

Abstract
DNA is the carrier of genetic information and the primary template from which all cellular information is ultimately derived. Changes in the DNA information content through mutation generate diversity for evolution through natural selection but are also a source of deleterious effects. It has since long been hypothesized that mutation accumulation in somatic cells of multicellular organisms could causally contribute to age-related cellular degeneration and death. Assays to detect different types of mutations, from base substitutions to large chromosomal aberrations, have been developed and show unequivocally that mutations accumulate in different tissues and cell types of ageing humans and animals. More recently, next-generation sequencing-based methods have been developed to accurately determine the complete landscape of base substitution mutations in single cells. The first results show that the somatic mutation rate is much higher than the germline mutation rate and that base substitution loads in somatic cells are high enough to potentially affect cellular function.
Introduction
As the carrier of genetic information, DNA is a very stable molecule. Yet, under physiological conditions, when DNA is continuously damaged by both endogenous and environmental factors, most of this stability is provided by a highly conserved system of genome maintenance mechanisms [1]. In the history of life, genes encoding DNA repair were probably among the first genetic traits selected in early life forms shortly after DNA replaced short RNA as a readily replicable and expandable repertoire of genetic instructions. DNA damage repair was critical in that process providing a major survival advantage [2]. Interestingly, through the inevitable errors occurring during repair and replication of damaged templates random changes in the genome’s DNA sequence-based code ensued, providing the substrate for evolution. This situation has in essence not changed since then and, also in metazoa, it is difficult to overemphasize the critical role of DNA repair and other genome maintenance systems in eliminating the many thousands of lesions generated each day in a typical cell through physicochemical mechanisms. Through errors during repair or replication, DNA damage is occasionally converted in mutations [3], which will accumulate with age. Also, DNA lesions themselves can accumulate with age because some may not be efficiently recognized by DNA repair systems or are irreparable (e.g. double-strand breaks of which the ends have diffused far apart). However, while there is indeed some evidence that DNA damage accumulates with age [4,5], spontaneous DNA damage levels are very low and extremely difficult to detect and characterize [6]. Most lesions are transient and efficiently repaired. By contrast, mutations cannot be repaired and their accumulation in various organs and tissues with age of humans or animals has now been amply confirmed. Here, we will discuss age-related mutations in the genome and the likelihood that these events are causally related to the age-related loss of function and increased susceptibility to disease that are the hallmarks of ageing (schematically depicted in Figure 1). While we will briefly discuss current evidence for the accumulation of various types of DNA mutations during ageing, the focus of this review will be on the recently emerged possibility to sequence the entire genome of single cells from ageing organisms. This will allow, for the first time, to quantitatively assess somatic mutations and their possible functional consequences.
Figure 1. DNA damage, mutations and ageing.

Schematic depiction of DNA damage and mutations in somatic tissues of ageing organisms and their possible consequences.
Genome instability and the logic of life
Mutations represent alterations in the DNA sequence while maintaining the same set of nucleotides. Mutant DNA is, therefore, chemically indistinguishable from unaltered sequences and cannot be recognized by repair enzymes. When a cell harboring a mutation is not eliminated, the mutation is permanent and in mitotically active cells all daughter cells will carry the same mutation. While most mutations have adverse effects or no effects at all, some can be beneficial under certain conditions. As briefly discussed above, this principle was co-opted by life itself, which is the product of DNA mutation and natural selection. Natural selection will typically drive mutation rates to the minimum possible level but never to zero, possibly due to factors such as fitness costs associated with excessive investment in replication fidelity [7,8] and genetic drift [9]. A complete absence of mutations would of course also prevent adaptability—the capacity to respond to environmental change—and eventually lead to extinction. Whichever of these factors is more important, there can be no doubt that it is the imperfection of replication and repair in coping with DNA damage that gave rise to the large diversity of life forms on our planet. Hence, genome instability in the germline is inherent to life and strictly necessary for its continuation. The germline mutation rate has been extensively studied and has been found to vary greatly between species. Indeed, as first noted by Sturtevant [8,10], the genetic material is mutable at a rate subject to natural selection.
With the emergence of multicellular organisms the possibility arose, at least in principle, to drive mutations to zero in only the somatic cells. Somatic cells are subject to the same imperfection of replication and repair as germ cells, and the selective pressures acting on germline and somatic mutation rates are likely connected [11]. Indirect evidence suggests that somatic mutation frequency is much higher than that in germ cells [11], but this has been difficult to test. With the emergence of next-generation sequencing, germline mutation frequencies can now be determined directly by whole genome sequencing of parents and offspring [12]. The frequency of de novo mutations in the offspring is a measure of the germline mutation frequency. By contrast, de novo somatic mutations cannot be determined by sequencing total genomic DNA due to the very low abundance of such mutations, which are unique to individual cells. This has been a major hindrance in attempts to test the somatic mutation theory of ageing, proposed as early as the 1950s [13,14].
Somatic mutations accumulate during ageing
For a long time, the only types of mutations readily detectable were chromosomal alterations, such as chromosomal aneuploidy [15]. More recently, cytogenetic methods gained accuracy and could be applied on a much larger scale due to the development of fluorescence in situ hybridization (FISH). Using these methods, it has been shown that lymphocytes with chromosomal aberrations increase with age in the blood of both humans and mice [16,17]. Interphase FISH can even be applied on non-dividing cells in tissues such as brain [18] for the detection of aneuploidy, i.e. gain or loss of entire chromosomes.
Chromosomal aneuploidy is a hallmark of pathological conditions and a causal factor of birth defects and cancer. Using advanced FISH technology, it has been shown that in the developing nervous system of humans and mice the frequency of aneuploid cells is as high as 33% for all chromosomes combined [19,20]. However, after completion of development, this extremely high level of aneuploidy was found to be significantly reduced suggesting selection against cells with high symptoms of genome instability. Indeed, this would be in keeping with the observed high levels of whole-chromosome aneuploidy and segmental deletions and duplications in individual blastomeres of cleavage-stage embryos after in vitro fertilization (IVF) [21]. Since IVF success rates are typically in the order of 25%, mutated blastomeres were most probably eliminated by extensive selection of non-mutated or less severely mutated blastomeres in the early embryo.
Still, even in adult organisms, aneuploidy levels of 4% per chromosome as observed in human brain for chromosome 21 [22] would mean that almost all cells in an organ such as brain or liver would be aneuploid for one chromosome. In mice, using a two-probe system with aneuploidies only called when indicated at both locations, we found that in the cerebral cortex the frequency of aneuploid cells can rise to a level as high as 5% per chromosome [23]. Overall, this would correspond to approximately half of all brain cells in old mice having at least one aneuploidy. Taking into consideration that aneuploidy is merely the tip of the iceberg, genome instability in cells during development and ageing may be very high. However, it should be noticed that single-cell sequencing results do not show high levels of aneuploidy in the human brain [24,25].
There is abundant evidence that in addition to chromosomal mutations, various other types of somatic mutations accumulate with age [26,27]. This became apparent once methods to detect such mutations, including base substitutions, deletions, and genome rearrangements, became available. First, methods using endogenous selectable marker genes, such as hypoxanthine-guanine phosphoribosyltransferase (HGPRT), were used to assess mutation frequencies in blood cells [28]. The results were similar to the aforementioned chromosomal studies in the sense that mutation frequency was found to increase with age, both in humans and rodents [29,30]. With the development of the first transgenic mice [31], and later flies [32], harboring reporter genes that can be recovered into Escherichia coli to study mutation frequency and spectrum in the animal host amply confirmed that mutations accumulate in essentially all organs and tissues [33–36], albeit at greatly different rates (Figure 2), and in both organisms [37].
Figure 2. Somatic mutations accumulate with age in four different tissues.
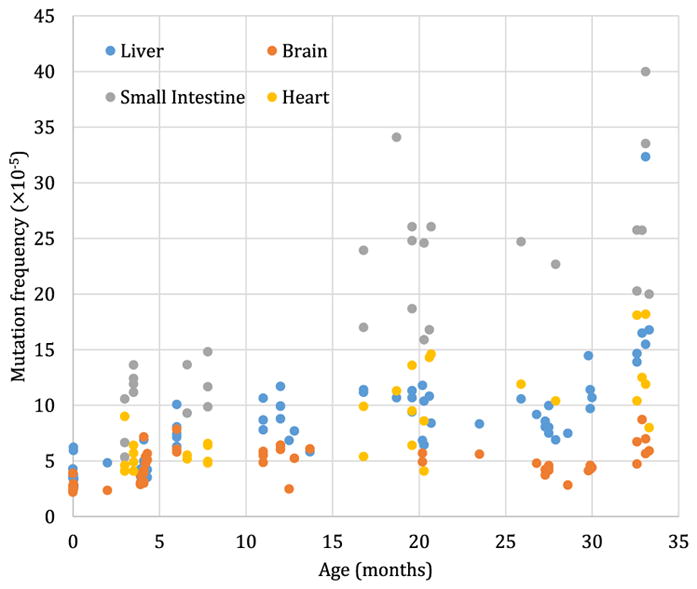
Using a transgenic mouse model harboring chromosomally integrated plasmids containing the lacZ reporter gene that can be excised and transferred into E. coli to select for mutants that inactivate the lacZ-encoded β-galactosidase, mutation frequency (y-axis) was determined as a function of the age of the animals. Each determination point is the average of at least five individual animals. Data were redrawn from [33] and [34].
There are other ways to gain information about mutation accumulation during ageing. Interestingly, tumors, as clonal expansions of single cells, can provide information about the somatic mutations present in these cells prior to tumorigenesis. Using data from The Cancer Genome Atlas (TCGA) to systematically study the frequency and spectrum of somatic mutations in thousands of cancer patients and different tumor types as a function of the age of the patient, we found that the number of identified somatic mutations increases exponentially with age [38]. Others have demonstrated ageing-specific signature mutations in human tumors [39].
Other types of mutations that have been studied include somatic L1 retrotranspositions, which are suppressed at young age but has been shown to strongly increase with age in several mouse tissues [40]. Also mutation frequency at microsatellite loci, which is already much higher than elsewhere in the genome increases with age in human blood [41]. Another type of repeat element, i.e. telomeres, regions of repetitive DNA protecting chromosome ends from deterioration, significantly shorten with age in mammalian cells and tissues [42]. Finally, an age-related accumulation of clonally expanded, copy number variants has been found [43,44].
Hence, the main question is no longer whether various types of mutations accumulate with age in different cell types but if these mutations have any functional impact. To predict functional impact, it is necessary to first fully characterize the landscape of somatic mutations in ageing humans or animals by sequence analysis.
Functional impact of somatic mutations
While it seems unlikely that genome instability is the only or even major cause of ageing, it is important to test if mutations at least causally contribute to cellular degeneration and death in addition to causing cancer, a process in which mutant cells can be selected. In other words, the question is if somatic mutations ever reach levels that are functionally detrimental to cells. To address this question, it is necessary to comprehensively characterize the total complement of mutations in individual cells across the genome during the lifetime. This can best be done by next-generation sequencing, which should allow the detection of a wide range of somatic mutations. However, since somatic mutations in normal tissues are unique for each individual cell (except for cells derived from the same ancestor cell), whole genome sequencing will simply provide the germline genotype rather than de novo somatic mutations. To analyze normal somatic tissues, it is necessary to analyze single cells or clones derived from single cells (Figure 3). As described above, to some extent tumors can serve as surrogates for single cells, but since mutations can also arise after neoplastic transformation, during tumor progression, it is difficult to draw definite conclusions other than that mutation frequency increases with age.
Figure 3. Single-cell analysis is required for detecting low-abundant, random mutations.
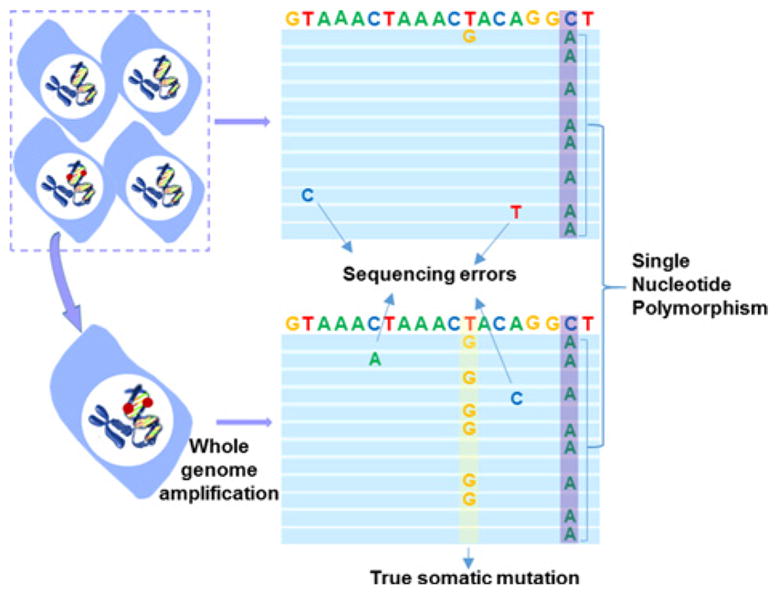
The T > G mutation (red dot ) can only be distinguished from sequencing errors after single-cell, whole genome amplification (WGA) when it shows up in ~50% of the reads (yellow; one mutated allele). Polymorphic variants (SNPs; blue) are also observed in the unamplified control DNA extracted from the bulk cell population. SNPs are variants between the genome of our cells and the reference genome. They are electronically discarded (after [65]).
More recently, whole genome sequencing of clonal organoid cultures derived from mouse or human primary multipotent cells revealed hundreds of base substitution mutations per genome increasing with age (Table 1) [45,46]. However, clonal amplification through organoid technology requires extensive cell culture and essentially limits analysis to stem or progenitor cells. Single cell technology allows direct analysis of all types of cells, including postmitotic cells, such as neurons and muscle fibers. However, analyzing mutations in single cells requires WGA, which suffers from artifacts. We recently developed a new protocol, i.e. single-cell multiple displacement amplification (SCMDA), with a single-cell variant caller (SCcaller), to accurately identify somatic mutations across the genome from a single cell after WGA [47]. The procedure was validated by directly comparing mutation frequency and spectrum between amplified single cells and unamplified clones derived from cells in the same population of early passage, human primary fibroblasts. We also sequenced SCMDA-amplified single cells and non-amplified clones derived from the same clone, reasoning that there should be significant overlap between the single cells and their kindred clone. The entire procedure is schematically depicted in Figure 4.
Table 1.
A summary of somatic and germline mutation frequencies
| Type | Species | Tissue/cell type | Method | Mutations per cell/clone (somatic) or generation (germline) | Reference |
|---|---|---|---|---|---|
| Germline | Human | Germline | Trio | 63.2 (n=78) | [12] |
| Human | Germline | Trio | 42 (n=2) | [66] | |
| Human | Germline | Trio | 63 (n=2) | [51] | |
| Human | Sperm | Single cell | 64–115 (n=8) | [50] | |
| Mouse | Germline | Multiple generations | 28 | [67] | |
| Mouse | Germline | Trio | 21 (n=12) | [68] | |
| Mouse | Germline | Trio | 32 ± 11 (s.d.; n=8) | [51] | |
| Somatic | Human | Multiple | Clone | 245–3516 (n=45) | [45] |
| Human | Fibroblast | Clone/single cell | 966 ± 248 (s.d.; n=10) | [51] | |
| Mouse | Multiple | Organoid | 179–1190 (n=25) | [46] | |
| Mouse | Fibroblast | Single cell | 1524 ± 259 (s.d.; n=5) | [51] |
Figure 4.
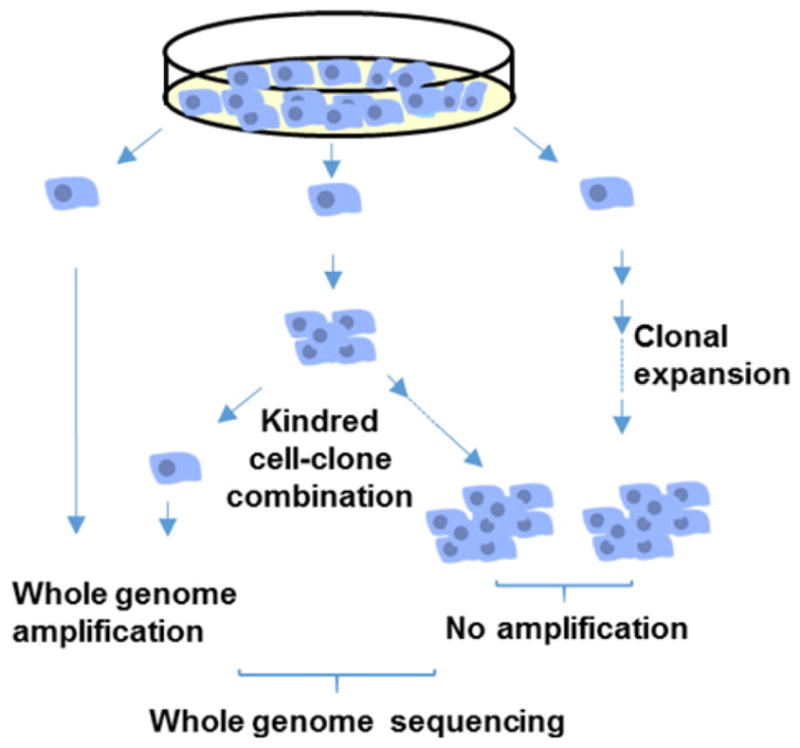
Schematic representation of whole genome sequence analysis of mutations in single human, primary fibroblasts after amplification and in unamplified clones from single fibroblasts taken from the same population.
The results shown in Figure 5 indicate approximately 1,000 somatic mutations in single human primary fibroblasts, in the same range as the numbers found in unamplified clones from the same population of cells (Table 1). Interestingly, these somatic mutation frequencies are much higher than the germline mutation frequency. In humans, the germline mutation frequency has been determined by whole genome sequencing of parents and children and calling de novo mutations in the offspring. This resulted in a germline mutation frequency of 1.2 × 10−8 [12], confirming earlier indirect estimates [48,49] as well as those derived from sequencing of single sperm cells [50]. When directly comparing the somatic mutation frequencies observed by us in primary human and mouse fibroblasts with the germline mutation frequencies obtained as described above, by sequencing parents and children in both mice and humans, we observed an almost two orders of magnitude higher somatic mutation frequency (Table 1) [51]. After correcting for the differences between these two species in the number of cell divisions per generation (errors during DNA replication are a major source of base substitutions), this difference remained (Figure 6). Of note, mouse cells now show a significantly higher somatic and germline mutation frequency than humans (Figure 6).
Figure 5. Frequency of somatic mutations in unamplified clones and amplified single cells.
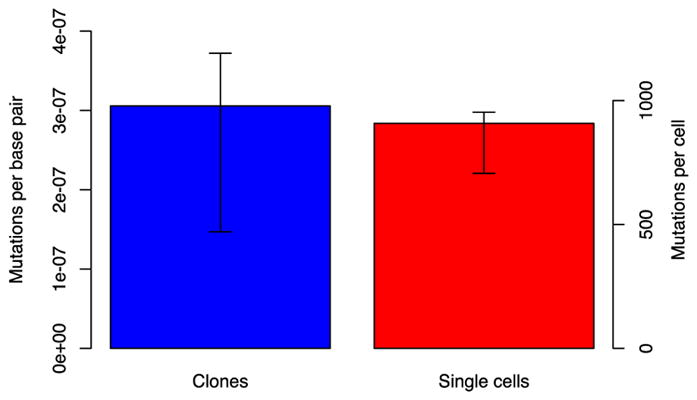
Clones and MDA amplicons derived from single human dermal fibroblasts were sequenced and the variants found compared with those in the bulk population to determine the somatic mutation frequency. There was no significant difference (P=0.76, Wilcoxon test) in the frequency of mutations observed, indicating that the single cell amplification protocol provides an accurate estimate of the somatic mutation frequency. The right-hand axis indicates the number of mutations per cell after adjustment for coverage. Error bars indicate 88% confidence intervals. From Milholland et al. [51].
Figure 6. Germline and somatic mutations in humans and mice.
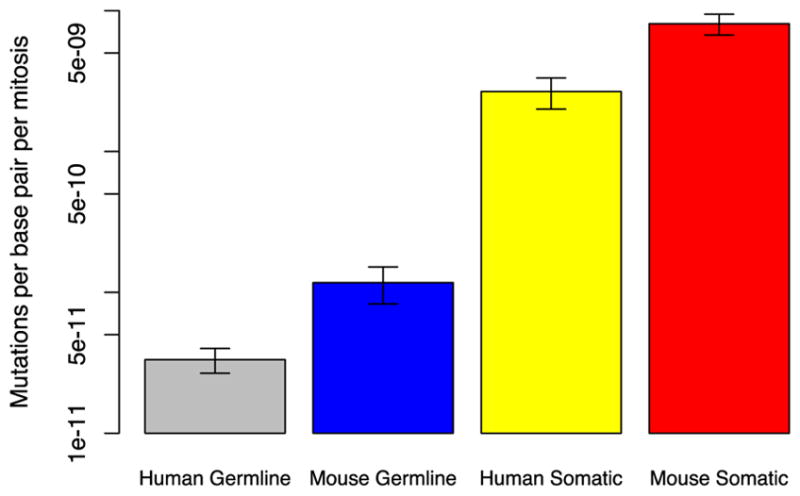
In humans and mice, germline mutation frequencies were measured by sequencing trios of parents and offspring, while somatic mutation frequencies were determined by sequencing single cells. Mutation rates were determined by dividing mutation frequencies observed by the estimated number of mitoses per generation. The heights of the bars reflect the median mutation rate (n=12, 8, 10, and 5) on a logarithmic scale. Error bars indicate +/−1 standard deviation. The somatic mutation rate was nearly two orders of magnitude higher than the germline mutation rate in both species; in mice, both the germline and somatic mutation rates were several times higher than their human equivalents. All differences were statistically significant (P<0.01, Wilcoxon test; after [51]).
When somatic mutation theories of ageing were first postulated in the 1950s by Failla [13] and Szilard [14], mutation rates were thought to be very low based on estimates of germline mutation rate in different species, which was well before next-generation sequencing allowed this to be assessed directly [49]. Hence, somatic mutation theories of ageing were almost immediately criticized for this reason, i.e. somatic mutation frequency was considered as too low to ever reach levels that could have functional consequences in ageing [52]. However, the results described above, coming from sequencing both single cells and clones derived from single cells indicate that the numbers of somatic mutations in cells can be as high as approximately 1,000 per genome (Table 1), and these are only base substitutions. Adding up small insertions and deletions (indels), copy number variations (CNVs), genome structural variations, and aneuploidies would yield levels of genome instability that could have functional consequences taking into account the significant age-related increases in somatic mutation frequencies that have been observed (see above). Application of methods such as SCMDA will soon reveal the entire landscape of somatic mutations in ageing tissues and organs of humans and experimental animals [53]. But how could a stochastic process of somatic mutation accumulation cause the functional changes that lead to ageing?
The simplest explanation as to how somatic mutations can lead to ageing phenotypes other than cancer is through loss of function of protein-coding genes. Indeed, in diploid yeast strains deletion of one gene is often enough to cause a growth disadvantage or a reduced replicative life span [54,55]. A major cause of ageing in yeast is the accumulation of circular forms of rDNA, which preferentially segregate to the mother during division and seem to exert their effect by interfering with cell growth [56]. Also, aged yeast cells display high rates of loss of heterozygosity, possibly due to impaired DNA double-strand break repair [57]. Base substitutions, however, are extremely rare in ageing yeast [58]. Mammals have much higher base substitution rates than yeast [11], and even the human germline, in spite of its much lower mutation rate than the soma, contains an average of 21 rare and several hundred common loss-of-function mutations (LOFs), i.e. genetic variants predicted to seriously disrupt the function of protein-coding genes [59]. This information was obtained by sequencing exomes of over 50,000 unrelated individuals. The rare LOFs are likely to have adverse phenotypic consequences and, indeed, as many as 3.5% of individuals harbored one or more variants deemed pathogenic [59]. Purifying selection ensures that such variants occur at very low frequency.
The above described LOFs were inherited and not a result of de novo mutations in the germline (although some could). However, of the approximately 60 de novo germline mutations in each newborn [12] as many as 10% are considered deleterious [49], probably mostly weakly deleterious. In contrast with germline mutations, de novo mutations in most somatic cells are much more difficult if not impossible to eliminate through selection. As we have seen above, a typical, mitotically active somatic cell from a young human individual can contain as many as 1,000 base substitutions. Of these base substitutions less than 10 are non-synonymous mutations in protein-coding sequences (Xiao Dong, unpublished). As mentioned above, mutation frequencies increase with age, possibly at least by a factor of two [34] essentially doubling the number of non-synonymous base substitutions. However, most of these mutations will either be not deleterious or only weakly deleterious. Hence, unless the number of small indels (which are most likely to cause loss of function) is surprisingly high, it seems unlikely that small deleterious effects on cell fitness due to non-synonymous mutations in the protein-coding part of the somatic genome can be a major cause of ageing.
In addition to mutations directly affecting proteins, the far majority of base substitutions affect parts of the genome that are not coding for proteins. While most of those mutations will have no effect at all, a fair number of the mare bound to affect the part of the genome that is involved in gene regulation. Indeed, instead of protein-coding gene sequences, the most likely targets for somatic mutations are DNA sequences involved in regulating gene activity. The human genome contains millions of gene regulatory sequences, most of which are distal, non-promoter sequences. The importance of these sequences is illustrated by the observation that the far majority of disease associations involve regulatory DNA, mostly enhancer sequences marked by DNase I hypersensitivity [60]. Enhancers drive the specific expression patterns of the several hundred cell types in a human body. They do that by serving as the specific DNA motifs that bind transcription factors, which recruit co-regulators to ultimately activate transcription. In each cell type or tissue, as many as 300 different transcription factors can be expressed [61]. However, the number of active enhancers in a given cell type has been predicted to be well over 10,000 [62], reflecting the promiscuous character of transcription factors in enhancer utilization. Other transcription factor binding sites are in promoters, silencers, insulators, and locus control regions. Hence, at a mutation load of approximately 1,000 base substitutions in a typical cell enhancer regions are more likely to be hit than protein-coding regions, which make up approximately 1–2% of the genome. This could affect normal patterns of gene expression and, for example, explain the observed age-related increase in transcriptional noise in mouse heart [63] and CD4+ T cells [64].
Conclusions and future directions
Driven by advances in genome technology, we have now reached a stage where for the first time it has become possible to directly test one of the oldest theories of ageing: the somatic mutation theory as first postulated by Failla [13] and Szilard [14]. Based on the first information on the actual numbers of base substitutions in a typical somatic cell and the observed age-related increase of various types of mutations, it is no longer reasonable to consider any age-related adverse effects of somatic mutagenesis limited to only cancer [65]. With the disposable nature of the ageing genome underscored by the dramatically higher frequency of mutations in the soma as compared with the germline (Table 1) [51,66,67,68], the observed base substitution loads in a typical single cell, in combination with as yet not quantified other types of mutations, such as CNV and genome structural variation, suggest that somatic mutations can adversely affect normal gene expression.
Importantly, the high level of integration of the many sequence features that encode specific cellular functions will amplify effects of multiple random mutations. Ironically, this same highly distributed functional organization of the genome provides it with robustness and a very high level of redundancy. This means that most changes caused by mutations will be mild and only gradually become more severe. Hence, in this respect somatic mutation accumulation could explain ageing very well. Indeed, ageing is not an all or nothing process but a gradual erosion of bodily functions. Meanwhile, it goes without saying that ageing is highly unlikely to have a single cause. Indeed, other effects of DNA damage, such as transcription interference and adverse cellular responses to DNA damage, such as apoptosis and cellular senescence, as well as changes at the protein level, are likely to also play major roles as cell autonomous mechanisms of ageing.
How to test a causal role of DNA mutations in ageing? A seemingly straightforward way is to experimentally increase mutagenesis either through treatment with mutagenic agents or by engineering defects that lead to increased mutation loads. Treatments with mutagenic agents, such as ionizing radiation, have since long been associated with premature ageing [69]. However, mutagenic agents do not exclusively cause mutations and lead to many other types of damage. Also DNA repair defective humans and mice have been studied for premature ageing and increased levels of mutations were observed sometimes but not always [27]. A problem with these studies is that DNA repair defects can accelerate DNA damage-induced cell death much more dramatically than mutation accumulation and never completely mimic the normal ageing process.
One way of testing for the possible functional effects of accumulating DNA mutations is through computational modeling. Once the complete tissue-specific landscape of somatic mutagenesis is known for a representative number of cells of a given type, it should be possible to map the mutation load of these cells onto a functional gene regulatory network specific for that particular cell type. This may allow accurate predictions of deleterious effects on cell function and the probability of functional loss for the tissue overall.
Summary.
DNA mutations are the substrate of evolution by natural selection and, therefore, an essential component of life.
In multicellular organisms, there is a decline in the force of natural selection to promote genome maintenance for long periods after the time of first reproduction.
Mutations, unlike DNA damage, cannot be repaired and their accumulation during ageing is both inevitable and irreversible.
Various types of mutations, from base substitutions to chromosomal aberrations have been found to accumulate with age.
Next-generation sequencing-based methods have become available to accurately determine the landscape of somatic mutations in different organs and tissues of humans and animals during ageing.
The somatic mutation rate is much higher than the germline mutation rate.
The first data on the load of base substitutions in a typical somatic cell suggest that mutation accumulation during ageing could affect both the protein-coding and the gene regulatory part of the genome.
Acknowledgments
Funding
Research in the Vijg lab was supported by grants from US National Institutes of Health [grant numbers AG017242, CA180126, AG047200, and AG038072]; and the Glenn Foundation for Medical Research.
Abbreviations
- CNV
copy number variation
- FISH
fluorescence in situ hybridization
- HGPRT
hypoxanthine-guanine phosphoribosyltransferase
- IVF
in vitro fertilization
- LOF
loss-of-function mutation
- MDA
multiple displacement amplification
- SCMDA
single-cell multiple displacement amplification
- SNP
single nucleotide polymorphism
- TCGA
The Cancer Genome Atlas
- WGA
whole genome amplification
Footnotes
Competing Interests
J.V., X.D., and L.Z. are three of the founders of SingulOmics Corp.
References
- 1.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 2.de Duve C. The onset of selection. Nature. 2005;433:581–582. doi: 10.1038/433581a. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 4.Beerman I, Seita J, Inlay MA, Weissman IL, Rossi DJ. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell. 2014;15:37–50. doi: 10.1016/j.stem.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol. 2004;6:168–170. doi: 10.1038/ncb1095. [DOI] [PubMed] [Google Scholar]
- 6.Collins AR, Cadet J, Moller L, Poulsen HE, Vina J. Are we sure we know how to measure 8-oxo-7,8-dihydroguanine in DNA from human cells? Arch Biochem Biophys. 2004;423:57–65. doi: 10.1016/j.abb.2003.12.022. [DOI] [PubMed] [Google Scholar]
- 7.Kimura M. Optimum mutation rate and degree of dominance as determined by the principle of minimum genetic load. J Genet. 1960;57:21–34. [Google Scholar]
- 8.Baer CF, Miyamoto MM, Denver DR. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. 2007;8:619–631. doi: 10.1038/nrg2158. [DOI] [PubMed] [Google Scholar]
- 9.Lynch M, Ackerman MS, Gout JF, Long H, Sung W, Thomas WK, et al. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet. 2016;17:704–714. doi: 10.1038/nrg.2016.104. [DOI] [PubMed] [Google Scholar]
- 10.Sturtevant AH. Essays on evolution. I. On the effects of selection on mutation rate. Quart Rev Biol. 1937;12:464–476. [Google Scholar]
- 11.Lynch M. Evolution of the mutation rate. Trends Genet. 2010;26:345–52. doi: 10.1016/j.tig.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475. doi: 10.1038/nature11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Failla G. The aging process and carcinogenesis. Ann NY Acad Sci. 1958;71:1124–1135. doi: 10.1111/j.1749-6632.1958.tb46828.x. [DOI] [PubMed] [Google Scholar]
- 14.Szilard L. On the nature of the aging process. Proc Natl Acad Sci USA. 1959;45:30–45. doi: 10.1073/pnas.45.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferguson-Smith MA. History and evolution of cytogenetics. Mol Cytogenet. 2015;8:19. doi: 10.1186/s13039-015-0125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramsey MJ, Moore DH, II, Briner JF, Lee DA, Olsen L, Senft JR, et al. The effects of age and lifestyle factors on the accumulation of cytogenetic damage as measured by chromosome painting. Mutat Res. 1995;338:95–106. doi: 10.1016/0921-8734(95)00015-x. [DOI] [PubMed] [Google Scholar]
- 17.Tucker JD, Spruill MD, Ramsey MJ, Director AD, Nath J. Frequency of spontaneous chromosome aberrations in mice: effects of age. Mutat Res. 1999;425:135–141. doi: 10.1016/s0027-5107(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 18.Faggioli F, Vijg J, Montagna C. Four-color FISH for the detection of low-level aneuploidy in interphase cells. Methods Mol Biol. 2014;1136:291–305. doi: 10.1007/978-1-4939-0329-0_14. [DOI] [PubMed] [Google Scholar]
- 19.Yurov YB, Iourov IY, Vorsanova SG, Liehr T, Kolotii AD, Kutsev SI, et al. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS ONE. 2007;2:e558. doi: 10.1371/journal.pone.0000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci USA. 2001;98:13361–13366. doi: 10.1073/pnas.231487398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanneste E, Voet T, Le Caignec C, Ampe M, Konings P, Melotte C, et al. Chromosome instability is common in human cleavage-stage embryos. Nat Med. 2009;15:577–583. doi: 10.1038/nm.1924. [DOI] [PubMed] [Google Scholar]
- 22.Rehen SK, Yung YC, McCreight MP, Kaushal D, Yang AH, Almeida BS, et al. Constitutional aneuploidy in the normal human brain. J Neurosci. 2005;25:2176–2180. doi: 10.1523/JNEUROSCI.4560-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faggioli F, Wang T, Vijg J, Montagna C. Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Hum Mol Genet. 2012;21:5246–5253. doi: 10.1093/hmg/dds375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van den Bos H, Spierings DC, Taudt AS, Bakker B, Porubsky D, Falconer E, et al. Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol. 2016;17:116. doi: 10.1186/s13059-016-0976-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knouse KA, Wu J, Whittaker CA, Amon A. Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc Natl Acad Sci USA. 2014;111:13409–13414. doi: 10.1073/pnas.1415287111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vijg J. Somatic mutations and aging: a re-evaluation. Mutat Res. 2000;447:117–135. doi: 10.1016/s0027-5107(99)00202-x. [DOI] [PubMed] [Google Scholar]
- 27.Vijg J. Aging of the Genome. Oxford; New York: 2007. [Google Scholar]
- 28.Albertini RJ, Castle KL, Borcherding WR. T-cell cloning to detect the mutant 6-thioguanine-resistant lymphocytes present in human peripheral blood. Proc Natl Acad Sci USA. 1982;79:6617–6621. doi: 10.1073/pnas.79.21.6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones IM, Thomas CB, Tucker B, Thompson CL, Pleshanov P, Vorobtsova I, et al. Impact of age and environment on somatic mutation at the hprt gene of T lymphocytes in humans. Mutat Res. 1995;338:129–139. doi: 10.1016/0921-8734(95)00018-2. [DOI] [PubMed] [Google Scholar]
- 30.Grist SA, McCarron M, Kutlaca A, Turner DR, Morley AA. In vivo human somatic mutation: frequency and spectrum with age. Mutat Res. 1992;266:189–196. doi: 10.1016/0027-5107(92)90186-6. [DOI] [PubMed] [Google Scholar]
- 31.Gossen JA, de Leeuw WJ, Tan CH, Zwarthoff EC, Berends F, Lohman PH, et al. Efficient rescue of integrated shuttle vectors from transgenic mice: a model for studying mutations in vivo. Proc Natl Acad Sci USA. 1989;86:7971–7975. doi: 10.1073/pnas.86.20.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Garcia AM, Derventzi A, Busuttil R, Calder RB, Perez E, Jr, Chadwell L, et al. A model system for analyzing somatic mutations in Drosophila melanogaster. Nat Methods. 2007;4:401–403. doi: 10.1038/NMETH1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dollé ME, Giese H, Hopkins CL, Martus HJ, Hausdorff JM, Vijg J. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat Genet. 1997;17:431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- 34.Dollé ME, Snyder WK, Gossen JA, Lohman PH, Vijg J. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc Natl Acad Sci USA. 2000;97:8403–8408. doi: 10.1073/pnas.97.15.8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dollé ME, Snyder WK, Dunson DB, Vijg J. Mutational fingerprints of aging. Nucleic Acids Res. 2002;30:545–549. doi: 10.1093/nar/30.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ono T, Ikehata H, Nakamura S, Saito Y, Hosoi Y, Takai Y, et al. Age-associated increase of spontaneous mutant frequency and molecular nature of mutation in newborn and old lacZ-transgenic mouse. Mutat Res. 2000;447:165–177. doi: 10.1016/s0027-5107(99)00200-6. [DOI] [PubMed] [Google Scholar]
- 37.Garcia AM, Calder RB, Dolle ME, Lundell M, Kapahi P, Vijg J. Age- and temperature-dependent somatic mutation accumulation in Drosophila melanogaster. PLos Genet. 2010;6:e1000950. doi: 10.1371/journal.pgen.1000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milholland B, Auton A, Suh Y, Vijg J. Age-related somatic mutations in the cancer genome. Oncotarget. 2015;6:24627–24635. doi: 10.18632/oncotarget.5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Cecco M, Criscione SW, Peterson AL, Neretti N, Sedivy JM, Kreiling JA. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging. 2013;5:867–883. doi: 10.18632/aging.100621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coolbaugh-Murphy MI, Xu J, Ramagli LS, Brown BW, Siciliano MJ. Microsatellite instability (MSI) increases with age in normal somatic cells. Mech Ageing Dev. 2005;126:1051–1059. doi: 10.1016/j.mad.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 42.Aubert G, Lansdorp PM. Telomeres and aging. Physiol Rev. 2008;88:557–579. doi: 10.1152/physrev.00026.2007. [DOI] [PubMed] [Google Scholar]
- 43.Jacobs KB, Yeager M, Zhou W, Wacholder S, Wang Z, Rodriguez-Santiago B, et al. Detectable clonal mosaicism and its relationship to aging and cancer. Nat Genet. 2012;44:651–658. doi: 10.1038/ng.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, et al. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet. 2012;44:642–650. doi: 10.1038/ng.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–264. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 2014;513:422–425. doi: 10.1038/nature13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong X, Zhang L, Milholland B, Lee M, Maslov AY, Wang T, et al. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat Methods. 2017;14:491–493. doi: 10.1038/nmeth.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keightley PD. Rates and fitness consequences of new mutations in humans. Genetics. 2012;190:295–304. doi: 10.1534/genetics.111.134668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kondrashov A. Genetics: the rate of human mutation. Nature. 2012;488:467–468. doi: 10.1038/488467a. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Fan HC, Behr B, Quake SR. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell. 2012;150:402–412. doi: 10.1016/j.cell.2012.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J. Differences between germline and somatic mutation rates in humans and mice. Nat Commun. 2017;8:15183. doi: 10.1038/ncomms15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maynard Smith J. A theory of ageing. Nature. 1959;184:956–957. [Google Scholar]
- 53.Gundry M, Vijg J. Direct mutation analysis by high-throughput sequencing: from germline to low-abundant, somatic variants. Mutat Res. 2012;729:1–15. doi: 10.1016/j.mrfmmm.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Delneri D, Hoyle DC, Gkargkas K, Cross EJ, Rash B, Zeef L, et al. Identification and characterization of high-flux-control genes of yeast through competition analyses in continuous cultures. Nat Genet. 2008;40:113–117. doi: 10.1038/ng.2007.49. [DOI] [PubMed] [Google Scholar]
- 55.McCormick MA, Delaney JR, Tsuchiya M, Tsuchiyama S, Shemorry A, Sim S, et al. A comprehensive analysis of replicative lifespan in 4,698 single-gene deletion strains uncovers conserved mechanisms of aging. Cell Metab. 2015;22:895–906. doi: 10.1016/j.cmet.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sinclair DA, Guarente L. Extrachromosomal rDNA circles–a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 57.McMurray MA, Gottschling DE. An age-induced switch to a hyper-recombinational state. Science. 2003;301:1908–1911. doi: 10.1126/science.1087706. [DOI] [PubMed] [Google Scholar]
- 58.Kaya A, Lobanov AV, Gladyshev VN. Evidence that mutation accumulation does not cause aging in Saccharomyces cerevisiae. Aging Cell. 2015;14:366–371. doi: 10.1111/acel.12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354:aaf6814. doi: 10.1126/science.aaf6814. [DOI] [PubMed] [Google Scholar]
- 60.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. A census of human transcription factors: function, expression and evolution. Nat Rev Genet. 2009;10:252–263. doi: 10.1038/nrg2538. [DOI] [PubMed] [Google Scholar]
- 62.Andersson R, Gebhard C, Miguel-Escalada I, Hoof I, Bornholdt J, Boyd M, et al. An atlas of active enhancers across human cell types and tissues. Nature. 2014;507:455–461. doi: 10.1038/nature12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dollé MET, et al. Increased cell-to-cell variation in gene expression in aging mouse heart. Nature. 2006;441:1011–1014. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- 64.Martinez-Jimenez CP, Eling N, Chen HC, Vallejos CA, Kolodziejczyk AA, Connor F, et al. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science. 2017;355:1433–1436. doi: 10.1126/science.aah4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vijg J. Somatic mutations, genome mosaicism, cancer and aging. Curr Opin Genet Dev. 2014;26:141–149. doi: 10.1016/j.gde.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Conrad DF, Keebler JE, DePristo MA, Lindsay SJ, Zhang Y, Casals F, et al. Variation in genome-wide mutation rates within and between human families. Nat Genet. 2011;43:712–714. doi: 10.1038/ng.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Uchimura A, Higuchi M, Minakuchi Y, Ohno M, Toyoda A, Fujiyama A, et al. Germline mutation rates and the long-term phenotypic effects of mutation accumulation in wild-type laboratory mice and mutator mice. Genome Res. 2015;25:1125–1134. doi: 10.1101/gr.186148.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adewoye AB, Lindsay SJ, Dubrova YE, Hurles ME. The genome-wide effects of ionizing radiation on mutation induction in the mammalian germline. Nat Commun. 2015;6:6684. doi: 10.1038/ncomms7684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Upton AC. Ionizing radiation and aging. Gerontologia. 1960;4:162–176. doi: 10.1159/000211008. [DOI] [PubMed] [Google Scholar]