

Summary
Lipid droplets (LDs) are ubiquitous fat storage organelles and play key roles in lipid metabolism and energy homeostasis; in addition, they contribute to protein storage, folding and degradation. Yet, a role for LDs in the nervous system remains largely unexplored. We discuss evidence supporting an intimate functional connection between LDs and Motor Neuron Disease (MND) pathophysiology, examining how LD functions in systemic energy homeostasis, in neuron-glia metabolic coupling, and in protein folding/clearance may affect or contribute to disease pathology. An integrated understanding of LD biology and neurodegeneration may open the way for new therapeutic interventions.
The Basics
Motor Neuron Diseases (MNDs) are characterized by the gradual degeneration and death of motor neurons (MNs), resulting in progressive paralysis and muscle atrophy. Amyotrophic Lateral Sclerosis (ALS) is a fatal disease and the most common MND, with a worldwide annual incidence of ~1.9 per 100,000 individuals (Arthur et al., 2016). A plethora of genes with a causative role in ALS have been identified (Renton et al., 2014). Among the cellular processes implicated in ALS pathophysiology are ER stress and protein clearance, neuron-glia metabolic coupling and energy homeostasis (Schmitt et al., 2014; Taylor et al., 2016). However, how alterations in these processes contribute to the disease remains poorly understood. Recently, connections have emerged between MNDs, including ALS, and lipid droplets (LDs), the cellular organelles for fat storage, possibly providing a new perspective for understanding disease-related mechanisms.
LDs are ubiquitous organelles with a unique structure: a core of neutral lipids (triacylglycerols (TAGs) and sterol esters) surrounded by a monolayer of phospholipids and proteins. LDs originate from the ER and, when lipids are needed, they are turned over by cytoplasmic lipases and autophagy. Mechanisms and regulation of LD biogenesis, growth, and turnover are active areas of investigation (Qi et al., 2017; Schulze et al., 2017; Walther et al., 2017).
LDs play critical roles in cellular processes also implicated in ALS pathophysiology. Foremost is their role in energy homeostasis, as they provide Fatty Acid (FA) fuel for ATP production. Emerging evidence also links LDs to neuron-glia metabolic coupling (Liu et al., 2017). Furthermore, LDs are involved in the resolution of ER stress and the clearing of protein aggregates (Welte, 2015; Welte and Gould, 2017). Genetic studies suggest that this overlap between LD functions and cellular processes implicated in ALS is not mere happenstance: many genes responsible for ALS, as well as for the MND Hereditary Spastic Paraplegia (HSP), play important roles in LD biology; on the other hand, disruption of lipid metabolism and energy homeostasis are prevalent in ALS (see below, Schmitt et al., 2014).
Here we discuss the role of LDs in energy homeostasis, glia-neuron metabolic coupling, and protein aggregation/clearance, and we formulate mechanistic hypotheses on how disruption of these LD-mediated processes could contribute to ALS pathophysiology. Although additional studies are needed to assess the specific role of LDs in ALS and other MNDs, it is conceivable that targeting LD-related processes might lead to new therapies for these devastating diseases.
LDs in Metabolic Organs and MNDs
Neurons have extremely high energy demands, and LDs are crucial for energy homeostasis in most cells. Might LD dysfunction promote neurodegeneration by disrupting the energy metabolism of neurons? At first glance, such a scenario seems unlikely because neurons derive much of their energy from glucose. However, when glucose is limiting, neurons use energy-rich substrates ultimately derived from the breakdown of FAs. Below, we discuss how LDs in various tissues across the organism could metabolically support MNs. We then contrast this systemic role with a local function in which LDs in glia provide fuel for neighbouring neurons. Finally, we consider whether LD functions beyond energy metabolism could directly affect MNs.
The brain lacks fuel stores and relies on a continuous supply of glucose from peripheral organs, especially the liver. When glucose is scarce, the liver coverts Acetyl-CoA produced during FA β-oxidation into ketone bodies (KBs). KBs are transported through the bloodstream to the brain where they are oxidized to produce energy. FAs metabolized in the liver are, in turn, replenished from dietary lipids in the gut or from lipids stored in adipose tissue. In all these tissues, FAs are stored as TAGs in LDs; thus LD dysfunction could indirectly impact MNs. This notion is attractive because many ALS genes are ubiquitously expressed, and non-cell autonomous processes can contribute to neuronal damage in ALS (Ilieva et al., 2009).
A link between ALS and disrupted organismal lipid metabolism is suggested by emerging data on TDP-43, a ubiquitous protein encoded by the ALS10 gene. TDP-43-mediated ALS may be caused by either loss of its normal function, gain of new toxic functions, or possibly a combination of both (Polymenidou and Cleveland, 2017; Robberecht and Philips, 2013). TDP-43 overexpressing mice display neurological symptoms, motor deficits, increased fat accumulation, and adipocyte hypertrophy (Stallings et al., 2013). Depleting TDP-43 postnatally in mice causes weight loss, body fat reduction, decreased adipocyte LD content, increased FA consumption, and rapid death (Chiang et al., 2010).
What is the mechanistic basis for these profound physiological changes? In skeletal muscles, TDP-43 depletion blocks insulin-induced trafficking of the glucose transporter Glut4 to the plasma membrane, by downregulating the Rab-GTPase-activating protein TBC1D1. Consequently, impairment in glucose uptake induces a metabolic switch towards lipids for energy production. Elevated FA consumption and oxidation by skeletal muscles presumably explain increased fat mobilization from adipose tissue (Chadt et al., 2008; Chiang et al., 2010). We speculate that TBC1D1 might even affect fat storage directly as the related isoform AS160/TBC1D4 regulates both Glut4 translocation and LD growth in adipocytes (Wu et al., 2014). In a yeast model for ALS, TDP-43 toxicity is enhanced by depletion of seipin (Armakola et al., 2012). Seipin has a prominent role in LD biogenesis and growth and is the causative gene of two MN disorders: dHMNV and Silver syndrome (Welte, 2015).
Intriguingly, the hVAPB/ALS8 gene also controls Glut4 trafficking, both in myoblasts and adipocytes, and affects LD size, number, and composition (Al-Anzi et al., 2015; Foster et al., 2000; Jansen et al., 2011). Mutations in genes controlling LD biogenesis/dynamics modify ALS8 phenotypes in flies (Sanhueza et al., 2015), and in C. elegans and mice, VAPB depletion induces LD accumulation and clustering in muscles (Han et al., 2013). In humans, mutations in spatacsin cause ALS and the MND HSP11, and its loss in mice induces MN degeneration, muscle atrophy, and a decrease in the steady-state number of LDs, possibly due to defective clearance of lipids by lysosomes (Branchu et al., 2017).
Evidence pointing to a metabolic switch from glucose towards lipids is also emerging in SOD1 mouse models of ALS: their spinal cord neurons display decreased glucose usage (Miyazaki et al., 2012), and a fat-rich diet restores normal body mass, delays disease onset and MN degeneration, and extends life expectancy (Schmitt et al., 2014). Consistent with a change in fuel preference in SOD1 mice, denervation of glycolytic muscle fibres, an early pre-symptomatic event, is preceded by increased expression of lipid-handling genes (Palamiuc et al., 2015). Increased FA requirements may explain why hyperlipidaemia positively correlates with survival in ALS patients and why they exhibit enhanced circulating levels of KBs (Schmitt et al., 2014).
Collectively, emerging evidence links ALS to systemic disruption of lipid metabolism (Figure 1A), though it remains to be established whether LD defects contribute to disease pathology or play more direct causative roles. We propose that LD dysfunction in various peripheral organs could throttle energy delivery to neurons and muscles, an energy supply these cells depend on under disease conditions.
Figure 1. How lipid droplets in peripheral organs and in glia could affect motor neuron diseases.
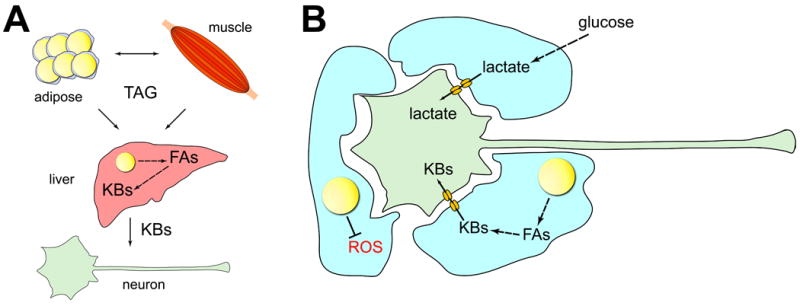
(A). TAGs are present in lipid droplets (LDs) in adipose tissue, muscle, and liver, and can be exchanged between these tissues via the bloodstream. In the liver, Fatty Acids (FAs) can be generated from TAGs and converted into Ketone Bodies (KBs). KBs travel through the blood to neurons where they are used as energy substrates. (B). Glia convert glucose into lactate that is then transferred to neurons as energy substrates. FAs in glial LDs are metabolized into KBs that are also transferred to neurons to be used as fuel. Finally, LDs in glia combat damage from Reactive Oxygen Species (ROS).
LDs in Glia-Neuron Metabolic Coupling
Neurons also derive fuel substrates from glia. According to the astrocyte-neuron lactate shuttle hypothesis, under conditions of intense neuronal activity, astrocytes a – type of glia – deliver glucose-derived lactate to neurons for ATP production (Belanger et al., 2011). Neurons can also receive lactate from oligodendrocytes (glial cells in the CNS that myelinate axons), via monocarboxylate transporters (MCTs). MCT1 ablation leads to axon damage and neuronal degeneration, and MCT1 levels are reduced in affected brain regions from ALS patients and in SOD1 mouse models of ALS (Lee et al., 2012). Intriguingly, MCTs can also transport KBs (Halestrap, 2012), and emerging evidence suggests that glia provide KBs as fuel to neurons, suggesting a link between LDs and ALS.
KBs are largely supplied by the liver; but like hepatocytes, astrocytes use FAs as primary metabolic fuel and produce large amount of KBs using a ketogenic machinery that is similar in both cell types (Guzman and Blazquez, 2001). This machinery requires the sequential action of three enzymes to transport FAs from the cytosol to the mitochondrial matrix. CPT1 (carnitine-palmitoyltransferase1) on the outer mitochondrial membrane conjugates carnitine with FAs; CACT (carnitine-acylcarnitine translocase) transfers acylcarnitine across the inner mitochondrial membrane where CPT2 conjugates FAs back to Coenzyme-A for subsequent β-oxidation. Inside mitochondria, acetyl moieties condense to generate KBs. In vivo, loss of CPT2 leads to increased starvation sensitivity and reduced life span of Drosophila adults. Because these phenotypes are reversed by selectively restoring CPT2 expression in glia, it was proposed that KB production and release from glia help power the brain (Schulz et al., 2015).
Together, these observations support a model in which disruption of KB production from LD-derived lipids or of their delivery may impact energy-dependent processes important for MN function and survival (Figure 1B). Consistent with this idea, inactivation of CTP2 in Drosophila leads to selective accumulation of LDs in glia, and glia-specific CPT2 expression reverses this phenotype (Schulz et al., 2015).
LDs in Neurons
Although it used to be controversial whether neuronal LDs exist at all, they have now been clearly identified in axons of Aplysia (Savage et al., 1987), neuronal cultures and brain sections of Huntington’s disease models (Martinez-Vicente et al., 2010), cerebral cortex neurons (Renvoise et al., 2016), MN axons of Drosophila larvae (Papadopoulos et al., 2015), and neuroblastoma cell lines (Holtta-Vuori et al., 2013).
Presence and abundance of neuronal LDs vary tremendously and may depend on variables such as developmental stage, nutrient or environmental conditions. In Drosophila glia, for example, LDs are induced by hypoxia in larvae (Bailey et al., 2015) and by neurodegeneration in adults (Liu et al., 2015). Sex-related differences in FA metabolism (Bakewell et al., 2006; Hoyenga and Hoyenga, 1982) could explain why cultured neurons isolated from females accumulate more LDs than neurons from males (Du et al., 2009). Neuronal LDs may also be rare or transient, making their identification particularly challenging. In adult mouse brain, LDs are rarely found, but they accumulate prominently inside neurons when their turnover is impaired by mutations in the TAG hydrolase DDHD2 (Inloes et al., 2014).
DDHD2 mouse knockouts exhibit motor and cognitive dysfunctions with extensive neurodegeneration, and in humans, DDHD2 mutations cause a MND known as HSP54 (Inloes et al., 2014). DDHD2-depleted neurons exhibit swelling of axons and dendrites, and Drosophila neuro-muscular junctions lacking DDHD2 display fewer active zones, the sites of neurotransmitter release (Schuurs-Hoeijmakers et al., 2012). We therefore speculate that excessive LDs may sequester and disrupt the trafficking of proteins and lipids required for synapse assembly. Indeed, LDs from mutant brains are associated with several proteins implicated in neuronal function (Figure 2) (Inloes et al., 2018).
Figure 2. How neuronal lipid droplets might affect motor neurons.
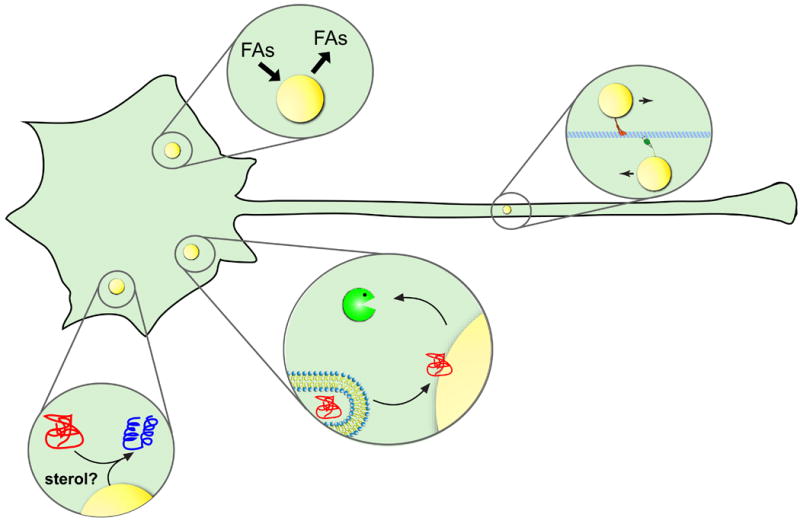
Lipid droplets (LDs) are present in neurons where they could act as organelles to sequester or release FAs, as platforms to refold or degrade misfolded proteins or as vehicles that traffic along axonal microtubules to deliver lipids or proteins.
Conversely, LDs in glia are larger and more easily detected than in neurons. Heterogeneity in LD content across a cell population was proposed to promote metabolic specialization. In the liver, some cells accumulate more LDs than their neighbours and, when needed, efficiently deliver FAs to surrounding cells (Herms et al., 2013). This heterogeneity may represent a strategy to alleviate the overall risk of lipotoxicity because cells with high LD content and higher levels of reactive oxygen species (ROS) express specialized defence mechanisms against oxidative damage. Might a similar division of labour also exist within the brain, with glia as the high-lipid content cells? Intriguingly, astrocytes contain higher levels of anti-oxidant molecules and ROS-detoxifying enzymes than neurons (Belanger et al., 2011).
There is direct evidence that LDs in glia protect neighbouring neurons from oxidative stress (Figure 1B). In Drosophila adults and in mice, mitochondrial dysfunction promotes - via ROS accumulation - increased synthesis of lipids, which are then transferred from neurons to glia and stored in LDs (Liu et al., 2017; Liu et al., 2015). In Drosophila larvae, increased glial LD content due to ROS accumulation (e.g. under hypoxia conditions) is protective because if glia are unable to make LDs, neurons exhibit increased damage from peroxidation (Bailey et al., 2015). It was proposed that glial LDs sequester lipids away from the plasma membrane, especially lipids containing polyunsaturated FAs (PUFAs), which are particularly vulnerable to peroxidation by ROS. PUFAs are more protected when incorporated into TAGs within LDs than when they are in phospholipids of cell membranes. During ROS-induced apoptosis, PUFAs of membrane phospholipids are peroxidated while their incorporation into LDs is thought to decrease their toxicity, suggesting that protection from oxidative stress is a general role of LDs (Li et al., 2017).
This protective role could be relevant for ALS as oxidative stress due to mitochondrial dysfunction is a leading cause of MN injury in ALS (Barber and Shaw, 2010). Consistently, markers of lipid peroxidation are found in the cerebrospinal fluid of ALS patients and in disease models, suggesting that accumulation of these toxic molecules might contribute to ALS (Simpson et al., 2004; Smith et al., 1998).
LDs in Protein Aggregation and Clearance
Protein turnover is critical for ALS, as a number of ALS-linked mutations affect genes directly involved in protein clearance and homeostasis (Taylor et al., 2016). Emerging evidence also suggests that LDs play a fundamental role in protein aggregation and clearance. Below we discuss whether and how LD-mediated control of these processes within neurons could affect their function and survival.
When misfolded or overabundant proteins stress the ER, an elaborate machinery is deployed to deliver them to the ubiquitin-proteasome system for degradation (ERAD or endoplasmic reticulum associated protein degradation). For ApoB-100, a major component of the very low density lipoproteins, proteasome-mediated degradation relies on LDs. Knockdown of Derlin-1, an ER protein coupling the transfer of proteins from the ER lumen to the ubiquitination system, decreases ApoB abundance on LDs. Conversely, knockdown of UBXD8, which binds ubiquitinated proteins and sends them for degradation, increases ubiquitinated ApoB levels on LDs. UBXD8 localizes to LDs and recruits VCP/p97, a member of the (AAA+) family which extracts ubiquitinated proteins from large complexes and delivers them to proteasomes. Various components of the ubiquitin-proteasome system also associate with LDs. These data suggest that Derlin dislocates ApoB to LDs where p97/VCP together with UBXD8 hands ApoB off to the proteasome for degradation. A similar mechanism likely operates for other proteins, for example, because a wide range of ubiquitinated proteins accumulates upon UBXD8 knockdown (Figure 2) (Ohsaki et al., 2006; Welte and Gould, 2017).
Of particular relevance to ALS are the findings that the ALS8 protein hVAPB associates with LDs (Cermelli et al., 2006) and controls the degradation of misfolded proteins by binding to a similar molecular cluster that includes Derlin1, VCP/p97 and the UBXD8-like protein FAF1 (Ernst et al., 2016). Specifically, hVAPB binds TTC39B (Huttlin et al., 2015), whose yeast orthologue, Ilm2, has a central role in LD-mediated clearance of misfolded proteins sequestered in inclusion bodies (IBs). Prior to IB clearance, IBs and LDs cluster in close proximity to each other, and the IB resident Ilm2 physically interacts with LD proteins. Without Ilm2, tethering between the two compartments fails and IBs are no longer efficiently cleared. Since IB clearing requires sterol ester containing LDs, LDs could be the source of a locally acting chemical chaperone that promotes refolding (Figure 2) (Moldavski et al., 2015). Relevant to ALS could be the observation that ALS8 causing proteins bind TTC39B more weakly than their wild-type counterpart (Huttlin et al., 2015).
A possible association between protein aggregates and LDs is also emerging in the nuclear compartment. The connection is through so-called promyelocytic leukemia (PML) nuclear bodies, membrane-less organelles of the nuclear matrix that control diverse processes including transcriptional activity, cell senescence, and protein degradation (Janer et al., 2006; Lallemand-Breitenbach and de The, 2010; Zhu and Brangwynne, 2015). PML bodies are present in close physical proximity to LDs within the nucleoplasm. This association is functionally important since knockdown of the PML isoform PMLII, a key organizer of PML bodies, results in fewer nuclear LDs, whereas its overexpression is correlated with more LDs (Ohsaki et al., 2016). In the brains of ALS patients, Nuclear Inclusions (NIs) have been found that contain ubiquitin, proteasome components, and PML (Seilhean et al., 2004). Furthermore, the ALS10 protein TDP-43 forms NIs that overlap with PML bodies (Wang et al., 2002). Finally, PML proteins selectively interact with several misfolded proteins, including TDP-43, and mark them through SUMOylation for proteasome degradation (Guo et al., 2014). Although the connection between the ability of the PML protein to degrade misfolded proteins and to control LD biogenesis remains to be determined, given the precedent in the cytoplasm, it is plausible that nuclear LDs are platforms for the turnover of aggregated proteins.
Mechanistic clues concerning LD-assisted protein clearance are also emerging. SigR1, the causative gene of a juvenile form of ALS (Al-Saif et al., 2011; Prause et al., 2013), encodes a protein localized at ER-LD contact sites where it is proposed to compartmentalize neutral lipids and regulate their export from the ER to LDs (Hayashi and Su, 2003). Here, SigR1 associates with lipid rafts, transiently stable membrane microdomains rich in cholesterol and sphingolipids (Rajendran and Simons, 2005) that are also sites where neurodegenerative proteins accumulate and aggregate (Burke et al., 2013; Di Paolo and Kim, 2011). Might LD-associated lipid rafts promote the sequestration of aggregated proteins on LDs for subsequent degradation? Although purely speculative, future critical tests of this notion seem worthwhile as LDs can host lipid raft proteins like caveolins, flotillins and stomatin (Fujimoto et al., 2001; Pol et al., 2004; Rajendran et al., 2007; Umlauf et al., 2004) and because the ER-associated lipid raft protein Erlin 2 is linked to both regulation of LD biogenesis (Wang et al., 2012) and certain MNDs (Al-Saif et al., 2012; Alazami et al., 2011).
Future Crucial Experiments
Going forward, it will be crucial to determine to what extent disease phenotypes in ALS models are altered by tissue-specific modulation of LD genes. Advanced microscopy and newly developed LD fluorescent markers (Kassan et al., 2013; Wang et al., 2016) will make possible the mapping of neuronal LD dynamics. Additionally, lipidomic and proteomic approaches associated with genetic analyses will lay the ground for a mechanistic understanding of the LD role in MN function and survival.
Acknowledgments
We thank Gareth Leng and Douglas Portman as well as three anonymous reviewers for insightful comments on a previous version of this manuscript. We apologize to the colleagues whose original research could not be cited due to space constraints. This work was supported by the Wellcome Trust (Pennetta8260) and Motor Neurone Disease Association (Pennetta6231) to GP and by the National Institutes of Health (NIH; R01GM102155) to MAW.
References
- Al-Anzi B, Arpp P, Gerges S, Ormerod C, Olsman N, Zinn K. Experimental and computational analysis of a large protein network that controls fat storage reveals the design principles of a signaling network. PLoS Comput Biol. 2015;11:e1004264. doi: 10.1371/journal.pcbi.1004264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Saif A, Al-Mohanna F, Bohlega S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol. 2011;70:913–919. doi: 10.1002/ana.22534. [DOI] [PubMed] [Google Scholar]
- Al-Saif A, Bohlega S, Al-Mohanna F. Loss of ERLIN2 function leads to juvenile primary lateral sclerosis. Ann Neurol. 2012;72:510–516. doi: 10.1002/ana.23641. [DOI] [PubMed] [Google Scholar]
- Alazami AM, Adly N, Al Dhalaan H, Alkuraya FS. A nullimorphic ERLIN2 mutation defines a complicated hereditary spastic paraplegia locus (SPG18) Neurogenetics. 2011;12:333–336. doi: 10.1007/s10048-011-0291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armakola M, Higgins MJ, Figley MD, Barmada SJ, Scarborough EA, Diaz Z, Fang X, Shorter J, Krogan NJ, Finkbeiner S, et al. Inhibition of RNA lariat debranching enzyme suppresses TDP-43 toxicity in ALS disease models. Nat Genet. 2012;44:1302–1309. doi: 10.1038/ng.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur KC, Calvo A, Price TR, Geiger JT, Chio A, Traynor BJ. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat Commun. 2016;7:12408. doi: 10.1038/ncomms12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, Postle AD, Gould AP. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell. 2015;163:340–353. doi: 10.1016/j.cell.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakewell L, Burdge GC, Calder PC. Polyunsaturated fatty acid concentrations in young men and women consuming their habitual diets. Br J Nutr. 2006;96:93–99. doi: 10.1079/bjn20061801. [DOI] [PubMed] [Google Scholar]
- Barber SC, Shaw PJ. Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med. 2010;48:629–641. doi: 10.1016/j.freeradbiomed.2009.11.018. [DOI] [PubMed] [Google Scholar]
- Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Branchu J, Boutry M, Sourd L, Depp M, Leone C, Corriger A, Vallucci M, Esteves T, Matusiak R, Dumont M, et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol Dis. 2017;102:21–37. doi: 10.1016/j.nbd.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke KA, Yates EA, Legleiter J. Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front Neurol. 2013;4:17. doi: 10.3389/fneur.2013.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermelli S, Guo Y, Gross SP, Welte MA. The lipid-droplet proteome reveals that droplets are a protein-storage depot. Curr Biol. 2006;16:1783–1795. doi: 10.1016/j.cub.2006.07.062. [DOI] [PubMed] [Google Scholar]
- Chadt A, Leicht K, Deshmukh A, Jiang LQ, Scherneck S, Bernhardt U, Dreja T, Vogel H, Schmolz K, Kluge R, et al. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nat Genet. 2008;40:1354–1359. doi: 10.1038/ng.244. [DOI] [PubMed] [Google Scholar]
- Chiang PM, Ling J, Jeong YH, Price DL, Aja SM, Wong PC. Deletion of TDP-43 down-regulates Tbc1d1, a gene linked to obesity, and alters body fat metabolism. Proc Natl Acad Sci U S A. 2010;107:16320–16324. doi: 10.1073/pnas.1002176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, Kochanek PM, Jenkins LW, Ren J, Gibson G, et al. Starving neurons show sex difference in autophagy. J Biol Chem. 2009;284:2383–2396. doi: 10.1074/jbc.M804396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst WL, Shome K, Wu CC, Gong X, Frizzell RA, Aridor M. VAMP-associated Proteins (VAP) as Receptors That Couple Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) Proteostasis with Lipid Homeostasis. J Biol Chem. 2016;291:5206–5220. doi: 10.1074/jbc.M115.692749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster LJ, Weir ML, Lim DY, Liu Z, Trimble WS, Klip A. A functional role for VAP-33 in insulin-stimulated GLUT4 traffic. Traffic. 2000;1:512–521. doi: 10.1034/j.1600-0854.2000.010609.x. [DOI] [PubMed] [Google Scholar]
- Fujimoto T, Kogo H, Ishiguro K, Tauchi K, Nomura R. Caveolin-2 is targeted to lipid droplets, a new “membrane domain” in the cell. J Cell Biol. 2001;152:1079–1085. doi: 10.1083/jcb.152.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Giasson BI, Glavis-Bloom A, Brewer MD, Shorter J, Gitler AD, Yang X. A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol Cell. 2014;55:15–30. doi: 10.1016/j.molcel.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M, Blazquez C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab. 2001;12:169–173. doi: 10.1016/s1043-2760(00)00370-2. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. The monocarboxylate transporter family--Structure and functional characterization. IUBMB Life. 2012;64:1–9. doi: 10.1002/iub.573. [DOI] [PubMed] [Google Scholar]
- Han SM, El Oussini H, Scekic-Zahirovic J, Vibbert J, Cottee P, Prasain JK, Bellen HJ, Dupuis L, Miller MA. VAPB/ALS8 MSP ligands regulate striated muscle energy metabolism critical for adult survival in caenorhabditis elegans. PLoS Genet. 2013;9:e1003738. doi: 10.1371/journal.pgen.1003738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Intracellular dynamics of sigma-1 receptors (sigma(1) binding sites) in NG108-15 cells. J Pharmacol Exp Ther. 2003;306:726–733. doi: 10.1124/jpet.103.051292. [DOI] [PubMed] [Google Scholar]
- Herms A, Bosch M, Ariotti N, Reddy BJ, Fajardo A, Fernandez-Vidal A, Alvarez-Guaita A, Fernandez-Rojo MA, Rentero C, Tebar F, et al. Cell-to-cell heterogeneity in lipid droplets suggests a mechanism to reduce lipotoxicity. Curr Biol. 2013;23:1489–1496. doi: 10.1016/j.cub.2013.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtta-Vuori M, Salo VT, Ohsaki Y, Suster ML, Ikonen E. Alleviation of seipinopathy-related ER stress by triglyceride storage. Hum Mol Genet. 2013;22:1157–1166. doi: 10.1093/hmg/dds523. [DOI] [PubMed] [Google Scholar]
- Hoyenga KB, Hoyenga KT. Gender and energy balance: sex differences in adaptations for feast and famine. Physiol Behav. 1982;28:545–563. doi: 10.1016/0031-9384(82)90153-6. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inloes JM, Hsu KL, Dix MM, Viader A, Masuda K, Takei T, Wood MR, Cravatt BF. The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proc Natl Acad Sci U S A. 2014;111:14924–14929. doi: 10.1073/pnas.1413706111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inloes JM, Kiosses WB, Wang H, Walther TC, Farese RV, Jr, Cravatt BF. Functional Contribution of the Spastic Paraplegia-Related Triglyceride Hydrolase DDHD2 to the Formation and Content of Lipid Droplets. Biochemistry. 2018;57:827–838. doi: 10.1021/acs.biochem.7b01028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janer A, Martin E, Muriel MP, Latouche M, Fujigasaki H, Ruberg M, Brice A, Trottier Y, Sittler A. PML clastosomes prevent nuclear accumulation of mutant ataxin-7 and other polyglutamine proteins. J Cell Biol. 2006;174:65–76. doi: 10.1083/jcb.200511045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen M, Ohsaki Y, Rega LR, Bittman R, Olkkonen VM, Ikonen E. Role of ORPs in sterol transport from plasma membrane to ER and lipid droplets in mammalian cells. Traffic. 2011;12:218–231. doi: 10.1111/j.1600-0854.2010.01142.x. [DOI] [PubMed] [Google Scholar]
- Kassan A, Herms A, Fernandez-Vidal A, Bosch M, Schieber NL, Reddy BJ, Fajardo A, Gelabert-Baldrich M, Tebar F, Enrich C, et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J Cell Biol. 2013;203:985–1001. doi: 10.1083/jcb.201305142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V, de The H. PML nuclear bodies. Cold Spring Harb Perspect Biol. 2010;2:a000661. doi: 10.1101/cshperspect.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Sancak Y, Frasor J, Atilla-Gokcumen GE. A Protective Role for Triacylglycerols during Apoptosis. Biochemistry. 2017 doi: 10.1021/acs.biochem.7b00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, MacKenzie KR, Putluri N, Maletic-Savatic M, Bellen HJ. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017;26:719–737. e716. doi: 10.1016/j.cmet.2017.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhang K, Sandoval H, Yamamoto S, Jaiswal M, Sanz E, Li Z, Hui J, Graham BH, Quintana A, et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–190. doi: 10.1016/j.cell.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. 2010;13:567–576. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Masamoto K, Morimoto N, Kurata T, Mimoto T, Obata T, Kanno I, Abe K. Early and progressive impairment of spinal blood flow-glucose metabolism coupling in motor neuron degeneration of ALS model mice. J Cereb Blood Flow Metab. 2012;32:456–467. doi: 10.1038/jcbfm.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldavski O, Amen T, Levin-Zaidman S, Eisenstein M, Rogachev I, Brandis A, Kaganovich D, Schuldiner M. Lipid Droplets Are Essential for Efficient Clearance of Cytosolic Inclusion Bodies. Dev Cell. 2015;33:603–610. doi: 10.1016/j.devcel.2015.04.015. [DOI] [PubMed] [Google Scholar]
- Ohsaki Y, Cheng J, Fujita A, Tokumoto T, Fujimoto T. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein B. Mol Biol Cell. 2006;17:2674–2683. doi: 10.1091/mbc.E05-07-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsaki Y, Kawai T, Yoshikawa Y, Cheng J, Jokitalo E, Fujimoto T. PML isoform II plays a critical role in nuclear lipid droplet formation. J Cell Biol. 2016;212:29–38. doi: 10.1083/jcb.201507122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamiuc L, Schlagowski A, Ngo ST, Vernay A, Dirrig-Grosch S, Henriques A, Boutillier AL, Zoll J, Echaniz-Laguna A, Loeffler JP, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med. 2015;7:526–546. doi: 10.15252/emmm.201404433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos C, Orso G, Mancuso G, Herholz M, Gumeni S, Tadepalle N, Jungst C, Tzschichholz A, Schauss A, Honing S, et al. Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet. 2015;11:e1005149. doi: 10.1371/journal.pgen.1005149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol A, Martin S, Fernandez MA, Ferguson C, Carozzi A, Luetterforst R, Enrich C, Parton RG. Dynamic and regulated association of caveolin with lipid bodies: modulation of lipid body motility and function by a dominant negative mutant. Mol Biol Cell. 2004;15:99–110. doi: 10.1091/mbc.E03-06-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Cleveland DW. Biological Spectrum of Amyotrophic Lateral Sclerosis Prions. Cold Spring Harb Perspect Med. 2017;7 doi: 10.1101/cshperspect.a024133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prause J, Goswami A, Katona I, Roos A, Schnizler M, Bushuven E, Dreier A, Buchkremer S, Johann S, Beyer C, et al. Altered localization, abnormal modification and loss of function of Sigma receptor-1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22:1581–1600. doi: 10.1093/hmg/ddt008. [DOI] [PubMed] [Google Scholar]
- Qi Y, Sun L, Yang H. Lipid droplet growth and adipocyte development: mechanistically distinct processes connected by phospholipids. Biochim Biophys Acta. 2017;1862:1273–1283. doi: 10.1016/j.bbalip.2017.06.016. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Le Lay S, Illges H. Raft association and lipid droplet targeting of flotillins are independent of caveolin. Biol Chem. 2007;388:307–314. doi: 10.1515/BC.2007.034. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Simons K. Lipid rafts and membrane dynamics. J Cell Sci. 2005;118:1099–1102. doi: 10.1242/jcs.01681. [DOI] [PubMed] [Google Scholar]
- Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci. 2014;17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoise B, Malone B, Falgairolle M, Munasinghe J, Stadler J, Sibilla C, Park SH, Blackstone C. Reep1 null mice reveal a converging role for hereditary spastic paraplegia proteins in lipid droplet regulation. Hum Mol Genet. 2016;25:5111–5125. doi: 10.1093/hmg/ddw315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- Sanhueza M, Chai A, Smith C, McCray BA, Simpson TI, Taylor JP, Pennetta G. Network analyses reveal novel aspects of ALS pathogenesis. PLoS Genet. 2015;11:e1005107. doi: 10.1371/journal.pgen.1005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage MJ, Goldberg DJ, Schacher S. Absolute specificity for retrograde fast axonal transport displayed by lipid droplets originating in the axon of an identified Aplysia neuron in vitro. Brain Res. 1987;406:215–223. doi: 10.1016/0006-8993(87)90785-2. [DOI] [PubMed] [Google Scholar]
- Schmitt F, Hussain G, Dupuis L, Loeffler JP, Henriques A. A plural role for lipids in motor neuron diseases: energy, signaling and structure. Front Cell Neurosci. 2014;8:25. doi: 10.3389/fncel.2014.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JG, Laranjeira A, Van Huffel L, Gartner A, Vilain S, Bastianen J, Van Veldhoven PP, Dotti CG. Glial beta-oxidation regulates Drosophila energy metabolism. Sci Rep. 2015;5:7805. doi: 10.1038/srep07805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze RJ, Sathyanarayan A, Mashek DG. Breaking fat: The regulation and mechanisms of lipophagy. Biochim Biophys Acta. 2017;1862:1178–1187. doi: 10.1016/j.bbalip.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurs-Hoeijmakers JH, Geraghty MT, Kamsteeg EJ, Ben-Salem S, de Bot ST, Nijhof B, van de V, II, van der Graaf M, Nobau AC, Otte-Holler I, et al. Mutations in DDHD2, encoding an intracellular phospholipase A(1), cause a recessive form of complex hereditary spastic paraplegia. Am J Hum Genet. 2012;91:1073–1081. doi: 10.1016/j.ajhg.2012.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seilhean D, Takahashi J, El Hachimi KH, Fujigasaki H, Lebre AS, Biancalana V, Durr A, Salachas F, Hogenhuis J, de The H, et al. Amyotrophic lateral sclerosis with neuronal intranuclear protein inclusions. Acta Neuropathol. 2004;108:81–87. doi: 10.1007/s00401-004-0855-x. [DOI] [PubMed] [Google Scholar]
- Simpson EP, Henry YK, Henkel JS, Smith RG, Appel SH. Increased lipid peroxidation in sera of ALS patients: a potential biomarker of disease burden. Neurology. 2004;62:1758–1765. doi: 10.1212/wnl.62.10.1758. [DOI] [PubMed] [Google Scholar]
- Smith RG, Henry YK, Mattson MP, Appel SH. Presence of 4-hydroxynonenal in cerebrospinal fluid of patients with sporadic amyotrophic lateral sclerosis. Ann Neurol. 1998;44:696–699. doi: 10.1002/ana.410440419. [DOI] [PubMed] [Google Scholar]
- Stallings NR, Puttaparthi K, Dowling KJ, Luther CM, Burns DK, Davis K, Elliott JL. TDP-43, an ALS linked protein, regulates fat deposition and glucose homeostasis. PLoS One. 2013;8:e71793. doi: 10.1371/journal.pone.0071793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Brown RH, Jr, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umlauf E, Csaszar E, Moertelmaier M, Schuetz GJ, Parton RG, Prohaska R. Association of stomatin with lipid bodies. J Biol Chem. 2004;279:23699–23709. doi: 10.1074/jbc.M310546200. [DOI] [PubMed] [Google Scholar]
- Walther TC, Chung J, Farese RV., Jr Lipid Droplet Biogenesis. Annu Rev Cell Dev Biol. 2017;33:491–510. doi: 10.1146/annurev-cellbio-100616-060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Zhang X, Lee JS, Wang X, Yang ZQ, Zhang K. Endoplasmic reticulum factor ERLIN2 regulates cytosolic lipid content in cancer cells. Biochem J. 2012;446:415–425. doi: 10.1042/BJ20112050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Becuwe M, Housden BE, Chitraju C, Porras AJ, Graham MM, Liu XN, Thiam AR, Savage DB, Agarwal AK, et al. Seipin is required for converting nascent to mature lipid droplets. Elife. 2016;5 doi: 10.7554/eLife.16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IF, Reddy NM, Shen CK. Higher order arrangement of the eukaryotic nuclear bodies. Proc Natl Acad Sci U S A. 2002;99:13583–13588. doi: 10.1073/pnas.212483099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA. Expanding roles for lipid droplets. Curr Biol. 2015;25:R470–481. doi: 10.1016/j.cub.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte MA, Gould AP. Lipid droplet functions beyond energy storage. Biochim Biophys Acta. 2017;1862:1260–1272. doi: 10.1016/j.bbalip.2017.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Xu D, Zhou L, Xie B, Yu L, Yang H, Huang L, Ye J, Deng H, Yuan YA, et al. Rab8a-AS160-MSS4 regulatory circuit controls lipid droplet fusion and growth. Dev Cell. 2014;30:378–393. doi: 10.1016/j.devcel.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Zhu L, Brangwynne CP. Nuclear bodies: the emerging biophysics of nucleoplasmic phases. Curr Opin Cell Biol. 2015;34:23–30. doi: 10.1016/j.ceb.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]