

Abstract
Fragile X syndrome (FXS) is the most common inherited cause of intellectual disabilities and a leading cause of autism. FXS is caused by a trinucleotide expansion in the gene FMR1 on the X chromosome. The neuroanatomical hallmark of FXS is an overabundance of immature dendritic spines, a factor thought to underlie synaptic dysfunction and impaired cognition. We showed that aberrantly increased activity of the Rho GTPase Rac1 inhibited the actin-depolymerizing factor cofilin, a major determinant of dendritic spine structure, and caused disease- associated spine abnormalities in the somatosensory cortex of FXS model mice. Increased cofilin phosphorylation and actin polymerization coincided with abnormal dendritic spines and impaired synaptic maturation. Viral delivery of a constitutively active cofilin mutant (cofilinS3A) into the somatosensory cortex of Fmr1-deficient mice rescued the immature dendritic spine phenotype and increased spine density. Inhibition of the Rac1 effector PAK1 with a small-molecule inhibitor rescued cofilin signaling in FXS mice, indicating a causal relationship between PAK1 and cofilin signaling. PAK1 inhibition rescued synaptic signaling (specifically the synaptic ratio of NMDA/AMPA in layer V pyramidal neurons) and improved sensory processing in FXS mice. These findings suggest a causal relationship between increased Rac1-cofilin signaling, synaptic defects, and impaired sensory processing in FXS and uncover a previously unappreciated role for impaired Rac1-cofilin signaling in the aberrant spine morphology and spine density associated with FXS.
INTRODUCTION
Fragile X syndrome (FXS) is the most common heritable form of intellectual disabilities and a leading cause of autism (1-3). The primary cause of FXS is an unstable trinucleotide CGG repeat expansion of the fragile X mental retardation 1 (FMR1) gene, leading to its hypermethylation and transcriptional silencing (4, 5). This results in the loss of the fragile X mental retardation protein (FMRP), an mRNA- binding protein that tightly controls the localization, stabilization, and translation of many mRNAs including those critical to neuronal development, plasticity, and dendritic spine architecture (6). FXS causes a complex and debilitating neurological phenotype including impaired cognition, social/language deficits, increased susceptibility to seizures, attentional deficits, hyperactivity, sleep disturbances, motor incoordination, and hypersensitivity to sensory stimuli (2, 7, 8). The neuroanatomical hallmark of FXS is an increase in the density of dendritic spines, which exhibit an immature (long and thin-shaped) morphology (9).
The Fmr1 knockout (KO) mouse is a well-established model of FXS that displays autistic-relevant behaviors, impaired cognition, sensory hypersensitivity, and aberrant spine morphology and density (10) in the brain, including the somatosensory cortex (11). The somatosensory cortex has been an area of intense study because of precisely coordinated events during its critical period that allow for proper development of spines, neurocortical circuitry, and synaptic plasticity (12, 13). It is thought that deficits in sensory processing, which arise in early childhood, occur as a result of altered developmental milestones during the critical period (14-16). Critical period defects have been observed in the developing somatosensory cortex of Fmr1 KO mice. For instance, an overabundance of filopodial protrusions, indicative of spine immaturity, is observed in 1-week-old Fmr1 KO dendrites of layer V pyramidal neurons in somatosensory cortex, which normalized at the close of the critical period (17). An increased proportion of silent synapses, indicative of immature glutamatergic synapses, was observed on 1-week-old Fmr1 KO thalamocortical projection neurons in layer IV of somatosensory cortex, which also normalized at the close of the critical period (18). Although abnormal spines and synaptic defects in the developing somatosensory cortex of Fmr1 KO mice have been observed, the molecular mechanisms underlying these phenotypes remain unknown.
Actin is the most abundant cytoskeletal protein in dendritic spines (19-21). Actin-binding proteins play a pivotal role in spine morphogenesis, synapse formation/elimination, and synaptic plasticity (22). Cofilin is an actin-depolymerizing factor that severs the filamentous actin (F-actin) at its pointed ends to regulate spine morphogenesis and synaptic plasticity (23, 24). Although cofilin typically severs F-actin and thereby blocks spine growth and maturation (25), it also promotes spine maturation (26) and reduces spine density (27). Upon phosphorylation at its Ser3 residue, cofilin becomes inactive and can no longer bind and sever F-actin (28, 29), whereby cofilin inactivation has been linked to spine immaturity (30). The Rho family of small guanosine triphosphatases (GTPases), which consist of Rac1, Cdc42, and RhoA, can promote cofilin phosphorylation and inactivation (31). Through its effector p21-activated kinase 1 (PAK1), Rac1 induces the phosphorylation of cofilin to promote actin polymerization and spine remodeling. However, whether Rac1-to-cofilin signaling is dysregulated in FXS and causally related to abnormal dendritic spines, synaptic dysfunction, and aberrant behaviors in a mouse model of FXS has not been investigated.
The present study was undertaken to examine a possible role for cofilin in the synaptic defects associated with FXS. Here, we show that Rac1-PAK1 signaling is increased in the somatosensory cortex of Fmr1 KO mice. Overexpression of constitutively active cofilin corrects aberrant spine morphology and density in the somatosensory cortex of Fmr1 KO mice. PAK inhibition corrects impaired cofilin signaling, glutamatergic signaling, and sensory processing in Fmr1 KO mice in vivo. These findings implicate dysregulation of Rac1-PAK1-cofilin signaling with dendritic spine defects, synaptic dysfunction, and impaired sensory processing associated with FXS.
RESULTS
Increased cofilin phosphorylation and actin polymerization coincide with FXS-associated spine defects
Cofilin plays a pivotal role in the regulation of spine morphology and density (26,32). We first examined the abundance of cofilin and its phosphorylation status at Ser3, a biochemical indicator of cofilin inactivity (28,29), in the somatosensory cortex of Fmr1 KO mice. We focused on the somatosensory cortex, because it affords a valuable system in which to investigate the formation of topographic maps and their plasticity during critical periods in development (12,13). Phosphorylation of cofilin at Ser3 was increased in whole-cell lysates (Fig. 1, A and B, and table S1) and isolated synaptosomes (fig. S1) of the somatosensory cortex of Fmr1 KO mice at 1 but not 4 weeks of age relative to control littermates (Fig. 1, C and D, and table S1). Total cofilin abundance did not differ significantly in Fmr1 KO versus wild-type mice at any age examined. Thus, the abundance of cofilin phosphorylated at Ser3 is increased, indicative of inactivation, in the somatosensory cortex of Fmr1 KO mice at 1 week of age, a critical window of development associated with marked synaptogenesis.
Fig. 1. Increased cofilin phosphorylation and actin polymerization coincide with spine defects in the developing somatosensory cortex of Fmr1 KO mice.
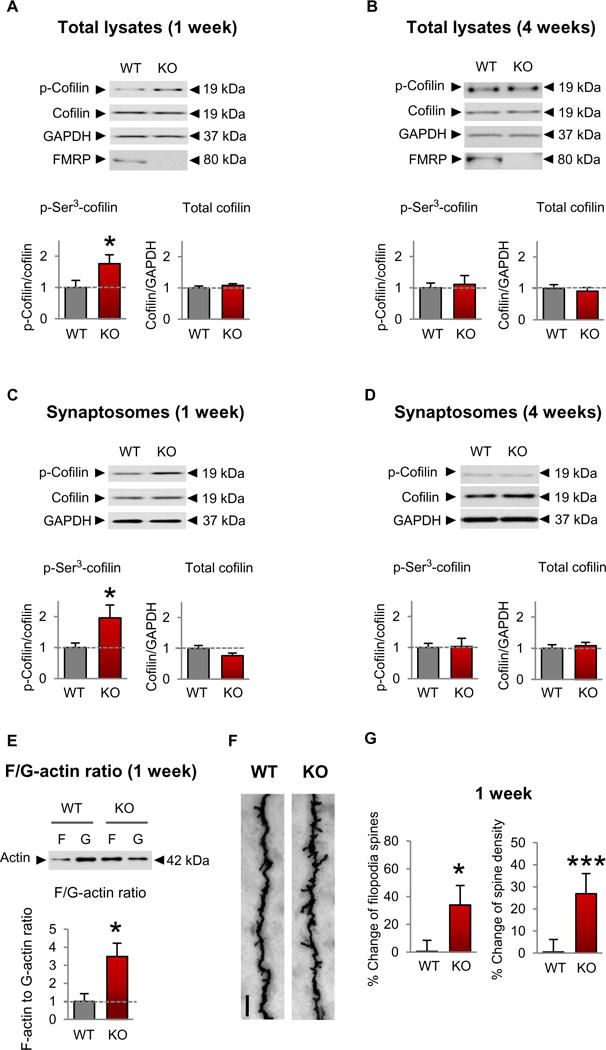
(A to D) Representative Western blots (top) and summary data (bottom) assessing the abundance of phosphorylated (p-) and total cofilin in lysates (A and B) and synaptosome fractions (C and D) of the somatosensory cortex of male Fmr1 KO and wild-type (WT) mice at 1 (A and C) and 4 (B and D) weeks of age. WT, n ≥ 12; KO, n ≥ 9 mice. (E) Representative Western blots (top) and summary data (bottom) for F-actin and G-actin in male Fmr1 KO animals and control littermates (WT, n = 5; KO, n = 6). (F) Representative Golgi staining images of the apical dendrites of layer V pyramidal neurons from WT (left) and Fmr1 KO (right) somatosensory cortices isolated from 1-week-old male mice. Scale bar, 5 μm. (G) Summary data showing percent change of immature spines and dendritic spine density (WT, n = 3 animals and 12 dendrites; KO, n = 3 animals and 13 dendrites). Data are means ± SEM. *P < 0.05, ***P < 0.001 (see table S1 for further details of the statistical tests).
Because phosphorylation status does not necessarily predict function, we used a functional assay of cofilin activity. Toward this end, we assessed the F-actin/G-actin (globular actin) ratio, which reflects the balance between actin polymerization and depolymerization, in somatosensory synaptosomes of 1-week-old Fmr1 KO mice and wildtype littermates. We reasoned that inactivation of cofilin would lead to an increase in actin polymerization and thus a higher F-actin/G-actin ratio in synaptosomes from the somatosensory cortex of 1-week-old Fmr1 KO than in those from wild-type littermates. Fmr1 KO mice exhibited a threefold increase in the F-actin/G-actin ratio relative to that of wild-type mice (Fig. 1E and table S1), indicative of a shift toward increased actin polymerization. These findings suggest that cofilin may be unable to depolymerize the actin cytoskeleton, and as a result, actin polymerization is increased.
We next assessed whether elevated cofilin phosphorylation and actin polymerization coincided with abnormal spine structure and density. We subjected brain slices at the level of the somatosensory cortex to Golgi impregnation and assessed spine structure and density in layer V pyramidal neurons at 1 week of age. We found a small, but significant, increase in spine density and an increase in long, spindly filopodial-type protrusions (Fig. 1, F and G, and table S1), a mark of immature spine morphology, in Fmr1 KO mice relative to wild-type littermates. Collectively, these findings show that aberrant cofilin phosphorylation coincides with elevated actin polymerization, which may give rise to spine defects in the somatosensory cortex of young Fmr1 KO mice.
Rac1 signaling is increased in Fmr1 KO mice
Cofilin is a central convergence point of signaling pathways, whereby kinases and phosphatases can modulate cofilin activity and thus the structure of the actin cytoskeleton (33-35). The actin cytoskeletal regulator and Rho GTPase, Rac1, plays a pivotal role in modulating cofilin activity and spine architecture, and its dysregulation is implicated in Fmr1 KO (36-39). Thus, we examined Rac1 activity in the somatosensory cortex of 1-week-old Fmr1 KO mice. Rac1-GTPase activity was increased in the somatosensory cortex of Fmr1 KO mice relative to wild-type littermates (Fig. 2A and table S2), with little or no detectable difference in total Rac1 abundance.
Fig. 2. Rac1 signaling is increased in the somatosensory cortex of FXS mice.
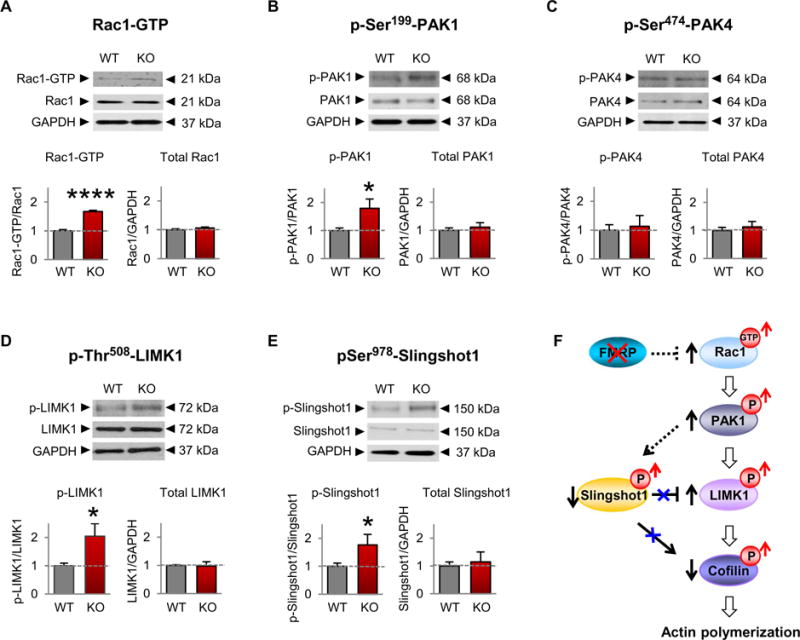
(A to E) Representative Western blotting of proteins in the Rac1 signaling pathway in somatosensory lysates (A) or synaptosomes (B to E) from 1-week-old male WT and Fmr1 KO mice (WT, n = 4 to 12;KO, n = 4 to 15). Data are means ± SEM. *P < 0.05, ****P <0.0001. (F) Model depicting a mechanism by which loss of FMRP leads to increased Rac1 signaling, cofilin phosphorylation (a measure of inactivation), and actin polymerization.
The PAK proteins are critical effectors that link the Rho family of GTPases to cytoskeleton remodeling (40). PAK1 serves as a target for the small guanosine triphosphate (GTP)–binding protein Rac1 and is thought to play a role in cell motility and cell morphology (41). However, whether PAK activity is dysregulated in FXS and affects cofilin phosphorylation and activity is currently unknown. We examined PAK1 activity in the somatosensory cortex of Fmr1 KO mice. Phosphorylation of PAK1 at Ser199, a phosphosite that undergoes autophosphorylation upon PAK1 activation (40), was greater in Fmr1 KO than in wild-type mice (Fig. 2B and table S2), with little or no difference in total PAK1 abundance. In contrast, phosphorylation of the Cdc42 effector PAK4 at Ser474, a site within its kinase domain (40), was unaltered in Fmr1 KO versus wild-type mice (Fig. 2C and table S2), consistent with increased Rac1-PAK1-cofilin signaling in FXS.
The small Rho-GTPase Rac1, through its effector PAK1, phosphorylates and activates the serine/threonine kinase Lim kinase 1 (LIMK1) and phosphorylates and inactivates the phosphatase Slingshot1 (SSH1), thereby promoting phosphorylation and inactivation of cofilin (22). We first examined the abundance and phosphorylation status of LIMK1. Upon phosphorylation at Thr508, LIMK1 is activated and modulates the actin cytoskeleton by promoting the phosphorylation and inactivation of its major substrate cofilin (28, 29). Phosphorylation of LIMK1 at Thr508 was increased in synaptosomes isolated from the somatosensory cortex of Fmr1 KO compared with those from wildtype mice (Fig. 2D and table S2), with no significant difference in total LIMK1 abundance. These findings indicate that increased phosphorylation of cofilin may be due, at least in part, to increased LIMK1 activity.
We next examined the abundance and phosphorylation of SSH1. Dephosphorylated/active SSH1 exhibits dual activity in that it dephosphorylates and activates cofilin and, in parallel, dephosphorylates and inactivates LIMK1. This releases the brake imposed by LIMK1 on cofilin (42). However, phosphorylation of SSH1 at Ser978 causes inactivation such that SSH1 can no longer dephosphorylate and inactivate LIMK1 or phosphorylate and activate cofilin (34). Phosphorylation of SSH1 at Ser978 was elevated in synaptosomes isolated from the somatosensory cortex of Fmr1 KO versus wild-type littermates at 1 week of age (Fig. 2E and table S2), with little or no difference in total SSH1 abundance. These findings are consistent with a model in which inactivation of cofilin may be a consequence of both overactivation of LIMK1 and inactivation of SSH1. Together, these findings document elevated Rac1-PAK1-cofilin signaling in a mouse model of FXS, which may cause an imbalance in actin polymerization at the synapse (Fig. 2F).
Inhibition of PAK rescues cofilin signaling and actin polymerization in Fmr1 KO mice
The findings thus far show that Rac1-PAK1 signaling, p-cofilin, and actin polymerization are increased in the somatosensory cortex of young Fmr1 KO mice but do not address a possible causal relationship between them. To address this issue, we treated Fmr1 KO mice with the small-molecule PAK inhibitor FRAX486, which is able to penetrate the blood-brain barrier (43), and examined the ability of PAK inhibition to correct aberrant cofilin signaling and actin polymerization. Administration of FRAX486 (single subcutaneous injection at 20 mg/kg for 8 hours) decreased the abundance of p-LIMK1 (Fig. 3A and table S3) and p-SSH1 (Fig. 3B and table S3) in the somatosensory cortex of 1-week-old Fmr1 KO mice relative to that of vehicle-treated KO mice, with no detectable effect on p-LIMK1 or p-SSH1 in wild-type mice. In addition, treatment with FRAX486 restored (reduced) the phosphorylation of cofilin (Fig. 3C and table S3) and the F-actin/G-actin ratio (Fig. 3D and table S3) in Fmr1 KO mice to near that of wild-type mice with little to no effect on cofilin or the F-actin/G-actin ratio in the corresponding region of wild-type mice. The lack of effect of FRAX486 on LIMK1-cofilin signaling in wildtype mice suggests a scenario whereby, in wild-type mice, LIMK-cofilin signaling is downstream not only of PAK1 but also of other kinases, such as ROCK1 (Rho-associated kinase 1) (44), which are not affected by FRAX486. Cofilin signaling did not differ detectably in vehicle-treated mice compared with untreated mice (fig. S2 and table S7). These data strongly suggest that increased Rac1-PAK1 activity causes increased cofilin phosphorylation and actin polymerization in the Fmr1 KO mouse (Fig. 3E).
Fig. 3. Inhibition of PAK restores cofilin signaling and actin polymerization in FXS model mice.
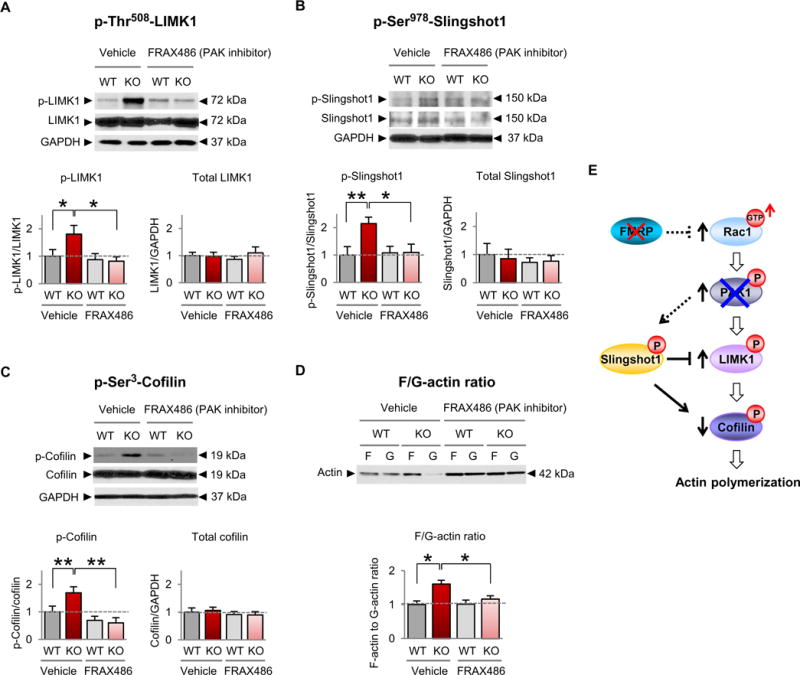
(A to D) Representative Western blots and summary data for p-Thr508-LIMK (A), p-Ser9780-Slinghot1 (B), p-Ser3-cofilin (C), and the F-actin/G-actin ratio (D) in somatosensory synaptosomes from 1-week-old male WT and Fmr1 KO mice given a single subcutaneous injection of FRAX486 or vehicle [20% (w/v) hydroxypropyl-β-cyclodextrin] 8 hours before sacrifice (WT vehicle, n ≥ 12; KO vehicle, n ≥ 10;WT FRAX486, n ≥ 8;KO FRAX486, n ≥ 9). We note that overall there was no significant difference in total actin abundance in synaptosomes from FRAX486- versus vehicle-treated mice. Data are means ± SEM. *P < 0.05, **P < 0.01. (E) Schematic of Rac-cofilin signaling depicting proposed mechanism by which inhibition of group 1 PAKs (PAK1, PAK2, and PAK3) restores the abundance of p-LIMK1, p-Slingshot1, p-cofilin, and actin polymerization in Fmr1 KO animals.
Adult Fmr1 KO mice exhibit normal Rac1-cofilin signaling
The results thus far show that LIMK-cofilin signaling is increased in total lysates and synaptosomes at 1 but not 4 weeks of age (Fig. 1, B and D), but they do not address signaling upstream or downstream of p-cofilin at 4 weeks of age. Rac1-GTPase, p-PAK1, p-LIMK1, and the F-actin/G-actin ratio were unaltered in samples of Fmr1 KO and wildtype littermates at 4 weeks of age (fig. S3, A to C and E, and table S8). By contrast, p-SSH1 was increased in synaptosomes of Fmr1 KO mice at 4 weeks of age (fig. S3D and table S8), consistent with the concept that SSH1 is phosphorylated by kinases other than PAK1, such as protein kinase D1 (34). By contrast, all members of the pathway (Rac1-GTP, p-PAK1, p-LIMK1, and p-SSH1) and the F-actin/G-actin ratio were comparable in the somatosensory cortex of Fmr1 KO and wildtype mice at 2 to 5 months of age (fig. S3, F to J, and table S8), and p-cofilin remained unaltered in Fmr1 KO mice relative to wild-type mice (fig. S4 and table S8). Thus, aberrant Rac1-cofilin signaling (with the exception of p-SSH1) is restricted to the critical period in the somatosensory cortex of Fmr1 KO mice.
CofilinS3A overexpression rescues spine defects in Fmr1 KO mice
The results thus far indicate that aberrant cofilin signaling in the somatosensory cortex coincides with defects in dendritic spines in Fmr1 KO mice, but do not address a causal relation between the two. We reasoned that if elevated actin polymerization (F-actin/G-actin ratio), due to cofilin inactivation, is driving the spine defects observed in the somatosensory cortex of Fmr1 KO mice during the critical period, then overexpression of a constitutively active nonphosphorylatable cofilin mutant in which the Ser3 residue is converted to an alanine (cofilinS3A) may depolymerize the actin cytoskeleton and correct spine morphology and spine density. To assess causality between increased abundance of p-cofilin and spine defects, we delivered green fluorescent protein (GFP)–tagged cofilinS3A (a constitutively active mutant), GFP-tagged wild-type cofilin, or GFP alone (negative control).
Before the delivery of cofilinS3A into the somatosensory cortex of Fmr1 KO and wild-type littermates, constructs were subcloned into a self-inactivating lentiviral vector, sequenced, and validated in cortical cultured neurons. To determine whether constitutively active cofilin could depolymerize the actin cytoskeleton, cofilinS3A was overexpressed in somatosensory cultured neurons at 4 to 5 days in vitro (DIV) and cultured for a further 5 to 7 days. The F-actin/G-actin ratio was significantly reduced in neurons overexpressing cofilinS3A relative to untreated cultures (Fig. 4, A and B, and table S4). In neurons overexpressing GFP, wild-type cofilin, or a constitutively inactive phosphomimetic cofilin mutant (cofilinS3D), the F-actin/G-actin ratio did not differ detectably from that of untreated cultures (Fig. 4B and table S4). These findings provide technical and biological validation that cofilinS3A depolymerizes the actin cytoskeleton in neurons when administered by means of the lentiviral expression system.
Fig. 4. Constitutively active cofilin rescues aberrant spine morphology and density In the somatosensory cortex of young Fmr1 KO mice.
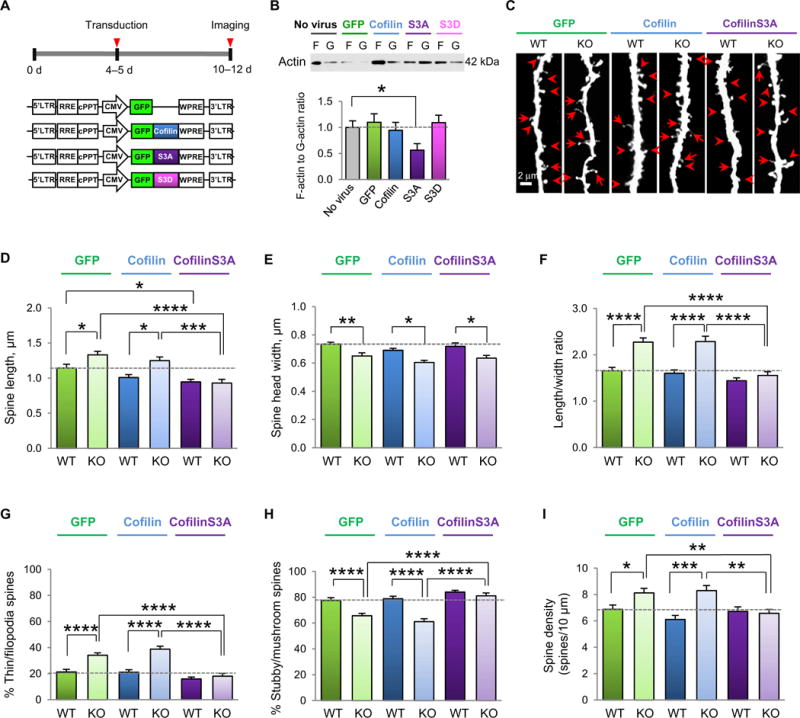
(A) Experimental timeline of viral application on cultured neurons or viral delivery and stereotaxic injection into the somatosensory cortex. (B) Representative Western blots and summary data of the F-actin/G-actin ratio in lysates from somatosensory cortical cultures (10 to 12 DIV) either unperturbed or infected with virus expressing GFP, WT cofilin, constitutively active coflinS3A, or phosphomimetic cofilinS3D (n = 13 to 15 culture wells per group from three independent experiments). (C) Representative fluorescent images assessing viral-mediated transduction of apical dendrites of layer V somatosensory cortex pyramidal neurons with GFP (left), GFP-WT cofilin (middle), and GFP-cofilinS3A (right) in male WT and Fmr1 KO mice at P10 to P12. Scale bar, 2 μm. Examples of mature spines (arrowheads) and immature protrusions (arrows) are indicated. (D to I) Summary data of average spine length (D), head width (E), spine length-to-width ratio (LWR) (F), % mature (stubby/mushroom) spines (G), % immature (thin/filopodia) spines (H), and spine density (I) of the samples imaged in (C) (n = 15 to 21 neurons pooled from 6 to 10 animals per group). Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We next assessed whether overexpression of cofilinS3A in the somatosensory cortex of Fmr1 KO mice in early postnatal life could correct aberrant dendritic spine morphology and density to wild-type levels. Lentiviral vector containing GFP-tagged cofilinS3A, GFP-tagged wild-type cofilin, or GFP alone was injected unilaterally into the somatosensory cortex of postnatal day 4 (P4) or P5 mice, followed by 5 to 7 days to allow for viral expression (Fig. 4C and table S4). Spine analysis was performed on layer V neurons of mice aged P10 to P12, a critical time windowwhen spine defects are prominent (17). Fmr1 KO neurons overexpressing GFP or wild-type cofilin showed a marked increase in average spine length relative to that of wild-type mice (Fig. 4, C and D, and table S4), indicative of an immature phenotype. In contrast, overexpression of wild-type cofilin had little or no effect on the F-actin/G-actin ratio or spine morphology in wild-type neurons (Fig. 4B), presumably because mature wild-type neurons normally exhibit short, stubby mature spines, indicative of normal actin depolymerization (26, 45). Overexpression of cofilinS3A modestly reduced the average spine length in wild-type neurons and rescued the increased spine length in Fmr1 KO neurons. Neurons from Fmr1 KO mice exhibited a marked decrease in average spine head width compared with those from wild-type mice (Fig. 4, C and E, and table S4). Whereas cofilinS3A did not alter spine head width, it rescued the increased spine length–to–head width ratio, a measure of spine immaturity, in Fmr1 KO neurons (Fig. 4, C and F, and table S4). This is consistent with the idea that actin-binding proteins located in the spine core away from the postsynaptic density (PSD), such as cortactin (46), can bind to cofilin independent of its Ser3 residue and presumably inhibit its depolymerizing activity in the spine core, thus not changing spine head shape. We next classified spines as mature (stubby and mushroom-shaped) and immature (long, thin, or spindly filopodial protrusions). Fmr1 KO neurons exhibited a marked increase in percent spindly or immature spines and a marked decrease in the percent mushroom or stubby spines, which were restored to wild-type levels by overexpression of cofilinS3A (Fig. 4, C, G, and H, and table S4). Last, Fmr1 KO neurons exhibited a small, but significant, increase in spine density relative to that of wild-type neurons (Fig. 4I and table S4), which was restored to wild-type levels by overexpression of cofilinS3A. Collectively, these findings document that inactivation of cofilin is causally linked to spine defects in the developing somatosensory cortex of Fmr1 KO mice during the critical period.
Altered glutamatergic signaling in Fmr1 KO mice is corrected by PAK inhibition
The results thus far demonstrate a causal relation between cofilin inactivation and dendritic spine defects in Fmr1 KO but do not address the functional consequences of elevated Rac1-PAK1-cofilin signaling in the somatosensory cortex of Fmr1 KO mice during the critical period. Excitatory intracortical circuitry in the somatosensory cortex develops primarily within the first 2 weeks of life (47). To investigate whether elevated Rac1-PAK1-cofilin signaling affects synaptic responses of intracortical synapses of layer V neurons, Fmr1 KO and wild-type littermates were treated with a single subcutaneous injection of the PAK inhibitor FRAX486 (20 mg/kg) or vehicle at P6 and again at P14. Voltage-clamp recordings were made from neurons at P7 or P15, respectively, in the presence of the γ-aminobutyric acid (GABA) inhibitor picrotoxin (50 μM) to isolate the excitatory postsynaptic current (EPSC). In vehicle-treated wild-type mice at P7, the N-methyl-D-aspartate (NMDA) component of synaptic transmission was small relative to the AMPA component (NMDA/AMPA ratio, 0.67 ± 0.06; Fig. 5, A and B, and table S5). In contrast, age-matched, vehicle-treated Fmr1 KO mice exhibited a significantly greater NMDA/AMPA ratio (1.53 ± 0.29; Fig. 5, A and B, and table S5). A single injection of the PAK inhibitor FRAX486 did not detectably alter the NMDA/AMPA ratio in wild-type animals (Fig. 5, A and B), but significantly reduced the NMDA/AMPA ratio in Fmr1 KO mice to a value indistinguishable from that of vehicle-treated wild-type animals.
Fig. 5. Inhibition of PAK corrects functional synaptic deficits in layer V of the somatosensory cortex.
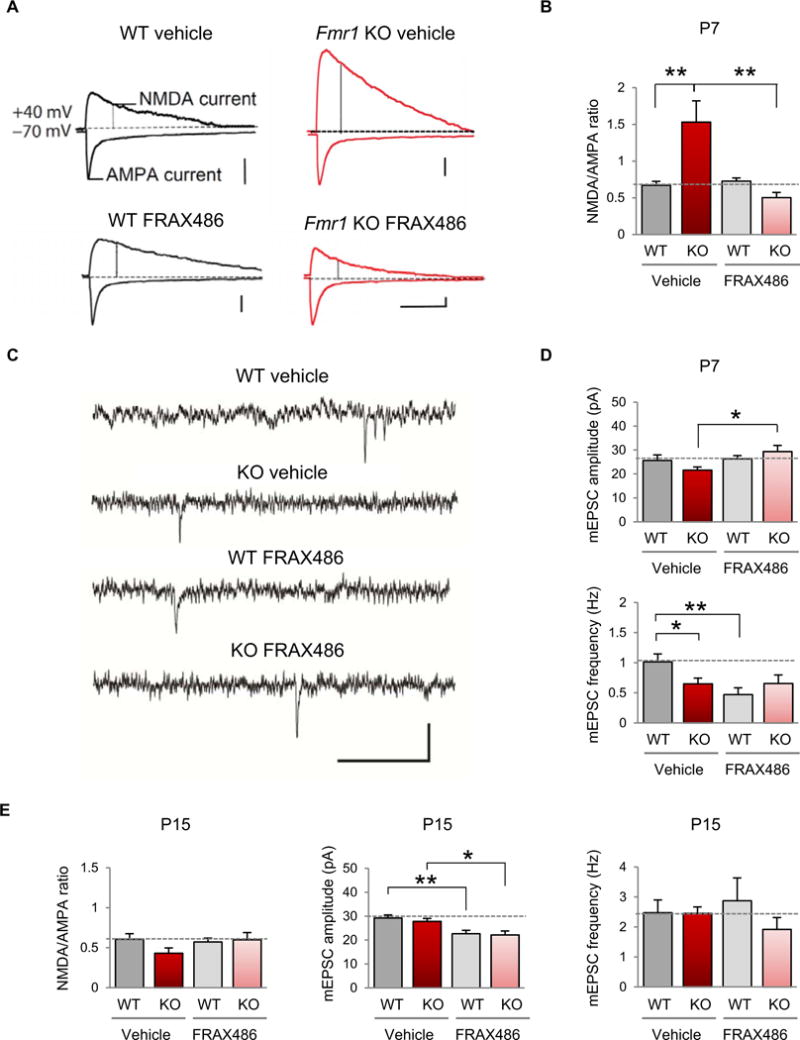
(A) Representative evoked EPSC recordings from male WT (left) and Fmr1 KO (right) mice, either vehicle-treated (top) or FRAX486-treated (bottom) at P7. AMPAR-mediated EPSCs were measured as the peak current at −70 mV, and the NMDA component was measured by depolarizing the cell to +40 mV and measuring the current 60 ms after the onset of the outward current in the presence of 50 μM picrotoxin. Calibration: 100 ms, 50 pA. (B) Summary data of the NMDA/AMPA ratio in all recordings (WT vehicle, n = 13; Fmr1 KO vehicle, n = 11; WT FRAX486, n = 11; Fmr1 KO FRAX486, n = 6). (C) Representative traces of mEPSC recordings from male WT and Fmr1 KO animals, vehicle- or FRAX486-treated at P7. Calibration: 500 ms, 50 pA. (D) Summary data of mEPSC amplitude and frequency in all recordings (WT vehicle, n = 10; Fmr1 KO vehicle, n = 10; WT FRAX486, n = 11; Fmr1 KO FRAX486, n = 9). (E) Summary data for all recordings at P15 (NMDA/AMPA ratio: WT vehicle, n = 15; Fmr1 KO vehicle, n = 7; WT FRAX486, n =11; Fmr1 KO FRAX486, n = 10; mEPSC amplitude and frequency: WT vehicle, n = 18; Fmr1 KO, n = 7; WT FRAX486, n = 16; Fmr1 KO FRAX486, n = 12). Data are means ± SEM. *P < 0.05, **P < 0.01.
To determine whether the alteration in the NMDA/AMPA ratio could be attributed to changes in the synaptic content of AMPA or NMDA receptors (NMDARs), we recorded miniature EPSCs (mEPSCs) in layer V neurons in the presence of picrotoxin and tetrodotoxin (TTX) (500 nM). The mEPSC amplitude was not significantly different between the vehicle-treated Fmr1 KO and wild-type littermate groups at P7, suggesting that there was no change in the AMPA content of synapses in KO animals. FRAX486 caused an increase in mEPSC amplitude in the KO animals, with little or no change in the mEPSC of wild-type mice. KO mice exhibited a reduction in mEPSC frequency relative to that of wild-type mice, which was not rescued by the administration of FRAX486. FRAX486-treated wild-type mice also had a significantly lower mEPSC frequency (Fig. 5, C and D, and table S5). These results demonstrate that the AMPA content of synapses is not altered in layer V neurons of Fmr1 KO mice; however, the reduced frequency of mEPSCs in the vehicle-treated KO could represent either a reduced release probability or postsynaptic processes such as loss of functional synapses, which are more prevalent in layer V neurons in Fmr1 KO mice.
We observed that Rac1-cofilin signaling is normalized after P14 in Fmr1 KO mice. Therefore, we assessed the NMDA/AMPA ratio in layer V in 2-week-old mice, a time when spine density is normal (17). At P15, the NMDA/AMPA ratio of vehicle-treated Fmr1 KO was not significantly different from that of FRAX486-treated Fmr1 KO mice or vehicle-treated wild-type littermates (Fig. 5E and table S5). The mEPSC amplitude and frequency did not differ detectably in KO and wild-type mice. However, the amplitude of mEPSCs was reduced in both drug-treated groups. It is possible that the reduction in mEPSC amplitude may be attributed to the distinct mechanisms of action of the drug on AMPA receptor (AMPAR) expression and synaptic targeting (48).
PAK inhibition rescues impaired sensory processing in Fmr1 KO mice
After the critical period of the barrel cortex is closed, Fmr1 KO mice exhibit a greater tactile response in the sensory cortex (49). Because the somatosensory cortex is critical for processing tactile information, we hypothesized that Fmr1 KO mice may not process tactile information properly. To test this, we used a whisker-dependent texture discrimination task (50). Three- to 4-week-old mice were used for this task (Fig. 6A and table S6), because after 3 to 4 weeks of age, the critical period closes, a period in which the hardwiring of synaptic circuits in the somatosensory cortex is generally established (18, 49, 51, 52). Wild-type mice could discriminate a novel texture (100-grit sandpaper of coarse texture) from a visually identical familiar textured object (80-grit sandpaper of fine texture; Fig. 6B and table S6), indicating normal sensory processing. In contrast, Fmr1 KO mice failed to discriminate a novel texture from a familiar texture (Fig. 6B and table S6), indicating impaired sensory processing. The texture discrimination task is dependent on sensory processing through intact mystacial vibrissae, given that wild-type mice with trimmed whiskers or introduced to “texture-less” objects (80- or 100-grit objects wrapped with parafilm) failed to discriminate the novel texture (Fig. 6, C and D, and table S6). Furthermore, the Fmr1 KO defect in texture discrimination was most likely not hippocampal-dependent, given that a 5-min retention time between the learning phase and the testing phase did not impair the ability of Fmr1 KO mice to distinguish a novel object in the novel object recognition task, which assesses hippocampal-dependent memory (Fig. 6E and table S6).
Fig. 6. Inhibition of PAK corrects impaired sensory processing in FXS model mice.
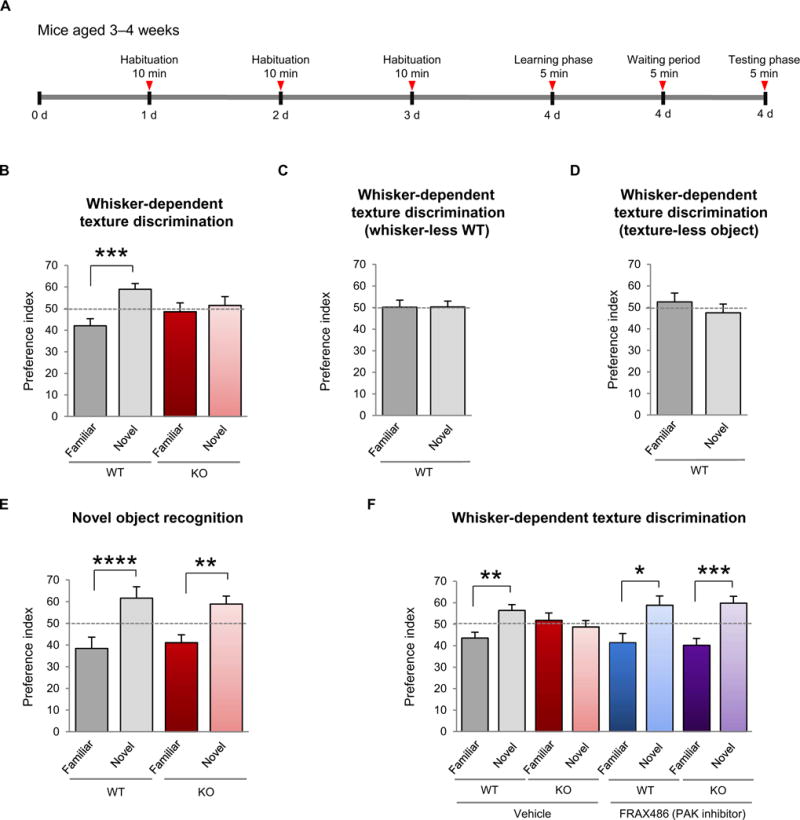
(A) Timeline of the whisker-dependent texture discrimination task. (B) Discrimination of a novel versus familiar texture in WT and Fmr1 KO mice, as assessed by a preference index at chance level indicating impaired sensory processing (WT, n = 13; KO, n = 7). (C and D) Discrimination of a novel texture versus a familiar texture WT mice subjected to (C) whisker trimming (n = 10 animals) or (D) a texture-less object (n = 7 animals), indicating the necessity for an intact mystacial vibrissae to discriminate textures. (E) Novel object recognition task in WT and Fmr1 KO mice (WT, n = 7; KO, n = 8 animals). (F) Sensory processing, as assessed by the ability to discriminate a novel texture versus a familiar texture, in Fmr1 KO animals subjected to chronic administration of FRAX486 (once at P7, once at P14, and once at 3 to 4 weeks of age 24 hours before testing) (WT vehicle, n = 15; KO vehicle, n = 9; WT FRAX486, n = 10;KO FRAX486, n =10). Data are means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Finally, we asked whether increased Rac1-PAK1-cofilin signaling caused the impaired sensory processing in Fmr1 KO mice. Wild-type and Fmr1 KO animals were given a single subcutaneous injection of the brain-penetrant PAK inhibitor FRAX486 or vehicle control once at P7, once at P14, and once at 3 to 4 weeks of age, 24 hours before the testing phase of the texture discrimination task. Vehicle-treated Fmr1 KO mice exhibited impaired sensory processing, which was rescued by FRAX486 (Fig. 6F and table S6). In separate control experiments, a single injection of FRAX486 at 3 to 4 weeks of age also rescued sensory processing, indicating that it is not essential to administer the inhibitor during the critical period. These findings link aberrant Rac1-PAK1-cofilin signaling to impaired sensory processing in FXS.
DISCUSSION
Here, we show that the actin-depolymerizing factor cofilin, a downstream target of the small Rho GTPase Rac1 and major determinant of dendritic spine structure, is dysregulated in the somatosensory cortex of young Fmr1 KO mice and causally related to spine abnormalities. We further show that Rac1-PAK1 signaling is elevated in the somatosensory cortex of Fmr1 KO mice and causally related to impaired cofilin signaling. Accordingly, Rac1-PAK1 signaling promotes phosphorylation and activation of LIMK1, an upstream inhibitor of cofilin, and in parallel promotes phosphorylation and inactivation of the phosphatase SSH1, an upstream activator of cofilin. This is significant in that LIMK1 phosphorylates and inactivates cofilin at Ser3, a highly conserved LIMK site, whereas SSH1 dephosphorylates and inactivates LIMK and dephosphorylates and activates cofilin at Ser3. These findings are consistent with a model whereby cofilin serves as a convergence point of signaling pathways that modulate the structure of the actin cytoskeleton (34). In the somatosensory cortex of wildtype mice, phosphorylation of cofilin at the LIMK site Ser3 is low and its activity is high. Depolymerization of actin by cofilin limits spine density and promotes maturation of spines. In the somatosensory cortex of Fmr1 KO mice, the translational brake imposed by FMRP is released, leading to elevated Rac1-PAK1 signaling. Accordingly, cofilin phosphorylation is high and its activity is low. This results in an imbalance in the actin cytoskeleton, which is shifted toward elevated actin polymerization (increased F-actin/G-actin ratio) and an immature spine phenotype. This, in turn, represses maturation of dendritic spines and allows spine numbers to go unchecked. The concept that increased Rac-PAK signaling coincides with increased spine density concurs with findings of others (53, 54). Constitutively active (nonphosphorylatable) cofilinS3A corrected aberrant spine morphology and density in the somatosensory cortex, indicating a causal relation between elevated cofilin phosphorylation and an overabundance of immature spines. These findings provide a novel and previously unappreciated mechanism, whereby FMRP regulates the activity of the actin-depolymerizing agent cofilin via aberrant Rac1-PAK1 signaling. When we were in the final stages of writing our paper, Kashima and colleagues (55) published a paper in which they showed that in fragile X mice, the abundance of the full-length isoform of bone morphogenetic protein type II receptor (BMPR2) is increased and activates LIMK1, which phosphorylates and inhibits cofilin to promote neurite outgrowth and dendritogenesis. Although not addressed by the paper, it is interesting to speculate that BMPR2 may activate LIMK via Rac1/PAK activation. In a sequel to this paper, Kashima and colleagues (56) go on to show that hyperactive locomotion in a Drosophila model of fragile X is a functional readout for the synaptic abnormalities (and increased BMPR2-LIMK signaling) associated with FXS.
Findings from the present study link aberrantly increased Rac1-PAK1 signaling to alterations in glutamatergic synaptic transmission. We show that the NMDA/AMPA ratio is increased at intracortical excitatory synapses in layer V of the somatosensory cortex of Fmr1 KO mice at P7. This is significant in that as glutamatergic synapses mature, activity-dependent incorporation of AMPARs increases, resulting in a reduction in the NMDA/AMPA ratio (57,58). Our finding that the NMDA/AMPA ratio is increased in Fmr1 KO mice at P7, but is normal at P15, is consistent with a delay in maturation of (rather than a permanent defect in) glutamatergic synapses in layer IV (18). Our finding of a reduction in mEPSC frequency, but not amplitude, at cortical synapses of Fmr1 KO mice early in development (P7) is consistent with the finding of elevated (NMDA only) “silent” synapses and a decrease in functional synapses at this age (57). The PAK inhibitor FRAX486 corrects the NMDA/AMPA ratio in layer V of Fmr1 KO mice to wild-type levels, indicating a causal relation between elevated Rac1-PAK1 signaling and aberrantly regulated glutamatergic transmission. This is significant in that cofilin drives AMPAR trafficking and synaptic incorporation during synaptic plasticity (48), a mechanism that may underlie the ability of FRAX486 to correct the NMDA/AMPAR ratio in Fmr1 KO mice. At the same time, the overabundance of immature filopodial protrusions observed in the somatosensory cortex of these FXS model mice may delay stabilization and maturation of synapses. Our finding that FRAX486 reduces the mEPSC frequency in wild-type animals suggests that PAK inhibition alters not only postsynaptic receptor dynamics and spine organization but also presynaptic release. An alternative interpretation is that PAK inhibition does not necessarily alter presynaptic release but rather the number of functional synapses.
Our finding that tactile learning, assessed by means of a whisker-dependent texture discrimination task (50), is impaired in Fmr1 KO mice is significant in that humans with FXS exhibit hypersensitivity to sensory stimuli such as touch (59, 60), and FXS model mice exhibit tactile hypersensitivity in response to whisker stimulation (49, 61). Tactile hypersensitivity may underlie the inability of Fmr1 KO mice to distinguish novel versus familiar textures. Our finding that activation of cofilin during the critical period corrects tactile hypersensitivity links aberrant Rac1-PAK1-cofilin signaling to impaired sensory processing in FXS model mice. However, our findings do not preclude the possibility that FRAX486 is acting independently of PAK. Previous studies show that pharmacological inhibition of PAK corrects spine density and hyperactive/repetitive behaviors (43). Moreover, crossing Fmr1 KO mice with mice expressing dominant-negative PAK corrects spine density, long-term potentiation at cortical synapses, and hyperactive/repetitive behaviors (62). Our study represents an advance over previous studies in that we demonstrate that activation of cofilin corrects spine morphology and number, and that PAK inhibition corrects aberrant cofilin signaling, actin polymerization (F-actin/G-actin ratio), NMDA/AMPA ratio, and a somatosensory cortex–specific behavior. Whereas our study shows rescue of the FXS phenotype in young mice at ages when the phenotype is strong, the earlier studies show rescue in adult mice, when the phenotype is thought to be more subtle.
In summary, the present study links aberrant Rac1-PAK-cofilin signaling to impaired spine morphology, synaptic transmission in layer V of the somatosensory cortex, and sensory perception in Fmr1 KO mice. These findings reveal a novel role for cofilin as a therapeutic target for FXS. Given that spine abnormalities are observed in different neuronal populations at different ages, the advent of cofilin as a therapeutic target for amelioration of FXS remains controversial. Dysregulation of cofilin signaling and its upstream regulators are implicated not only in FXS but also in Alzheimer’s, Parkinson’s, and Huntington’s diseases and schizophrenia (63-66). Thus, findings in the present study may have profound implications not only for patients with FXS but also for those with other neurobehavioral and neuropsychiatric disorders.
MATERIALS AND METHODS
Animals
FVB.129P2-Pde6b+ Tyrc-ch Fmr1tm1Cgr/J (Fmr1 KO) and FVB.129P2-Pde6b+ Tyrc-ch/AntJ (wild-type) mice were obtained from the Jackson Laboratory, maintained in a temperature- and light-controlled environment with a 12-hour/12-hour light/dark cycle, and treated in accordance with the principles and procedures of the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. Protocols were approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine. Because FXS is X-linked, all experiments were performed on male mice.
Genotyping
Animals were genotyped using standard polymerase chain reaction (PCR) techniques as previously described (67). To detect the Fmr1 wild-type allele [527-base pair (bp) product], PCR was performed on DNA from mouse tails using primers for Fmr1_S1 (5′-GTGGTTAGC-TAAAGTGAGGATGAT-3′) andFmr1_S2 (5′-CAGGTTTGTTGGG-ATTAACAGATC-3′). To detect the Fmr1 KO allele (501-bp product), primers for Fmr1_S1 and Fmr1_N2 (5′-TGGGCTCTATGGCTTCT-GA-3′), which bind to the Neo cassette that replaced exon 5 from the Fmr1 gene, were used. The PCR conditions were identical for both Fmr1 S1/S2 and S1/N2 combinations: 94°C for 2 min, 35× (94°C for 30 s, 60°C for 40 s, 72°C for 90 s) and 5 min at 72°C using standard PCR reagents.
Golgi staining and neuronal morphology
FD Rapid GolgiStain Kit (FD NeuroTechnologies) was used as the staining procedure. One-week-old Fmr1 KO or wild-type littermate control animals were anesthetized with isoflurane, and whole brains were removed from each animal. Brains were quickly rinsed with Milli-Q water, immersed in Golgi impregnation solution (solutions A and B), and stored in the dark at room temperature for 2 weeks. Brains were then transferred to solution C and stored in the dark at 4°C for 1 to 3 days. Brains were then snap-frozen in cold 2-methylbutane on dry ice for 2 min and stored in −80°C. Thick sections (200 mm) were cut using a Leica cryostat at −20°C from brains embedded in Tissue Freezing Medium (Triangle Biomedical Sciences). Sections were then transferred to cold SuperFrost Plus microscope slides. To complete the staining procedure, microscope slides containing the brains sections were rinsed in Milli-Q water twice for 2 min each, placed in a mixture (one part solution D, one part solution E, and two parts of Milli-Q water) for 10 min and rinsed again in Milli-Q water twice for 4 min each. Slides were dehydrated with 50, 75, and 95% ethanol for 4 min followed by absolute ethanol two times for 8 min each. Slides were cleared with xylene three times for 4 min each and coverslipped with Permount. Six animals (three animals and 12 to 13 neurons per genotype) were used for dendritic spine analysis. Dendritic spines of layer V pyramidal neurons from the primary somatosensory cortex were imaged on an Olympus brightfield microscope using Neurolucida software with a 100× oil immersion lens. Dendritic spine density was determined by counting the total number of spines along the apical dendrite from the soma to 130 μm whether it was a primary, secondary, or tertiary branch. All neurons per genotype were pooled, and the average spine number per unit length (10 μm) was established. The classification of dendritic spine morphology was based on previous findings (68, 69) and determined in all imaged neurons using a categorization macro in Neurolucida software. All neurons for a given genotype were pooled, and fractions of all of the individually classified spines were assessed for statistical significance. All experiments were performed with an examiner blinded to genotype. Distracting artifacts were digitally removed in Adobe Photoshop for display purposes only.
Synaptosomal preparation and Western blotting
Synaptosomes were prepared as described (70). Briefly, Fmr1 KO and wild-type littermate controls aged 1 week, 4 weeks, or 2 to 5 months were anesthetized with isoflurane and decapitated, and whole brains were removed. Brains were quickly rinsed with Milli-Q water, and the somatosensory cortex was isolated and homogenized in gradient buffer [1 ml of 1.28 M sucrose, 4 mM EDTA, 50 mM dithiothreitol (DTT), 20 mM tris (pH 7.4) with protease and phosphatase inhibitors (Sigma-Aldrich)]. The homogenate was centrifuged at 1000g for 10 min. The pellet was discarded, and the supernatant was loaded on a Percoll discontinuous gradient (3, 10, 15, and 23%) and centrifuged at 31,000g for 6 min. Synaptosome fractions were removed from the 15 to 23% interface. Phosphate-buffered saline was added, and synaptosomes were centrifuged at 20,000g for 10 min. Supernatants were discarded, and the pellets were resuspended and lysed in 50 mM tris, 150 mM NaCl, 1% Triton X-100,0.5% deoxycholate (pH 7.2) for Western blotting. Protein concentrations of synaptosome fractions were measured by means of the bicinchoninic acid assay (Pierce). Aliquots of equal protein samples (between 2 and 15 μg) were run on SDS–polyacrylamide gel electrophoresis (4 to 12%), transferred to a nitrocellulose membrane, and probed with a primary antibody overnight at 4°C and a secondary antibody for 1 hour at room temperature. Membranes were washed and reacted with enhanced chemiluminescence reagent (Pierce). Band densities were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Lentiviral vectors
For overexpression of wild-type and mutant cofilin in neurons of intact animals, we subcloned pEGFP-N1, pcDNA3-EGFP-cofilin, pcDNA3- EGFP-cofilinS3A, and pcDNA3-EGFP-cofilinS3D (gift of I. Ethell, University of California at Riverside) into a self-inactivating lentiviral vector pRRLsin.cPPT.CMV.eGFP.Wpre to generate pRRLsin.cPPT.CMV.eGFP.Wpre, pRRLsin.cPPT.CMV.eGFP-WTcofilin.Wpre, pRRLsin. cPPT.CMV.eGFP-cofilinS3A.Wpre, and pRRLsin.cPPT.CMV.eGFP-cofilinS3D.Wpre transfer constructs. High-titer vesicular stomatitis virus-pseudotyped lentiviral stocks were produced in human embryonic kidney (HEK)–293T cells (71–73). Briefly, cells were transfected with the pRRLsin.cPPT.CMV.eGFP.Wpre, pRRLsin.cPPT.CMV.eGFP- WTcofilin.Wpre, pRRLsin.cPPT.CMV.eGFP-cofilinS3A.Wpre, or pRRLsin.cPPT.CMV.eGFP-cofilinS3D.Wpre transfer constructs; the packaging constructs pMDLg-pRRE and pRSV-REV; and pMD2.G envelope protein construct by means of calcium phosphate (73, 74). Viral titers were determined by transducing HeLa cells with serial dilutions of lentivirus concentrated by ultracentrifugation as previously described (75). GFP fluorescence was evaluated by flow cytometry (Becton Dickinson LSR II flow cytometer) 72 hours after transduction. Titer was 3 × 108 transducing units/ml after 200-fold concentration of vector supernatant.
In vivo delivery of viral constructs
A self-inactivation lentivirus encoding either GFP, wild-type cofilin, or cofilinS3A was delivered into the right somatosensory cortex of P4 or P5 Fmr1 KO mice and wild-type littermates by stereotaxic injection. Briefly, animals were placed on a stereotaxic frame, anesthetized with 4% isoflurane, and maintained at 1.5% isoflurane anesthesia. Throughout the injection procedure, supplemental heat was provided using a heating lamp. The incision site was scrubbed with Betadine (Purdue) antiseptic. A small burr hole was drilled in the skull overlaying the right somatosensory cortex. Concentrated viral solution (1.0 ml) was injected into the right somatosensory cortex by means of a 10-ml Hamilton syringe with a 26-gauge needle driven by a Quintessential Stereotaxic Injector (Stoelting Company) at a flow rate of 0.1 ml/min. The injection site was defined by the following coordinates: at P5, 2.5 to 3 mm posterior to bregma, 2 mm lateral to bregma, and 0.3 mm ventral from dura. The needle was left in place for an additional 2 min and then gently withdrawn. The incision was closed with cyanoacrylate glue. After injection, animals were placed in a clean-heated cage to recover. The animals were anesthetized again 5 to 7 days after surgery and decapitated, and brains were put in a cold 4% paraformaldehyde/4% sucrose solution to sit overnight at 4°C in the dark. Brains were snap- frozen and cut into 100-mm-thick sections in the coronal plane with a Leica CM30505 cryostat. Sections were mounted, and labeled neurons were identified and imaged using an LSM 510 Meta confocal microscope.
Confocal microscopy imaging analysis and quantification
An LSM 510 Meta confocal microscope was used to image GFP-labeled sections, whereby GFP was excited using the argon 488-nm laser line. Layer V somatosensory neurons were imaged using a series of high-resolution optical sections (1024 pixel × 1024 pixel format) that were acquired using a 63× oil immersion objective (numerical aperture, 1.4), with 1× to 1.5× zoom at 0.38-mm step intervals (z-stack). Three-dimensional image stacks were processed and analyzed using ImageJ (NIH). The segmented line tool was used to assess spine length and head width. Spine length was defined as the distance from the tip of the spine head to the base of the dendrite, whereas the head width was defined as the largest diameter across the spine head perpendicular to the spine length. The LWR, which reflects spine morphology (76), was assessed by dividing the length of each individual spine by the head width of that spine (the higher the LWR, the more immature the spine, and the lower the LWR, the more mature the spine). Dendritic spine density was determined by counting the total number of spines along the apical dendrite from the soma to 100 μm whether it was a primary, secondary, or tertiary branch. Last, dendritic spines were grouped according to morphology, and the percentage of spines based on morphology was quantified, whereby morphology included (i) mature spines (mushroom spines defined as a short protrusion with a thick head and a thin neck, and stubby spines defined as a short protrusion with a thick head and no well-defined neck) and (ii) immature spines (thin/filopodia spines defined as a long protrusion with small heads). Six to 10 animals were used per group, and one to seven dendrites were analyzed per animal. In some images, distracting fluorescent processes were digitally removed in Adobe Photoshop for display purposes only.
PAK inhibition
The small-molecule inhibitor FRAX486 (Tocris Bioscience), which provides good potency and selectivity of group I PAKs (PAK1/2/3) over group II PAKs (PAK4/5/6) (43), was dissolved in 20% (w/v) hydroxypropyl-β-cyclodextrin vehicle (Sigma-Aldrich). At P7 in both wild-type and KO animals, a solution (2 mg/ml) of drug or vehicle control was administered by subcutaneous injection at 20 mg/kg in a volume proportional to the animal’s weight. Eight hours after drug administration, whereby the maximum concentration of FRAX486 is reached in the brain (43), mice were sacrificed, synaptosomes from mouse brains were isolated, and Western analysis was conducted to assess cofilin signaling and F-actin/G-actin ratio. For electrophysiology experiments, animals were injected at P6 or P14, and experiments were performed at P7 or P15, respectively.
Electrophysiological recordings
Coronal slices (400 μm) were prepared from Fmr1 KO mice and littermate wild-type mice at 1 or 2 weeks of age. The investigator was blind to the genotype, and post hoc genotyping was completed after experiments and analysis. Acute sections were prepared for electrophysiological recordings as previously described (18, 77). Whole-cell patch recordings were made from upper layer V pyramidal neurons in somatosensory cortex using a Multiclamp 700B patch clamp amplifier (Axon Instruments) at 31°C. Borosilicate glass electrodes were made to have resistances of 4 to 5 megohms when filled with cerebrospinal fluid (CSF) internal solution containing 95 mM CSF, 25 mM CsCl, 10 mM Cs-Hepes, 10 mM Cs-EGTA, 2 mM NaCl, 2 mM Mg-adenosine triphosphate (ATP), 10 mM QX-314,5 mM TEA-Cl (tetraethylammonium chloride), 5 mM 4-AP, pH adjusted to 7.3 with CsOH. For measurement of the NMDA/AMPA ratio, extracellular stimulation was delivered with a unipolar stimulating electrode placed in lower layer IV. Only stable evoked EPSC recordings with constant latency were accepted as monosynaptic EPSCs. Series resistance was continuously monitored using hyperpolarizing voltage steps generated by pClamp 10 software (Axon Instruments), and recordings were discarded if there was a >15% change during the course of the experiment. All recordings were made in the presence of the GABA type A (GABAA) receptor antagonist picrotoxin (50 μM). Recordings of epileptic discharge, voltage-activated currents, or polysynaptic currents were discarded. In a single trial, an AMPAR-mediated current was evoked first while holding the cell at −70 mV before stepping to +40 mV, where an NMDAR- mediated current was recorded. Twenty-five sweeps were used to calculate the average AMPAR current amplitude measured as the peak current at −70 mV and NMDAR currents measured during a 2.5-ms window, 60 ms after the response onset at +40 mV. To record mEPSC, whole-cell recordings were performed by holding the cell at −70 mV with K-gluconate internal solution containing 120 mM K-gluconate, 9 mM KCl, 10 mM KOH, 3.48 mM MgCl2, 10 mM Hepes, 4 mM NaCl, 4 mM Na2ATP, 0.4 mM Na3GTP, and 17.5 mM sucrose (pH 7.25 to 7.35). Ten-minute epochs of mEPSCs recordings were performed in the presence of picrotoxin (50 μM) and TTX (500 nM). mEPSCs were analyzed using MiniAnalysis (Synaptosoft), and amplitude and frequency were averaged from the 10-min recording.
Rac1 activation assay
Total somatosensory cortex lysates from wild-type and KO animals were homogenized with Mg2+ lysis buffer (Millipore) with a complete protease inhibitor cocktail. The expression level of Rac1-GTP was then assessed using a Rac/Cdc42 pull-down kit (Millipore). Samples (100 mg) were incubated and rocked with 10 mg of glutathione S-transferase (GST)-tagged PAK-PBD agarose beads (Millipore) for 2 hours. Beads were pelleted by centrifugation (14,000g for 15 s at 4 °C), and the supernatant was discarded. The pelleted beads were washed with lysis buffer three times, resuspended in 50 ml of 2× Laemmli buffer, boiled for 5 min, and subjected to Western blot analysis. GTP and guanosine diphosphate (GDP) loading controls were incubated with 100 μM GTP-gS or 1 mM GDP for 30 min at 30°C.
F-actin/G-actin ratio
F-actin/G-actin ratio was assessed as previously described (27). Briefly, once synaptosome fractions from wild-type and KO animals were isolated, fractions were resuspended in cold lysis buffer [10 mM K2HPO4, 100 mM NaF, 50 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM DTT, 0.5% Triton X-100, 1 mM sucrose (pH 7.0)] and centrifuged at 15,000g for 30 min. Separation of F-actin to G-actin was achieved in that F-actin is insoluble (pellet) to this buffer, whereas G-actin is soluble (supernatant). The G-actin supernatant was transferred to a fresh tube, and the F-actin pellet was resuspended in lysis buffer plus an equal volume of a second buffer [1.5 mM guanidine hydrochloride, 1 mM sodium acetate, 1 mM CaCl2, 1 mM ATP, 20 mM tris-HCl (pH 7.5)] and then incubated on ice for 1 hour to convert F-actin into soluble G-actin with gentle mixing every 15 min. Samples were centrifuged at 15,000g for 30 min, and the supernatant (containing the F-actin, which was converted to G-actin) was transferred to a fresh tube. F-actin and G-actin samples were loaded with equal volumes and analyzed by Western blotting.
Whisker-dependent texture discrimination task
The whisker-dependent discrimination task was performed as previously described (50). Wild-type and Fmr1 KO mice aged 3 to 4 weeks were used in this study because this marks a window after the critical period closes when the hardwiring of synaptic circuits in the somatosensory cortex is generally established. A plastic rectangular arena was used that consisted of transparent walls and a transparent floor. Objects used for the texture discrimination task were constructed with metal blocks wrapped with red-colored sandpaper. Different grades of red sandpaper (80 and 100 grit) were used to create objects that could be distinguished through texture. To minimize object recognition by olfactory cues, at least three identical objects with a certain grade of sandpaper were used. For three consecutive days before the testing day, each animal was allowed to explore the empty arena for 10 min. The testing day was consisted of two sessions, which were recorded with a video camera centered above the arena. A mouse was placed in the center of the arena between two identical objects (80-grit blocks) equidistant from each other and allowed to explore for 5 min. The mouse was then placed in a holding cage for 5 min. During this waiting period, an 80-grit block was replaced with a new identical 80-grit block and the other 80-grit block was replaced with a 100-grit block (the position of this block was switched from mouse to mouse). The mouse was placed back in the arena and allowed to explore for 5 min. Investigation was defined as directing the nose toward the object or touching the object with a distance of less than 2 cm. Resting, grooming, or sitting on the object was not considered investigation. Mice that did not investigate both objects for a minimum of 10 s were excluded from the study. Time (in seconds) exploring the novel texture divided by the total time exploring both objects yielded a preference index, where a preference index above 50 indicated an ability to discriminate the novel texture from the familiar texture. For “whisker-less” control experiments, whiskers from wild-type animals were completely trimmed before habituation. For texture-less control experiments, wild-type animals were subjected to objects wrapped with parafilm to eliminate the ability to palpate the sandpaper. For PAK inhibition experiments, a solution (2 mg/ml) of drug or vehicle control was administered by subcutaneous injection at 20 mg/kg in a volume proportional to the animal’s weight once at P7 (during the critical period), once at P14 (toward the close of the critical period), and once more at 3 to 4 weeks (after the critical period closes) of age on the third day of habituation 24 hours before the testing phase. All experiments were recorded and scored by two observers blind to the genotype.
Antibodies
The following antibodies were used in this study: rabbit anti-phospho-cofilin (Ser3; 1:1000; Cell Signaling), rabbit anti-cofilin (1:1000; Cell Signaling Technology), rabbit anti-phospho-LIMK1 (Thr508; 1:1000; Abcam), rabbit anti-LIMK1 (1:1000; Cell Signaling Technology), rabbit anti-phospho-Slingshot1 (Ser978; 1:1000; ECM Biosciences), rabbit anti-Slingshot1 (1:1000; Abcam), rabbit anti–phospho-PAK1 (Ser199; 1:1000; Abcam), rabbit anti–phospho-PAK1 (Thr423; 1:1000; Cell Signaling Technology), rabbit anti-PAK1 (1:1000; Cell Signaling Technology), mouse anti-phospho-PAK4 (Ser474; 1:1000; Santa Cruz Biotechnology), rabbit anti-PAK4 (1:1000; Cell Signaling Technology), mouse anti-Rac1 (1:1000; Millipore), mouse anti-GAPDH (1:50,000; Fitzgerald), mouse anti-actin (1:10,000; Sigma-Aldrich), rabbit anti-FMRP (1:1000; Abcam), rabbit anti-PSD95 (1:1000; Cell Signaling Technology), mouse anti-SV2 (1:2000; Developmental Studies Hybridoma Bank), mouse anti-VAMP2 (1:1000; Thermo Fisher Scientific), rabbit histone H3 (1:1000; Cell Signaling Technology), horseradish peroxidase (HRP)-linked rabbit anti-immunoglobulin G (IgG) (1:5000; Cell Signaling Technology), and HRP-linked mouse anti-IgG (1:5000; Cell Signaling Technology).
Cell culture and viral transduction
Primary cultures of somatosensory cortical neurons were prepared from embryonic day 18 (E18) rats, and cortical cells were cultured in Neurobasal medium with B-27 supplement and GlutaMAX. Neuronal cultures were maintained 4 or 5 DIV before application of lentivirus containing GFP, wild-type cofilin, cofilinS3A, or cofilinS3D. Five to 7 days were allowed for transduction of the virus to occur. Cultures were used to assess transduction efficiency in primary neuronal cultures and F-actin/G-actin ratio by Western blotting analysis.
Statistical analysis
Data are presented as means ± SEM. For Western blot experiments, Golgi staining experiments, and behavioral experiments, significance was assessed by means of Student’s t test (unpaired, two-tailed). For PAK inhibition experiments, electrophysiology experiments, F-actin/G-actin ratio experiments to validate cofilin constructs, and for cofilin overexpression dendritic spine rescue experiments, significance was assessed by means of a two-way analysis of variance (ANOVA) followed by a Tukey’s test. P < 0.05 was considered to be statistically significant.
Supplementary Material
Fig. S1. Validation of somatosensory cortex synapse enrichment.
Fig. S2. Subcutaneous administration of hydroxypropyl-α-cyclodextrin (vehicle) has little to no effect on cofilin signaling.
Fig. S3. Mature Fmr1 KO mice exhibit normal Rac1 signaling at somatosensory cortex synapses.
Fig. S4. Mature Fmr1 KO mice exhibit normal cofilin phosphorylation and activity at somatosensory cortex synapses.
Table S1. Summary of statistics for Fig. 1.
Table S2. Summary of statistics for Fig. 2.
Table S3. Summary of statistics for Fig. 3.
Table S4. Summary of statistics for Fig. 4.
Table S5. Summary of statistics for Fig. 5.
Table S6. Summary of statistics for Fig. 6.
Table S7. Summary of statistics for fig. S2.
Table S8. Summary of statistics for figs. S3 and S4.
Acknowledgments
We thank J. Crawley and M. Porch for invaluable help with the texture discrimination assay and F. Pontarelli and B. Court-Vasquez for technical assistance. We thank I. Ethell (University of California at Riverside) for providing cofilin constructs. We thank members of the Zukin laboratory for their helpful comments and suggestions on the manuscript.
Funding: This work was supported by NIH grants MH092877, NS45693, and HD083828 and grants from the McKnight Foundation, National Alliance for Research on Schizophrenia and Depression (NARSAD), and F.M. Kirby Foundation (to R.S.Z.) and AHA Scientist Development Grant and NARSAD Young Investigator Grant (to J.-Y.H.). R.S.Z. is the F.M. Kirby Professor in Neural Repair and Protection. A.P. was partially supported in part by R36 MH108362 and T32 GM007288.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencesignaling.org/cgi/content/full/10/504/eaan0852/DC1
Author contributions: A.P., Q.H., J.-Y.H., A.C., and R.S.Z. conceived and designed the experiments. A.P. performed behavioral, molecular, and dendritic spine experiments and analyzed the results. Q.H. performed all electrophysiology experiments and analyzed the results. M.P. performed behavioral experiments. A.C. and R.S.Z. helped guide the research. A.P., Q.H., J.-Y.H., A.C., and R.S.Z. interpreted all the results and wrote the paper.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: Causes, diagnosis, mechanisms, and therapeutics. J Clin Invest. 2012;122:4314–4322. doi: 10.1172/JCI63141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhakar AL, Dölen G, Bear MF. The pathophysiology of fragile X (and what it teaches us about synapses) Annu Rev Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huber KM, Klann E, Costa-Mattioli EM, Zukin RS. Dysregulation of mammalian target of rapamycin signaling in mouse models of autism. J Neurosci. 2015;41:13836–13842. doi: 10.1523/JNEUROSCI.2656-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gross C, Hoffmann A, Bassell GJ, Berry-Kravis EM. Therapeutic strategies in fragile X syndrome: From bench to bedside and back. Neurotherapeutics. 2015;12:584–608. doi: 10.1007/s13311-015-0355-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: Mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darnell JC, Klann E. The translation of translational control by FMRP: Therapeutic targets for FXS. Nat Neurosci. 2013;16:1530–1536. doi: 10.1038/nn.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heulens I, Kooy F. Fragile X syndrome: From gene discovery to therapy. Front Biosci. 2011;16:1211–1232. doi: 10.2741/3785. [DOI] [PubMed] [Google Scholar]
- 8.Santoro MR, Bray SM, Warren ST. Molecular mechanisms of fragile X syndrome: A twenty-year perspective. Annu Rev Pathol. 2012;7:219–245. doi: 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 9.Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM. Adult fragile X syndrome. Acta Neuropathol. 1985;67:289–295. doi: 10.1007/BF00687814. [DOI] [PubMed] [Google Scholar]
- 10.Zukin RS, Richter JD, Bagni C. Signals, synapses, and synthesis: How new proteins control plasticity. Front Neural Circuits. 2009;14 doi: 10.3389/neuro.04.014.2009. eCollection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He CX, Portera-Cailliau C. The trouble with spines in fragile X syndrome: Density, maturity and plasticity. Neuroscience. 2013;251:120–128. doi: 10.1016/j.neuroscience.2012.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erzurumlu RS, Gaspar P. Development and critical period plasticity of the barrel cortex. Eur J Neurosci. 2012;35:1540–1553. doi: 10.1111/j.1460-9568.2012.08075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu C-S, Ballester Rosado CJ, Lu H-C. What can we get from ‘barrels’: The rodent barrel cortex as a model for studying the establishment of neural circuits. Eur J Neurosci. 2011;34:1663–1676. doi: 10.1111/j.1460-9568.2011.07892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LeBlanc JJ, Fagiolini M. Autism: A “critical period” disorder? Neural Plast. 2011;2011:921680. doi: 10.1155/2011/921680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meredith RM. Sensitive and critical periods during neurotypical and aberrant neurodevelopment: A framework for neurodevelopmental disorders. Neurosci Biobehav Rev. 2015;50:180–188. doi: 10.1016/j.neubiorev.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Baranek GT, Roberts JE, David FJ, Sideris J, Mirrett PL, Hatton DD, Bailey DB., Jr Developmental trajectories and correlates of sensory processing in young boys with fragile X syndrome. Phys Occup Ther Pediatr. 2008;28:79–98. doi: 10.1300/j006v28n01_06. [DOI] [PubMed] [Google Scholar]
- 17.Nimchinsky EA, Oberlander AM, Svoboda K. Abnormal development of dendritic spines in FMRI knock-out mice. J Neurosci. 2001;21:5139–5146. doi: 10.1523/JNEUROSCI.21-14-05139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harlow EG, Till SM, Russell TA, Wijetunge LS, Kind P, Contractor A. Critical period plasticity is disrupted in the barrel cortex of FMR1 knockout mice. Neuron. 2010;65:385–398. doi: 10.1016/j.neuron.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cingolani LA, Goda Y. Actin in action: The interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- 20.Hotulainen P, Hoogenraad CC. Actin in dendritic spines: Connecting dynamics to function. J Cell Biol. 2010;189:619–629. doi: 10.1083/jcb.201003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamprecht R. The actin cytoskeleton in memory formation. Prog Neurobiol. 2014;117:1–19. doi: 10.1016/j.pneurobio.2014.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Pontrello CG, Ethell IM. Accelerators, brakes, and gears of actin dynamics in dendritic spines. Open Neurosci J. 2009;3:67–86. doi: 10.2174/1874082000903020067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penzes P, Cahill ME. Deconstructing signal transduction pathways that regulate the actin cytoskeleton in dendritic spines. Cytoskeleton. 2012;69:426–441. doi: 10.1002/cm.21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirao T, González-Billault C. Actin filaments and microtubules in dendritic spines. J Neurochem. 2013;126:155–164. doi: 10.1111/jnc.12313. [DOI] [PubMed] [Google Scholar]
- 25.Rust MB. ADF/cofilin: A crucial regulator of synapse physiology and behavior. Cell Mol Life Sci. 2015;72:3521–3529. doi: 10.1007/s00018-015-1941-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ, Sabatini BL. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci. 2005;8:1727–1734. doi: 10.1038/nn1566. [DOI] [PubMed] [Google Scholar]
- 27.Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, Krnjević K, Roman G, Costa-Mattioli M. mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat Neurosci. 2013;16:441–448. doi: 10.1038/nn.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 29.Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 30.Hotulainen P, Llano O, Smirnov S, Tanhuanpää K, Faix J, Rivera C, Lappalainen P. Defining mechanisms of actin polymerization and depolymerization during dendritic spine morphogenesis. J Cell Biol. 2009;185:323–339. doi: 10.1083/jcb.200809046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bamburg JR, Bernstein BW. Roles of ADF/cofilin in actin polymerization and beyond. F1000 Biol Rep. 2010;2:62. doi: 10.3410/B2-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pontrello CG, Sun MY, Lin A, Fiacco TA, DeFea KA, Ethell IM. Cofilin under control of β-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci USA. 2012;109:E442–E451. doi: 10.1073/pnas.1118803109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agnew BJ, Minamide LS, Bamburg JR. Reactivation of phosphorylated actin depolymerizing factor and identification of the regulatory site. J Biol Chem. 1995;270:17582–17587. doi: 10.1074/jbc.270.29.17582. [DOI] [PubMed] [Google Scholar]
- 34.Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013;25:457–469. doi: 10.1016/j.cellsig.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 35.Moriyama K, Iida K, Yahara I. Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells. 1996;1:73–86. doi: 10.1046/j.1365-2443.1996.05005.x. [DOI] [PubMed] [Google Scholar]
- 36.Castets M, Schaeffer C, Bechara E, Schenck A, Khandjian EW, Luche S, Moine H, Rabilloud T, Mandel JL, Bardoni B. FMRP interferes with the Rac1 pathway and controls actin cytoskeleton dynamics in murine fibroblasts. Hum Mol Genet. 2005;14:835–844. doi: 10.1093/hmg/ddi077. [DOI] [PubMed] [Google Scholar]
- 37.de Diego-Otero Y, Romero-Zerbo Y, el Bekay R, Decara J, Sanchez L, Rodriguez-de Fonseca F, del Arco-Herrera I. α-tocopherol protects against oxidative stress in the fragile X knockout mouse: An experimental therapeutic approach for the Fmr1 deficiency. Neuropsychopharmacology. 2009;34:1011–1026. doi: 10.1038/npp.2008.152. [DOI] [PubMed] [Google Scholar]
- 38.Bongmba OYN, Martinez LA, Elhardt ME, Butler K, Tejada-Simon MV. Modulation of dendritic spines and synaptic function by Rac1: A possible link to Fragile X syndrome pathology. Brain Res. 2011;1399:79–95. doi: 10.1016/j.brainres.2011.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen LY, Rex CS, Babayan AH, Kramár EA, Lynch G, Gall CM, Lauterborn JC. Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J Neurosci. 2010;30:10977–10984. doi: 10.1523/JNEUROSCI.1077-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao ZS, Manser E. PAK family kinases: Physiological roles and regulation. Cell Logist. 2012;2:59–68. doi: 10.4161/cl.21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rane CK, Minden A. P21 activated kinases: Structure, regulation, and functions. Small GTPases. 2014;5:e28003. doi: 10.4161/sgtp.28003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- 43.Dolan BM, Duron SG, Campbell DA, Vollrath B, Shankaranarayana Rao BS, Ko H-Y, Lin GG, Govindarajan A, Choi S-Y, Tonegawa S. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc Natl Acad Sci USA. 2013;110:5671–5676. doi: 10.1073/pnas.1219383110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39:1071–1076. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 45.Shi Y, Pontrello CG, DeFea KA, Reichardt LF, Ethell IM. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci. 2009;29:8129–8142. doi: 10.1523/JNEUROSCI.4681-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Racz B, Weinberg RJ. The subcellular organization of cortactin in hippocampus. J Neurosci. 2004;24:10310–10317. doi: 10.1523/JNEUROSCI.2080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Micheva KD, Beaulieu C. Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. J Comp Neurol. 1996;373:340–354. doi: 10.1002/(SICI)1096-9861(19960923)373:3<340::AID-CNE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 48.Gu J, Lee CW, Fan Y, Komlos D, Tang X, Sun C, Yu K, Hartzell HC, Chen G, Bamburg JR, Zheng JQ. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat Neurosci. 2010;13:1208–1215. doi: 10.1038/nn.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Bonnan A, Bony G, Ferezou I, Pietropaolo S, Ginger M, Sans N, Rossier J, Oostra B, LeMasson G, Frick A. Dendritic channelopathies contribute to neocortical and sensory hyperexcitability in Fmr1−/y mice. Nat Neurosci. 2014;17:1701–1709. doi: 10.1038/nn.3864. [DOI] [PubMed] [Google Scholar]
- 50.Wu HPP, Ioffe JC, Iverson MM, Boon JM, Dyck RH. Novel, whisker-dependent texture discrimination task for mice. Behav Brain Res. 2013;237:238–242. doi: 10.1016/j.bbr.2012.09.044. [DOI] [PubMed] [Google Scholar]
- 51.Bureau I, Shepherd GMG, Svoboda K. Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J Neurosci. 2008;28:5178–5188. doi: 10.1523/JNEUROSCI.1076-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patel AB, Loerwald KW, Huber KM, Gibson JR. Postsynaptic FMRP promotes the pruning of cell-to-cell connections among pyramidal neurons in the L5A neocortical network. J Neurosci. 2014;34:3413–3418. doi: 10.1523/JNEUROSCI.2921-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the Rho family of small GTPases: Antagonistic roles of Rac and Rho. Cereb Cortex. 2000;10:927–938. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- 54.Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: Evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 55.Kashima R, Roy S, Ascano M, Martinez-Cerdeno V, Ariza-Torres J, Kim S, Louie J, Lu Y, Leyton P, Bloch KD, Kornberg TB, Hagerman PJ, Hagerman R, Lagna G, Hata A. Augmented noncanonical BMP type II receptor signaling mediates the synaptic abnormality of fragile X syndrome. Sci Signal. 2016;9:ra58. doi: 10.1126/scisignal.aaf6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kashima R, Redmond PL, Ghatpande P, Roy S, Kornberg TB, Hanke T, Knapp S, Lagna G, Hata A. Hyperactive locomotion in a Drosophila model is a functional readout for the synaptic abnormalities underlying fragile X syndrome. Sci Signal. 2017;10:eaai8133. doi: 10.1126/scisignal.aai8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Isaac JTR, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- 58.Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- 59.Rogers SJ, Hepburn S, Wehner E. Parent reports of sensory symptoms in toddlers with autism and those with other developmental disorders. J Autism Dev Disord. 2003;33:631–642. doi: 10.1023/b:jadd.0000006000.38991.a7. [DOI] [PubMed] [Google Scholar]
- 60.Miller LJ, McIntosh DN, McGrath J, Shyu V, Lampe M, Taylor AK, Tassone F, Neitzel K, Stackhouse T, Hagerman RJ. Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: A preliminary report. Am J Med Genet. 1999;83:268–279. [PubMed] [Google Scholar]
- 61.Arnett MT, Herman DH, McGee AW. Deficits in tactile learning in a mouse model of fragile X syndrome. PLOS ONE. 2014;9:e109116. doi: 10.1371/journal.pone.0109116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi ML, Rao BSS, Seo JS, Choi HS, Dolan BM, Choi SY, Chattarji S, Tonegawa S. Inhibition of p21-activated kinase rescues symptoms of fragile X syndrome in mice. Proc Natl Acad Sci USA. 2007;104:11489–11494. doi: 10.1073/pnas.0705003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ma QL, Yang F, Frautschy SA, Cole GM. PAK in Alzheimer disease, Huntington disease and X-linked mental retardation. Cell Logist. 2012;2:117–125. doi: 10.4161/cl.21602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye DZ, Field J. PAK signaling in cancer. Cell Logist. 2012;2:105–116. doi: 10.4161/cl.21882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayashi-Takagi A, Araki Y, Nakamura M, Vollrath B, Duron SG, Yan Z, Kasai H, Huganir RL, Campbell DA, Sawa A. PAKs inhibitors ameliorate schizophrenia-associated dendritic spine deterioration in vitro and in vivo during late adolescence. Proc Natl Acad Sci USA. 2014;111:6461–6466. doi: 10.1073/pnas.1321109111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim H, Oh JY, Choi SL, Nam YJ, Jo A, Kwon A, Shin EY, Kim EG, Kim HK. Down-regulation of p21-activated serine/threonine kinase 1 is involved in loss of mesencephalic dopamine neurons. Mol Brain. 2016;9:45. doi: 10.1186/s13041-016-0230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spencer CM, Serysheva E, Yuva-Paylor LA, Oostra BA, Nelson DL, Paylor R. Exaggerated behavioral phenotypes in Fmr1/Fxr2 double knockout mice reveal a functional genetic interaction between Fragile X-related proteins. Hum Mol Genet. 2006;15:1984–1994. doi: 10.1093/hmg/ddl121. [DOI] [PubMed] [Google Scholar]
- 68.Grossman AW, Aldridge GM, Weiler IJ, Greenough WT. Local protein synthesis and spine morphogenesis: Fragile X syndrome and beyond. J Neurosci. 2006;26:7151–7155. doi: 10.1523/JNEUROSCI.1790-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Su T, Fan HX, Jiang T, Sun WW, Den WY, Gao MM, Chen SQ, Zhao QH, Yi YH. Early continuous inhibition of group 1 mGlu signaling partially rescues dendritic spine abnormalities in the Fmr1 knockout mouse model for fragile X syndrome. Psychopharmacology. 2011;215:291–300. doi: 10.1007/s00213-010-2130-2. [DOI] [PubMed] [Google Scholar]
- 70.Dunkley PR, Jarvie PE, Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- 71.Follenzi A, Sabatino G, Lombardo A, Boccaccio C, Naldini L. Efficient gene delivery and targeted expression to hepatocytes in vivo by improved lentiviral vectors. Hum Gene Ther. 2004;13:243–260. doi: 10.1089/10430340252769770. [DOI] [PubMed] [Google Scholar]
- 72.Miyawaki T, Ofengeim D, Noh KM, Latuszek-Barrantes A, Hemmings BA, Follenzi A, Zukin RS. The endogenous inhibitor of Akt, CTMP, is critical to ischemia-induced neuronal death. Nat Neurosci. 2009;12:618–626. doi: 10.1038/nn.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MVL, Zukin RS. Repressor element-1 silencing transcription factor (REST)- dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proc Nati Acad Sci USA. 2012;109:E962–E971. doi: 10.1073/pnas.1121568109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hwang JY, Kaneko N, Noh KM, Pontarelli F, Zukin RS. The gene silencing transcription factor REST represses miR-132 expression in hippocampal neurons destined to die. J Mol Biol. 2014;426:3454–3466. doi: 10.1016/j.jmb.2014.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Follenzi A, Naldini L. Generation of HIV-1 derived lentiviral vectors. Methods Enzymol. 2002;346:454–465. doi: 10.1016/s0076-6879(02)46071-5. [DOI] [PubMed] [Google Scholar]
- 76.Risher WC, Ustunkaya T, Singh Alvarado J, Eroglu C. Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLOS ONE. 2014;9:e107591. doi: 10.1371/journal.pone.0107591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.He Q, Nomura T, Xu J, Contractor A. The developmental switch in GABA polarity is delayed in fragile X mice. J Neurosci. 2014;34:446–450. doi: 10.1523/JNEUROSCI.4447-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Validation of somatosensory cortex synapse enrichment.
Fig. S2. Subcutaneous administration of hydroxypropyl-α-cyclodextrin (vehicle) has little to no effect on cofilin signaling.
Fig. S3. Mature Fmr1 KO mice exhibit normal Rac1 signaling at somatosensory cortex synapses.
Fig. S4. Mature Fmr1 KO mice exhibit normal cofilin phosphorylation and activity at somatosensory cortex synapses.
Table S1. Summary of statistics for Fig. 1.
Table S2. Summary of statistics for Fig. 2.
Table S3. Summary of statistics for Fig. 3.
Table S4. Summary of statistics for Fig. 4.
Table S5. Summary of statistics for Fig. 5.
Table S6. Summary of statistics for Fig. 6.
Table S7. Summary of statistics for fig. S2.
Table S8. Summary of statistics for figs. S3 and S4.