondue A, et al. Early lineage restriction in temporally distinct populations of Mesp1 progenitors during mammalian heart development. Nat Cell Biol. 2014;16:829–840. doi: 10.1038/ncb3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Pater E, Clijsters L, Marques SR, Lin YF, Garavito-Aguilar ZV, Yelon D, Bakkers J. Distinct phases of cardiomyocyte differentiation regulate growth of the zebrafish heart. Development. 2009;136:1633–1641. doi: 10.1242/dev.030924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bressan M, Liu G, Mikawa T. Early mesodermal cues assign avian cardiac pacemaker fate potential in a tertiary heart field. Science. 2013;340:744–748. doi: 10.1126/science.1232877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- 13.Paige SL, Plonowska K, Xu A, Wu SM. Molecular regulation of cardiomyocyte differentiation. Circ Res. 2015;116:341–353. doi: 10.1161/CIRCRESAHA.116.302752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olson EN, Schneider MD. Sizing up the heart: development redux in disease. Genes Dev. 2003;17:1937–1956. doi: 10.1101/gad.1110103. [DOI] [PubMed] [Google Scholar]
- 15.Leu M, Ehler E, Perriard JC. Characterisation of postnatal growth of the murine heart. Anat Embryol (Berl) 2001;204:217–224. doi: 10.1007/s004290100206. [DOI] [PubMed] [Google Scholar]
- 16.Hirschy A, Schatzmann F, Ehler E, Perriard JC. Establishment of cardiac cytoarchitecture in the developing mouse heart. Dev Biol. 2006;289:430–441. doi: 10.1016/j.ydbio.2005.10.046. [DOI] [PubMed] [Google Scholar]
- 17.Lin YF, Swinburne I, Yelon D. Multiple influences of blood flow on cardiomyocyte hypertrophy in the embryonic zebrafish heart. Dev Biol. 2012;362:242–253. doi: 10.1016/j.ydbio.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopaschuk GD, Collins-Nakai RL, Itoi T. Developmental changes in energy substrate use by the heart. Cardiovasc Res. 1992;26:1172–1180. doi: 10.1093/cvr/26.12.1172. [DOI] [PubMed] [Google Scholar]
- 19.Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. 2014;157:565–579. doi: 10.1016/j.cell.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foglia MJ, Poss KD. Building and re-building the heart by cardiomyocyte proliferation. Development. 2016;143:729–740. doi: 10.1242/dev.132910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van den Hoogenhof MM, Pinto YM, Creemers EE. RNA Splicing: Regulation and Dysregulation in the Heart. Circ Res. 2016;118:454–468. doi: 10.1161/CIRCRESAHA.115.307872. [DOI] [PubMed] [Google Scholar]
- 22.Yin Z, Ren J, Guo W. Sarcomeric protein isoform transitions in cardiac muscle: a journey to heart failure. Biochim Biophys Acta. 2015;1852:47–52. doi: 10.1016/j.bbadis.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giudice J, Xia Z, Wang ET, Scavuzzo MA, Ward AJ, Kalsotra A, Wang W, Wehrens XH, Burge CB, Li W, et al. Alternative splicing regulates vesicular trafficking genes in cardiomyocytes during postnatal heart development. Nat Commun. 2014;5:3603. doi: 10.1038/ncomms4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Porrello ER, Olson EN. A neonatal blueprint for cardiac regeneration. Stem Cell Res. 2014;13:556–570. doi: 10.1016/j.scr.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szibor M, Poling J, Warnecke H, Kubin T, Braun T. Remodeling and dedifferentiation of adult cardiomyocytes during disease and regeneration. Cell Mol Life Sci. 2014;71:1907–1916. doi: 10.1007/s00018-013-1535-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sylva M, van den Hoff MJ, Moorman AF. Development of the human heart. Am J Med Genet A. 2014;164A:1347–1371. doi: 10.1002/ajmg.a.35896. [DOI] [PubMed] [Google Scholar]
- 27.Dirkx E, da Costa Martins PA, De Windt LJ. Regulation of fetal gene expression in heart failure. Biochim Biophys Acta. 2013;1832:2414–2424. doi: 10.1016/j.bbadis.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 28.Kikuchi K. Advances in understanding the mechanism of zebrafish heart regeneration. Stem Cell Res. 2014;13:542–555. doi: 10.1016/j.scr.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Uygur A, Lee RT. Mechanisms of Cardiac Regeneration. Dev Cell. 2016;36:362–374. doi: 10.1016/j.devcel.2016.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bootman MD, Smyrnias I, Thul R, Coombes S, Roderick HL. Atrial cardiomyocyte calcium signalling. Biochim Biophys Acta. 2011;1813:922–934. doi: 10.1016/j.bbamcr.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 31.Spater D, Hansson EM, Zangi L, Chien KR. How to make a cardiomyocyte. Development. 2014;141:4418–4431. doi: 10.1242/dev.091538. [DOI] [PubMed] [Google Scholar]
- 32.Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- 33.Yelon D, Horne SA, Stainier DY. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev Biol. 1999;214:23–37. doi: 10.1006/dbio.1999.9406. [DOI] [PubMed] [Google Scholar]
- 34.Rohr S, Otten C, Abdelilah-Seyfried S. Asymmetric involution of the myocardial field drives heart tube formation in zebrafish. Circ Res. 2008;102:e12–19. doi: 10.1161/CIRCRESAHA.107.165241. [DOI] [PubMed] [Google Scholar]
- 35.de Bold AJ. Atrial natriuretic factor: a hormone produced by the heart. Science. 1985;230:767–770. doi: 10.1126/science.2932797. [DOI] [PubMed] [Google Scholar]
- 36.Fawcett DW, McNutt NS. The ultrastructure of the cat myocardium. I. Ventricular papillary muscle. J Cell Biol. 1969;42:1–45. doi: 10.1083/jcb.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McNutt NS, Fawcett DW. The ultrastructure of the cat myocardium. II. Atrial muscle. J Cell Biol. 1969;42:46–67. doi: 10.1083/jcb.42.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bass A, Stejskalova M, Ostadal B, Samanek M. Differences between atrial and ventricular energy-supplying enzymes in five mammalian species. Physiol Res. 1993;42:1–6. [PubMed] [Google Scholar]
- 39.Lin FJ, You LR, Yu CT, Hsu WH, Tsai MJ, Tsai SY. Endocardial cushion morphogenesis and coronary vessel development require chicken ovalbumin upstream promoter-transcription factor II. Arterioscler Thromb Vasc Biol. 2012;32:e135–146. doi: 10.1161/ATVBAHA.112.300255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40**.Wu SP, Cheng CM, Lanz RB, Wang T, Respress JL, Ather S, Chen W, Tsai SJ, Wehrens XH, Tsai MJ, et al. Atrial identity is determined by a COUP-TFII regulatory network. Dev Cell. 2013;25:417–426. doi: 10.1016/j.devcel.2013.04.017. Using genetic analysis combined with gene expression profiling and ChiP-seq Wu et al. reveal that COUP-TFII restricts atrial cell identity, by simultaneously promoting atrial genes and inhibiting ventricular genes. COUP-TFII appears to directly binds a large range of both atrial and ventricular genes, indicating that much of COUP-TFII’s activity may be due to its ability to directly affect the transcriptional network regulating atrial and ventricular identity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruneau BG, Logan M, Davis N, Levi T, Tabin CJ, Seidman JG, Seidman CE. Chamber-specific cardiac expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev Biol. 1999;211:100–108. doi: 10.1006/dbio.1999.9298. [DOI] [PubMed] [Google Scholar]
- 42.Bruneau BG, Nemer G, Schmitt JP, Charron F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman CE, et al. A murine model of Holt-Oram syndrome defines roles of the T-box transcription factor Tbx5 in cardiogenesis and disease. Cell. 2001;106:709–721. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 43.Liberatore CM, Searcy-Schrick RD, Yutzey KE. Ventricular expression of tbx5 inhibits normal heart chamber development. Dev Biol. 2000;223:169–180. doi: 10.1006/dbio.2000.9748. [DOI] [PubMed] [Google Scholar]
- 44.Xin M, Small EM, van Rooij E, Qi X, Richardson JA, Srivastava D, Nakagawa O, Olson EN. Essential roles of the bHLH transcription factor Hrt2 in repression of atrial gene expression and maintenance of postnatal cardiac function. Proc Natl Acad Sci U S A. 2007;104:7975–7980. doi: 10.1073/pnas.0702447104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bodmer R, Jan LY, Jan YN. A new homeobox-containing gene, msh-2, is transiently expressed early during mesoderm formation of Drosophila. Development. 1990;110:661–669. doi: 10.1242/dev.110.3.661. [DOI] [PubMed] [Google Scholar]
- 46.Prall OW, Menon MK, Solloway MJ, Watanabe Y, Zaffran S, Bajolle F, Biben C, McBride JJ, Robertson BR, Chaulet H, et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schott JJ, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 48.Scott IC. Life before Nkx2.5: cardiovascular progenitor cells: embryonic origins and development. Curr Top Dev Biol. 2012;100:1–31. doi: 10.1016/B978-0-12-387786-4.00001-4. [DOI] [PubMed] [Google Scholar]
- 49.McCulley DJ, Black BL. Transcription factor pathways and congenital heart disease. Curr Top Dev Biol. 2012;100:253–277. doi: 10.1016/B978-0-12-387786-4.00008-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Targoff KL, Colombo S, George V, Schell T, Kim SH, Solnica-Krezel L, Yelon D. Nkx genes are essential for maintenance of ventricular identity. Development. 2013;140:4203–4213. doi: 10.1242/dev.095562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.George V, Colombo S, Targoff KL. An early requirement for nkx2.5 ensures the first and second heart field ventricular identity and cardiac function into adulthood. Dev Biol. 2015;400:10–22. doi: 10.1016/j.ydbio.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foglia MJ, Cao J, Tornini VA, Poss KD. Multicolor mapping of the cardiomyocyte proliferation dynamics that construct the atrium. Development. 2016 doi: 10.1242/dev.136606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R, Pradervand S, Evans SM, Clark B, Feramisco JR, Giles W, et al. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004;117:373–386. doi: 10.1016/s0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- 54.Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev. 1995;9:1654–1666. doi: 10.1101/gad.9.13.1654. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka M, Chen Z, Bartunkova S, Yamasaki N, Izumo S. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development. 1999;126:1269–1280. doi: 10.1242/dev.126.6.1269. [DOI] [PubMed] [Google Scholar]
- 56.Bruneau BG, Bao ZZ, Fatkin D, Xavier-Neto J, Georgakopoulos D, Maguire CT, Berul CI, Kass DA, Kuroski-de Bold ML, de Bold AJ, et al. Cardiomyopathy in Irx4-deficient mice is preceded by abnormal ventricular gene expression. Mol Cell Biol. 2001;21:1730–1736. doi: 10.1128/MCB.21.5.1730-1736.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bao ZZ, Bruneau BG, Seidman JG, Seidman CE, Cepko CL. Regulation of chamber-specific gene expression in the developing heart by Irx4. Science. 1999;283:1161–1164. doi: 10.1126/science.283.5405.1161. [DOI] [PubMed] [Google Scholar]
- 58.Warren SA, Terada R, Briggs LE, Cole-Jeffrey CT, Chien WM, Seki T, Weinberg EO, Yang TP, Chin MT, Bungert J, et al. Differential role of Nkx2-5 in activation of the atrial natriuretic factor gene in the developing versus failing heart. Mol Cell Biol. 2011;31:4633–4645. doi: 10.1128/MCB.05940-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruneau BG, Bao ZZ, Tanaka M, Schott JJ, Izumo S, Cepko CL, Seidman JG, Seidman CE. Cardiac expression of the ventricle-specific homeobox gene Irx4 is modulated by Nkx2-5 and dHand. Dev Biol. 2000;217:266–277. doi: 10.1006/dbio.1999.9548. [DOI] [PubMed] [Google Scholar]
- 60.Dyson E, Sucov HM, Kubalak SW, Schmid-Schonbein GW, DeLano FA, Evans RM, Ross J, Jr, Chien KR. Atrial-like phenotype is associated with embryonic ventricular failure in retinoid X receptor alpha −/− mice. Proc Natl Acad Sci U S A. 1995;92:7386–7390. doi: 10.1073/pnas.92.16.7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang GF, Nikovits W, Jr, Bao ZZ, Stockdale FE. Irx4 forms an inhibitory complex with the vitamin D and retinoic X receptors to regulate cardiac chamber-specific slow MyHC3 expression. J Biol Chem. 2001;276:28835–28841. doi: 10.1074/jbc.M103716200. [DOI] [PubMed] [Google Scholar]
- 62*.Chen J, Kubalak SW, Minamisawa S, Price RL, Becker KD, Hickey R, Ross J, Jr, Chien KR. Selective requirement of myosin light chain 2v in embryonic heart function. J Biol Chem. 1998;273:1252–1256. doi: 10.1074/jbc.273.2.1252. Mlc2a is initially expressed throughout the heart and then restricted to the atrium. However, in Mlc2v mutants, Chen et al. show that Mlc2a continues to be expressed in the ventricle. [DOI] [PubMed] [Google Scholar]
- 63.Attenhofer Jost CH, Connolly HM, Dearani JA, Edwards WD, Danielson GK. Ebstein’s anomaly. Circulation. 2007;115:277–285. doi: 10.1161/CIRCULATIONAHA.106.619338. [DOI] [PubMed] [Google Scholar]
- 64.Kapoor N, Liang W, Marban E, Cho HC. Direct conversion of quiescent cardiomyocytes to pacemaker cells by expression of Tbx18. Nat Biotechnol. 2013;31:54–62. doi: 10.1038/nbt.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rentschler S, Yen AH, Lu J, Petrenko NB, Lu MM, Manderfield LJ, Patel VV, Fishman GI, Epstein JA. Myocardial Notch signaling reprograms cardiomyocytes to a conduction-like phenotype. Circulation. 2012;126:1058–1066. doi: 10.1161/CIRCULATIONAHA.112.103390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66*.Schumacher JA, Bloomekatz J, Garavito-Aguilar ZV, Yelon D. Tal1 Regulates the formation of intercellular junctions and the maintenance of identity in the endocardium. Dev Biol. 2013;383:214–226. doi: 10.1016/j.ydbio.2013.09.019. These three studies reveal that one of Scl/Tal1 roles is to inhibit endocardial and endothelial cells from transforming into a myocardial cell. ChIP-Seq studies reveal that Scl1/Tal1 binds to a large range of primed (H3K4me1) endothelial and myocardial enhancers. Repression of the myocardial gene regulatory program appears to occur through the occupation of cardiac enhancers used by inductive factors. Like COUP-TFII, Scl/Tal1 is only required during transitory period, after which time the epigenetic landscape changes and myocardial enhancers are no longer primed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67**.Van Handel B, Montel-Hagen A, Sasidharan R, Nakano H, Ferrari R, Boogerd CJ, Schredelseker J, Wang Y, Hunter S, Org T, et al. Scl represses cardiomyogenesis in prospective hemogenic endothelium and endocardium. Cell. 2012;150:590–605. doi: 10.1016/j.cell.2012.06.026. These three studies reveal that one of Scl/Tal1 roles is to inhibit endocardial and endothelial cells from transforming into a myocardial cell. ChIP-Seq studies reveal that Scl1/Tal1 binds to a large range of primed (H3K4me1) endothelial and myocardial enhancers. Repression of the myocardial gene regulatory program appears to occur through the occupation of cardiac enhancers used by inductive factors. Like COUP-TFII, Scl/Tal1 is only required during transitory period, after which time the epigenetic landscape changes and myocardial enhancers are no longer primed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68**.Org T, Duan D, Ferrari R, Montel-Hagen A, Van Handel B, Kerenyi MA, Sasidharan R, Rubbi L, Fujiwara Y, Pellegrini M, et al. Scl binds to primed enhancers in mesoderm to regulate hematopoietic and cardiac fate divergence. EMBO J. 2015;34:759–777. doi: 10.15252/embj.201490542. These three studies reveal that one of Scl/Tal1 roles is to inhibit endocardial and endothelial cells from transforming into a myocardial cell. ChIP-Seq studies reveal that Scl1/Tal1 binds to a large range of primed (H3K4me1) endothelial and myocardial enhancers. Repression of the myocardial gene regulatory program appears to occur through the occupation of cardiac enhancers used by inductive factors. Like COUP-TFII, Scl/Tal1 is only required during transitory period, after which time the epigenetic landscape changes and myocardial enhancers are no longer primed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69*.Du Z, Santella A, He F, Shah PK, Kamikawa Y, Bao Z. The Regulatory Landscape of Lineage Differentiation in a Metazoan Embryo. Dev Cell. 2015;34:592–607. doi: 10.1016/j.devcel.2015.07.014. In systematically determining the efect of 204 essential genes on the entire C. elegans lineages, Du and colleagues found that fate change was dependent on the proximity of the different lineages. Such that fate changes most frequently occurred between proximal fates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70*.Martinotti T. Suggli effetti degli ferite de cuore. G Accad Med Torino. 1888;7:404–414. The ability of the heart to respond to injury has fascinated scientists for a very long time. This is exemplified by these classic studies by Italian scientists at the end of the 19th century, investigating the effects of heart injury in adult rats. [Google Scholar]
- 71*.Mircoli ST. Sulle alterationi acuto del miocardio per stimoli semplici e specifici. Archs Sci med. 1889;13:1–20. The ability of the heart to respond to injury has fascinated scientists for a very long time. This is exemplified by these classic studies by Italian scientists at the end of the 19th century, investigating the effects of heart injury in adult rats. [Google Scholar]
- 72*.Oberpriller JO, Oberpriller JC. Response of the adult newt ventricle to injury. J Exp Zool. 1974;187:249–253. doi: 10.1002/jez.1401870208. These studies were the first to identify a genetically tractable model organism that could regenerate their heart. Opening up the possibility of elucidating the molecules underlying heart regeneration. [DOI] [PubMed] [Google Scholar]
- 73.Polezhaev LV. Loss and restoration of regenerative capacity in tissues and organs of animals. Cambridge, Mass: Harvard University Press; 1972. [Google Scholar]
- 74*.Witman N, Murtuza B, Davis B, Arner A, Morrison JI. Recapitulation of developmental cardiogenesis governs the morphological and functional regeneration of adult newt hearts following injury. Dev Biol. 2011;354:67–76. doi: 10.1016/j.ydbio.2011.03.021. L.V. Polezhaev and P.P. Rumyantsev were among a group of Russian scientists during the early 20th century who investigated ability of variety of tissues in a number of different organisms to regenerate after injury. Their studies of heart regeneration in Urodele amphibans along with J.O. and J.C. Oberpriller in the 1970-80s in the United States laid the ground work for modern studies in regeneration today. [DOI] [PubMed] [Google Scholar]
- 75.Bader D, Oberpriller J. Autoradiographic and electron microscopic studies of minced cardiac muscle regeneration in the adult newt, notophthalmus viridescens. J Exp Zool. 1979;208:177–193. doi: 10.1002/jez.1402080206. [DOI] [PubMed] [Google Scholar]
- 76.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–2190. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 77.Poss KD. Advances in understanding tissue regenerative capacity and mechanisms in animals. Nat Rev Genet. 2010;11:710–722. doi: 10.1038/nrg2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78**.Jopling C, Sleep E, Raya M, Marti M, Raya A, Izpisua Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. 2010;464:606–609. doi: 10.1038/nature08899. By genetically marking mature cardiomyocytes, using Cre-lox recombination these seminal studies revealed that new cardiomyocytes created during heart regeneration are derived from mature pre-existing cardiomyocytes. They also confirmed previous finding by Oberpriller, Polezhaev and Rumyantsev that myocadial cells appear to go through a process of de-differentiation during regeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79**.Kikuchi K, Holdway JE, Werdich AA, Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY, Poss KD. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature. 2010;464:601–605. doi: 10.1038/nature08804. By genetically marking mature cardiomyocytes, using Cre-lox recombination these seminal studies revealed that new cardiomyocytes created during heart regeneration are derived from mature pre-existing cardiomyocytes. They also confirmed previous finding by Oberpriller, Polezhaev and Rumyantsev that myocadial cells appear to go through a process of de-differentiation during regeneration. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang J, Panakova D, Kikuchi K, Holdway JE, Gemberling M, Burris JS, Singh SP, Dickson AL, Lin YF, Sabeh MK, et al. The regenerative capacity of zebrafish reverses cardiac failure caused by genetic cardiomyocyte depletion. Development. 2011;138:3421–3430. doi: 10.1242/dev.068601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schindler YL, Garske KM, Wang J, Firulli BA, Firulli AB, Poss KD, Yelon D. Hand2 elevates cardiomyocyte production during zebrafish heart development and regeneration. Development. 2014;141:3112–3122. doi: 10.1242/dev.106336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sallin P, de Preux Charles AS, Duruz V, Pfefferli C, Jazwinska A. A dual epimorphic and compensatory mode of heart regeneration in zebrafish. Dev Biol. 2015;399:27–40. doi: 10.1016/j.ydbio.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 83.Holtzinger A, Evans T. Gata5 and Gata6 are functionally redundant in zebrafish for specification of cardiomyocytes. Dev Biol. 2007;312:613–622. doi: 10.1016/j.ydbio.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gajewski K, Fossett N, Molkentin JD, Schulz RA. The zinc finger proteins Pannier and GATA4 function as cardiogenic factors in Drosophila. Development. 1999;126:5679–5688. doi: 10.1242/dev.126.24.5679. [DOI] [PubMed] [Google Scholar]
- 85.Zhao R, Watt AJ, Battle MA, Li J, Bondow BJ, Duncan SA. Loss of both GATA4 and GATA6 blocks cardiac myocyte differentiation and results in acardia in mice. Dev Biol. 2008;317:614–619. doi: 10.1016/j.ydbio.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reiter JF, Alexander J, Rodaway A, Yelon D, Patient R, Holder N, Stainier DY. Gata5 is required for the development of the heart and endoderm in zebrafish. Genes Dev. 1999;13:2983–2995. doi: 10.1101/gad.13.22.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Peterkin T, Gibson A, Patient R. Redundancy and evolution of GATA factor requirements in development of the myocardium. Dev Biol. 2007;311:623–635. doi: 10.1016/j.ydbio.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yelon D, Ticho B, Halpern ME, Ruvinsky I, Ho RK, Silver LM, Stainier DY. The bHLH transcription factor hand2 plays parallel roles in zebrafish heart and pectoral fin development. Development. 2000;127:2573–2582. doi: 10.1242/dev.127.12.2573. [DOI] [PubMed] [Google Scholar]
- 89.Srivastava D, Thomas T, Lin Q, Kirby ML, Brown D, Olson EN. Regulation of cardiac mesodermal and neural crest development by the bHLH transcription factor, dHAND. Nat Genet. 1997;16:154–160. doi: 10.1038/ng0697-154. [DOI] [PubMed] [Google Scholar]
- 90.Yamagishi H, Yamagishi C, Nakagawa O, Harvey RP, Olson EN, Srivastava D. The combinatorial activities of Nkx2.5 and dHAND are essential for cardiac ventricle formation. Dev Biol. 2001;239:190–203. doi: 10.1006/dbio.2001.0417. [DOI] [PubMed] [Google Scholar]
- 91.Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MA, He D, Small EM, Mileikovsky M, et al. Complex Interdependence Regulates Heterotypic Transcription Factor Distribution and Coordinates Cardiogenesis. Cell. 2016;164:999–1014. doi: 10.1016/j.cell.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599–604. doi: 10.1038/nature11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–598. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lou X, Deshwar AR, Crump JG, Scott IC. Smarcd3b and Gata5 promote a cardiac progenitor fate in the zebrafish embryo. Development. 2011;138:3113–3123. doi: 10.1242/dev.064279. [DOI] [PubMed] [Google Scholar]
- 96.Takeuchi JK, Bruneau BG. Directed transdifferentiation of mouse mesoderm to heart tissue by defined factors. Nature. 2009;459:708–711. doi: 10.1038/nature08039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gupta V, Gemberling M, Karra R, Rosenfeld GE, Evans T, Poss KD. An injury-responsive gata4 program shapes the zebrafish cardiac ventricle. Curr Biol. 2013;23:1221–1227. doi: 10.1016/j.cub.2013.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jopling C, Sune G, Faucherre A, Fabregat C, Izpisua Belmonte JC. Hypoxia induces myocardial regeneration in zebrafish. Circulation. 2012;126:3017–3027. doi: 10.1161/CIRCULATIONAHA.112.107888. [DOI] [PubMed] [Google Scholar]
- 99.Karra R, Knecht AK, Kikuchi K, Poss KD. Myocardial NF-kappaB activation is essential for zebrafish heart regeneration. Proc Natl Acad Sci U S A. 2015;112:13255–13260. doi: 10.1073/pnas.1511209112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kikuchi K, Holdway JE, Major RJ, Blum N, Dahn RD, Begemann G, Poss KD. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev Cell. 2011;20:397–404. doi: 10.1016/j.devcel.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim J, Wu Q, Zhang Y, Wiens KM, Huang Y, Rubin N, Shimada H, Handin RI, Chao MY, Tuan TL, et al. PDGF signaling is required for epicardial function and blood vessel formation in regenerating zebrafish hearts. Proc Natl Acad Sci U S A. 2010;107:17206–17210. doi: 10.1073/pnas.0915016107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Parente V, Balasso S, Pompilio G, Verduci L, Colombo GI, Milano G, Guerrini U, Squadroni L, Cotelli F, Pozzoli O, et al. Hypoxia/reoxygenation cardiac injury and regeneration in zebrafish adult heart. PLoS One. 2013;8:e53748. doi: 10.1371/journal.pone.0053748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang J, Cao J, Dickson AL, Poss KD. Epicardial regeneration is guided by cardiac outflow tract and Hedgehog signalling. Nature. 2015;522:226–230. doi: 10.1038/nature14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu CC, Kruse F, Vasudevarao MD, Junker JP, Zebrowski DC, Fischer K, Noel ES, Grun D, Berezikov E, Engel FB, et al. Spatially Resolved Genome-wide Transcriptional Profiling Identifies BMP Signaling as Essential Regulator of Zebrafish Cardiomyocyte Regeneration. Dev Cell. 2016;36:36–49. doi: 10.1016/j.devcel.2015.12.010. [DOI] [PubMed] [Google Scholar]
- 105.Han P, Zhou XH, Chang N, Xiao CL, Yan S, Ren H, Yang XZ, Zhang ML, Wu Q, Tang B, et al. Hydrogen peroxide primes heart regeneration with a derepression mechanism. Cell Res. 2014;24:1091–1107. doi: 10.1038/cr.2014.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106**.Peng X, He Q, Li G, Ma J, Zhong TP. Rac1 PAK2 pathway is essential for zebrafish heart regeneration. Biochem Biophys Res Commun. 2016 doi: 10.1016/j.bbrc.2016.03.011. A large number of genes and signaling pathways have been shown to be required for cardiac regeneration. However, the ability of Neuregulin signaling to trigger processes of cardiac regeneration, such as de-differentiation and proliferation, without injury distinguishes this study. Revealing that this signaling event maybe be the chemical cue triggered during injury to stimulate regeneration. This study mirrors the work by D’Uva et al. on Neureguling signaling in mouse heart. [DOI] [PubMed] [Google Scholar]
- 107**.Zhao L, Borikova AL, Ben-Yair R, Guner-Ataman B, MacRae CA, Lee RT, Burns CG, Burns CE. Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc Natl Acad Sci U S A. 2014;111:1403–1408. doi: 10.1073/pnas.1311705111. This is the first paper to reveal a unique plasticity within larval atrial cells. Upon ventricular injury in the larval stage atrial cells respond by migrating to the ventricle and trans-differentiating to a ventricle fate to help compensate for the loss of the ventricular cells. This event appear to be age specific as atrial cells respond to adult ventricular injury by proliferating but not by transdifferentiating. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huang Y, Harrison MR, Osorio A, Kim J, Baugh A, Duan C, Sucov HM, Lien CL. Igf Signaling is Required for Cardiomyocyte Proliferation during Zebrafish Heart Development and Regeneration. PLoS One. 2013;8:e67266. doi: 10.1371/journal.pone.0067266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gemberling M, Karra R, Dickson AL, Poss KD. Nrg1 is an injury-induced cardiomyocyte mitogen for the endogenous heart regeneration program in zebrafish. Elife. 2015;4 doi: 10.7554/eLife.05871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang R, Han P, Yang H, Ouyang K, Lee D, Lin YF, Ocorr K, Kang G, Chen J, Stainier DY, et al. In vivo cardiac reprogramming contributes to zebrafish heart regeneration. Nature. 2013;498:497–501. doi: 10.1038/nature12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Curado S, Anderson RM, Jungblut B, Mumm J, Schroeter E, Stainier DY. Conditional targeted cell ablation in zebrafish: a new tool for regeneration studies. Dev Dyn. 2007;236:1025–1035. doi: 10.1002/dvdy.21100. [DOI] [PubMed] [Google Scholar]
- 112.Flink IL. Cell cycle reentry of ventricular and atrial cardiomyocytes and cells within the epicardium following amputation of the ventricular apex in the axolotl, Amblystoma mexicanum: confocal microscopic immunofluorescent image analysis of bromodeoxyuridine-labeled nuclei. Anat Embryol (Berl) 2002;205:235–244. doi: 10.1007/s00429-002-0249-6. [DOI] [PubMed] [Google Scholar]
- 113.McDonnell TJ, Oberpriller JO. The response of the atrium to direct mechanical wounding in the adult heart of the newt, Notophthalmus viridescens. An electron-microscopic and autoradiographic study. Cell Tissue Res. 1984;235:583–592. doi: 10.1007/BF00226956. [DOI] [PubMed] [Google Scholar]
- 114.Oberpriller JO, Oberpriller JC, Aafedt BC. Changes in binucleation and cellular dimensions of rat left atrial myocytes after induced left ventricular infarction. Am J Anat. 1987;179:285–290. doi: 10.1002/aja.1001790310. [DOI] [PubMed] [Google Scholar]
- 115.McDonnell TJ, Oberpriller JO. The atrial proliferative response following partial ventricular amputation in the heart of the adult newt. A light and electron microscopic autoradiographic study. Tissue Cell. 1983;15:351–363. doi: 10.1016/0040-8166(83)90068-x. [DOI] [PubMed] [Google Scholar]
- 116.Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C, Stein JI, Penninger JM. Functional Recovery of a Human Neonatal Heart After Severe Myocardial Infarction. Circ Res. 2016;118:216–221. doi: 10.1161/CIRCRESAHA.115.307017. [DOI] [PubMed] [Google Scholar]
- 117.Boulton J, Henry R, Roddick LG, Rogers D, Thompson L, Warner G. Survival after neonatal myocardial infarction. Pediatrics. 1991;88:145–150. [PubMed] [Google Scholar]
- 118.Peeters S, Vandenplas Y, Jochmans K, Bougatef A, De Waele M, De Wolf D. Myocardial infarction in a neonate with hereditary antithrombin III deficiency. Acta Paediatr. 1993;82:610–613. doi: 10.1111/j.1651-2227.1993.tb12770.x. [DOI] [PubMed] [Google Scholar]
- 119.Saker DM, Walsh-Sukys M, Spector M, Zahka KG. Cardiac recovery and survival after neonatal myocardial infarction. Pediatr Cardiol. 1997;18:139–142. doi: 10.1007/s002469900133. [DOI] [PubMed] [Google Scholar]
- 120.Murugan SJ, Gnanapragasam J, Vettukattil J. Acute myocardial infarction in the neonatal period. Cardiol Young. 2002;12:411–413. doi: 10.1017/s1047951100013068. [DOI] [PubMed] [Google Scholar]
- 121.Cesna S, Eicken A, Juenger H, Hess J. Successful treatment of a newborn with acute myocardial infarction on the first day of life. Pediatr Cardiol. 2013;34:1868–1870. doi: 10.1007/s00246-012-0417-2. [DOI] [PubMed] [Google Scholar]
- 122.Deutsch MA, Cleuziou J, Noebauer C, Eicken A, Vogt M, Hoerer J, Lange R, Schreiber C. Successful management of neonatal myocardial infarction with ECMO and intracoronary r-tPA lysis. Congenit Heart Dis. 2014;9:E169–174. doi: 10.1111/chd.12117. [DOI] [PubMed] [Google Scholar]
- 123.Sturzu AC, Rajarajan K, Passer D, Plonowska K, Riley A, Tan TC, Sharma A, Xu AF, Engels MC, Feistritzer R, et al. Fetal Mammalian Heart Generates a Robust Compensatory Response to Cell Loss. Circulation. 2015;132:109–121. doi: 10.1161/CIRCULATIONAHA.114.011490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Drenckhahn JD, Schwarz QP, Gray S, Laskowski A, Kiriazis H, Ming Z, Harvey RP, Du XJ, Thorburn DR, Cox TC. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev Cell. 2008;15:521–533. doi: 10.1016/j.devcel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 125.Villa del Campo C, Claveria C, Sierra R, Torres M. Cell competition promotes phenotypically silent cardiomyocyte replacement in the mammalian heart. Cell Rep. 2014;8:1741–1751. doi: 10.1016/j.celrep.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 126**.Darehzereshki A, Rubin N, Gamba L, Kim J, Fraser J, Huang Y, Billings J, Mohammadzadeh R, Wood J, Warburton D, et al. Differential regenerative capacity of neonatal mouse hearts after cryoinjury. Dev Biol. 2015;399:91–99. doi: 10.1016/j.ydbio.2014.12.018. This was the first paper to show that mouse neonates can regenerate their hearts after a ventricular resection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Strungs EG, Ongstad EL, O’Quinn MP, Palatinus JA, Jourdan LJ, Gourdie RG. Cryoinjury models of the adult and neonatal mouse heart for studies of scarring and regeneration. Methods Mol Biol. 2013;1037:343–353. doi: 10.1007/978-1-62703-505-7_20. [DOI] [PubMed] [Google Scholar]
- 128**.Polizzotti BD, Ganapathy B, Haubner BJ, Penninger JM, Kuhn B. A cryoinjury model in neonatal mice for cardiac translational and regeneration research. Nat Protoc. 2016;11:542–552. doi: 10.1038/nprot.2016.031. Similar to Gemberling M et al. this study reveals the role of Neureguiling signaling in being necessary and sufficient to trigger a regenerative response in the mammalian heart. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331:1078–1080. doi: 10.1126/science.1200708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yu W, Huang X, Tian X, Zhang H, He L, Wang Y, Nie Y, Hu S, Lin Z, Zhou B, et al. GATA4 regulates Fgf16 to promote heart repair after injury. Development. 2016;143:936–949. doi: 10.1242/dev.130971. [DOI] [PubMed] [Google Scholar]
- 131.D’Uva G, Aharonov A, Lauriola M, Kain D, Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D, Yifa O, et al. ERBB2 triggers mammalian heart regeneration by promoting cardiomyocyte dedifferentiation and proliferation. Nat Cell Biol. 2015;17:627–638. doi: 10.1038/ncb3149. [DOI] [PubMed] [Google Scholar]
- 132.Driesen RB, Verheyen FK, Debie W, Blaauw E, Babiker FA, Cornelussen RN, Ausma J, Lenders MH, Borgers M, Chaponnier C, et al. Re-expression of alpha skeletal actin as a marker for dedifferentiation in cardiac pathologies. J Cell Mol Med. 2009;13:896–908. doi: 10.1111/j.1582-4934.2008.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dispersyn GD, Mesotten L, Meuris B, Maes A, Mortelmans L, Flameng W, Ramaekers F, Borgers M. Dissociation of cardiomyocyte apoptosis and dedifferentiation in infarct border zones. Eur Heart J. 2002;23:849–857. doi: 10.1053/euhj.2001.2963. [DOI] [PubMed] [Google Scholar]
- 134.Izumo S, Nadal-Ginard B, Mahdavi V. Protooncogene induction and reprogramming of cardiac gene expression produced by pressure overload. Proc Natl Acad Sci U S A. 1988;85:339–343. doi: 10.1073/pnas.85.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Chen MC, Chang JP, Huang SC, Chang HW, Chen CJ, Yang CH, Liu WH. Dedifferentiation of atrial cardiomyocytes in cardiac valve disease: unrelated to atrial fibrillation. Cardiovasc Pathol. 2008;17:156–165. doi: 10.1016/j.carpath.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 136.Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A, Polyakova V, Bauer EP, Klovekorn WP, Schaper J. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107:984–991. doi: 10.1161/01.cir.0000051865.66123.b7. [DOI] [PubMed] [Google Scholar]
- 137.Heling A, Zimmermann R, Kostin S, Maeno Y, Hein S, Devaux B, Bauer E, Klovekorn WP, Schlepper M, Schaper W, et al. Increased expression of cytoskeletal, linkage, and extracellular proteins in failing human myocardium. Circ Res. 2000;86:846–853. doi: 10.1161/01.res.86.8.846. [DOI] [PubMed] [Google Scholar]
- 138.Ausma J, Wijffels M, van Eys G, Koide M, Ramaekers F, Allessie M, Borgers M. Dedifferentiation of atrial cardiomyocytes as a result of chronic atrial fibrillation. Am J Pathol. 1997;151:985–997. [PMC free article] [PubMed] [Google Scholar]
- 139.Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104:2923–2931. doi: 10.1161/hc4901.100526. [DOI] [PubMed] [Google Scholar]
- 140.Black FM, Packer SE, Parker TG, Michael LH, Roberts R, Schwartz RJ, Schneider MD. The vascular smooth muscle alpha-actin gene is reactivated during cardiac hypertrophy provoked by load. J Clin Invest. 1991;88:1581–1588. doi: 10.1172/JCI115470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lowes BD, Gilbert EM, Abraham WT, Minobe WA, Larrabee P, Ferguson D, Wolfel EE, Lindenfeld J, Tsvetkova T, Robertson AD, et al. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med. 2002;346:1357–1365. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- 142.Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA. Titin isoform switch in ischemic human heart disease. Circulation. 2002;106:1333–1341. doi: 10.1161/01.cir.0000029803.93022.93. [DOI] [PubMed] [Google Scholar]
- 143.Hannenhalli S, Putt ME, Gilmore JM, Wang J, Parmacek MS, Epstein JA, Morrisey EE, Margulies KB, Cappola TP. Transcriptional genomics associates FOX transcription factors with human heart failure. Circulation. 2006;114:1269–1276. doi: 10.1161/CIRCULATIONAHA.106.632430. [DOI] [PubMed] [Google Scholar]
- 144.Hasegawa K, Lee SJ, Jobe SM, Markham BE, Kitsis RN. Cis-Acting sequences that mediate induction of beta-myosin heavy chain gene expression during left ventricular hypertrophy due to aortic constriction. Circulation. 1997;96:3943–3953. doi: 10.1161/01.cir.96.11.3943. [DOI] [PubMed] [Google Scholar]
- 145.Herzig TC, Jobe SM, Aoki H, Molkentin JD, Cowley AW, Jr, Izumo S, Markham BE. Angiotensin II type1a receptor gene expression in the heart: AP-1 and GATA-4 participate in the response to pressure overload. Proc Natl Acad Sci U S A. 1997;94:7543–7548. doi: 10.1073/pnas.94.14.7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Thompson JT, Rackley MS, O’Brien TX. Upregulation of the cardiac homeobox gene Nkx2-5 (CSX) in feline right ventricular pressure overload. Am J Physiol. 1998;274:H1569–1573. doi: 10.1152/ajpheart.1998.274.5.H1569. [DOI] [PubMed] [Google Scholar]
- 147.Nadruz W, Jr, Kobarg CB, Constancio SS, Corat PD, Franchini KG. Load-induced transcriptional activation of c-jun in rat myocardium: regulation by myocyte enhancer factor 2. Circ Res. 2003;92:243–251. doi: 10.1161/01.res.0000053184.94618.97. [DOI] [PubMed] [Google Scholar]
- 148.Oka T, Xu J, Molkentin JD. Re-employment of developmental transcription factors in adult heart disease. Semin Cell Dev Biol. 2007;18:117–131. doi: 10.1016/j.semcdb.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD. Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res. 2006;98:837–845. doi: 10.1161/01.RES.0000215985.18538.c4. [DOI] [PubMed] [Google Scholar]
- 150.Hautala N, Tokola H, Luodonpaa M, Puhakka J, Romppanen H, Vuolteenaho O, Ruskoaho H. Pressure overload increases GATA4 binding activity via endothelin-1. Circulation. 2001;103:730–735. doi: 10.1161/01.cir.103.5.730. [DOI] [PubMed] [Google Scholar]
- 151.Liang Q, De Windt LJ, Witt SA, Kimball TR, Markham BE, Molkentin JD. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J Biol Chem. 2001;276:30245–30253. doi: 10.1074/jbc.M102174200. [DOI] [PubMed] [Google Scholar]
- 152.Molkentin JD, Markham BE. Myocyte-specific enhancer-binding factor (MEF-2) regulates alpha-cardiac myosin heavy chain gene expression in vitro and in vivo. J Biol Chem. 1993;268:19512–19520. [PubMed] [Google Scholar]
- 153.Konno T, Chen D, Wang L, Wakimoto H, Teekakirikul P, Nayor M, Kawana M, Eminaga S, Gorham JM, Pandya K, et al. Heterogeneous myocyte enhancer factor-2 (Mef2) activation in myocytes predicts focal scarring in hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2010;107:18097–18102. doi: 10.1073/pnas.1012826107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154**.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. This important study links inflammatory response cytokines occurring during acute and chronic cardiac disease to myocardial de-differentiation during cardiac disease. OSM, an inflammatory cytokine, is shown to stimulate de-differentiation in the absence of injury. The opposing effects OSM has on acute cardiac injury (reparative) and chronic cardiac disease (detrimental) suggests that de-differentiation may function as a initial survival and proliferative mechanism that contributes to a maladaptive response during the later stages of cardiac disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Xu J, Gong NL, Bodi I, Aronow BJ, Backx PH, Molkentin JD. Myocyte enhancer factors 2A and 2C induce dilated cardiomyopathy in transgenic mice. J Biol Chem. 2006;281:9152–9162. doi: 10.1074/jbc.M510217200. [DOI] [PubMed] [Google Scholar]
- 156.Pereira AH, Clemente CF, Cardoso AC, Theizen TH, Rocco SA, Judice CC, Guido MC, Pascoal VD, Lopes-Cendes I, Souza JR, et al. MEF2C silencing attenuates load-induced left ventricular hypertrophy by modulating mTOR/S6K pathway in mice. PLoS One. 2009;4:e8472. doi: 10.1371/journal.pone.0008472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kubin T, Poling J, Kostin S, Gajawada P, Hein S, Rees W, Wietelmann A, Tanaka M, Lorchner H, Schimanski S, et al. Oncostatin M is a major mediator of cardiomyocyte dedifferentiation and remodeling. Cell Stem Cell. 2011;9:420–432. doi: 10.1016/j.stem.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 158.Kolattukudy PE, Quach T, Bergese S, Breckenridge S, Hensley J, Altschuld R, Gordillo G, Klenotic S, Orosz C, Parker-Thornburg J. Myocarditis induced by targeted expression of the MCP-1 gene in murine cardiac muscle. Am J Pathol. 1998;152:101–111. [PMC free article] [PubMed] [Google Scholar]
- 159.Bersell K, Arab S, Haring B, Kuhn B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell. 2009;138:257–270. doi: 10.1016/j.cell.2009.04.060. [DOI] [PubMed] [Google Scholar]
- 160.Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, Bruneau BG. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- 161.Zhou Y, Wang L, Vaseghi HR, Liu Z, Lu R, Alimohamadi S, Yin C, Fu JD, Wang GG, Liu J, et al. Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell. 2016;18:382–395. doi: 10.1016/j.stem.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, Zhou B, Chang CP. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. 2010;466:62–67. doi: 10.1038/nature09130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Han P, Li W, Yang J, Shang C, Lin CH, Cheng W, Hang CT, Cheng HL, Chen CH, Wong J, et al. Epigenetic response to environmental stress: Assembly of BRG1-G9a/GLP-DNMT3 repressive chromatin complex on Myh6 promoter in pathologically stressed hearts. Biochim Biophys Acta. 2016 doi: 10.1016/j.bbamcr.2016.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]